
Improvement of D–π–A organic dye-based dye-sensitized solar cell performance by simple triphenylamine donor substitutions on the π-linker of the dye†
N.
Prachumrak
a,
T.
Sudyoadsuk
a,
A.
Thangthong
a,
P.
Nalaoh
 a,
S.
Jungsuttiwong
b,
R.
Daengngern
c,
S.
Namuangruk
c,
P.
Pattanasattayavong
a and
V.
Promarak
*a
a,
S.
Jungsuttiwong
b,
R.
Daengngern
c,
S.
Namuangruk
c,
P.
Pattanasattayavong
a and
V.
Promarak
*a
aDepartment of Material Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Wangchan, Rayong 21210, Thailand. E-mail: vinich.p@vistec.ac.th
bDepartment of Chemistry, Faculty of Science, Ubon Ratchathani University, Ubon Ratchathani 34190, Thailand
cNANOTEC, National Science and Technology Development Agency, Pathum Thani 12120, Thailand
First published on 22nd December 2016
Abstract
We report a new simple way to improve the performance of a basic D–π–A organic dye in dye-sensitized solar cells (DSSCs) by donor substitutions on the π-linker of the dye. Three new molecularly engineered D–π–A dyes, namely T2-4, comprising triphenylamine (TPA) as a donor, terthiophene containing different numbers of TPA substitutions as a π-conjugated linker and cyanoacrylic acid as an acceptor, were synthesized and characterized. A detailed study on the effect of different dye structures on the performance of the DSSCs was conducted systematically using theoretical, photophysical, photovoltaic as well as photoelectrochemical methods and compared with that of the traditional D–π–A dye, namely T1. The introduction of electron donating TPA substitutes on the π-linker of the D–π–A dye were beneficial for a decrease of the electron recombination between redox electrolyte and the TiO2 surface as well as an increase of the electron correction efficiency, leading to improved open-circuit voltage (VOC) and short-circuit current (JSC). Consequently, DSSCs sensitized by dye T2 bearing one extra TPA substitution on the terminal thiophene ring of the π-linker delivered the best power conversion efficiency, reaching 8.08% at AM 1.5 simulated sunlight, a remarkable improvement of about 41% compared with 5.72% of T1 reference cells, owing to its high JSC and VOC. With the addition of CDCA as a coadsorbent, the best performing cells based on dye T2 exhibited an impressive conversion efficiency of 9.02% (JSC = 16.91 mA cm−2, VOC = 754 mV, FF = 0.705), exceeding that of the N719-based standard cell (8.20%). This renders the facile dye modification very promising to acquire simple effective organic dyes for high-efficiency DSSCs.
Introduction
Since the initial report by O'Regan and Grätzel in 1991,1 in the recent decades, dye-sensitized solar cells (DSSCs) have received massive attention as an alternative to conventional inorganic silicon based solar cells because of their possible low cost and ease of large-scale production and being environmentally friendly.2 In DSSCs, the dyes always have a significant influence on the power conversion efficiency (η) as well as device stability and, therefore, has been largely developed.3 Ruthenium complexes and zinc porphyrin based dyes are currently the most efficient dyes with DSSCs showing certified η values of 11.9%4 and 13.0%,5 respectively. These dyes, however, are costly and hard to prepare in high yields, which has led to the evolution of metal-free organic dyes. Attention has been paid to organic dyes due to their low cost, high molecular extinction coefficient (ε) and better flexibility for the structural control of optical and light-harvesting properties, indicating their brighter future in commercial applications.6 To date many organic dyes have been intensively investigated and most are designed basically based on a donor–π-linker–acceptor (D–π–A) structure.7 The η of DSSCs based on these dyes has quickly increased to up to over 10.0%.8 One of the key areas hindering the enhancement in DSSC performance is the degree of light-harvesting of D–π–A organic dyes, which can be adjusted by the exchange and variation of the donor (D), π-linker (π), and acceptor (A) parts, and expansion of the π-conjugation system. These modifications will directly influence the absorption properties as well as the energy levels of the HOMO (highest occupied molecular orbital) and the LUMO (lowest unoccupied molecular orbital) of the dyes. D–π–A dyes with various kinds of electron donating moieties including triarylamine,9 carbazole,10 coumarin,11 perylene,12 indoline,13 phenothiazine,14 tetrahydroquinoline,15 and phenoxazine16 have been assembled and moderate to high efficiency DSSCs have been realized. Among them arylamine based organic dyes are promising candidates for highly efficient DSSCs.9,17 Different electron donating molecular architectures including D–D,18 2D,19 (D)2D,20 (D–π)–D21 and dendritic structures22 have also been explored aiming to boost the electron donating ability of the donor as well as to minimize dye aggregation on the surface of semiconducting oxide. Cyanoacrylic acid is commonly used as an acceptor in D–π–A dyes, owing to its relatively strong electronic coupling/anchoring with the semiconducting oxide electrode and capability of inducing an intense charge transfer transition (ICT) resulting in the red-shift of the dye's maximum absorption. To ease charge transfer and later charge separation between the donor and acceptor parts in the excited state, several π-conjugated systems including oligothiophenes,23 fused thiophenes,17,24 polyenes,25 polyynes,26 furanes,27 pyrroles,28 and fused aromatic rings29 have been incorporated into D–π–A dye molecules as π-linkers. Thiophene-based π-conjugated systems are broadly employed as π-linkers in D–π–A dyes because thiophene rings can excellently form a planar π-conjugated linkage with simple chemistry for ring connection and easy ring functionalization. The π-linkers with quasi-planar conformation are not only capable of conducting electrons from the donor to acceptor parts, but also effectively expand the absorption range of the dyes to the visible region. Conversely, increasing the coplanarity or lengthening of the π-linker is likely to initiate π-stacked aggregation of the dyes on the semiconducting oxide surface, causing a drop in electron-injection yield.30 Therefore, the π-linker should be designated wisely to avoid this problem. Modification or substitution of the π-linkers with hydrophobic long alkyl chains and aromatic units is one of the most effective approaches to suppress dye aggregation and has been widely studied.30,31 Furthermore, due to the hydrophobic effect these structural adaptations can effectually prevent the semiconducting oxide surface from the oxidized redox mediator (e.g., I−/I3−) in the electrolyte and thus decrease dark current, resulting in better device performance,32 as well as increase cell stability by commendably subduing water-induced dye desorption phenomena.33 Lately, amendment of the π-linker part of D–π–A dyes by inserting an electron-deficient moiety as an additional acceptor such as benzothiadiazole,34 diketopyrrolopyrrole,35 isoindigo,36 benzotriazole,37 quinoxaline38 and pyrido[3,4-b]pyrazine39 forming molecular structures of D–π–A–π–A, D–A–π–A and D–π–A–A was introduced and some of these dyes exhibited intense improvement in the light-to-electrical-energy conversion efficiency.In this contribution, we present a new simple strategy for molecular modification of the π-linker of the D–π–A dye in an attempt to additionally improve its performance in DSSCs. In our design of the dyes T2-4 with the electron donor triphenylamine (TPA) and the electron-acceptor/anchoring group cyanoacrylic acid, the terthiophene π-linker is substituted by different numbers of sterically hindered electron donating TPA units along the backbone as illustrated in Scheme 1. To the best of our knowledge, this is the first detailed study of tuning the photovoltaic performance of DSSCs through the structural variation of the π-linker of the D–π–A dye by electron donor substitutions. The primary tests reveal that this new molecular alteration allowed us, in a few steps and with good yields, to achieve dyes with remarkable device performances. These new dyes especially dye T2 display a notably enhanced power conversion efficiency of 8.08%, which is superior to the performance of the basic D–π–A dye T1 (5.72%). The details are provided in the following Results and discussion section.
 | ||
| Scheme 1 Synthesis of dyes T1-4. Reagents and conditions: (i) Br2, CHCl3; (ii) 4-(diphenylamino)phenylboronic acid, Pd(PPh3)4, 2 M Na2CO3 (aq), THF, heat; (iii) cyanoacetic acid, piperidine, CHCl3, heat. | ||
Results and discussion
Synthesis and characterization of dyes
The dyes T1-4 were synthesized as depicted in Scheme 1. Bromination of terthiophene-5-carboxaldehyde 1 with different amounts of Br2/CHCl3 resulted in bromo-substituted terthiophene-5-carbaldehydes 2, 3, 4, 5 in good yields. Their molecular masses were evaluated by APCI-Q-TOF. The extensive 2D-NMR analysis including COSY, HMQC and HMBC correctly confirmed the regioselectivity of the bromo groups substituted on the terthiophene backbone for each compound (see the ESI†). The Suzuki cross-coupling reaction of compounds 2–5 with an excess of 4-(diphenylamino)phenylboronic acid gave the corresponding aldehydes 6–9 in 76–83% yields. Finally, dyes T1-4 were obtained via Knoevenagel condensation of aldehydes 6–9 with cyanoacetic acid using piperidine in CHCl3, respectively. These newly synthesized dyes are dark red in color and soluble in most organic solvents. All key intermediates and dyes were characterized by their 1H NMR, 13C NMR, and APCI-Q-TOF/MALDI-TOF mass spectral data.Photophysical, thermal and electrochemical properties of dyes
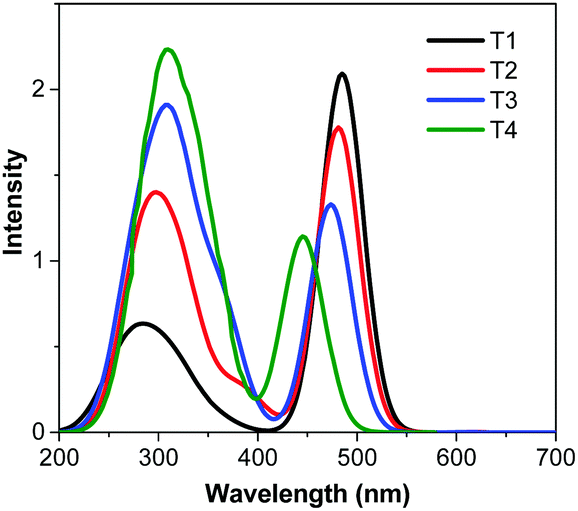
The UV-visible absorption spectra of the dyes in CH2Cl2 solution display two major absorption bands at λmax ∼ 300 nm, one of which is assigned to the π–π* transition of the TPA chromophore, at a longer wavelength λmax of 490–495 nm, and the other is associated with an intramolecular charge transfer (ICT) excitation from the TPA donor group to the cyanoacrylic acid acceptor unit (Fig. 1a). The latter band exhibits a strong solvent effect in polar solvents, proving its ICT character (Fig. 1b). The introduction of TPA donors on the π-linker in T2-4 results in a gradual hypsochromic shift of the ICT band from the molar absorption coefficient (ε) of 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 860 M−1 cm−1 for T1 to 31690 M−1 cm−1 for T2, 20890 M−1 cm−1 for T3 and 14000 M−1 cm−1 for T4. The most pronounced hypsochromic shift (lowest ε) is observed when three TPA units are substituted on the π-linker in T4. Herein the decrease of the ε value is more important which could be understood by the low oscillator strength (f) of the ICT according to the Franck–Condon principle or the poor orbital overlap of the HOMO and the LUMO (see DFT calculations, Table 1 and Table S1, ESI†).17,40 This may be explained by the steric repulsion induced by the bulky TPA moiety that leads to a decrease of planarity of the π-conjugated linker especially in T4. This twisted conformation also impedes good delocalization of the π electrons and slightly blue-shifts the position of the ICT band of T2-4 with respect to T1. A higher amount of energy is required to undertake an intramolecular charge transfer once the π-linker part adopts a twist conformation as observed in many cases of alkyl substituted oligothiophene π-linkers.30,41 The absorption bands of the dyes chemisorbed onto the surface of TiO2 films display a blue shift compared with those in solution (Fig. 1c). Such a phenomenon has been recognized before, and was attributed to the reduction of the electron accepting ability of the cyanoacrylate group.42 The absorption spectrum of T2 shows a slight red shift compared to those of T1, T3 and T4, as similarly observed in the absorption spectra of these dyes in thin films (Fig. 1d). However, the absorption band edges of all spectra extend beyond 650 nm, indicating a certain degree of J-aggregation of the dyes.43
860 M−1 cm−1 for T1 to 31690 M−1 cm−1 for T2, 20890 M−1 cm−1 for T3 and 14000 M−1 cm−1 for T4. The most pronounced hypsochromic shift (lowest ε) is observed when three TPA units are substituted on the π-linker in T4. Herein the decrease of the ε value is more important which could be understood by the low oscillator strength (f) of the ICT according to the Franck–Condon principle or the poor orbital overlap of the HOMO and the LUMO (see DFT calculations, Table 1 and Table S1, ESI†).17,40 This may be explained by the steric repulsion induced by the bulky TPA moiety that leads to a decrease of planarity of the π-conjugated linker especially in T4. This twisted conformation also impedes good delocalization of the π electrons and slightly blue-shifts the position of the ICT band of T2-4 with respect to T1. A higher amount of energy is required to undertake an intramolecular charge transfer once the π-linker part adopts a twist conformation as observed in many cases of alkyl substituted oligothiophene π-linkers.30,41 The absorption bands of the dyes chemisorbed onto the surface of TiO2 films display a blue shift compared with those in solution (Fig. 1c). Such a phenomenon has been recognized before, and was attributed to the reduction of the electron accepting ability of the cyanoacrylate group.42 The absorption spectrum of T2 shows a slight red shift compared to those of T1, T3 and T4, as similarly observed in the absorption spectra of these dyes in thin films (Fig. 1d). However, the absorption band edges of all spectra extend beyond 650 nm, indicating a certain degree of J-aggregation of the dyes.43
 | ||
| Fig. 1 Plots of absorption spectra of (a) dyes T1-4 in CH2Cl2 solution, (b) dye T2 in different solvents and (c) dyes T1-4 adsorbed on TiO2 films coated on fused silica substrates and (d) dyes T1-4 as thin films coated on fused silica substrates. | ||
| Dye | λ soluabs (ε)a (nm, M−1 cm−1) |
|
λ em (nm) | T 5d (°C) | E 1/2 vs. SCEd (V) | HOMOe (V vs. SCE) | E 0–0 (eV) | LUMOg (V vs. SCE) | f |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in CH2Cl2 solutions. b Measured as dyes adsorbed on TiO2 films. c Obtained from TGA measured at 10 °C min−1 under N2. d Obtained from CV measured in CH2Cl2 solutions at a scan rate of 50 mV s−1 and n-Bu4NPF6 as electrolyte. e Estimated from HOMO = Eoxonset. f Estimated from the intercept of the normalized absorption and emission spectra in each solvent used (E0–0 = 1240/λ). g Calculated from LUMO = HOMO − E0–0. h Oscillator strength (f) of the ICT transition obtained from DFT calculation. | |||||||||
| T1 | 304 (15650), 495 (34860) |
455 | 629 | 315 | −1.56, 0.82, 1.02, 1.17 | 0.74 | 2.16 | −1.42 | 2.09 |
| T2 | 305 (38130), 493 (31690) |
487 | 618 | 328 | −1.54, 0.85, 1.05, 1.20 | 0.77 | 2.15 | −1.38 | 1.88 |
| T3 | 302 (54000), 494 (20890) |
490 | 619 | 342 | −1.51, 0.85, 1.06, 1.27 | 0.78 | 2.14 | −1.36 | 1.33 |
| T4 | 301 (67420), 490 (14000) |
486 | 621 | 335 | −1.56, 0.83, 1.04 | 0.77 | 2.18 | −1.41 | 1.14 |
The thermal properties of the dyes examined by thermogravimetric analysis (TGA) reveal that they are thermally stable compounds, with temperatures at 5% weight loss (T5d) well over 315 °C (Table 1). The high thermal stability of a dye is crucial for the lifetime of a solar cell.44
Cyclic voltammetry (CV) analyses were performed to determine the electrochemical behaviour and the energy levels of the dyes, and all data are shown in Fig. 2 and Table 1. Upon the application of positive potentials, the dyes display multiple quasi-reversible oxidation waves, while upon the application of negative potentials they undergo one quasi-reversible reduction process. The redox potentials (Eoxonset) of the dyes T1-4 corresponding to the HOMO energy level, are located at 0.74, 0.79, 0.78 and 0.77 V vs. SCE, respectively. Thus these dyes have comparable HOMO energy levels (0.74–0.79 V vs. SCE), which are more positive than the I−/I3− redox potential (∼0.2 V vs. SCE), assuring efficient dye regeneration by electron transfer from the I−/I3− redox couple to the oxidized dyes. The lowest transition energies (E0–0) of the dyes T1-4 estimated from the intercept of normalized absorption and emission spectra (Fig. S2, ESI†) are 2.16, 2.15, 2.14 and 2.18 eV, respectively. Therefore, the LUMO energy levels of T1-4 calculated by subtracting E0–0 from the HOMO are −1.42, −1.38, −1.36 and −1.41 V vs. SCE, respectively (Table 1). The LUMO levels of these dyes are much more negative than the level of the conduction band edge of TiO2 (−0.7 V vs. SCE), ensuring that the electron injection process from the excited state of the dyes into the TiO2 working electrode is thermodynamically favorable. These results confirm that the newly synthesized dyes T2-4 have sufficient energetic driving force for both the injection of electrons from the excited state of the dyes into the TiO2 and the oxidized dyes to recapture an electron from the I−/I3− electrolyte.
 | ||
| Fig. 2 CV curves measured in CH2Cl2 solution containing n-Bu4NPF6 at a scan rate of 50 mV s−1. | ||
Quantum chemical calculations
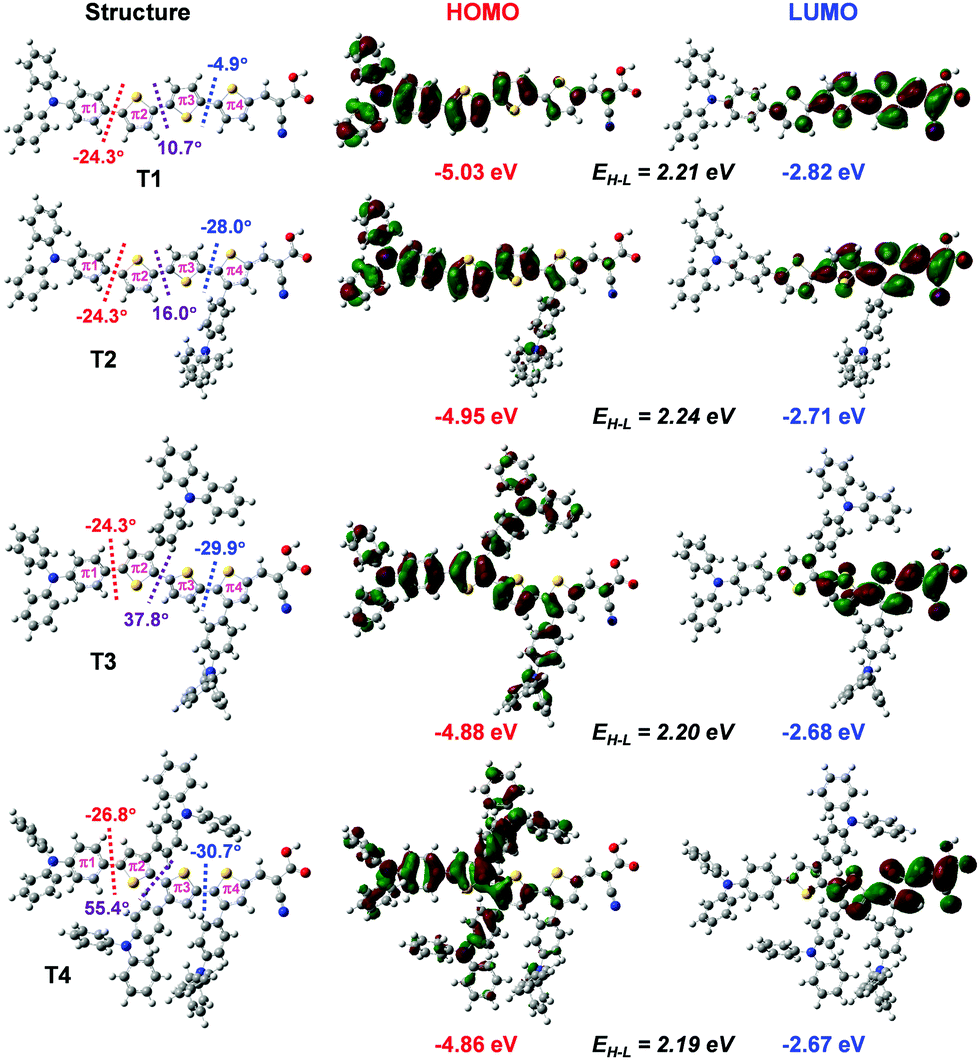 | ||
| Fig. 3 Optimized structures, dihedral angles between moieties (left), HOMO (middle) and LUMO (right) distributions of the dyes and their energies calculated by at the B3LYP/6-31G(d,p) level. | ||
 | ||
| Fig. 4 Density difference between the ground (ρ−) and excited (ρ+) states of the dye. Distance of the charge transfer (DCT) upon excitation is also given. | ||
 | ||
| Fig. 5 Simulated absorption spectra of the dyes calculated by TD-CAM-B3LYP/6-31G(d,p) in CH2Cl2 solvent. | ||
 | ||
| Fig. 6 MOs relevant to the lowest excitation of dye–TiO2 systems. | ||
| Dye |
μ
⊥a (D) |
E ex eV (nm) | f | Compositionb |
|---|---|---|---|---|
| a Dipole moment in the perpendicular direction to the TiO2 surface. b H = HOMO, L = LUMO. | ||||
| T1–TiO2 | 18.25 | 2.74 (452) | 2.0996 | 0.20 (H−1 → L+22) + 0.18 (H−1 → L+25) + 0.14 (H → L+33) |
| T2–TiO2 | 23.22 | 2.71 (456) | 1.7801 | 0.13 (H−1 → L+24) + 0.14 (H−1 → L+21) + 0.14 (H → L+24) |
| T3–TiO2 | 13.65 | 2.39 (519) | 1.1675 | 0.20 (H → L+22) + 0.19 (H → L + 25) + 0.16 (H → L+33) |
| T4–TiO2 | 11.74 | 2.36 (525) | 1.1726 | 0.20 (H → L+22) + 0.18 (H → L+25) + 0.21 (H → L+33) |
Overall, the calculated results clearly explain the experimental evidence of why T2 is the most efficient dye among its sister dyes. Adding one TPA unit to the dye could improve the electron injection driving force in the device. Adding more TPA units, in turn, decreases the dye performance because they make the dye structures bulky resulting in decreased charge transfer ability and obviously they drastically decrease electron injection driving force in the device systems. Dye T2 is therefore a well-rationalized adjustment and is the dye with the highest efficiency.
Photovoltaic performance of dyes
To investigate the photovoltaic performance of the newly synthesized dyes T1-4 the DSSCs with T1-4 sensitizers and an E4 liquid electrolyte were fabricated and studied under standard AM 1.5 illumination (100 mW cm−2) with a metal mask surrounding the active area to prevent inflated photocurrents arising from stray light. The current–voltage (J–V) characteristic plots and incident photon to current conversion efficiency (IPCE) spectra of the DSSCs are shown in Fig. 7 and the detailed cell performances are listed in Table 3. All the devices exhibited good power conversion efficiencies (η) ranging from 5.72 to 8.08%. To validate the obtained results the performance of the solar cells based on the known dye T1 of η (JSC, VOC and FF) of 5.72% (12.40 mA cm−2, 659 mV, 0.700) is compared to the reported results, and it is found that the outcomes from this study some consistent with those results: 5.41% (15.2 mA cm−2, 610 mV, 0.58)50 and 3.21% (7.68 mA cm−2, 600 mV, 0.696).51 Of all the dyes tested, the cell based on T2 displays the highest η of 8.08% (JSC = 16.17 mA cm−2, VOC = 746 mV, FF = 0.670), reaching ∼98% of the standard N719-based cell (8.20%) fabricated under the same conditions. Also, other cells sensitized with dyes T3 and T4 exhibit attractive performances with η values of 7.21%, and 5.79%, respectively. Accordingly, substitution of the TPA substituents on the π-linker does seem to improve the efficiency of the DSSCs, and the performance of the device is strongly dependent on the number of TPA substituents. Once TPA units are substituted as additional electron-donating units on the π linker of T1, giving T2-4, the improved JSC and VOC are observed. When compared with T1, the new dyes T2-4 show a significant higher VOC of about 69–104 mV. The increase in the VOC of T2-4 can be explained by two reasons. Firstly, the hydrophobic properties of the extra TPA group may act as a barrier to impede the diffusion of the electrolyte on the surface of the TiO2, resulting in the suppression of the dark current and blocking of the hydrophilic ions from approaching the TiO2 surface, hence increasing the electron lifetime and VOC. Similar results were observed in the cases of 2,3-diarylamine substituted thiophene-based organic dyes,50 Ru-based dyes52 and other organic dyes.53 Secondly, relatively large dihedral angles between each thiophene ring in the π-linker that are caused by the introduction of the TPA group (see theoretical calculation) may lead to a reduction of electron recombination and an increase of VOC. Such a phenomenon has also been observed in other dyes.36,54 | ||
| Fig. 7 (a) IPCE spectra and (b) J–V curves measured under illumination of AM 1.5G simulated sunlight (100 mW cm−2) of the DSSCs sensitized by dyes T1-4. | ||
| Dye | Dye loadinga (molecule cm−2) | J SC (mA cm−2) | V OC (mV) | FF | η (%) | R CT (ohm) at −0.70b V | τ rec (ms) at −0.70b V |
|---|---|---|---|---|---|---|---|
|
a Measured by desorption method.30
b Obtained EIS measurement.
c Dye:CDCA of 1:20 mole ratio.
|
|||||||
| T1 | 9.69 × 1016 | 12.40 | 659 | 0.709 | 5.72 | 18.01 | 7.93 |
| T2 | 5.58 × 1016 | 16.17 | 746 | 0.670 | 8.08 | 184.06 | 45.22 |
| T3 | 4.47 × 1016 | 14.07 | 763 | 0.671 | 7.21 | 321.96 | 60.54 |
| T4 | 2.17 × 1016 | 10.81 | 728 | 0.736 | 5.79 | 73.90 | 27.78 |
| T1:CDCAc | 7.49 × 1016 | 14.49 | 661 | 0.723 | 6.94 | 22.00 | 9.14 |
| T2:CDCAc | 4.02 × 1016 | 17.05 | 751 | 0.706 | 9.02 | 192.26 | 46.46 |
| T3:CDCAc | 3.17 × 1016 | 15.11 | 767 | 0.707 | 8.19 | 366.41 | 69.50 |
| T4:CDCAc | 1.35 × 1016 | 12.22 | 731 | 0.745 | 6.67 | 83.37 | 28.66 |
| N719 | 5.83 × 1016 | 16.81 | 730 | 0.690 | 8.20 | 108.48 | 34.45 |
To understand the high VOC observed for dyes T2-4, electrochemical impedance spectroscopy (EIS) was performed. Fig. 8 shows EIS results for devices based on T1-4 under forward bias (∼0.7 V) in the dark. The larger semicircles in the Nyquist plots (Fig. 8a) are related to the resistance of charge-transfer processes occurring at the TiO2/dye/electrolyte interface. Clearly, the charge-transfer resistances (RCT) at the interfaces of the DSSCs based on T2-4 are increased in comparison to the cell based on T1, indicating a smaller dark current (Fig. 8c and Table 3). The RCT values of the DSSCs are increased in the order T1 (18.01 Ω) < T4 (73.90 Ω) < T2 (184.06 Ω) < T3 (321.96 Ω), which are in agreement with the order of the VOC values. The electron lifetime in the DSSCs is an essential measure to define the dynamics of charge recombination (dark current), which is related to the VOC of the cell. It has been reported that the VOC relies on charge recombination reactions rather than on the molecular structures of the dyes.55 From the Bode phase plot (Fig. 8b), the electron recombination lifetime (τrec) was also extracted using τrec = 1/ωmin (Fig. 8d and Table 3).56 The order of increasing τrec values, T1 (7.93 ms) < T4 (27.78 ms) < T2 (45.22 ms) < T3 (60.54 ms), was consistent with that of the RCT values. The much longer electron lifetime for the newly designed dyes T2-4 compared to a reference D–π–A dye T1 is observed. This result suggests that the charge recombination rate between the TiO2 film and the electrolyte is indeed suppressed by introduction of the extra TPA electron donor groups on the thiophene units of the π-linker efficiently, resulting in an increase of both the electron lifetime in TiO2 and the photovoltage. It is also noticed that the electron lifetime is longer for the cells of T3 and T2 than that for N719 (27.78 ms). To further investigate the reason for the differences between the VOC values, the electron lifetimes of the DSSCs were also measured under open-circuit conditions by means of intensity-modulated photovoltage spectroscopy (IMVS). Fig. 9a plots the electron lifetime (τn) of the DSSCs as a function of light intensity. When at the same light intensity, the electron lifetimes for the DSSCs sensitized by dyes T2-4 are much longer than that for T1, which are in line with the order of the VOC values. This suggests that the electron injected from the excited dyes T2-4 can survive for a longer time and hence enable electron transport without undergoing losses. Therefore, both EIS and IMVS measurements approve that substitutions of the TPA unit on the π-linker of the D–π–A dye are able to reduce the photoelectron recombination rate effectively due to the longer electron lifetime.
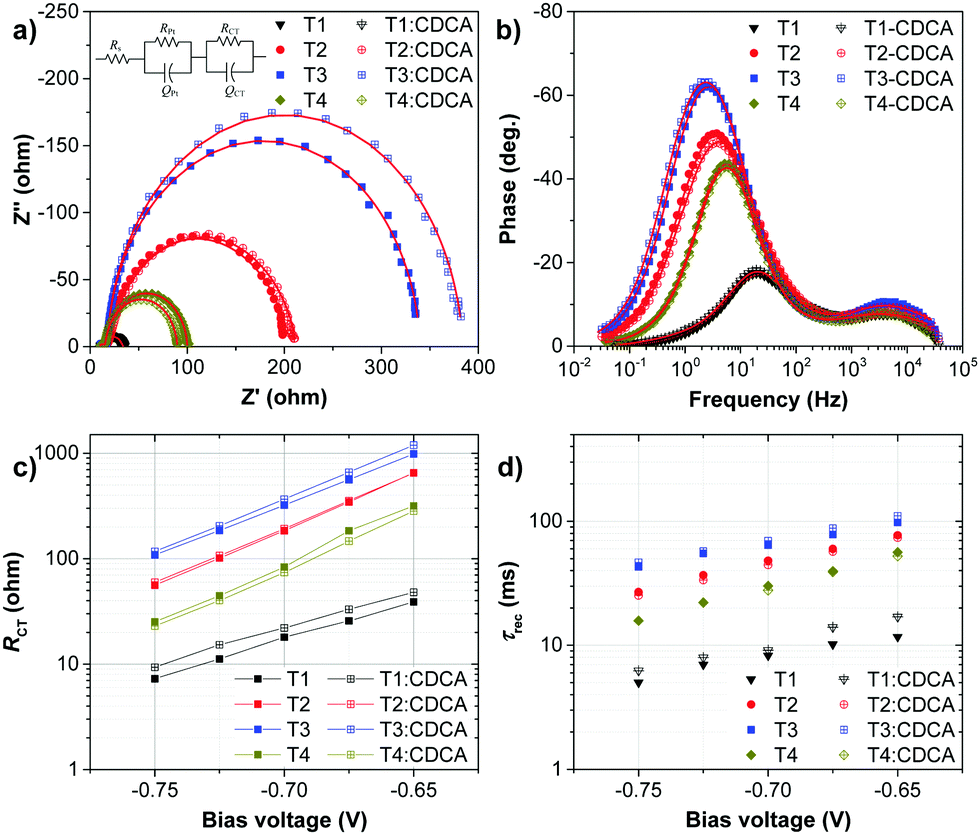 | ||
| Fig. 8 Plots of (a) Nyquist (inset: equivalent circuit for the DSSC) and (b) Bode-phase measured at 0.7 V bias voltage, and (c) charge-transfer resistance (RCT) and (d) electron recombination lifetime (τrec) as a function of bias voltages of the DSSCs in the dark. | ||
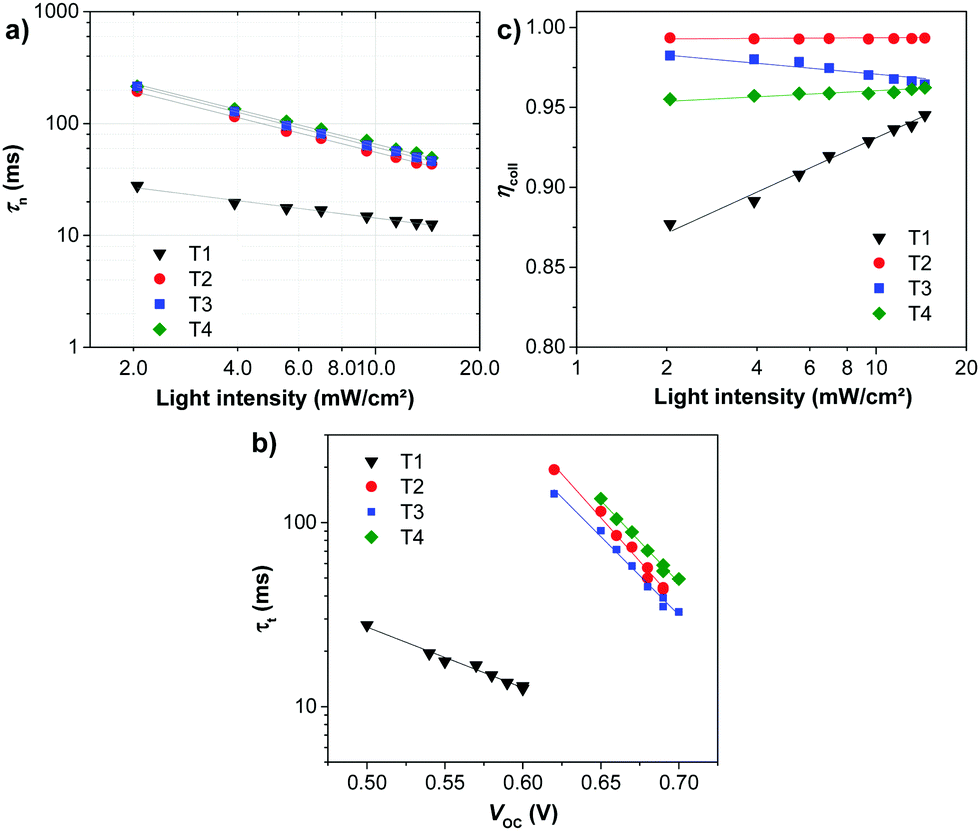 | ||
| Fig. 9 Plots of (a) the electron lifetime (τn) as a function of light intensity, (b) electron transport time (τt) as a function of open-circuit voltage and (c) electron collection efficiency (ηcoll) as a function of incident light intensity of the DSSCs. | ||
As mentioned in the theoretical calculation part, the presence of the TPA group seems to not only provide a hydrophobic blocking barrier, but also builds up bulkiness of the dye molecule, which could hinder the aggregation of dyes. It has been reported that too much close π-stacked aggregation of the dyes on the TiO2 electrode leads to inefficient electron injection because of intermolecular energy transfer processes between the dyes, thus resulting in low cell efficiency.57 Conversely, too low amount of dye adsorbed on TiO2 films decreases the ability of the dye to harvest light, which also results in a low power conversion efficiency.23,58 The amount of the molecules of dyes T1-4 adsorbed on the TiO2 films of the same surface area, thickness and porosity was analyzed and found to be 9.69 × 1016, 5.58 × 1016, 4.47 × 1016, and 2.17 × 1016, molecule cm−2, respectively (Table 3). The results reveal that the amount of dye adsorbed is steadily decreased with the increasing number of extra TPA substituents on the molecule or the size of the dye molecule, suggesting that the resulting overall light harvesting efficiency (LHE) or JSC of the devices will be strongly affected. LHE or absorptance is the fraction of light intensity absorbed by the dye in the DSSCs and strongly depends on the ε value of the dye and its adsorption capacity (Γ) onto the TiO2 film as given by the following expression:59
| LHE(λ) = 1 − 10−ε(λ)Γ | (1) |
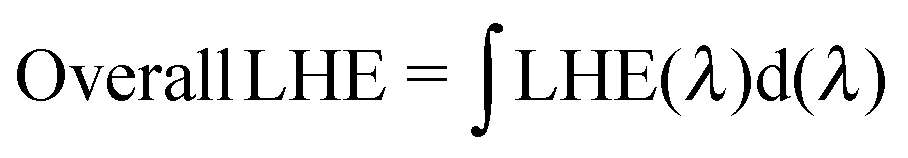 | (2) |
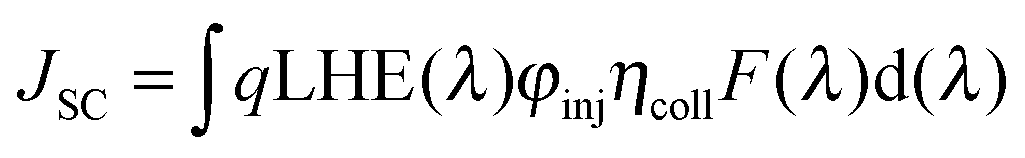 | (3) |
Device performance of dyes coadsorbed with CDCA
From our previous study61 and other reports,43,51,62 the delicate balance between dye density and dye deaggregation is crucial when coadsorbents are used. An appropriate amount of chenodeoxycholic acid (CDCA) was added as the coadsorbent to alleviate dye aggregation and lessen the adverse impact of dye aggregation on electron injection while still keeping an optimum amount of dye adsorbed on the TiO2 film. The J–V curves and IPCE spectra are shown in Fig. 10 and the relevant data are listed in Table 3. The data show that the conversion efficiencies for all the DSSCs are improved upon adding CDCA of 20:1 mole ratio, and they follow the same order as the CDCA-free cells: T4 (6.67%) < T1 (6.94%) < T3 (8.12%) < T2 (9.02%). The T2:CDCA-based cell exhibits an impressively high cell performance with a η of 9.02% (JSC = 17.05 mA cm−2; VOC = 751 mV; FF = 0.706) surpassing that of N719 (8.20%). This improvement originates from an increase in all photovoltaic parameters (JSC, VOC and FF) of the DSSCs (Table 3). The increase in the JSC upon the addition of CDCA can be ascribed to the relief of dye aggregation and the subsequent destruction of the quenching processes of the energy transfer,63 while the increase in VOC can be attributed to the suppression of electron recombination via the formation of a thin hydrophobic layer on the free TiO2 surface, thereby keeping charged species coming from the electrolyte away from bare TiO2.64 The increase in the fill factor (FF) may partly result from the increase in the VOC and JSC of the DSSCs. The FF signifies the deflection of the device from ideal (resistance-free) solar cells, therefore, it may also be influenced by many other parameters such as the structure of the dye film (adsorption), the TiO2, the ITO film, electrolyte, built-in voltage etc.65 It is noticed that by co-adsorption with CDCA, the T1:CDCA-based DSSC exhibits a larger improvement in the device performance (∼20% increment of η, ∼14% increment of JSC) compared to that of T2-4 (∼11–15% increment of η, ∼5–11% increment of JSC). As mentioned earlier, the skeletal structure of dye T1 is smaller and less bulky than that of dyes T2-4, obviously, the insertion of the co-adsorbents would have a stronger effect in decreasing dye aggregation in dye T1 molecules more than in dyes T2-4, accounting for the large increase in the photocurrent (JSC) of T1. However, in all cases co-adsorption of CDCA lowers the surface tension of TiO2 surface resulting in efficient suppression of dark current at the TiO2/electrolyte interface, resulting in an increase of VOC. Based on the EIS data in the dark (Fig. 8 and Table 3), upon the addition of CDCA to the dyes, the charge-transfer resistance (T1:CDCA = 22.00 Ω; T2:CDCA = 192.26 Ω; T3:CDCA = 366.41 Ω; T4:CDCA = 83.37 Ω;) at the TiO2/electrolyte interface and the electron recombination lifetime (T1:CDCA = 9.14 ms; T2:CDCA = 46.46 ms; T3:CDCA = 69.50 ms; T4:CDCA = 28.66 ms) of the DSSCs are improved, accounting for the increase of VOC, which benefits from a better dark-current suppression.
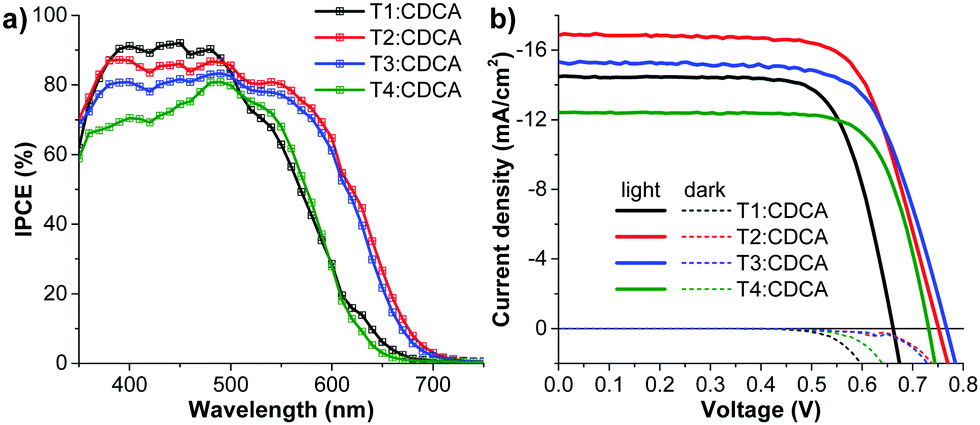 | ||
| Fig. 10 (a) IPCE spectra and (b) J−V curves measured under illumination of AM 1.5G simulated sunlight (100 mW cm−2) of the DSSCs sensitized by T1-4:CDCA. | ||
Experimental
Materials and instrumentation
All chemicals were used as received. THF and CH2Cl2 were distilled according to the standard method. 1H and 13C NMR spectra were recorded using a Brüker AVANCE 600 MHz spectrometer. Infrared (IR) spectra were measured using a Perkin-Elmer Frontier FT-IR spectrometer. UV-Vis spectra of CH2Cl2 were recorded using a Perkin-Elmer UV Lambda 1050 spectrometer. The absorption spectra of the dye adsorbed on TiO2 were measured at room temperature using a Perkin-Elmer Lambda 1050 spectrometer and an integrating sphere system. The transparent TiO2 film (∼3 μm thickness) was coated on the fused silica (JGS3) substrate using a screen-printing technique. Cyclic voltammetry (CV) measurements were performed on an Autolab potentiostat PGSTAT 101 equipped with a platinum counter electrode, a glassy carbon working electrode, an Ag/AgCl reference electrode and n-Bu4NPF6 as a supporting electrolyte in CH2Cl2 under an Ar atmosphere at a scan rate of 50 mV s−1. The concentration of analytical materials and the electrolyte were 10−3 and 0.1 M, respectively. Melting points were measured using a Krüss KSP1N melting point meter and were uncorrected. High resolution MALDI-TOF mass spectra were recorded on a Brüker Daltonics (Bremen, Germany) Autoflex Speed Matrix-Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometer (BIFEX).Computational details
The calculations of free dye structures were carried out using a Gaussian 09 program package.66 Ground-state geometries were optimized without any constraints using density functional theory (DFT) at the B3LYP/6-31G(d,p) basis set. The solvent effect of CH2Cl2 was taken into account by a polarizable Continuum Model (PCM). The prototype systems were modelled by dye adsorbed on the (TiO2)38 cluster and the calculations were performed using the DMol3 code in Material Studio 5.5™. The dye-(TiO2)38 systems were optimized using the Perdew–Burke–Ernzerhof (PBE) functional with the double numerical polarization (DNP) basis set. The excited state calculations were done by time-dependent DFT using the CAM-B3LYP functional with the same basis set in the ground state.Fabrication of DSSCs and measurements
Fluorine-doped SnO2 (FTO) conducting glasses (15 ohm sq−1, TEC15, Pilkington) were coated with a double nanostructure thick TiO2 film (∼9 + 5 μm thickness) consisting of transparent (18NR-T, Dyesol) and scattering (PST-400C, JGC Catalysts and Chemical Ltd) TiO2 layers. Prior to dye sensitization, the TiO2 electrodes with a cell geometry of 0.5 × 0.5 cm2 were treated with an aqueous solution of 4 × 10−2 M TiCl4 at 70 °C, then heated at 450 °C for 30 min and cooled to 80 °C. The TiO2 electrodes were immersed in the dye solution (5 × 10−4 M dye or dye:CDCA (1:20 mole ratio) in the 1:1 mixture of CHCl3 and EtOH) in the dark at room temperature for 24 h. Excess dye was removed by rinsing with an appropriate solvent. The Pt counter electrode was prepared on a predrilled 8 ohm sq−1, TEC8, FTO glass (Pilkington) via the thermal decomposition of 7 × 10−3 M H2PtCl6 in isopropanol solution at 385 °C. The dye-adsorbed TiO2 photoanode and the Pt counter electrode were assembled into a sealed cell by heating a gasket Meltonix 1170-25 film (25 μm thicknesses, Solaronix) as a spacer between the electrodes. An E4 electrolyte comprising of 0.05 M I2, 0.1 M LiI, 0.6 M tetrapropyl ammonium iodide (TPAI) and 0.4 M tert-butylpyridine (4-TBP) in a mixed solvent of acetonitrile and valeronitrile (85/15, v/v) was filled through a predrilled hole using a vacuum backfilling method. The hole was capped by using a hot-melt sealing film (Meltonix 1170-25, 25 μm thickness, Solaronix) and a thin glass cover. Finally, the Scotch 3M conducting tape and silver paint (SPI supplies) were coated on the electrodes to enhance the electric contact. For each dye, five devices were fabricated and measured for consistency and the averaged cell data were reported. The reference cells with the same device configuration based on the Ru-complex dye N719, as the sensitizer, were also fabricated for comparison.
The current density–voltage of the DSSCs was measured by using a Keithley 2400 source meter unit in a 4-terminal sense configuration. The data were averaged from forward and backward scans with a bias step and a delay time of 10 mV and 40 ms, respectively, according to the method of Koide and Han.67 Simulated sunlight was provided by a Newport sun simulator 96000 equipped with an AM 1.5G filter. To minimize the error of measurements, the irradiation intensity of 100 mW cm−2 was approximated using a calibrated BS-520 Si photodiode (Bunnkoukeiki Co., Ltd, Japan), whose spectral response was very similar to that of the DSSCs. The spectral output of the lamp was also matched to the standard AM 1.5G solar spectrum in the region of 350–750 nm by using a KG-5 filter with spectral mismatch less than 2% as reported by Ito et al.68 Incident photon to electron conversion efficiency (IPCE) of the devices under short-circuit conditions was evaluated by means of an Oriel 150W Xe lamp fitted with a Cornerstone TM 130 1/8 m monochromator as a monochromatic light source, a Newport 818-UV silicon photodiode for power density calibration and a Keithley 6485 picoammeter. All measurements were performed using a black plastic mask with an aperture area of 0.25 cm2 and no mismatch correction for the efficiency conversion data.
Electrochemical impedance spectra (EIS) were analyzed using an EA163 eDAQ potentiostat integrated with an ERZ100 eDAQ Z100 electrochemical impedance analyzer as a function of bias potential under dark conditions. Nyquist plots of all DSSCs were recorded over a frequency range of 50 mHz–100 kHz with an amplitude of 10 mV and fitted using ZMAN software (WonTech Co. Ltd) and equivalent circuit Rs − RPt||QPt − RCT||QCT. The Rs denotes the ohmic series resistance of the cell, RPt stands for charge resistance at the Pt/electrolyte electrode and RCT represents the charge recombination resistance at the TiO2/electrolyte interface. The Q parameters are constant phase elements. The electron collection efficiency (ηcoll) was calculated according to the relation; ηcoll = 1 − (τt/τn) where τt is the electron transport time at short circuit and τn is the electron lifetime at open circuit.69 The lifetime constants τt and τn were measured by intensity-modulated photocurrent spectroscopy (IMPS) and intensity-modulated photovoltage spectroscopy (IMVS), respectively, using an Autolab PGSTAT302N/FRA32M electrochemical workstation combined with an Autolab LED driver. A 530 nm green light-emitting diode was employed as the light source. The amplitude of the modulated intensity was 10% of the DC intensity. The modulation frequencies were varied between 0.1 Hz and 100 kHz.
Synthesis
Compounds 1,70271 and T151 were synthesized according to the described procedures.:4) as the eluent followed by recrystallization in a mixture of CH2Cl2 and MeOH.
3: orange solids (69%). FTIR (KBr): δ 1661 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1; 1H NMR (600 MHz, DMSO-d6) δ 9.88 (1H, s), 8.14 (1H, s), 7.72 (1H, d, J = 3.6 Hz), 7.44 (1H, d, J = 4.2 Hz), 7.32 (1H, d, J = 3.6 Hz), 7.28 (1H, d, J = 3.6 Hz) ppm; 13C NMR (150 MHz, DMSO-d6) δ 183.82, 141.96, 140.42, 140.08, 138.60, 137.40, 132.41, 131.98, 130.92, 126.43, 125.75, 112.17, 108.67 ppm; APCI-Q-TOF (m/z) calcd for C13H6Br2OS3: 433.7948; found 433.7736 (M+).
O) cm−1; 1H NMR (600 MHz, DMSO-d6) δ 9.88 (1H, s), 8.14 (1H, s), 7.72 (1H, d, J = 3.6 Hz), 7.44 (1H, d, J = 4.2 Hz), 7.32 (1H, d, J = 3.6 Hz), 7.28 (1H, d, J = 3.6 Hz) ppm; 13C NMR (150 MHz, DMSO-d6) δ 183.82, 141.96, 140.42, 140.08, 138.60, 137.40, 132.41, 131.98, 130.92, 126.43, 125.75, 112.17, 108.67 ppm; APCI-Q-TOF (m/z) calcd for C13H6Br2OS3: 433.7948; found 433.7736 (M+).
4: orange solids (80%). FTIR (KBr): ν 1661 (CO) cm−1; 1H NMR (600 MHz, DMSO-d6) δ 9.89 (1H, s), 8.16 (1H, s), 7.77 (1H, d, J = 4.2 Hz), 7.49 (1H, s) ppm; 13C NMR (150 MHz, DMSO-d6) δ 183.93, 141.94, 140.79, 139.78, 135.48, 133.67, 132.88, 130.14, 128.70, 113.04, 109.05, 108.61 ppm; APCI-Q-TOF (m/z) calcd for C13H5Br3OS3: 513.7012; found 513.6880 (M+).
5: orange solids (82%). FTIR (KBr): ν 1661 (CO) cm−1; 1H NMR (600 MHz, DMSO-d6) δ 9.91 (1H, s), 8.18 (1H, s), 7.89 (1H, s), 7.56 (1H, s) ppm; 13C NMR (150 MHz, DMSO-d6) δ 184.12, 141.8, 141.66, 138.03, 135.38, 134.07, 132.08, 129.95, 129.35, 115.82, 114.19, 113.12, 110.18 ppm; APCI-Q-TOF (m/z) calcd for C13H4Br4OS3: 591.9700; found 591.6261 (M+).
:4) as the eluent followed by recrystallization in a mixture of CH2Cl2 and MeOH.
7: purple solids (77%); m.p. > 250 °C; FTIR (KBr): ν 1661 (CO), cm−1; 1H NMR (600 MHz, CDCl3): δ 9.85 (1H, s), 7.65 (1H, d, J = 3.6 Hz), 7.46 (2H, d, J = 8.4 Hz), 7.25–7.27 (10H, m), 7.02–7.20 (20H, m) ppm; 13C NMR (150 MHz, CDCl3) δ 182.34, 147.80, 147.78, 147.59, 147.37, 147.10, 124.84, 141.48, 140.60, 138.57, 137.27, 135.14130.20, 129.75, 129.38, 129.35, 128.98, 127.34, 126.69, 126.43, 126.16, 125.76, 124.76, 124.58, 123.74, 123.53, 123.36, 123.31, 123.15 ppm; APCI-Q-TOF (m/z) calcd for C49H34N2OS3: 762.1833; found 762.1567 (M+).
8: purple solids (69%); m.p. > 250 °C; FTIR (KBr): ν 1661 (CO) cm−1; 1H NMR (600 MHz, CDCl3): δ 9.79 (1H, s), 7.57 (1H, s), 7.39 (2H, d, J = 9.0 Hz), 7.22–7.15 (14H, m), 7.12 (2H,d, J = 8.4 Hz), 7.09 (1H, s), 7.06 (12H, d, J = 7.8 Hz), 7.00–6.93 (12H, m), 6.81 (1H, d, J = 4.2 Hz) ppm; 13C NMR (150 MHz, CDCl3) δ 182.46, 147.56, 147.44, 147.38, 141.65, 139.99, 139.42, 139.37, 130.13, 129.37, 129.34, 127.92, 126.43, 125.94, 125.83, 124.72, 124.71, 124.62, 123.37, 123.32, 123.28, 123.30, 123.14, 124.58, 123.74, 123.53, 123.36, 123.31, 123.15; APCI-Q-TOF (m/z) calcd for C67H47N3OS3: 1005.2881; found 1005.3111 (M+).
9: purple solids (78%); m.p. > 250 °C; FTIR (KBr): ν 1669 (CO) cm−1. 1H NMR (600 MHz, CDCl3): δ 9.78 (1H, s), 7.58 (1H, s), 7.60 (2H, d, J = 8.4 Hz), 7.18 (6H, d, J = 7.8 Hz), 6.78–7.11 (50H, m) ppm; 13C NMR (150 MHz, CDCl3) δ 182.31, 148.39, 147.86, 147.69, 147.61, 147.54, 146.91, 146.79, 144.55, 141.76, 141.67, 140.38, 140.22, 139.81, 139.37, 134.95, 132.29, 130.53, 130.24, 129.91, 129.58, 129.49, 129.35, 129.33, 129.00, 128.92, 128.69, 127.74, 127.00, 126.56, 124.75, 124.64, 124.58, 124.54, 124.42, 124.31, 124.17, 123.91, 123.68, 123.61, 123.52, 123.39, 123.28, 123.01, 122.95; APCI-Q-TOF (m/z) calcd for C85H60N4OS3: 1248.3929; found 1248.4203 (M+).
:20) as the eluent followed by recrystallization in a mixture of CH2Cl2 and MeOH.
T2: dark red solids (79%); m.p. > 250 °C; FTIR (KBr): ν 3350 (OH), 2216 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1590 (CO) cm−1; 1H NMR (600 MHz, DMSO-d6): δ 8.24 (1H, s), 7.59 (1H, s), 7.35–7.44 (4H, m), 7.04–7.12 (28H, m) ppm; 13C NMR (150 MHz, DMSO-d6): δ 163.70, 147.84, 147.80, 147.45, 142.49, 140.86, 137.30, 135.27, 135.20, 130.81, 130.17, 130.10, 130.03, 129.90, 129.50, 128.60, 127.88, 127.19, 126.96, 126.94, 126.69, 125.14, 125.03, 124.88, 124.80, 124.47, 124.14, 123.88, 123.35, 123.03, 118.56 ppm; MALDI-TOF (m/z) calcd for C52H35N3O2S3: 829.1891; found 829.1410 (M+).
N), 1590 (CO) cm−1; 1H NMR (600 MHz, DMSO-d6): δ 8.24 (1H, s), 7.59 (1H, s), 7.35–7.44 (4H, m), 7.04–7.12 (28H, m) ppm; 13C NMR (150 MHz, DMSO-d6): δ 163.70, 147.84, 147.80, 147.45, 142.49, 140.86, 137.30, 135.27, 135.20, 130.81, 130.17, 130.10, 130.03, 129.90, 129.50, 128.60, 127.88, 127.19, 126.96, 126.94, 126.69, 125.14, 125.03, 124.88, 124.80, 124.47, 124.14, 123.88, 123.35, 123.03, 118.56 ppm; MALDI-TOF (m/z) calcd for C52H35N3O2S3: 829.1891; found 829.1410 (M+).
T3: dark red solid (83%); m.p. > 250 °C; FTIR (KBr): ν 3350 (OH), 2216 (CN), 1590 (CO) cm−1; 1H NMR (600 MHz, DMSO-d6): δ 8.11 (1H, s), 7.67 (1H, s), 7.56 (2H, d, J = 8.4 Hz), 7.40 (1H, s), 7.32 (4H, t, J = 8.4 Hz), 7.23–7.29 (10H, m), 7.20 (2H, d, J = 8.4 Hz), 7.09 (2H, t, J = 7.8 Hz) 6.98–6.95 (8H, m) ppm; 13C NMR (150 MHz, DMSO-d6): δ 163.88, 147.81, 147.70, 147.53, 147.35, 147.32, 147.20, 142.18, 140.44, 139.22, 137.45, 134.47, 130.70, 130.13, 129.49, 128.63, 128.52, 128.19, 127.03, 126.89, 126.72, 124.94, 124.76, 124.68, 124.08, 123.84, 123.80, 123.25, 123.09, 123.01, 118.87 ppm; MALDI-TOF (m/z) calcd for C70H48N4O2S3: 1072.2939; found 1072.1646 (M+).
T4: red solids (69%); m.p. > 250 °C; FTIR (KBr): ν 3350 (OH), 2215 (CN), 1587 (CO) cm−1; 1H NMR (600 MHz, DMSO-d6): δ 8.38 (1H, s), 7.92 (1H, s), 7.60 (2H, d, J = 8.4 Hz), 7.51 (1H, s), 7.34 (8H, t, J = 7.8 Hz) 7.18–7.25 (16H, m), 6.88–7.11 (40H, m), 6.77 (4H, t, J = 8.4 Hz) ppm; 13C NMR (150 MHz, DMSO-d6): δ 163.61, 148.12, 147.76, 147.41, 147.39, 147.23, 146.81, 146.63, 144.16, 141.68, 140.33, 139.98, 134.69, 134.08, 131.45, 131.07, 130.13, 129.98, 129.92, 129.84, 129.31, 129.25, 129.10, 128.75, 127.17, 126.93, 126.18, 125.44, 124.85, 124.35, 124.28, 124.15, 124.04, 123.91, 123.63, 123.55, 123.49, 123.41, 123.30, 118.83 ppm; MALDI-TOF (m/z) calcd for C88H61N5O2S3: 1315.3987; found 1315.6084 (M+).
Conclusions
We have demonstrated the successful development of new efficient organic dyes by means of simple substitution of a donor moiety on the π-linker of the basic D–π–A dye for dye-sensitized solar cells (DSSCs). The three dyes T2-4 feature terthiophene as a π-linker with different numbers of triphenylamine (TPA) substituents on thiophene rings of the terthiophene segment. The dyes present reasonably good power conversion efficiencies (5.79–8.08%) in DSSCs, which are higher than that of the traditional D–π–A dye T1 (5.72%). Detailed studies reveal that substitution of TPA on the π-linker of the D–π–A dye can not only decrease the recombination process between the injected electrons and the electrolytes, but may also hinder the back electron transfer and slow a recombination reaction, thus yielding high photovoltage and high photocurrent, respectively. An efficiency of 9.02% (JSC = 16.91 mA cm−2, VOC = 754 mV, FF = 0.705), surpassing that of the N719-based cell (8.20%), has been realized at the AM 1.5 irradiation (100 mW cm−2) by dye T2 (coadsorbed with CDCA) having one TPA unit substitution on the terminal thiophene ring of the π-linker. We believe that this approach of structural modification will open very promising perspectives to develop more effective organic dyes for DSSCs in the future.Acknowledgements
This work was financially supported by the Thailand Research Fund (Grant No. BRG5980011).Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- S. Zhang, X. Yang, Y. Numata and L. Han, Energy Environ. Sci., 2013, 6, 1443 Search PubMed; M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef CAS PubMed; A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef PubMed.
- J.-F. Yin, M. Velayudham, D. Bhattacharya, H.-C. Lin and K.-L. Lu, Coord. Chem. Rev., 2012, 256, 3008 CrossRef CAS; M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330 CrossRef PubMed.
- M. Grätzel, J. Photochem. Photobiol., A, 2014, 164, 3 CrossRef.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- Y.-Z. Wu and W.-H. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC.
- C.-P. Lee, R. Y.-Y. Lin, L.-Y. Lin, C.-T. Li, T.-C. Chu, S.-S. Sun, J. T. Lin and K.-C. Ho, RSC Adv., 2015, 5, 23810 RSC; Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734 RSC.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799 CrossRef CAS PubMed; M. Zhang, Y. Wang, M. Xu, W. Ma, R. Li and P. Wang, Energy Environ. Sci., 2013, 6, 2944 Search PubMed; K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379 RSC.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC.
- T. Sudyoadsuk, S. Pansay, S. Morada, R. Rattanawan, S. Namuangruk, T. Kaewin, S. Jungsuttiwong and V. Promarak, Eur. J. Org. Chem., 2013, 5051 CrossRef CAS; D. Kim, J. K. Lee, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 1913 CrossRef; N. Koumura, Z. S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2008, 130, 4202 CrossRef; A. Venkateswararao, K. R. J. Thomas, C.-P. Lee, C.-T. Li and K.-C. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 2528 Search PubMed.
- K. Hara, K. Sayama, Y. Ohga, A. Shinpo, S. Suga and H. Arakawa, Chem. Commun., 2001, 569 RSC; Z. S. Wang, Y. Cui, K. Hara, Y. Dan-Oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138 CrossRef CAS.
- Z. Yao, H. Wu, Y. Ren, Y. Guo and P. Wang, Energy Environ. Sci., 2015, 8, 1438 Search PubMed; Y. Jin, J. Hua, W. Wu, X. Ma and F. Meng, Synth. Met., 2008, 158, 64 CrossRef CAS; Y. Shibano, T. Umeyama, Y. Matano and H. Imahori, Org. Lett., 2007, 9, 1971 CrossRef PubMed; T. Edvinsson, C. Li, N. Pschirer, J. Schoneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137 Search PubMed.
- T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036 RSC; T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218 CrossRef CAS PubMed; D. Kim, M.-S. Kang, K. Song, S. O. Kang and J. Ko, Tetrahedron, 2008, 64, 10417 CrossRef.
- D. Muenmart, N. Prachumrak, R. Tarsang, S. Namuangruk, S. Jungsuttiwong, T. Sudyoadsuk, P. Pattanasattayavong and V. Promarak, RSC Adv., 2016, 6, 38481 RSC; H. N. Tian, X. C. Yang, R. K. Chen, A. Hagfeldt and L. C. Sun, Energy Environ. Sci., 2009, 2, 674 Search PubMed; Y. Hua, S. Chang, J. He, C. Zhang, J. Zhao, T. Chen, W.-Y. Wong, W.-K. Wong and X. Zhu, Chem. – Eur. J., 2014, 20, 6300 CrossRef CAS PubMed.
- R. Chen, X. Yang, H. Tian, X. Wang, A. Hagfeldt and L. Sun, Chem. Mater., 2007, 19, 4007 CrossRef CAS.
- H. Tan, C. Pan, G. Wang, Y. Wu, Y. Zhang, Y. Zou, G. Yu and M. Zhang, Org. Electron., 2013, 14, 2795 CrossRef CAS; Z. Wan, C. Jia, L. Zhou, W. Huo, X. Yao and Y. Shi, Dyes Pigm., 2012, 95, 41 CrossRef.
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, Y. Pan and P. Wang, Chem. Mater., 2010, 22, 1915 CrossRef CAS.
- B. Liu, Q. B. Liu, D. You, X. Y. Li, Y. Naruta and W. H. Zhu, J. Mater. Chem., 2012, 22, 13348 RSC; C. J. Yang, Y. J. Chang, M. Watanabe, Y. S. Hon and T. J. Chow, J. Mater. Chem., 2012, 22, 4040 RSC; S. Namuangruk, R. Fukuda, M. Ehara, J. Meeprasert, T. Khanasa, S. Morada, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, J. Phys. Chem. C, 2012, 116, 25653 Search PubMed; T. Sudyoadsuk, J. Khunchalee, S. Pansay, P. Tongkasee, S. Morada, T. Kaewin, S. Jungsuttiwong and V. Promarak, Tetrahedron Lett., 2013, 54, 4903 CrossRef CAS.
- Z. Ning, Q. Zhang, H. Pei, J. Luan, C. Lu, Y. Cui and H. Tian, J. Phys. Chem. C, 2009, 113, 10307 Search PubMed; S. H. Lin, Y. C. Hsu, J. T. Lin, C. K. Lin and J. S. Yang, J. Org. Chem., 2010, 75, 7877 CrossRef CAS PubMed; Y. Numata, A. Islam, H. Chen and L. Han, Energy Environ. Sci., 2012, 5, 8548 Search PubMed.
- T. Khanasa, N. Jantasing, S. Morada, N. Leesakul, R. Tarsang, S. Namuangruk, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2013, 2608 CrossRef CAS; J. Tang, J. Hua, W. Wu, J. Li, Z. Jin, Y. Long and H. Tian, Energy Environ. Sci., 2010, 3, 1736 Search PubMed; Z. Q. Wan, C. Y. Jia, Y. D. Duan, L. L. Zhou, J. Q. Zhang, Y. Lin and Y. Shi, RSC Adv., 2012, 2, 4507 RSC.
- T. Kaewpuang, N. Prachumrak, S. Namuangruk, S. Jungsuttiwong, T. Sudyoadsuk, P. Pattanasattayavong and V. Promarak, Eur. J. Org. Chem., 2016, 2528 CrossRef CAS.
- P. Thongkasee, A. Thangthong, N. Janthasing, T. Sudyoadsuk, S. Namuangruk, T. Kaewin, S. Jungsuttiwong and V. Promarak, ACS Appl. Mater. Interfaces, 2014, 6, 8212 Search PubMed; B.-K. An, P. L. Burn and P. Meredith, Chem. Mater., 2009, 21, 3315 CrossRef CAS.
- J. Liu, R. Li, X. Si, D. Zhou, Y. Shi, Y. Wang, X. Jing and P. Wang, Energy Environ. Sci., 2010, 3, 1924 Search PubMed; K. Hara, Z.-S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476 CrossRef CAS PubMed; X. H. Zhang, Z. S. Wang, Y. Cui, N. Koumura, A. Furube and K. Hara, J. Phys. Chem. C, 2009, 113, 13409 Search PubMed; E. Miyazaki, T. Okanishi, Y. Suzuki, N. Ishine, H. Mori, K. Takimiya and Y. Harima, Bull. Chem. Soc. Jpn., 2011, 84(5), 459 CrossRef.
- G. L. Zhang, H. Bala, Y. M. Cheng, D. Shi, X. J. Lv, Q. J. Yu and P. Wang, Chem. Commun., 2009, 2198 RSC.
- K. Hara, M. Kurashige, S. Ito, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Chem. Commun., 2003, 252 RSC; K. Hara, T. Sato, R. Katoh, A. Furube, T. Yoshihara, M. Murai, M. Kurashige, S. Ito, A. Shinpo, S. Suga and H. Arakawa, Adv. Funct. Mater., 2005, 15, 246 CrossRef CAS; B. Liu, W. H. Zhu, Q. Zhang, W. J. Wu, M. Xu, Z. J. Ning, Y. S. Xie and H. Tian, Chem. Commun., 2009, 1766 RSC.
- C. Teng, X. Yang, C. Yang, S. Li, M. Cheng, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 9101 CAS.
- S. H. Kim, H. W. Kim, C. Sakong, J. Namgoong, S. W. Park, M. J. Ko, C. H. Lee, W. I. Lee and J. P. Kim, Org. Lett., 2011, 13, 5784 CrossRef CAS PubMed.
- Y.-S. Yen, Y.-C. Hsu, J. T. Lin, C.-W. Chang, C.-P. Hsu and D.-J. Yin, J. Phys. Chem. C, 2008, 112, 12557 Search PubMed; Q. Li, L. Lu, C. Zhong, J. Huang, Q. Huang, J. Shi, X. Jin, T. Peng, J. Qin and Z. Li, Chem. – Eur. J., 2009, 15, 9664 CrossRef CAS PubMed.
- X. Zhu, H. Tsuji, A. Yella, A. S. Chauvin, M. Grätzel and E. Nakamura, Chem. Commun., 2013, 49, 582 RSC; J. Zheng, K. Zhang, Y. Fang, Y. Zuo, Y. Duan, Z. Zhuo, X. Chen, W. Yang, Y. Lin, M. S. Wong and F. Pan, ACS Appl. Mater. Interfaces, 2015, 7, 25341 Search PubMed; J.-H. Chen, C.-H. Tsai, S.-A. Wang, Y.-Y. Lin, T.-W. Huang, S.-F. Chiu, C.-C. Wu and K.-T. Wong, J. Org. Chem., 2011, 76, 8977 CrossRef CAS PubMed; D. Bi, A. Mishra, P. Gao, M. Franckevičius, C. Steck, S. M. Zakeeruddin, M. K. Nazeeruddin, P. Bäuerle, M. Grätzel and A. Hagfeldt, ChemSusChem, 2016, 9, 433 CrossRef PubMed.
- J. Nishida, T. Masuko, Y. Cui, K. Hara, H. Shibuya, M. Ihara, T. Hosoyama, R. Goto, S. Mori and Y. Yamashita, J. Phys. Chem. C, 2010, 114, 17920 Search PubMed; H. Choi, S. O. Kang, J. Ko, G. Gao, H. S. Kang, M. S. Kang, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2009, 48, 5938 CrossRef CAS PubMed.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J. H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS PubMed; K. J. Jiang, N. Masaki, J. B. Xia, S. Noda and S. Yanagida, Chem. Commun., 2006, 2460 RSC; C. Y. Chen, S. J. Wu, C. G. Wu, J. G. Chen and K. C. Ho, Angew. Chem., Int. Ed., 2006, 45, 5822 CrossRef PubMed.
- N. Koumura, Z. S. Wang, M. Miyashita, Y. Uemura, H. Sekiguchi, Y. Cui, A. Mori, S. Mori and K. Hara, J. Mater. Chem., 2009, 19, 4829 RSC.
- X. H. Zhang, Y. Cui, R. Katoh, N. Koumura and K. Hara, J. Phys. Chem. C, 2010, 114, 18283 CAS.
- S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J.-E. Moser, R. H. Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291 CrossRef CAS; J. J. Kim, H. Choi, J. W. Lee, M. S. Kang, K. Song, S. O. Kang and J. Ko, J. Mater. Chem., 2008, 18, 5223 RSC; Z.-M. Tang, T. Lei, K.-J. Jiang, Y.-L. Song and J. Pei, Chem. – Asian J., 2010, 5, 1911 CrossRef PubMed; R. Grisorio, L. De Marco, R. Giannuzzi, G. Gigli and G. P. Suranna, Dyes Pigm., 2016, 131, 282 CrossRef; H. Huang, H. Chen, J. Long, G. Wang and S. Tan, J. Power Sources, 2016, 326, 438 CrossRef.
- S. Y. Qu, C. J. Qin, A. Islam, Y. Z. Wu, W. H. Zhu, J. L. Hua, H. Tian and L. Y. Han, Chem. Commun., 2012, 48, 6972 RSC; S. Y. Qu, W. J. Wu, J. L. Hua, C. Kong, Y. T. Long and H. Tian, J. Phys. Chem. C, 2010, 114, 1343 Search PubMed.
- W. Ying, F. Guo, J. Li, Q. Zhang, W. Wu, H. Tian and J. Hua, ACS Appl. Mater. Interfaces, 2012, 4, 4215 CAS.
- J. X. He, F. L. Guo, X. Li, W. J. Wu, J. B. Yang and J. L. Hua, Chem. – Eur. J., 2012, 18, 7903 CrossRef CAS PubMed.
- D. W. Chang, H. J. Lee, J. H. Kim, S. Y. Park, S.-M. Park, L. M. Dai and J.-B. Baek, Org. Lett., 2011, 13, 3880 CrossRef CAS PubMed; K. Pei, Y. Z. Wu, Q. Zhang, B. Chen, H. Tian and W. H. Zhu, Chem. – Eur. J., 2012, 18, 8190 CrossRef PubMed.
- W. Ying, J. Yang, M. Wielopolski, T. Moehl, J.-E. Moser, P. Comte, J. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, Chem. Sci., 2014, 5, 206 RSC.
- D. Joly, L. Pellejà, S. Narbey, F. Oswald, T. Meyer, Y. Kervella, P. Maldivi, J. N. Clifford, E. Palomares and R. Demadrille, Energy Environ. Sci., 2015, 8, 2010 Search PubMed; Z. Shen, X. Zhang, F. Giordano, Y. Hu, J. Hua, S. M. Zakeeruddin, H. Tian and M. Grätzel, Mater. Chem. Front., 2017, 1, 181 RSC.
- N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256 CrossRef CAS PubMed.
- H. Choi, J. K. Lee, K. Song, S. O. Kang and J. Ko, Tetrahedron, 2007, 63, 3115 CrossRef CAS.
- R. Y.-Y. Lin, H.-W. Lin, Y.-S. Yen, C.-H. Chang, H.-H. Chou, P.-W. Chen, C.-Y. Hsu, Y.-C. Chen, J. T. Lin and K.-C. Ho, Energy Environ. Sci., 2013, 6, 2477 Search PubMed.
- M.-S. Tsai, Y.-C. Hsu, J. T. Lin, H.-C. Chen and C.-P. Hsu, J. Phys. Chem. C, 2007, 111, 18785 CAS.
- Z. J. Ning, Y. C. Zhou, Q. Zhang, D. G. Ma, J. J. Zhang and H. Tian, J. Photochem. Photobiol., A, 2007, 192, 8 CrossRef CAS.
- Q. Feng, Q. Zhang, X. Lu, H. Wang, G. Zhou and Z.-S. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8982 CAS.
- W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z.-S. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756 CrossRef CAS.
- K.-F. Chen, Y.-C. Hsu, Q. Wu, M.-C. P. Yeh and S.-S. Sun, Org. Lett., 2009, 11, 377 CrossRef CAS PubMed; Y. J. Chang and T. J. Chow, Tetrahedron, 2009, 65, 4726 CrossRef.
- R. Rattanawan, V. Promarak, T. Sudyoadsuk, S. Namuangruk, N. Kungwan, S. Yuan and S. Jungsuttiwong, J. Photochem. Photobiol., A, 2016, 322, 16 CrossRef.
- K. R. J. Thomas, Y.-C. Hsu, J. T. Lin, K.-M. Lee, K.-C. Ho, C.-H. Lai, Y.-M. Cheng and P.-T. Chou, Chem. Mater., 2008, 20, 1830 CrossRef CAS.
- P. Shen, Y. Liu, X. Huang, B. Zhao, N. Xiang, J. Fei, L. Liu, X. Wang, H. Huang and S. Tan, Dyes Pigm., 2009, 83, 187 CrossRef CAS.
- L. Schmidt-Mende, J. E. Kroeze, J. R. Durrant, M. K. Nazeeruddin and M. Grätzel, Nano Lett., 2005, 5, 1315 CrossRef CAS PubMed.
- J. E. Kroeze, N. Hirata, S. Koops, M. K. Nazeeruddin, L. Schmidt-Mende, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2006, 128, 16376 CrossRef CAS PubMed.
- H. Tian, I. Bora, X. Jiang, E. Gabrielsson, K. M. Karlsson, A. Hagfeldt and L. Sun, J. Mater. Chem., 2011, 21, 12462 RSC.
- A. Islam, M. Akhtaruzzaman, T. H. Chowdhury, C. Qin, L. Han, I. M. Bedja, R. Stalder, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2016, 8, 4616 CAS.
- J. Van de Lagemaat, N.-G. Park and A. J. Frank, J. Phys. Chem. B, 2000, 104, 2044 CrossRef CAS.
- L. Luo, C.-J. Lin, C.-Y. Tsai, H.-P. Wu, L.-L. Li, C.-F. Lo, C.-Y. Lin and E. W.-G. Diau, Phys. Chem. Chem. Phys., 2010, 12, 1064 RSC; M. Pastore and F. D. Angelis, J. Phys. Chem. Lett., 2013, 4, 956 CrossRef CAS PubMed.
- Z. S. Wang, Y. Cui, Y. Danoh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224 CAS.
- M. Grätzel, Platinum Met. Rev., 1994, 38, 151 Search PubMed.
- A. Mihi, F. J. López-Alcaraz and H. Míguez, Appl. Phys. Lett., 2006, 88, 193110 CrossRef.
- K. Sirithip, N. Prachumrak, R. Rattanawan, T. Keawin, T. Sudyoadsuk, S. Namuangruk, S. Jungsuttiwong and V. Promarak, Chem. – Asian J., 2015, 10, 882 CrossRef CAS PubMed.
- J.-S. Ni, Y.-C. Yen and J. T. Lin, J. Mater. Chem. A, 2016, 4, 6553 CAS.
- J. H. Yum, S. J. Moon, R. Humphry-Baker, P. Walter, T. Geiger, F. Nüesch, M. Grätzel and M. K. Nazeeruddin, Nanotechnology, 2008, 19, 424005 CrossRef CAS PubMed.
- A. C. Khazraji, S. Hotchandani, S. Das and P. V. Kamat, J. Phys. Chem. B, 1999, 103, 4693 CrossRef CAS.
- G. Kron, U. Rau and J. H. Werner, J. Phys. Chem. B, 2003, 107, 13258 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Wallingford CT, 2009 Search PubMed.
- N. Koide and L. Han, Rev. Sci. Instrum., 2004, 75, 2828 CrossRef CAS.
- S. Ito, H. Matsui, K. Okada, S. Kusano, T. Kitamura, Y. Wada and S. Yanagida, Sol. Energy Mater. Sol. Cells, 2004, 82, 421 CrossRef CAS.
- G. Schlichthorl, N. G. Park and A. J. Frank, J. Phys. Chem. B, 1999, 103, 782 CrossRef.
- J. Cheng, X. Liang, Y. Cao, K. Guo and W.-Y. Wong, Tetrahedron, 2015, 71, 5634 CrossRef CAS.
- H.-H. Gao, X. Qian, W.-Y. Chang, S.-S. Wang, Y.-Z. Zhu and J.-Y. Zheng, J. Power Sources, 2016, 307, 866 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: More DFT calculations, fluorescence, IMPS and IMVS data, and NMR and MALDI-TOF spectra of the intermediates and dyes. See DOI: 10.1039/c6qm00271d |
| This journal is © the Partner Organisations 2017 |