
Graphene oxide coupled carbon nitride homo-heterojunction photocatalyst for enhanced hydrogen production†
Mohammad Ziaur
Rahman
a,
Jun
Zhang
b,
Youhong
Tang
 c,
Kenneth
Davey
a and
Shi-Zhang
Qiao
*a
c,
Kenneth
Davey
a and
Shi-Zhang
Qiao
*a
aSchool of Chemical Engineering, The University of Adelaide, SA 5005, Australia. E-mail: s.qiao@adelaide.edu.au
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China
cSchool of Computer Science, Engineering and Mathematics, Flinders University, SA 5001, Australia
First published on 11th November 2016
Abstract
This contribution reports the synthesis, characterization and application of a new ternary homo-heterojunction photocatalyst for improved hydrogen production via water-splitting. The heterostructure is constructed by soft-grafting of graphitic carbon nitride (GCN) and graphene oxide (GO) into an amorphous carbon nitride (ACN) substrate. In this ternary hybrid, a cascaded redox-junction is formed that significantly facilitates the separation of photogenerated electron–hole pairs (EHPs), retards EHP recombination and shuttles electrons to the photocatalyst/liquid interface for proton reduction reactions. When deposited with 3 wt% Pt as a cocatalyst, this new photocatalyst exhibits hydrogen production of 251 μmol h−1 from 10 vol% aqueous triethanolamine solution under visible light (420 nm) irradiation with an apparent quantum efficiency of 6.3%. This ternary photocatalyst therefore outperforms stand-alone/binary photocatalysts and promises to be a viable alternative to metal-based photocatalysts.
Introduction
Photocatalytic hydrogen production is a potential alternative to dwindling carboniferous fossil fuels.1,2 In photocatalytic hydrogen production, because water and sunlight are, essentially, zero-cost materials, production cost therefore depends on judicial selection of materials for the photocatalyst. For hydrogen production to be cost-competitive with present fossil-fuel based energy generation, an ideal catalyst needs to be stable, nontoxic, earth-abundant and inexpensive.3,4 Significantly, these criteria could be met simultaneously if the catalyst was metal-free.5–7In this regard, single elementals (e.g. cyclo-octasulfur and red-phosphorous)8,9 or binary compounds (e.g. graphitic carbon nitride,5 amorphous carbon nitride,10 boron carbide11 and graphene oxide12) and ternary compounds (e.g. carbon doped boron nitride13) have been developed as metal-free photocatalysts and demonstrated for hydrogen generation in recent years. Among these, graphitic carbon nitride (GCN), amorphous carbon nitride (ACN) and graphene oxide (GO) are the most notable metal-free photocatalysts due to their unique physicochemical properties and high durability during catalytic reactions. However, the quantum yield of hydrogen production on pristine GCN, ACN and GO is significantly low. Attempts have been made to increase the efficiency of these three metal-free photocatalysts. For example, heteroatom doping, morphological fine tuning and addition of other semiconductor(s) as heterostructures have been tried to augment the efficiency of GCN.14–16 The improvement in the efficiency of ACN was investigated by selective breaking of hydrogen bonds,17 and, for GO, the enhancement of efficiency was achieved through architecting nitrogen functionalities and morphological fine tuning.18,19 Despite these efforts, however, their photocatalytic performance remains moderate. This suggests that stand-alone GCN, ACN or GO is not sufficient for high performance.
Interestingly, single elemental or compound photocatalysts in joint-venture forms as heterostructures were shown to be more effective because of the excellent charge separation in the heterostructures.20 As a result, a series of heterostructure photocatalysts were developed by coupling both metal and metal-free semiconductors.20,21 MoS2/GCN, WoC3/GCN, CdS/GCN, NiS/GCN, ZnO/GCN, and SnO/GCN are examples of binary heterostructure photocatalysts.22–30 Apart from these metal-based heterostructures, recently, metal-free GCN/graphene, GCN/GCN, CN-CNS (CNS stands for sulfur mediated CN) and CN-CNB (CNB stands for boron doped CN) binary hybrids were reported for photocatalytic hydrogen evolution as well.31–34 As can be gleaned, GCN has been used predominantly (a more complete list of the reported carbon nitride and graphene based photocatalysts is available in the literature35–37). Notably, a metal-based binary heterostructure exhibited a superior performance over a metal-free binary heterostructure, even with the inclusion of precious metal Pt as a co-catalyst in the metal-free systems. However, because of the instability and toxicity of most of the metal-based systems, a metal-free system with a comparable performance to that of metal-based systems is urgently needed. Whilst metal-free binary systems clearly have shown a limitation, the research question arose as to: ‘What about a metal-free ternary structure as a hydrogen evolution photocatalyst?’ To address this research gap, we have developed a new ternary homo-heterojunction photocatalyst by coupling GCN, ACN and GO (we denote it as GCN/ACN/GO), and demonstrated its photocatalytic hydrogen evolution performance.
For an improved hydrogen evolution, a photocatalyst should have three fundamental attributes: extended visible light absorption, efficient transfer and migration of photogenerated charge carriers to reaction sites, and utilization of the charge carriers for redox reactions. We have taken these criteria into account when designing the proposed ternary hybrid photocatalyst. Our hypothesis was based on the fact that the visible light photon absorption ability of the participating GCN, ACN and GO will provide a larger absorption overlap with the solar spectrum when they join as a tandem structure. Additionally, in a GCN/ACN/GO ternary homo-heterojunction, the homojunction is formed between GCN and ACN whilst coupling of GO adds a heterojunction with the GCN/ACN homojunction. The junction forms at the interface due to band alignment of GCN and ACN where one functions as an electron-depletion region whilst the other as an electron-accumulation region. Therefore, an internal electric field should be created at the interface with a potential difference between the two sides that facilitates charge separation. Incorporation of GO to form an additional heterojunction will promote interfacial charge transfer as well as reducing recombination by preventing the back transfer of electrons across the junction,38,39 thereby, boosting the charge carrier collection efficiency for redox reactions. In addition, the large surface area of GCN/ACN/GO facilitates the utilization of charge carriers for the desired redox reaction. As a result, this ternary hybrid is expected to be a highly efficient hydrogen evolution photocatalyst under simulated solar irradiation.
To the best of our knowledge, this is the first report of a ternary hybrid where a dual homo-heterojunction concept was applied and demonstrated as a hydrogen evolution photocatalyst (see Table S1 (ESI†) for a comprehensive survey of recently reported ternary hybrids).
Experimental
Preparation of GCN/ACN/GO
GO was synthesized from graphite by following a modified Hummers' method (see the ESI†).40,41 ACN was synthesized through calcination of 12 g thiourea in a tube furnace under N2 flow. The initial temperature was 450 °C (with a 10 °C ramp) for 1.5 h, then 550 °C (5 °C ramp) for 2.0 h, and finally 620 °C (2.0 °C ramp) for 1 h under a N2 flow. The furnace was allowed to cool to room temperature (RT) and ACN was collected. When thiourea undergoes polycondensation, there is a possibility that sulfur residues remain in the final product. However, after elevated temperature heating (620 °C), no trace of sulfur was found in the corresponding X-ray Photoelectron spectroscopy (XPS) of ACN (see Fig. S1, ESI†). For GCN/ACN/GO synthesis, 3 g cyanamide and the as-prepared 3 g ACN were dispersed in GO solution (0.5 mg mL−1). The ratio of GO to ACN was 1 wt%. This mixture was stirred in a water-bath at 90 °C for 24 h. After which the product was dried in an oven at 70 °C. The solid samples were then placed in a ceramic-boat and heated at 550 °C (2.3 °C ramp) for 4 h under a N2 flow in a tube-furnace. After the furnace had cooled to RT, the desired GCN/ACN/GO composite was collected and characterized. In a similar preparation method, ACN/GO and GCN/ACN samples were synthesized without adding cyanamide and GO, respectively. Bulk GCN was synthesized from cyanamide following the procedures described elsewhere.5,16Physicochemical characterization
The crystal structure was characterized by X-ray diffraction (XRD) using a powder X-ray diffractometer (Miniflex, Rigaku) at 40 kV and 15 mA with Cu Kα radiation (λ = 0.154178 nm). Transmission electron microscope (TEM) images were obtained with Tecnai G2 spirit. Scanning electron microscope (SEM) images were obtained using a Quanta 450 SEM. Raman spectra were collected using an iHR550 Raman microscope (HORIBA scientific) with 600 g mm−1 gratings and 532 nm solid lasers for excitation. The Fourier transform infrared (FTIR) spectrum was recorded on a FTIR spectrometer (Nicolet 6700). Optical properties were analyzed from the UV-Vis diffuse reflectance spectra (DRS) using a UV-Vis spectrophotometer (UV2600, Shimadzu, Japan) in the wavelength of 200 to 800 nm at RT. A Micromeritics Tristar II 3020 nitrogen adsorption apparatus (USA) was used to analyse the BET specific surface area. All samples were degassed at 150 °C before N2 adsorption measurements. Photoluminescence (PL) spectra were measured at RT using a fluorescence spectrometer (RF-5301PC, Shimadzu, Japan). XPS analysis was carried out on an AXIS ultra-spectrometer (Kratos Analytical Ltd, GB) with monochromatic Al Kα radiation at a pressure of ca. 5 × 10−9 Pa. Thermogravimetric analysis (TGA) was performed in TGA/DSC 2 (Mettler-Toledo) over 25–1000 °C at 10 °C min−1 under a N2 flow of 50 mL min−1.Photocatalytic H2 production test
In a 100 mL Pyrex flask, 50 mg of the photocatalyst was dispersed in 80 mL of 10 vol% triethanolamine (TEOA) and Pt was loaded onto the surface of the catalyst by an in situ photodeposition method using H2PtCl6 as a precursor. To maintain anaerobic conditions, the openings of the flask were sealed with silicone rubber-septa and the suspension was thoroughly degassed with Ar for 30 min. A xenon (Xe) arc lamp (with an intensity 300 W cm−2) with a cut-off filter (λ > 420 nm) was used to achieve visible-light irradiation. The rate of H2 evolution was determined by sampling 0.4 mL of gas intermittently each hour through the septum of an online gas chromatograph (GC) (Clarus 480, PerkinElmer, USA). The GC was equipped with a thermal conductive detector (TCD) with a 5 Å molecular sieve column and Ar as the carrier gas. The reactor was evacuated each 5 h of a 15 h recycling period and kept under continuous stirring to prevent sedimentation of the photocatalyst during H2 production. Deionized water was used to rinse glassware prior to experiments. Experiments were carried out at ambient temperature and pressure. The apparent quantum efficiency (AQE) was measured under the same photocatalytic reaction conditions. Four (4) low-power 420 nm LEDs (3 W), which were positioned 1 cm from the reactor in four (4) different directions, were used as light sources to trigger the photocatalytic reaction. The focused intensity for each 420 nm LED was ca. 6.0 mW cm−2. The AQE was calculated using eqn (1).42 | (1) |
Photo-electrochemical measurements
The working electrode was prepared by grinding 0.1 g of the photocatalyst with 0.03 g of polyethylene glycol (PEG) in 0.5 mL of ethanol to make a slurry. Using a doctor-blade method, the slurry was then coated on the fluorine-doped tin oxide (FTO) glass electrode (3 cm2) and dried in an oven at 350 °C for 30 min under a N2 gas flow. Electrodes coated with slurry had a measured film thickness of around 10 to 11 μm. The active area of the electrode was about 1.35 cm2. The photocurrent, electro-impedance spectroscopy (EIS) measurements and linear sweep voltammetry (LSV) experiments were performed in a three-electrode electrochemical system (CHI 650D instruments) where the sample coated FTO was used as a working-electrode whilst Pt and Ag/AgCl were used as a counter- and a reference-electrode, respectively. During photocurrent measurements, light was produced by a 300 W Xe arc lamp and 0.2 M Na2S + 0.05 M Na2SO3 aqueous solution was used as the electrolyte. EIS was recorded over a 0.005–105 Hz frequency with an ac amplitude of 10 mV at 0.5 V bias. LSV curves were obtained in the sweep range −0.3 to −2.0 V with a scan rate of 5 mV s−1. In both cases, 0.5 M Na2SO4 was used as the electrolyte. A Mott–Schottky plot was also recorded in the same electrochemical system using 0.5 M Na2SO4 as the electrolyte.Results and discussion
Insight into dual homo-heterojunction formation mechanisms
ACN was synthesized from thiourea through a series of temperature step-and-hold processes. ACN was then used as the substrate on which GCN and GO were added through simultaneous polymerization of cyanamide and graphene oxide (GO). Polycondensation of thiourea monomers left some defect sites that contained amino groups on the surfaces of ACN.10,17,43 Due to this structural imperfection, when cyanamide was calcined together with the parent ACN, the carbon atoms in cyanamide attacked the amino groups in ACN electrophilically to produce cyanamide grafted ACN hybrids.16 This hybrid turned into a GCN/ACN homojunction when heated at 550 °C due to polymerization of cyanamide into GCN on the surface of the parent ACN. When cyanamide was mixed with GO solution, polycondensation of cyanamide released nitrogen-containing species at 550 °C that led to the partial reduction of GO.18,44,45 This then protected the GO against oxidation in air during the reaction at 550 °C, and helped in making firm contact between the surfaces of GCN/ACN and GO.46 As a result, a tightly bound GCN/ACN/GO layered structure was obtained.Structure, morphology and composition
A combined study of XRD, TEM and SEM was carried out to reveal the structure and morphology of the as-prepared GCN/ACN/GO.The XRD patterns of GCN, ACN, GO and GCN/ACN/GO are shown in Fig. 1a. It is seen that GCN, ACN and GCN/ACN/GO showed two peaks, one at around 27.0° and the other at around 13°. However, in the case of ACN, the peak at ∼13° is relatively flat – a finding that is consistent with previous results.10 The GO shows one prominent peak at ∼11° and a minor peak at ∼25°, these are also comparable with previous studies.47 The GCN/ACN/GO retains identical peaks to that of ACN, such as a flatter peak at ∼13° (which describes the in-plane structural packing motif48) and a broader inter-planar stacking peak at ∼27.0°. This indicates that neither GCN nor GO is connected to the lattice of the ACN; rather they are overlapped and connected to surface terminal groups.46,49 Therefore, there were no characteristic diffraction peaks observed for GO on the XRD patterns of the composite samples. Additional reasons for the absence of GO peaks in GCN/ACN/GO's XRD patterns may be due to (i) high dispersion and the low amounts of GO (1 wt%) used in the composite and (ii) the destruction of regular stacking of GO through added nitrogen functionalities which is released from ammonia during polycondensation of cyanamide into GCN.18,45 At an elevated temperature, the weak hydrogen bonds in ACN become vulnerable and break. This hydrogen then initiates the reduction of ammonia to release nitrogen. The breaking of hydrogen bonds is indicated by gradual weakening of the 13° peak in ACN. Due to the breaking of hydrogen bonds, a structural fluctuation is expected that disturbs the periodic stacking and results in widening of the 27° peak.17 Following the peak broadening, the peak at 27° in GCN/ACN/GO is also left-shifted compared with that of GCN and ACN which indicates a decrease of d-spacing and compactness of the overlapped layers. This reflects the strong coupling between GCN, ACN and GO that enables the new ternary hybrid to have a synergistic effect.31,36,37
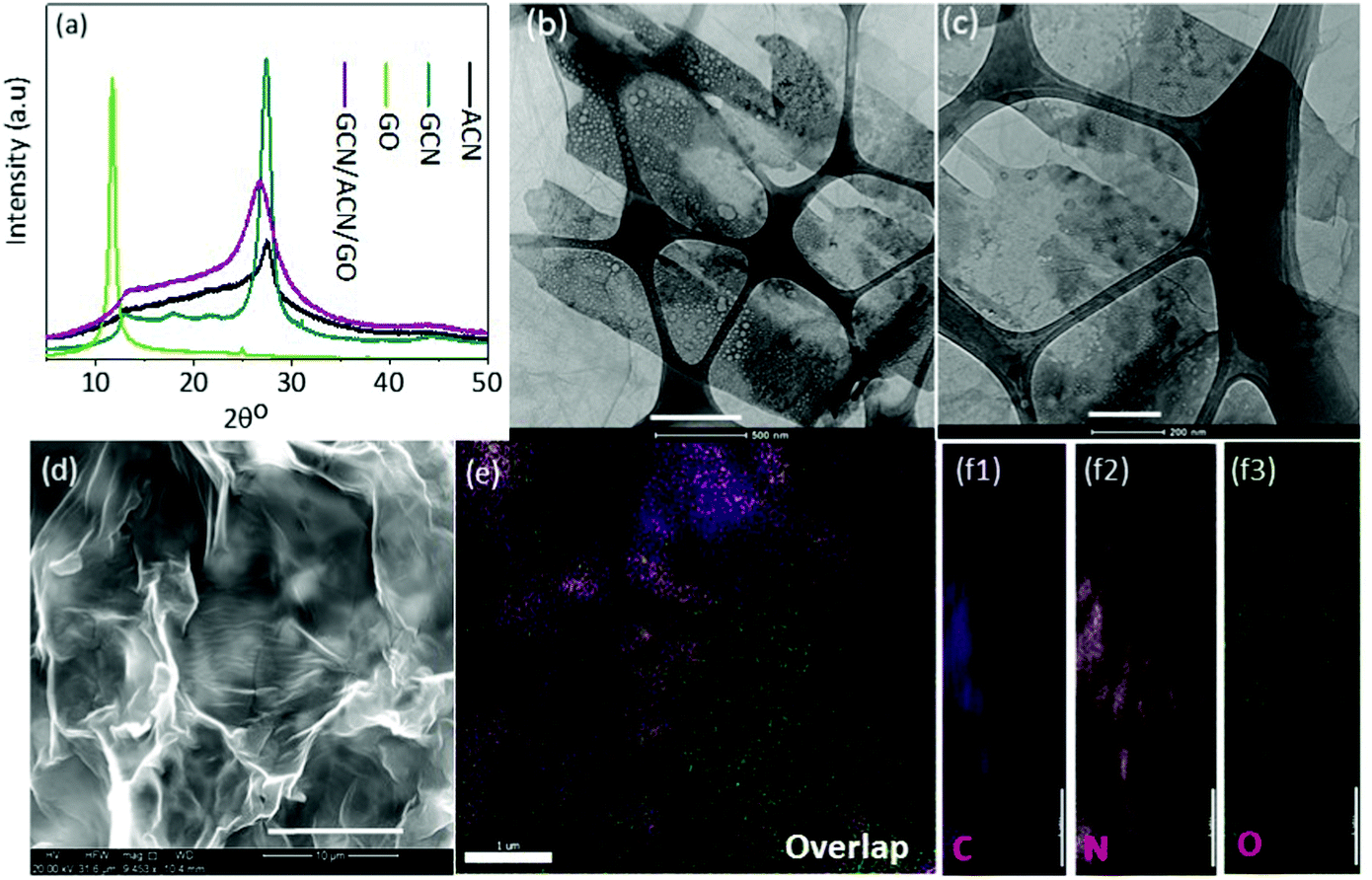 | ||
| Fig. 1 Structure, morphology and composition. (a) XRD patterns, (b and c) TEM images. Scale bar 500 nm and 200 nm, respectively, (d) SEM image, scale bar 10 μm and (e and f) EDS elemental mapping of GCN/ACN/GO. | ||
The overlapped layers of GCN, ACN and GO in GCN/ACN/GO are also supported by the electron-microscope images. In the represented TEM images (Fig. 1b, c and Fig. S2, ESI†), a layered interfacial contact between GO and GCN/ACN can be observed. The GO can be seen as transparent sheets spread over the entire surface. In the backdrop of the GO sheets, a conglomerate of both porous-hollow sheets and plain-sheets can be seen as a black-ash-white painted canvas. These nanosheets are mounted on each other, forming a layered structure. However, although GO can be clearly identified, a discernible fine-distinguish between GCN and ACN is less clear. It is because both ACN and GCN are of the same phase materials (nevertheless, the presence of ACN and GCN in GCN/CAN/GO is confirmed from the combined analysis of XPS, FTIR and TGA analysis. See Fig. S3 and the associated text in the ESI†). The curved-nanosheets shown in Fig. 1d appear to be the result of interconnected and overlapping layers of GCN, ACN and GO, and therefore, no bulk phase separation with GO, GCN and ACN is seen in these SEM images. This intimate contact of GCN, ACN and GO is ideal for vectorial charge transfer synergistically and leads to enhanced photocatalytic performance.50 The distribution of constituting elements for GO, GCN, ACN and GCN/ACN/GO have been explored through energy dispersive spectroscopy (EDS). The resulting elemental mapping as depicted in Fig. 1e, f and Fig. S4 (ESI†) shows the distribution of C, N and O in both the composite structure GCN/ACN/GO as well as in the individual participating semiconductors (GO, GCN and ACN). The XPS survey revealed that the chemical composition of GCN/ACN/GO was composed entirely of C, N and adventitious O (Fig. S5a, ESI†). This was also confirmed from the EDS analysis where, along with C and N, very little oxygen was noticed (Fig. S5b, ESI†).
The chemical states were studied through FTIR, XPS and Raman spectra. In the FTIR spectra of GO, there are C–O stretching vibrations of epoxy groups at 1096 cm−1 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of COOH groups at 1725 cm−1 (Fig. S6, ESI†).12,51,52 Notably, however, these C–O and CO vibration modes, except typical C–N heterocycles (C–N, CN) at 1000 to 1500 cm−1, are absent in the GCN/ACN/GO hybrid.45 This is also supported by the XPS spectra. In deconvoluted XPS C1s spectra (Fig. 2a), there are CC–C and NC–N bonds, but non-sp2 carbon bonds such as C–O, CO and O–CO of GO are absent.53 The absence of epoxy, carboxyl and carboxylic groups indicates the functionalization of the GO periphery with pyridinic and pyrrolic nitrogen during copolymerization of N-rich cyanamide and GO on the ACN substrate (see deconvoluted XPS N1s spectra as shown in Fig. 2b for different nitrogen species in GCN/ACN/GO).18 This is also consistent with the XRD results. The presence of tertiary nitrogen and amino groups in the deconvoluted XPS N1s spectra of GCN/ACN/GO suggests that the cyanamide is decomposed into GCN, but incompletely condensed.43 Following the prolonged heat treatment at 550 °C, the pre-condensed cyanamide is diffused further into the ACN network by elimination of significant amounts of ammonia. The cyanamide decomposition might incorporate nitrogen atoms into the edges and defect sites of GO.45,54 These N-atoms can be further translated into the graphitic-N which possibly serves as a link between GCN/ACN and GO.41 This process leads to the reduction of electrical resistivity of ternary hybrids.41 As a result, the GCN/ACN/GO hybrid is expected to be an excellent photocatalyst. A more detailed study of the deconvoluted XPS C1s and N1s spectra are presented in Fig. S7 and Tables S2, S3 in the ESI.† Moreover, in the FTIR spectra as shown in Fig. S6 (ESI†), the pronounced, but shifted peak at 809 cm−1 (which originates from the triazine unit) confirms the presence of GCN in the GCN/ACN/GO skeleton.55 This result suggests that hybridization occurs between GCN/ACN and GO. This is also supported by the Raman analysis and is discussed later.
O stretching vibrations of COOH groups at 1725 cm−1 (Fig. S6, ESI†).12,51,52 Notably, however, these C–O and CO vibration modes, except typical C–N heterocycles (C–N, CN) at 1000 to 1500 cm−1, are absent in the GCN/ACN/GO hybrid.45 This is also supported by the XPS spectra. In deconvoluted XPS C1s spectra (Fig. 2a), there are CC–C and NC–N bonds, but non-sp2 carbon bonds such as C–O, CO and O–CO of GO are absent.53 The absence of epoxy, carboxyl and carboxylic groups indicates the functionalization of the GO periphery with pyridinic and pyrrolic nitrogen during copolymerization of N-rich cyanamide and GO on the ACN substrate (see deconvoluted XPS N1s spectra as shown in Fig. 2b for different nitrogen species in GCN/ACN/GO).18 This is also consistent with the XRD results. The presence of tertiary nitrogen and amino groups in the deconvoluted XPS N1s spectra of GCN/ACN/GO suggests that the cyanamide is decomposed into GCN, but incompletely condensed.43 Following the prolonged heat treatment at 550 °C, the pre-condensed cyanamide is diffused further into the ACN network by elimination of significant amounts of ammonia. The cyanamide decomposition might incorporate nitrogen atoms into the edges and defect sites of GO.45,54 These N-atoms can be further translated into the graphitic-N which possibly serves as a link between GCN/ACN and GO.41 This process leads to the reduction of electrical resistivity of ternary hybrids.41 As a result, the GCN/ACN/GO hybrid is expected to be an excellent photocatalyst. A more detailed study of the deconvoluted XPS C1s and N1s spectra are presented in Fig. S7 and Tables S2, S3 in the ESI.† Moreover, in the FTIR spectra as shown in Fig. S6 (ESI†), the pronounced, but shifted peak at 809 cm−1 (which originates from the triazine unit) confirms the presence of GCN in the GCN/ACN/GO skeleton.55 This result suggests that hybridization occurs between GCN/ACN and GO. This is also supported by the Raman analysis and is discussed later.
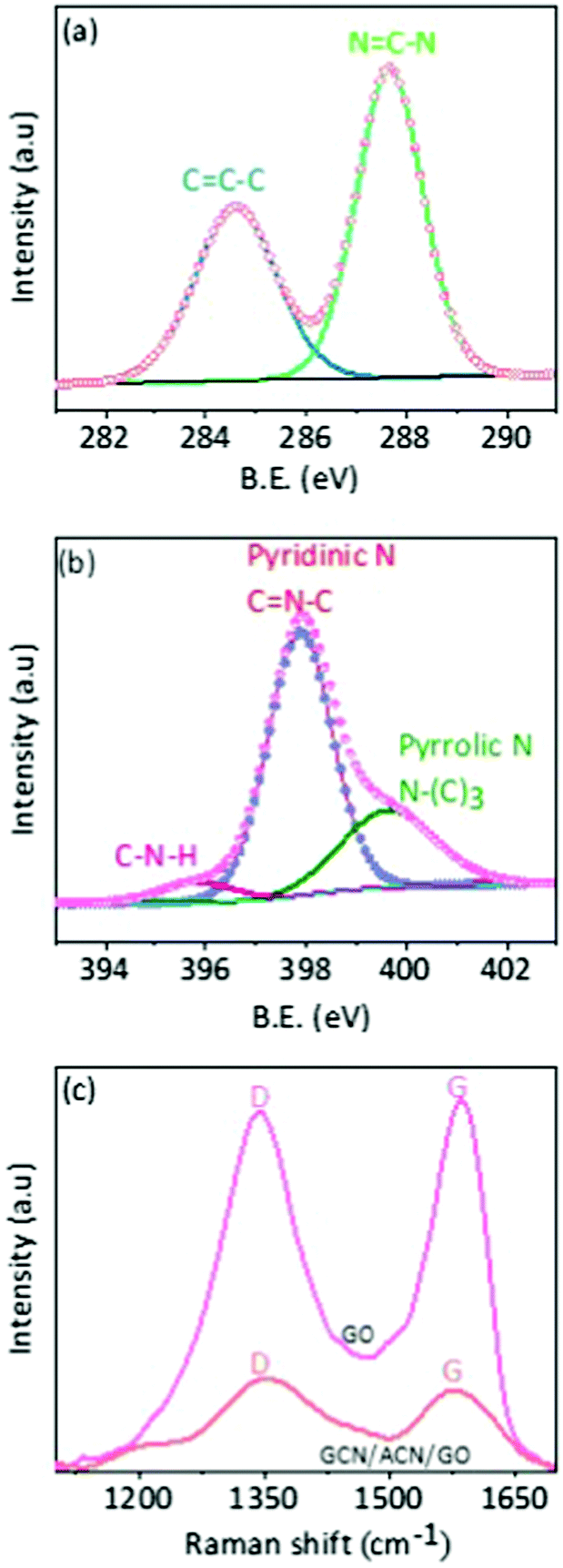 | ||
| Fig. 2 Chemical properties. (a) Deconvoluted XPS C1s spectra of GCN/ACN/GO. (b) Deconvoluted XPS N1s spectra of GCN/ACN/GO, and (c) Raman spectra of GO and GCN/ACN/GO. | ||
The elimination of self-agglomeration of constituent semiconductors is crucial for sustaining the heterostructure.56 The UV-Vis diffuse reflectance spectra of the GCN/ACN/GO exhibits almost an identical absorption edge as that of pure ACN (Fig. S8, ESI†). This is the consequence of the fact that neither GCN nor GO has restacked itself or incorporated itself into the lattice of the parent CN, but is holding onto the surfaces in an overlapped-layered fashion.12,19,46 Additionally, UV-Vis shows a typical graphene-like featureless absorption in the 650–800 nm range, which highlights the true likelihood of the postulated GO in the GCN/CN/GO composite network.37
Pore sizes were distributed in a broad range from 10 nm to more than 100 nm (Fig. S9a, ESI†). This reflects the non-restacking of GCN and GO.41,46 Moreover, the BET surface area of the ternary composite is much larger than that of pure ACN or GCN/ACN (Fig. S9b, ESI†), indicating the inclusion of GO.41 The relatively higher surface area of the ternary hybrid provides larger reaction sites, and consequently is beneficial for enhanced hydrogen production. An alternative technique that can provide proof of inclusion of GO is Raman spectroscopy. For graphitic-like structures (i.e. GO), two dominant vibrational modes D (associated with the order/disorder of the systems) and G (indicator of the stacking structure) can be observed in Raman spectra.41 In a pure phase, GO is essentially a monolayer of graphene terminated with oxide epoxy groups and exhibits a higher D/G ratio.57 When GO undergoes a partial reduction due to coupling with GCN/ACN to form a composite structure, a segregated pure phase of GO cannot be found. Therefore, a decrease in the D/G ratio would be expected, implying a structural disorder of GO on coupling with the GCN/ACN network.58
Previous studies revealed that the structural disorder of GO was indicative of a decrease in the size of sp2 domains on reduction.59 Our experimental work shows that typical D and G band peaks are present in GCN/ACN/GO at 1350 and 1600 cm−1, respectively (Fig. 2c), but with a decreased D/G ratio.60 This result confirms the presence of non-pure phase GO,50 and a coupling between GCN/ACN and GO via defect sites in parent ACN.41
Generation, separation and collection of charge carriers
Enhanced hydrogen production can be achieved if a photocatalyst has (i) an extended absorption range in the visible light spectrum, (ii) high charge separation efficiency by means of reducing electron–hole recombination, (iii) high charge transfer efficiency through low charge transfer resistance, and (iv) large active surface area and high mass-transfer through the pores. The experimental evaluation of these is discussed in the following.For a given photocatalyst, extended visible light absorption and the separation of photogenerated charge carriers are the most important parameters. UV-Vis measurements showed that the spreading of photon absorption for GCN/ACN/GO is more pronounced in 400 to 800 nm (Fig. S8, ESI†).
As the UV-Vis spectra of GCN/ACN follows the identical trend of the parent ACN, it can be inferred that the intense optical absorption of GCN/ACN/GO in visible light is caused when GO is overlaid on the surface of GCN/ACN networks, indicating the photosensitizing capability of GO. In a recent study conducted by Xu et al.,61 it was shown that when GO coupled with GCN in a composite the bandgap of the composite decreases over that of pristine GCN. This is due to the dominant effect of O atoms in the electronic structure of the composite. The decrease in bandgap leads to a red-shift in optical absorption. Alternatively, we can say that the optical absorption of GCN is enhanced by coupling GO in a composite form. The UV-Vis spectra of GCN/ACN/GO also showed a red-shift over that of both GCN and ACN, implying that the red-shift is due to the incorporation of GO in the ternary network. Moreover, as shown in Fig. S10 (ESI†), GO has an acceptable bandgap to capture visible light. Therefore, the light sensitizing ability of GO in our study is consistent with the study of Xu et al. In addition, due to interfacial interaction, a change in electronic and optical properties is expected.62 This extended photon absorption could result in abundant photogenerated charge carriers which is highly desirable for enhanced photocatalytic activity.
The increase in light absorption alone is partially accounted for the substantial enhancement in photocatalytic activities. The efficiency of hydrogen production depends primarily on the dissociation of excitons (excited electrons and holes), spatial separation of photoinduced electron–hole and suppression of backward recombination. In GCN/ACN/GO, cascaded redox-junctions form due to band alignment of GCN, ACN and GO. In this cascaded junction, the different electronic levels of the donor and acceptor drive the spatial separation of charges. Moreover, partially reduced GO can serve as an electron sink and retard the back electron transfer and lower electron–hole recombination.18,38 This conjecture is strongly supported by photoluminescence (PL) studies. The intensity of PL is highly reduced in the case of GCN/ACN/GO (Fig. 3a). This is attributed to the reduced radiative recombination of electron–hole pairs.15 A thorough mechanistic understanding of charge separation and reduced recombination is related to the fluctuations in charge distribution and creation of a built-in potential at the interface. The lowest unoccupied molecular orbitals (LUMO) of both carbon nitrides and graphene oxide are composed of p states of C atoms, whereas the highest occupied molecular orbital (HOMO) is composed of p states of N atoms and O atoms, respectively.5,61 Electrons from C atoms of carbon nitrides were transferred to the highly electronegative O atoms of GO when GO interacted with the carbon nitrides. This created hole-rich and electron-rich regions simultaneously in the opposite interface. This interfacial charge redistribution modulated the electrostatic potential in the whole system that led to the formation of a potential well (generally known as built-in potential).61,63,64 This potential well is beneficial for migration of the photo-excited carrier. Together with the built-in potential, the difference in momentum in the k-space due to the indirect bandgap nature of the composite was favorable to hinder the electron–hole pair recombination.
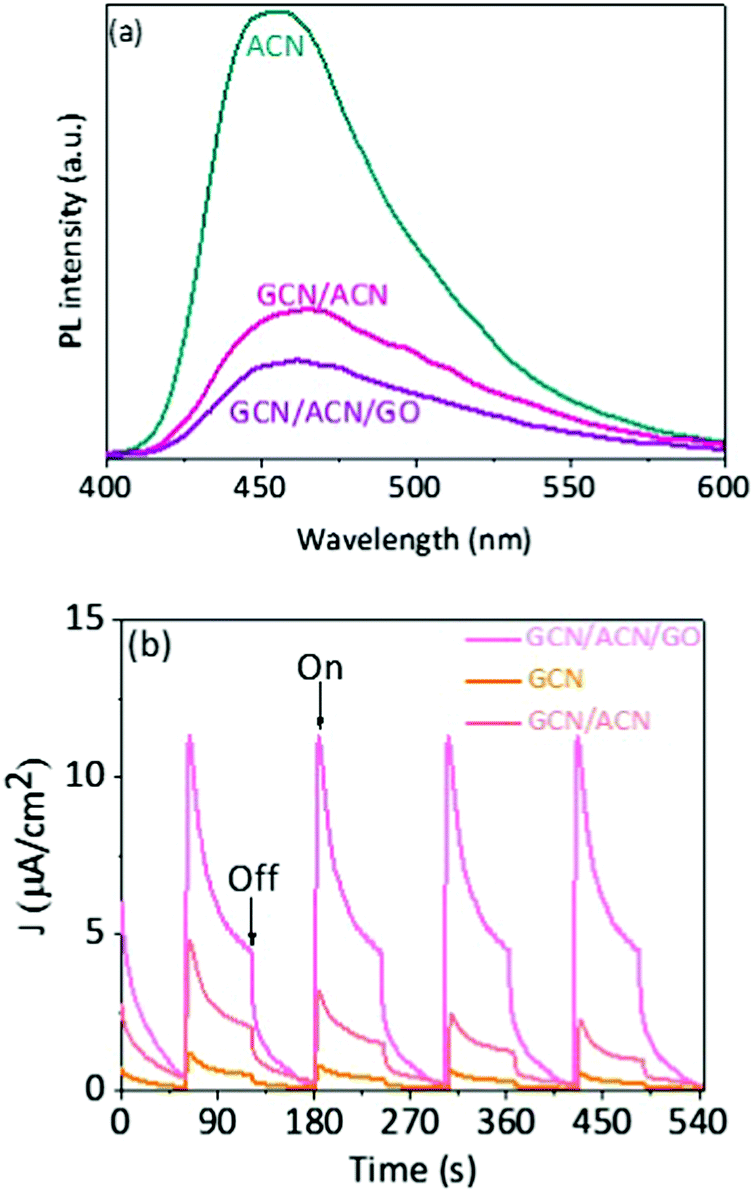 | ||
| Fig. 3 (a) Photoluminescence spectra. (b) Transient photocurrent response obtained in a 0.2 M Na2S + 0.05 M Na2SO3 mixed aqueous solution showing typical on–off cycles of intermittent visible-light irradiation at a 0.5 V bias. | ||
The rapid electron transfer across the GCN/ACN homojunction and carrier sweeping by GO can lead to an increase in photocurrent.65 Indeed, the photocurrent generated by GCN/ACN/GO is noticeably greater than that with ACN and GCN/ACN (Fig. 3b). This attests efficient separation of the photogenerated charge carriers.42 Faster quenching of strong photoluminescence and high photocurrent have been reported, respectively, to be strongly correlated with suppressed recombination and efficient charge separation.15,42
To initiate proton reduction and oxidation of sacrificial agents, efficient collection and faster transport of spatially separated charge carriers are crucial rate-determining steps. Nyquist plot, a widely accepted technique, was adopted to determine charge transfer efficiency. A good photocatalyst usually shows short-radius semicircles in the Nyquist plot. Generally, this short-radius semicircle is related to scaled-up charge transfer efficiency.42 With reference to Fig. 4a, the radius of the Nyquist semicircle for GCN/CAN/GO is about half that of GCN/ACN, and almost negligible in comparison with ACN, implying that the charge transfer efficiency is meaningfully increased (see also Fig. S11, ESI†).45 A reduction in the charge transfer resistance of the GCN/ACN nano-junction in comparison with that of singlet ACN can be observed in Fig. 4a. This finding indicates the superiority of the heterostructure over a single semiconductor. The charge transfer efficiency of GCN/ACN/GO significantly improved over GCN/ACN after incorporation of GO into the GCN/ACN framework. This is because partially reduced GO possesses an excellent electronic conductivity and a high mobility of charge carriers that promote transportation of injected carriers along the graphene surface.66,67
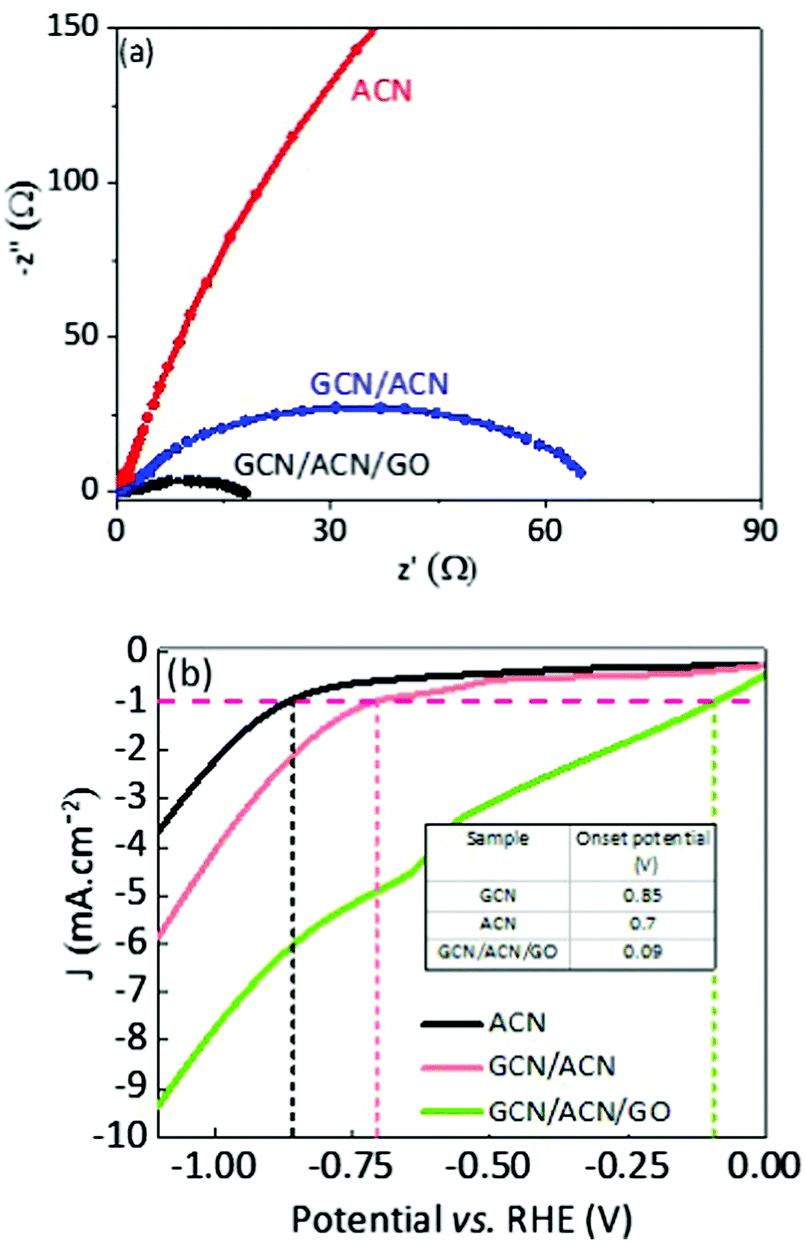 | ||
| Fig. 4 (a) Nyquist plot obtained in a 0.5 M Na2SO4 aqueous solution, and (b) linear sweep voltammetry curves for hydrogen evolution reactions (HERs) which were taken at a scan rate of 5 mV s−1 in a 0.5 M Na2SO4 aqueous solution. | ||
From a thermodynamic standpoint, a reduction in overpotential for the water-splitting reaction is an additive crucial step. Pristine GCN suffers from a high overpotential because of its non-conductive nature. The highly porous surface of GCN/ACN/GO ensures a low reflectance, higher light trapping, and consequently, a high absorption of visible light photons. The hierarchical porous structure induces greater mass transport during hydrogen evolution reaction (HER) and the reduced charge transfer resistance can significantly enhance the level of current for hydrogen production.68 In fact, the voltammetry test shows that the HER onset potential of GCN/ACN/GO is the lowest among ACN and GCN/ACN (Fig. 4b), confirming its superior HER performance.
Because of extended photon absorption, and spatial separation, collection and transportation of charge carriers, the graphene oxide coupled carbon nitride homojunction is supposed to be a highly efficient hydrogen evolution photocatalyst under visible light irradiation.
Photocatalytic activities and stability
The photocatalytic activities of the GCN/ACN/GO were evaluated by assessing the hydrogen production rate under visible light irradiation (λ = 420 nm). Fig. S12 (ESI†) shows the average hydrogen generation rate in a reaction system consisting of 10 vol% triethanolamine as a sacrificial agent without Pt as a co-catalyst. Without this noble-metal co-catalyst, the hydrogen production rate (33.5 μmol h−1) of GCN/ACN/GO outperformed a number of elemental, compound and binary metal-free photocatalysts.35,37,69,70 This is a highly significant result.Pt is a widely used co-catalyst to augment photocatalytic performance. When the electronic, optical and thermodynamics attributes of a photocatalyst are in favour of an enhanced hydrogen evolution rate, inclusion of Pt can instigate evolution by lowering the energy required for redox reactions. For pristine GCN, ACN or GO, the fundamental electro-optical properties are not optimized. Therefore, even with incorporation of Pt, the photocatalytic performance remains low.5,10,12 In contrast, GCN/ACN/GO is endowed with strong visible light absorption, greatly enhanced charge separation and transport dynamics. Therefore, it is expected that addition of Pt will have a multiplying impact on hydrogen generation. To test this, we in situ photodeposited 3 wt% Pt. When Pt was used as a co-catalyst the average hydrogen evolution rate of GCN/ACN/GO increased from 33.5 to 251 μmol h−1 (Fig. 5a). The GCN/ACN/GO consistently showed increased hydrogen production over ACN, ACN/GO and GCN/ACN binary structures, irrespective of the presence of Pt as a co-catalyst. This production rate is also greater than that reported for other metal-free binary combinations such as GCN-GCN, GCN/graphene, etc.5,31,71 The activity of the GCN/ACN/GO was quantified by measuring the apparent quantum efficiency (AQE). The calculated AQE for GCN/ACN/GO at λ = 420 nm is 6.3%. To confirm that the hydrogen evolution reaction proceeds through light absorption, we initiated the measurement of AQE for a given wavelength of the incident light. The wavelength dependent AQE (Fig. 5b) is in good agreement with the optical absorption spectra, confirming that the evolution of hydrogen is solely due to the photocatalysis process.5,10 Indeed, no hydrogen was detected when the experiment was carried out in the dark. However, a decrease in AQE with an increase in the wavelength, which is usual for a photocatalyst, may be due to the lower probability of valence electrons to be excited to the conduction band under longer wavelength irradiation.18
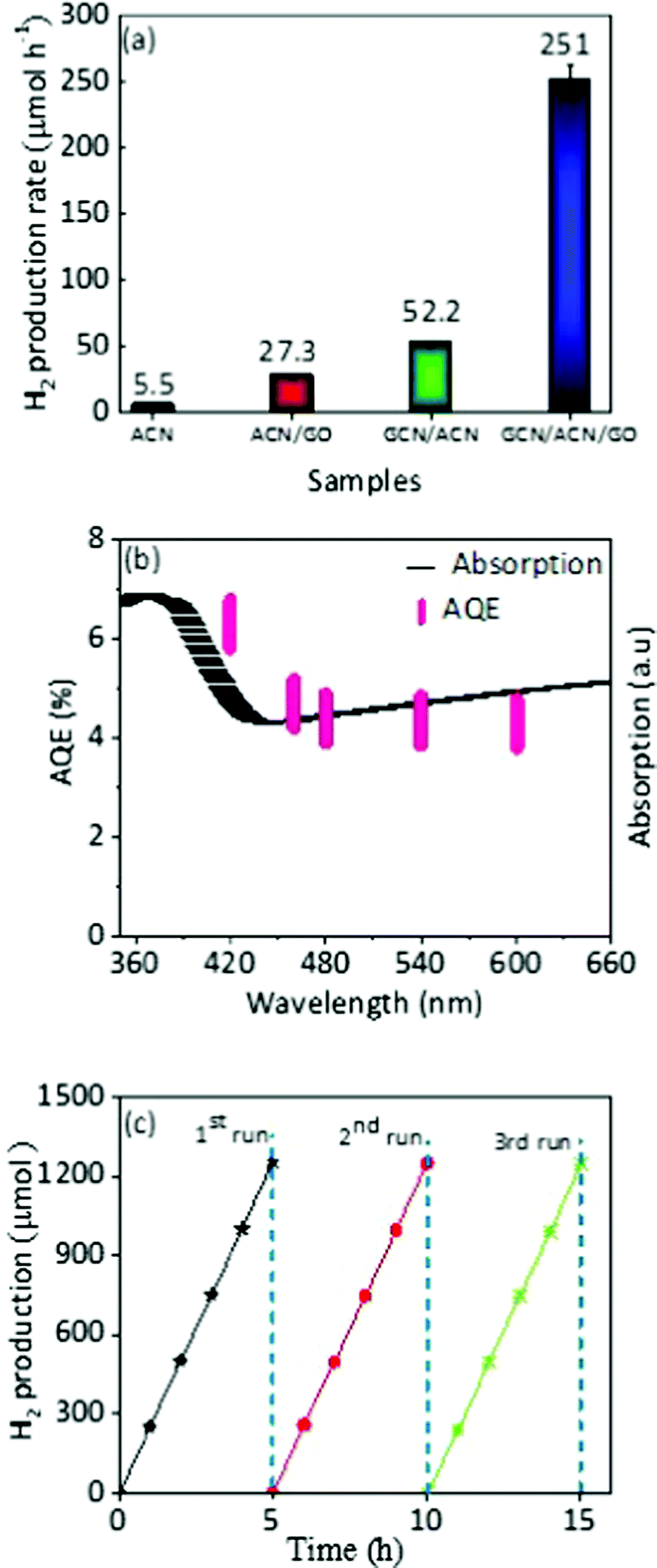 | ||
| Fig. 5 Visible light HER activity, when a Xe arch lamp (300 W) with a 420 nm cut-off filter was used to irradiate a reaction system consisting of 10 vol% triethanolamine, 50 mg of photocatalyst and 3 wt% Pt as a co-catalyst. (a) Average rate of hydrogen production with Pt as a co-catalyst, (b) wavelength dependent AQE, and (c) cyclic hydrogen production on GCN/ACN/GO over 15 h. | ||
With high photocatalytic activity, stability is an important issue for a photocatalyst. To this end, we conducted cyclic hydrogen production for a period of 15 h with intermittent evacuation in each 5 h. As shown in Fig. 5c, there was no distinct degradation observed during cyclic hydrogen production; it is evident that the GCN/ACN/GO is not only photocatalytically highly efficient, but durable too. The estimation of hydrogen production stability was also tested in which we measured hydrogen evolution for the first cycle of a 5 h reaction, and stored the samples in the reaction solutions for 10 days before the second cycle. After 10 days, we re-irradiated the reaction systems for 5 h and found that the hydrogen production was almost identical (Fig. S13, ESI†). This result confirmed the excellent stability of hydrogen production on GCN/ACN/GO. The structural and chemical stability is discussed in the ESI† (Fig. S14).
Interestingly, the hydrogen production rate with GCN/ACN/GO is comparable with a number of metal-based ternary hybrid photocatalysts.50,72,73 In this new ternary hybrid, the advantages of enhanced charge separation (owing to homo-heterojunction) and low-resistance electron transport capabilities (due to strong interfacial interactions at the GCN/ACN and GO interfaces) are combined. It is therefore perhaps no surprise that the GCN/ACN/GO hybrid produced a relatively high rate of hydrogen.
Mechanistic insight into the photocatalytic mechanisms
The charge transfer processes and the overall photocatalytic mechanisms of GCN/ACN/GO can be tentatively explained using the scheme shown in Scheme 1. Cyanamide derived GCN has a bandgap of ca. 2.6 eV whereas thiourea derived ACN has a bandgap of 2.55 eV (Fig. S15, ESI†). The corresponding band positions can be calculated using a Mott–Schottky plot (see Fig. S16 and Table S4, ESI†). When these are combined, they form a homojunction that results in a band-offset in relative CB and VB positions due to the band alignment effect.16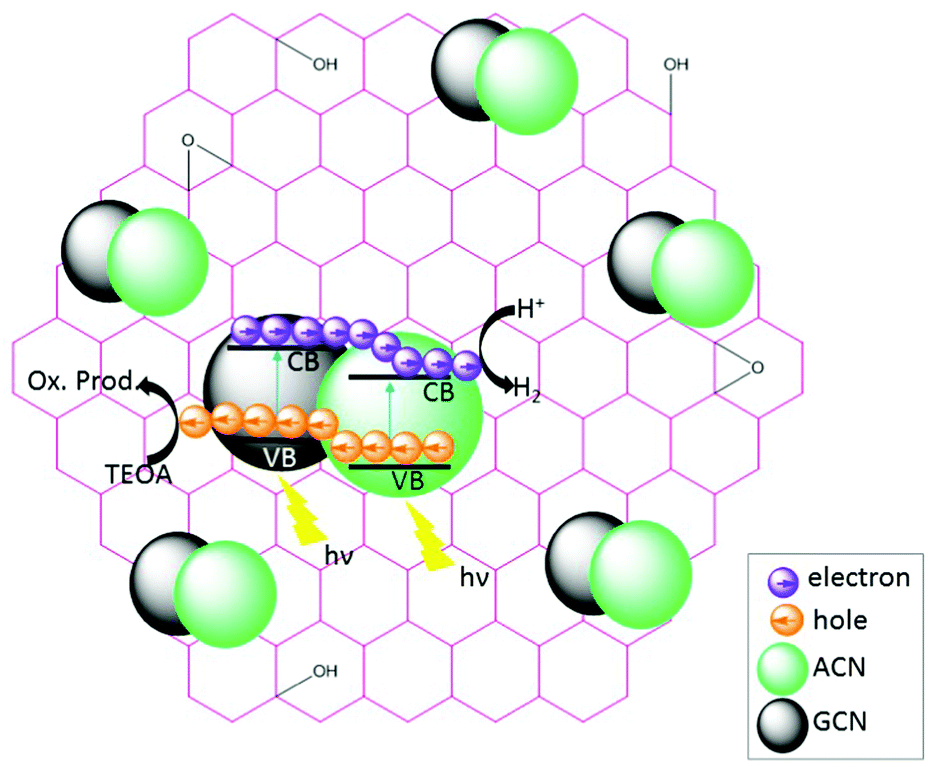 | ||
| Scheme 1 Hypothetical sketch of the photocatalytic mechanisms. | ||
When irradiated by a Xe arc lamp, photons of visible light are absorbed by both GCN and ACN semiconductors due to different band gaps. Upon absorption of photons, which have energy equal to or greater than the respective band gap energy, electrons from the VB of both GCN and ACN are excited to their respective CB. As the CB of ACN lies below that of GCN, photo-excited electrons from the CB of GCN are transferred to that of ACN. For photo-generated holes, the process is reversed. Accumulating electrons in the CB of ACN and holes in the VB of GCN render a significant reduction in electron–hole recombination. The redox potential of partially reduced GO nanosheets is lower than the CB of ACN,50 and ACN is firmly connected to the GO surfaces. Therefore, electrons from the CB of ACN will be injected into the CB of GO. The GO acts as an electron collector and transporter to reaction sites where H+ is reduced to H2 molecules (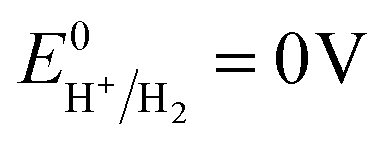 vs. SHE, pH = 0). Inclusion of Pt during photocatalytic testing further promotes the migration of electrons to reaction sites. Accumulated holes in the VB of GCN are scavenged by the sacrificial agent TEOA, completing the overall photocatalytic reactions.
vs. SHE, pH = 0). Inclusion of Pt during photocatalytic testing further promotes the migration of electrons to reaction sites. Accumulated holes in the VB of GCN are scavenged by the sacrificial agent TEOA, completing the overall photocatalytic reactions.
Conclusions
Graphene oxide coupled carbon nitride homojunction (GCN/ACN/GO) as a new ternary hybrid has been demonstrated for photocatalytic hydrogen evolution for the first time. The ternary composite exhibited an exceptional rate (251 μmol h−1) of H2 production from water with an AQE of 6.3% under visible light irradiation when 10 vol% triethanolamine was used as a hole scavenger and 3 wt% Pt as a co-catalyst. This hydrogen evolution rate outperformed that of metal-free compound semiconductors or binary composite photocatalysts irrespective of the inclusion of Pt as a co-catalyst. Experimental results confirmed that this new ternary heterostructure enhanced the performance for photocatalytic hydrogen production, because of: (i) participating semiconductors of different bandgaps that absorb multiple wavelength photons of visible light that increase photogeneration of EHPs; (ii) a built-in potential as a means to separate photogenerated EHPs effectively, which in turn, prevent electron–hole annihilation; and (iii) inclusion of GO to promote the collection and migration of photogenerated electrons that expand the reduction reaction surface through apparent synergistic effects of GO. It is concluded that these new findings will promote future synthesis of feasible metal-free composites combining elemental photocatalysts (i.e. P and S) to the CN framework for enhanced hydrogen production via water splitting. Due to its optimized electro-optical-chemical properties, this ternary hybrid is also promising for CO2 reduction, electrocatalysis, energy storage, photovoltaics and many more applications.Acknowledgements
This work was financially supported by the Australian Research Council (ARC) through the Discovery Project program (DP130104459, DP140104062 and DP160104866).References
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Z. Rahman, C. W. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709–728 CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- G. Liu, P. Niu, L. Yin and H.-M. Cheng, J. Am. Chem. Soc., 2012, 134, 9070–9073 CrossRef CAS PubMed.
- F. Wang, W. K. H. Ng, J. C. Yu, H. Zhu, C. Li, L. Zhang, Z. Liu and Q. Li, Appl. Catal., B, 2012, 111–112, 409–414 CrossRef CAS.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, G. Liu and H.-M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- J. Liu, S. Wen, Y. Hou, F. Zuo, G. J. O. Beran and P. Feng, Angew. Chem., Int. Ed., 2013, 52, 3241–3245 CrossRef CAS PubMed.
- T.-F. Yeh, J.-M. Syu, C. Cheng, T.-H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- J. Ran, T. Y. Ma, G. Gao, X.-W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 CAS.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, R.-Q. Sun and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, L. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- L.-C. Chen, C.-Y. Teng, C.-Y. Lin, H.-Y. Chang, S.-J. Chen and H. Teng, Adv. Energy Mater., 2016, 1600719 CrossRef.
- T.-F. Yeh, C.-Y. Teng, S.-J. Chen and H. Teng, Adv. Mater., 2014, 26, 3297–3303 CrossRef CAS PubMed.
- Y.-P. Yuan, L.-W. Ruan, J. Barber, S. C. Joachim Loo and C. Xue, Energy Environ. Sci., 2014, 7, 3934–3951 CAS.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 CAS.
- H. J. Yun, H. Lee, N. D. Kim, D. M. Lee, S. Yu and J. Yi, ACS Nano, 2011, 5, 4084–4090 CrossRef CAS PubMed.
- M. Yang, S. Hu, F. Li, Z. Fan, F. Wang, D. Liu and J. Gui, Ceram. Int., 2014, 40, 11963–11969 CrossRef CAS.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- G. Dukovic, M. G. Merkle, J. H. Nelson, S. M. Hughes and A. P. Alivisatos, Adv. Mater., 2008, 20, 4306–4311 CrossRef CAS.
- D. Chen, K. Wang, T. Ren, H. Ding and Y. Zhu, Dalton Trans., 2014, 43, 13105–13114 RSC.
- Z. Chen, P. Sun, B. Fan, Z. Zhang and X. Fang, J. Phys. Chem. C, 2014, 118, 7801–7807 CAS.
- J. Fu, B. Chang, Y. Tian, F. Xi and X. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 CAS.
- L. Ge, C. Han, X. Xiao and L. Guo, Int. J. Hydrogen Energy, 2013, 38, 6960–6969 CrossRef CAS.
- R. Yin, Q. Luo, D. Wang, H. Sun, Y. Li, X. Li and J. An, J. Mater. Sci., 2014, 49, 6067–6073 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- J. Zhang, M. Zhang, R.-Q. Sun and X. Wang, Angew. Chem., 2012, 124, 10292–10296 CrossRef.
- H. Li, Y. Liu, X. Gao, C. Fu and X. Wang, ChemSusChem, 2015, 8, 1189–1196 CrossRef CAS PubMed.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W.-K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS.
- X. An and J. C. Yu, RSC Adv., 2011, 1, 1426–1434 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed.
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- Y.-C. Chen, Y.-C. Pu and Y.-J. Hsu, J. Phys. Chem. C, 2012, 116, 2967–2975 CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- M. Z. Rahman, J. Ran, Y. Tang, M. Jaroniec and S. Z. Qiao, J. Mater. Chem. A, 2016, 4, 2445–2452 CAS.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC.
- Y.-S. Jun, W. H. Hong, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 4270–4274 CrossRef CAS.
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, L. Niu and J. Ye, Energy Environ. Sci., 2011, 4, 4517–4521 CAS.
- Z. Hu, Y. Chen, Q. Hou, R. Yin, F. Liu and H. Chen, New J. Chem., 2012, 36, 1373 RSC.
- P. Niu, L.-C. Yin, Y.-Q. Yang, G. Liu and H.-M. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- J. Zhang, L. Qi, J. Ran, J. Yu and S. Z. Qiao, Adv. Energy Mater., 2014, 4, 1301925 CrossRef.
- C. Hontoria-Lucas, A. J. López-Peinado, J. d. D. López-González, M. L. Rojas-Cervantes and R. M. Martín-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- B. V. Lotsch, Annu. Rev. Mater. Res., 2015, 45, 85–109 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- L. Xu, W.-Q. Huang, L.-L. Wang, Z.-A. Tian, W. Hu, Y. Ma, X. Wang, A. Pan and G.-F. Huang, Chem. Mater., 2015, 27, 1612–1621 CrossRef CAS.
- L. Xu, W.-Q. Huang, L.-L. Wang and G.-F. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 20350–20357 CAS.
- R. Long, J. Phys. Chem. Lett., 2013, 4, 1340–1346 CrossRef CAS PubMed.
- R. Long, Y. Dai and B. Huang, J. Phys. Chem. Lett., 2013, 4, 2223–2229 CrossRef CAS.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- A. Naumov, F. Grote, M. Overgaard, A. Roth, C. E. Halbig, K. Norgaard, D. M. Guldi and S. Eigler, J. Am. Chem. Soc., 2016, 138, 11445–11448 CrossRef CAS PubMed.
- C. H. Lui, Z. Li, K. F. Mak, E. Cappelluti and T. F. Heinz, Nat. Phys., 2011, 7, 944–947 CrossRef CAS.
- U. Sim, J. Moon, J. An, J. H. Kang, S. E. Jerng, J. Moon, S.-P. Cho, B. H. Hong and K. T. Nam, Energy Environ. Sci., 2015, 8, 1329–1338 CAS.
- M. Reza Gholipour, C.-T. Dinh, F. Beland and T.-O. Do, Nanoscale, 2015, 7, 8187–8208 RSC.
- G. Liu, P. Niu and H.-M. Cheng, ChemPhysChem, 2013, 14, 885–892 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- Q. Gu, J. Long, H. Zhuang, C. Zhang, Y. Zhou and X. Wang, Phys. Chem. Chem. Phys., 2014, 16, 12521–12534 RSC.
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem., 2008, 18, 2379–2385 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00241b |
| This journal is © the Partner Organisations 2017 |