
Influence of protein adsorption on the cellular uptake of AuNPs conjugated with chiral oligomers†
Jun
Deng
,
Honghao
Zheng
and
Changyou
Gao
 *
*
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: cygao@mail.hz.zj.cn; Fax: +86-571-87951108
First published on 15th September 2016
Abstract
Upon nanoparticles's (NPs) encounter with biological fluids, proteins shall be inevitably adsorbed on their surface to form a protein corona. Although its importance is widely accepted, information on the influence of the surface chirality of NPs on the protein corona and thereby cellular uptake is still missing. Herein, poly(acryloyl-L(D)-valine (L(D)-PAV) and poly(acryloyl-L(D)-valine)-b-poly(2-hydroxyethyl methylacrylate) (L(D)-PAV-b-PHEMA) chiral molecules were conjugated on a gold NP (AuNP) surface. Their interaction with serum proteins and A549 and HepG2 cancer cells in a medium containing concentrated serum was studied. The proteins in the serum-rich medium largely adsorbed onto PAV–AuNP NPs regardless of their optical activity, and shielded the chiral-selectivity in terms of cellular uptake. The introduction of a hydrophilic PHEMA block underneath the PAV block could significantly reduce the adsorption of serum proteins, and maintain the chiral-selective cellular uptake of PAV-b-PHEMA–AuNPs, indicating efficient performance by combining non-specific repelling and chiral-targeting effects.
1. Introduction
Nanotechnology enables materials to be specifically designed with unique physical, chemical, and biological properties according to the requirements.1 Nowadays the use of nanomaterials (NMs) for therapeutic purposes is one of the most exciting fields in nanotechnology.2 Due to their small size, NMs can cross biological barriers and permeate into organs and tissues.3,4 Thus, various nanoparticles (NPs) are prepared, and are currently under investigation for applications in drug delivery,5 diagnostics,6,7 imaging,8,9 and medical products.10,11Most applications of NPs in drug delivery, diagnostics and imaging currently concentrate on specific targeting.12,13 Localized treatment enabled by site-specific drug delivery and diagnostics can improve therapeutic efficiency. Thus, various targeting ligands have been employed. One of these is folic acid, which has been coupled to the NP surface to target the cargo at cancer cells overexpressing the folic acid receptor.14 Moreover, an anti-HER2 antibody and a CD8 antibody have been applied for the targeting of NPs towards cells overexpressing the EGFR-2 (HER2) receptor15 and expressing CD8,16 respectively. Mannose is also used to target dendritic cells.17
Although in simplified model systems the targeting performance is well demonstrated, it is dramatically weakened and even totally lost in real application. It is known that upon contact with biological systems, a number of proteins will adsorb on the NP surface rapidly and form a protein layer, namely a “protein corona”.18,19 This “protein corona” dramatically alters the physicochemical properties of NPs, including the hydrodynamic size,20 surface charge,21 and aggregation behavior.20 Moreover, the protein corona gives NPs a new biological identity that determines NP–cell interactions, rather than their initial synthetic identity.22,23 For example, surface-bound proteins promote or inhibit cellular uptake,24,25 activate intracellular signaling pathways,26,27 and influence cytotoxicity28 and body distribution.29 Very recently, the effect of the protein corona on specific cell targeting has been pointed out. For example, Anna Salvati et al. observed that transferrin-functionalized NPs lose their targeting capabilities when a protein corona forms on the surface.30 Kang et al. found that carbohydrate-based nanocarriers exhibit specific dendritic cell targeting with minimum influence from the protein corona.18 Although its importance is widely accepted, information of the influence of protein adsorption on cell targeting is still very limited, especially the influence of the protein corona on the targeting effect and the way to overcome the negative impact.
Chirality is an important and general phenomenon in living systems, and plays a significant role in biological systems. Chirality-selective biological behaviors including protein adsorption,31–33 cell adhesion,31,34 and cell differentiation35 are widely studied. Indeed, studies on nanoscale chirality have attracted much interest.36–38 For example, quantum dots (QDs) capped with different chiral forms of the tripeptide glutathione (GSH) was examined with respect to cytotoxicity, in which QDs modified with D-GSH, a nonbiologically active form of GSH, showed less cytotoxicity than L-GSH-coated ones.39 More recently, we have found that molecular chirality on gold nanoparticles (AuNPs) functions as a direct regulator for uptake by cancer cells.40 Right handed poly(acryloyl-valine) functionalized AuNPs (D-PAV–AuNPs) are much more specific for cancer cell targeting.40 However, preliminary results show that the chiral targeting effect is weakened significantly in the presence of a particular biological milieu. This suggests a future need to design and characterize the NP–corona interface in a more sophisticated way.
In this study, poly(acryloyl-L(D)-valine) (L(D)-PAV) and poly(acryloyl-L(D)-valine)-block-poly(2-hydroxyethyl methylacrylate) (L(D)-PAV-b-PHEMA) molecules are synthesized, and grafted onto AuNPs (Fig. 1). Modification with hydrophilic polymers such as PHEMA is one of the state-of-the-art approaches to reduce nonspecific interactions with serum proteins,27,41,42 which is often recognized as the “stealth effect”.43 The combination of “stealth” behavior with “atop” attachment of chiral groups has not been studied. For the first time, the interaction of serum proteins with PAV–AuNPs and PAV-b-PHEMA–AuNPs is compared, and their uptake by A549 and HepG2 cells in a serum-rich (50% (v/v)) medium is quantified.
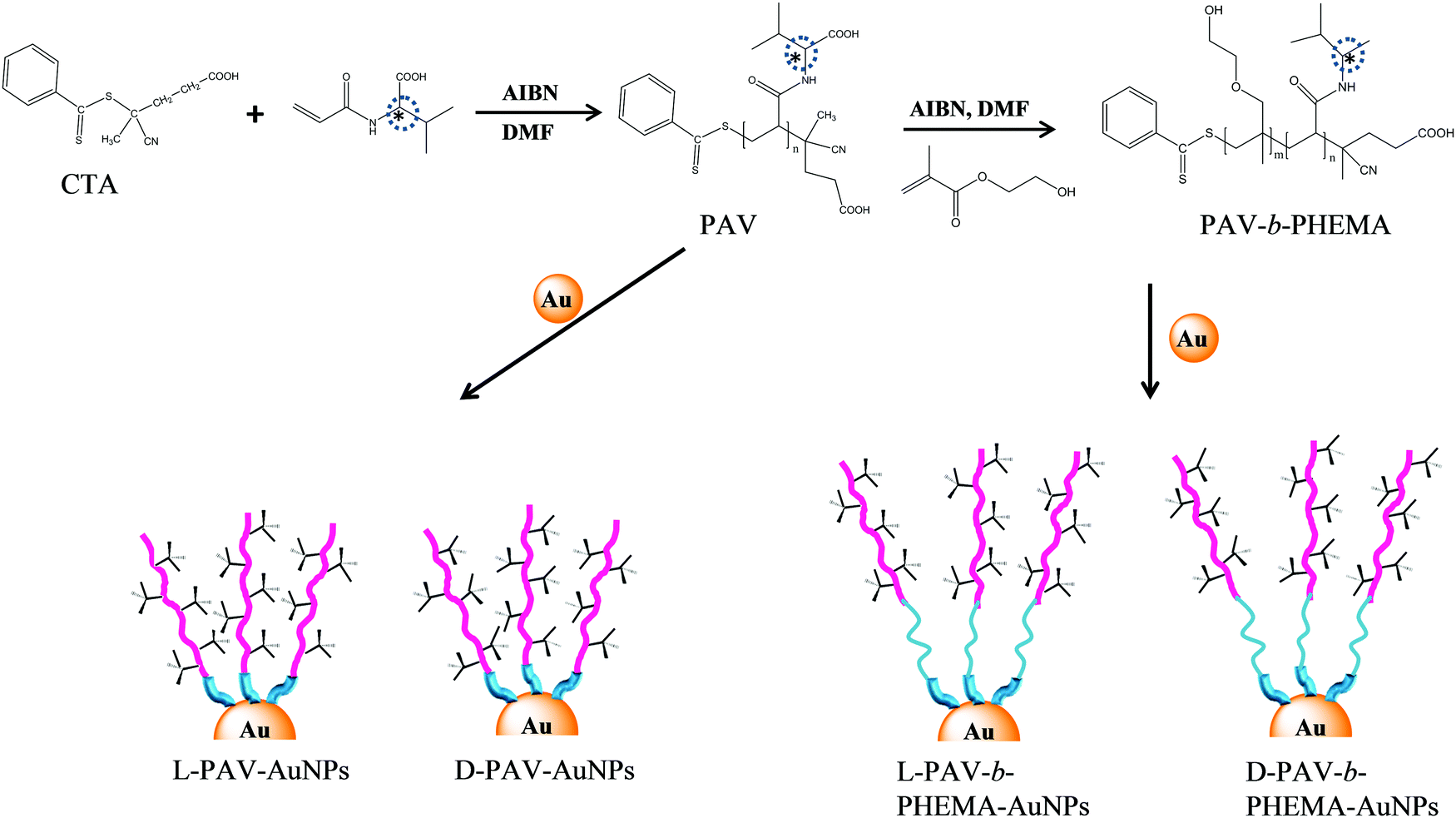 | ||
| Fig. 1 Schematic illustration of the synthesis of poly(acryloyl-L(D)-valine) (L(D)-PAV) and poly(acryloyl-L(D)-valine)-b-poly(2-hydroxyethyl methacrylate) (L(D)-PAV-b-PHEMA), and their conjugation to the AuNP surface. | ||
2. Materials and methods
2.1 Materials
Gold(III) chloride hydrate (HAuCl4) and sodium citrate were purchased from Sinopharm group Co. Ld. Poly(acryloyl-L-valine) (L-PAV) (Mw 4926 Da, polydispersity 1.24), poly(acryloyl-D-valine) (D-PAV) (Mw 4997 Da, polydispersity 1.15), poly(2-hydroxyethyl methacrylate) (PHEMA) (Mw 3965 Da, polydispersity 1.27), L-PAV-b-PHEMA) (Mw 8891 Da) and D-PAV-b-PHEMA (Mw 8962 Da) were synthesized and are characterized in the ESI.† Milli Q water was used throughout the experiments. 15 nm AuNPs were synthesized using a citrate reduction method.21 All other chemicals were of analytical grade and used without further treatment if not specially mentioned.2.2 Immobilization of L(D)-PAV and L(D)-PAV-b-PHEMA on AuNPs
The surface of AuNPs was conjugated to obtain L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs by using a ligand exchange protocol. Briefly, the stock AuNP solution was incubated with a large amount of 2 mg mL−1L(D)-PAV or L(D)-PAV-b-PHEMA solution under vigorous shaking at 37 °C overnight. The obtained solution was centrifuged (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g min−1, 30 min) to remove free PAV or PAV-b-PHEMA molecules. To ensure complete exchange, the NPs were re-incubated with 2 mg mL−1L(D)-PAV or L(D)-PAV-b-PHEMA, and the solution was allowed to stir for another 12 h. Then the NP solution was centrifuged, followed by dialysis in water for at least 3 days to remove free PAV or PAV-b-PEHMA molecules.
000 g min−1, 30 min) to remove free PAV or PAV-b-PHEMA molecules. To ensure complete exchange, the NPs were re-incubated with 2 mg mL−1L(D)-PAV or L(D)-PAV-b-PHEMA, and the solution was allowed to stir for another 12 h. Then the NP solution was centrifuged, followed by dialysis in water for at least 3 days to remove free PAV or PAV-b-PEHMA molecules.
2.3 Characterization of NPs
The gold concentration (m, g L−1) of the AuNP solution and the diameter of NPs in the dry state were measured by using inductively coupled plasma mass spectrometry (ICP-MS, Thermo Elemental Corporation of USA, XSeries II) and transmission electron microscopy (TEM, H-7650), respectively.26The circular dichroism (CD) spectrum was recorded by using a JASCO-820 spectropolarimeter equipped with a thermostatically controlled cell holder. The temperature of the sample was controlled at 25 °C. The UV region was scanned between 190 and 400 nm with an average of 3 scans. The concentration of AuNPs in L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs was kept at 50 nM. The final spectrum (Δθ) of L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs was obtained by subtracting the spectrum of the same concentration of AuNP (without PAV or PAV-b-PHEMA grafting) solution.
2.4 Dynamic light scattering (DLS) and zeta potential measurement
The Z-average hydrodynamic diameters of L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs were measured at 37 °C in water and 50% fetal bovine serum (FBS)/high-glucose Dulbecco's modified Eagle medium (FBS/DMEM) by using DLS with a high performance particle analyzer (Zetasizer Nano, Malvern) equipped with a 633 nm wavelength laser. The scattering intensity was recorded at a 173° angle in kilo counts per second.The zeta potentials of L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs were measured at pH 7.4 and 37 °C by using the same machine (Zetasizer Nano, Malvern) in water and 50% FBS/DMEM, respectively.
2.5 Cellular uptake of NPs
The amount of L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs internalized by A549 or HepG2 cells was determined by ICP-MS. Briefly, the cells were seeded on a 24-well plate at a density of 5 × 104 cells per cm2, and allowed to attach for 24 h. The medium was replaced with 50% FBS/DMEM containing L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs with an Au concentration of 50 μg mL−1, respectively. After 24 h, the plates were washed 5 times with phosphate buffered saline (PBS) to remove unassociated NPs. After the cells were harvested by trypsinization, their numbers were quantified by a cell counter. The cells were treated with aqua regia (HCl:HNO3, 3:1, volume ratio) for 2 h. The obtained solution was diluted to determine the Au concentration by ICP-MS. The Au amount per 104 cells from ICP-MS analysis is presented as the mean ± standard deviation (n = 4).
2.6 Protein adsorption
The amount of serum proteins adsorbed on the NPs was measured by using the micro bicinchoninic acid (μBCA) protein assay kit according to the manual instruction. Briefly, the NPs were incubated in the cell culture medium containing different concentrations of FBS (0.1–50% (v/v)). The samples were shaken at 37 °C for 12 h. After being centrifuged at 10 °C for 30 min, the supernatant was discarded. The NPs were re-dispersed in PBS by gentle shaking, and centrifuged again. In total 3 washes were performed to remove loosely adsorbed proteins. The protein-adsorbed NPs were then treated with 5% sodium dodecylsulphate (SDS) to release the serum proteins, whose amount was quantified by using μBCA assay reagent kits. The concentration of AuNPs used here was 1 mg mL−1 to ensure a large enough amount of adsorbed proteins for μBCA measurement.3. Results and discussion
3.1 Properties of NPs
Transmission electron microscopy (TEM) showed that both PAV–AuNPs (Fig. 2a and b) and PAV-b-PHEMA–AuNPs (Fig. 2c and d) were approximately spherical in shape and narrowly distributed (Fig. S1, ESI†). The density of PAV and PAV-b-PHEMA molecules on the AuNPs calculated from thermogravimetric analysis (TGA) (Fig. S2, ESI†) was nearly 0.26 chains per nm2 and 0.15 chains per nm2 (Table 1), respectively. This result suggests that the density of the chiral center on PAV–AuNPs was significantly larger than that on PAV-b-PHEMA–AuNPs. However, the density of chains on L-PAV–AuNPs and D-PAV–AuNPs or L-PAV-b-PHEMA–AuNPs and D-PAV-b-PHEMA–AuNPs showed almost no difference (Fig. S2, ESI†). The circular dichroism (CD) signal of PAV–AuNPs was significantly larger than that of PAV-b-PHEMA–AuNPs (Fig. 2e), which is consistent with the larger chiral units on PAV–AuNPs. Importantly, L-PAV–AuNPs and D-PAV–AuNPs or L-PAV-b-PHEMA–AuNPs and D-PAV-b-PHEMA–AuNPs show the essential mirror image of the CD spectra in the region of 190−400 nm (Fig. 2e), demonstrating their reverse optical activity.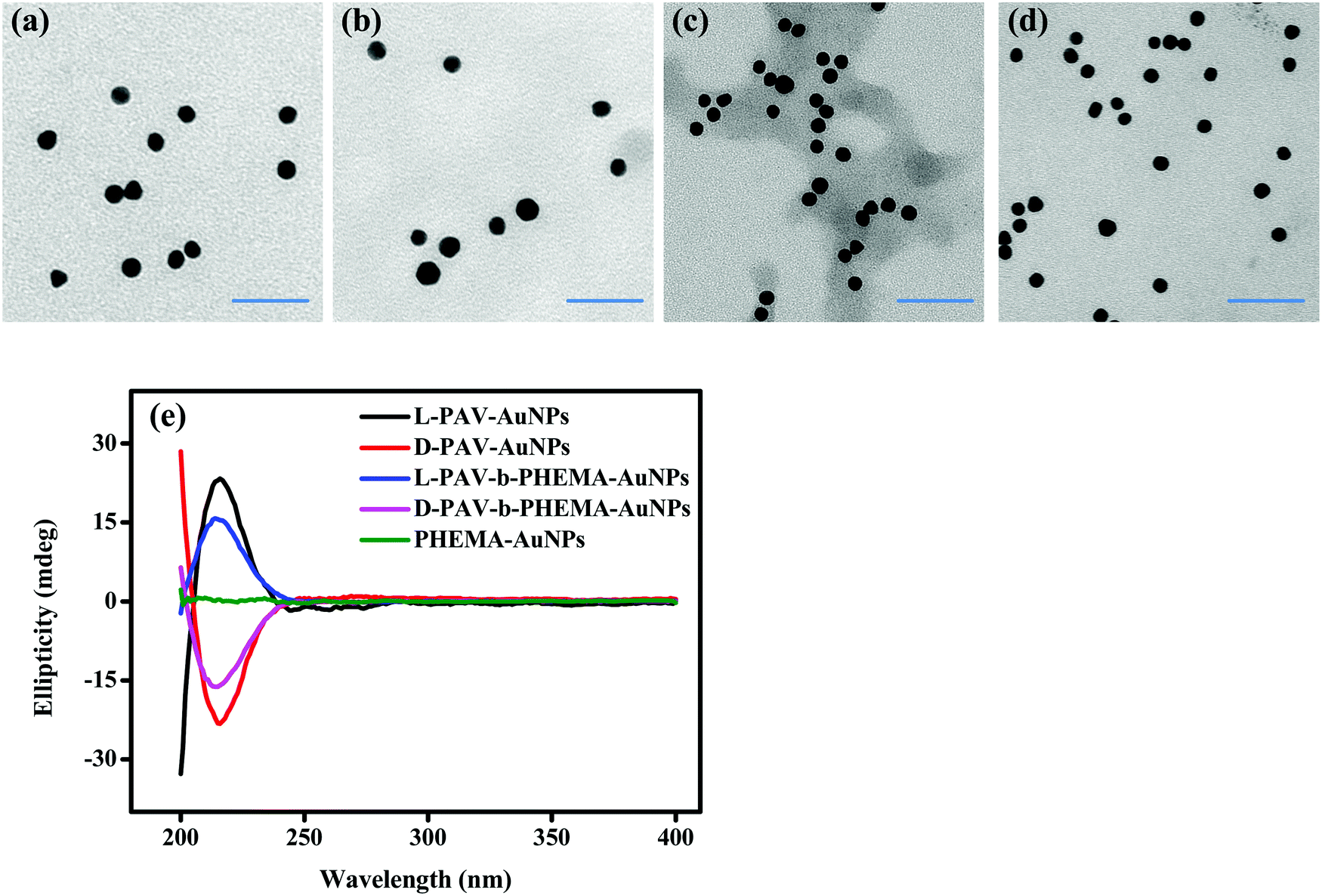 | ||
| Fig. 2 Nanoparticle characterization. TEM images of (a) L-PAV–AuNPs, (b) D-PAV–AuNPs, (c) L-PAV-b-PHEMA–AuNPs and (d) D-PAV-b-PHEMA–AuNPs. (e) CD spectra of L(D)-PAV–AuNPs and L(D)-PAV-b-PHEMA–AuNPs with an AuNP concentration of 50 nM. | ||
| Sample | Ligand density (TGA, chains nm−2) | Diameter (TEM, nm) | Hydrodynamic diameter (DLS, nm) | Surface zeta potential (mV) | ||
|---|---|---|---|---|---|---|
| Water | 50% FBS | Water | 50% FBS | |||
| L-PAV–AuNPs | 0.262 | 14.5 ± 1.1 | 20.3 ± 0.4 | 24.3 ± 0.2 | −31.3 ± 2.8 | −7.7 ± 0.3 |
| D-PAV–AuNPs | 0.268 | 14.1 ± 1.4 | 20.5 ± 0.3 | 24.5 ± 0.2 | −29.9 ± 3.8 | −7.1 ± 0.9 |
| L-PAV-b-PHEMA–AuNPs | 0.145 | 14.7 ± 0.8 | 22.3 ± 0.3 | 25.5 ± 0.3 | −20.3 ± 0.7 | −8.8 ± 0.5 |
| D-PAV-b-PHEMA–AuNPs | 0.146 | 14.8 ± 1.2 | 22.7 ± 0.2 | 25.7 ± 0.2 | −20.3 ± 1.4 | −8.8 ± 0.7 |
Other physical properties of PAV–AuNPs and PAV-b-PHEMA–AuNPs are summarized in Table 1. The more negative surface zeta potential of PAV–AuNPs in water (∼ −30 mV) than that of PAV-b-PHEMA–AuNPs (∼ −20 mV) is consistent with the larger number of carboxyl groups (more chiral units) on PAV–AuNPs (Table 1). Besides, the hydrodynamic diameter of PAV-b-PHEMA–AuNPs (∼22 nm) was slightly larger than that of PAV–AuNPs (∼20 nm) due to their longer chain length, leading to a thicker hydration layer. Furthermore, the stability of the AuNPs during the ligand exchanging process was monitored by measuring the AuNP surface plasmon resonance (SPR), a parameter that is extremely sensitive to NP size and interparticle spacing.44 A slight red-shift (4 nm) was observed in the SPR peaks of PAV–AuNPs (522 nm) and PAV-b-PHEMA–AuNPs (522 nm) compared to that of citrate–AuNPs (518 nm) without significant peak broadening (Fig. S3, ESI†). No shift and significant peak broadening were observed between the SPR peaks of L-PAV–AuNPs and D-PAV–AuNPs (or L-PAV-b-PHEMA–AuNPs and D-PAV-b-PHEMA–AuNPs) (Fig. S3, ESI†). The intensity difference was caused by the slight difference in concentrations used here (Fig. S3, ESI†). These results indicate that all the NPs are very stable during the ligand exchange process.
The stability of the AuNPs in high-glucose DMEM containing 50% FBS was further characterized. Compared to that in water, the SPR peak of PAV–AuNPs and PAV-b-PHEMA–AuNPs showed a slight red-shift (∼6 nm) in 50% FBS/DMEM without significant peak broadening (Fig. S3, ESI†). The hydrodynamic diameters of PAV–AuNPs (∼24.5 nm) and PAV-b-PHEMA–AuNPs (∼25.5 nm) were increased to 4 nm and 3 nm, respectively due to adsorption of proteins (Table 1). Their surface became less negative too (Table 1). Therefore, both PAV–AuNPs and PAV-b-PHEMA–AuNPs possess excellent colloidal stability in 50% FBS/DMEM, and their surface charge (∼1 mV difference) and hydrodynamic diameters (∼1 nm difference) showed no significant difference.
With these characterizations, one can conclude that L-PAV–AuNPs and D-PAV–AuNPs, or the L-PAV-b-PHEMA–AuNPs and D-PAV-b-PHEMA–AuNPs have identical physicochemical properties except for reverse molecular chirality.
3.2 Cellular uptake in medium with concentrated serum
In a recent study, we found that D-PAV–AuNPs were internalized with a significantly larger amount than L-PAV–AuNPs by A549 and HepG2 cancer cells when these NPs were co-incubated in a normal culture medium with 10% FBS or a low concentration of FBS.40 This chirality-dependent uptake effect is likely attributed to the preferable interaction between the L-phospholipid-based cell membrane and the D-enantiomers.40 However, the realistic biological environment in which the NPs interact in vivo is more complicated, especially with very high concentration of serum proteins, than that in the cell culture medium in vitro. In this regard, herein the cellular uptake was performed in DMEM containing 50% FBS. To verify the suitability of the culture conditions, the cytoviability was assessed by MTT assay, which reflects the cell metabolic activity based on the ability of the mitochondrial succinate/tetrazolium reductase system in living cells.45 Fig. S4 (ESI†) shows that the viability of both HepG2 and A549 cells was maintained nearly 100% after they were incubated with PAV–AuNPs or PAV-b-PHEMA–AuNPs with an Au concentration of 50 μg mL−1, respectively, revealing no cytotoxicity of the NPs at this concentration in 50% FBS/DMEM.Next, the amount of cellular uptake of the NPs (including minor amount of cell-surface attached) was quantified by ICP-MS with high detection sensitivity. As shown in Fig. 3, unlike in the normal culture medium, in the 50% FBS/DMEM the internalized amount of L-PAV–AuNPs and D-PAV–AuNPs showed no significant difference (p > 0.05). This result demonstrates that PAV–AuNPs in a serum-rich medium lose their chiral-specificity completely. The most probable scenario is that a large amount of proteins may adsorb onto the surface of PAV–AuNPs from the 50% FBS/DMEM to fully cover the PAV molecules, hence the chiral effect is screened.
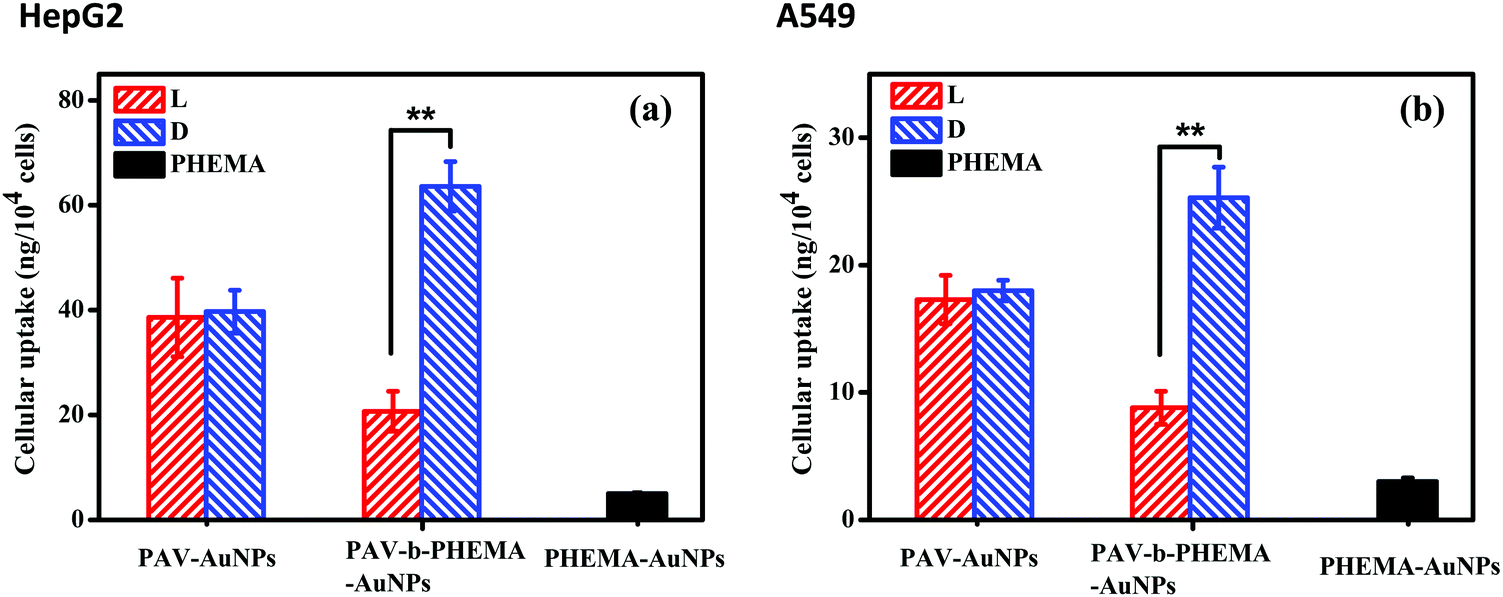 | ||
| Fig. 3 Internalized amount of PAV–AuNPs and PAV-b-PHEMA–AuNPs by (a) HepG2 and (b) A549 cells at an Au concentration of 50 μg mL−1. The cells were cultured with the NPs for 24 h in 50% FBS/DMEM. ** Indicates a significant difference at the p < 0.01 level. | ||
PHEMA is a hydrophilic polymer which demonstrates the so-called antifouling effect.46–48 A suitably designed PHEMA surface can modulate protein adsorption well and thereby cell behaviors.27,48 Thus, AuNPs were further surface-modified with PAV-b-PHEMA molecules, with PEHMA underneath the PAV block to modulate unspecific protein adsorption. Fig. 3 shows that the immobilization of control PHEMA molecules could significantly decrease the uptake of PHEMA–AuNPs than both PAV–AuNPs and PAV-b-PHEMA–AuNPs. This effect is understood as the repelling function of the very hydrophilic polymers immobilized onto the particle surface,49 as frequently observed for poly(ethylene glycol) (PEG) modified ones,50 which can resist protein adsorption and thereby cellular interaction. By contrast, when AuNPs were conjugated with PAV-b-PHEMA molecules, the interplay between the repulsion of non-specific adsorption contributed by PHEMA and the specific cell-affinity contributed by the D-PAV molecules resulted in a significantly higher uptake of D-PAV-b-PHEMA–AuNPs than L-PAV-b-PHEMA–AuNPs (p < 0.01) by HepG2 cells (Fig. 3a) and A549 cells (Fig. 3b) even in 50% FBS/DMEM. These results clearly demonstrate that the introduction of the underneath PHEMA block can effectively maintain the chiral selectivity of the upper PAV block on the AuNPs in terms of cellular uptake performance.
3.3 Protein adsorption behaviors
Upon contact with physiological fluids, various proteins adsorb on the surface of NPs to rapidly form a protein shell.51,52 This rapidly formed protein corona can alter the synthetic identity of NPs, such as the size, shape, surface charge and the agglomeration state.23,53 More and more studies certify that it is the protein corona that determines the biological fate of NPs including their cellular internalization, trafficking, opsonization and eventually the pattern of biodistribution.54,55 As discovered in this study, protein adsorption may play a key role in the cellular uptake of PAV–AuNPs or PAV-b-PHEMA–AuNPs especially in a culture medium containing concentrated FBS. Therefore, a micro bicinchoninic acid (μBCA) assay was used to determine protein adsorption on the NPs. As shown in Fig. 4a, the amount of serum proteins adsorbed on PAV–AuNPs from 50% FBS/DMEM was significantly higher than that from 10% FBS/DMEM (p < 0.01). In particular, unlike the significantly lower adsorption amount of proteins in 10% FBS by D-PAV–AuNPs, the adsorption amount of proteins became insignificant between L-PAV–AuNPs and D-PAV–AuNPs. Upon incubation in 50% FBS/DMEM, PAV-b-PHEMA–AuNPs reduced serum protein adsorption significantly compared with the PAV-coated ones (p < 0.01) (Fig. 4a). Therefore, PAV-b-PHEMA–AuNPs show low overall protein adsorption compared to PAV–AuNPs.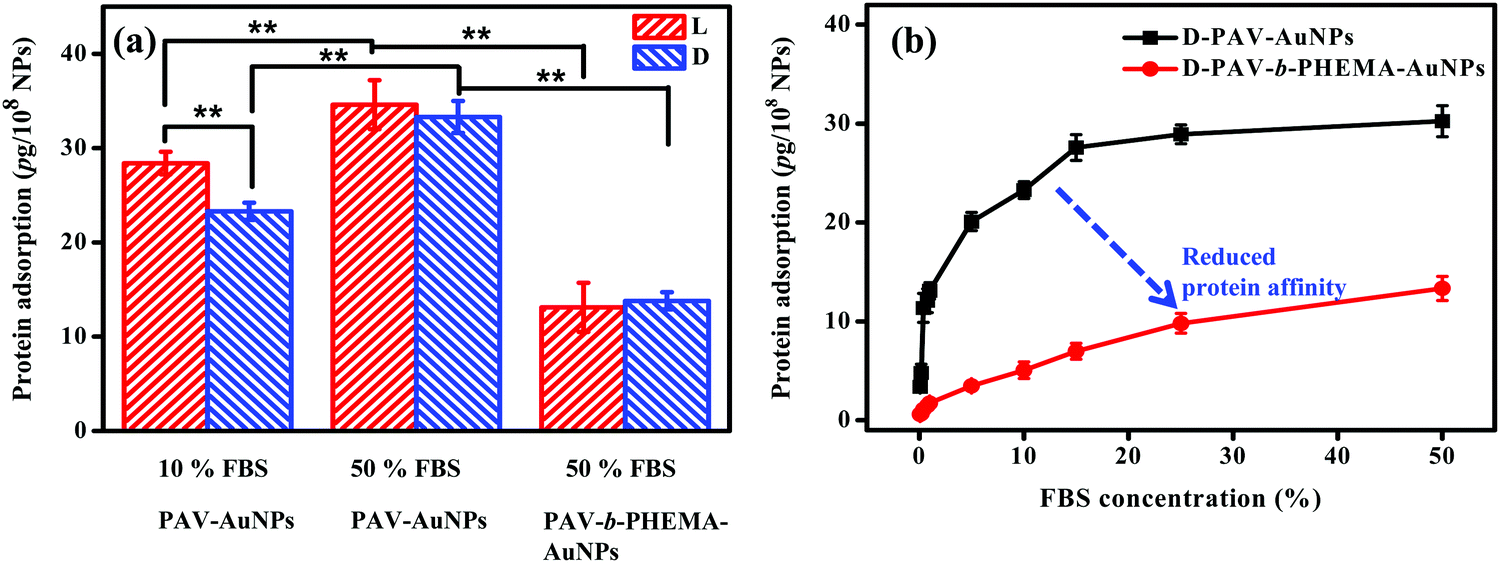 | ||
| Fig. 4 (a) The amount of serum proteins being adsorbed on PAV–AuNPs and PAV-b-PHEMA–AuNPs in different concentrations of FBS/DMEM, respectively. (b) Concentration-dependent adsorption of serum proteins to D-PAV–AuNPs and D-PAV-b-PHEMA–AuNPs. | ||
To further substantiate the interaction of serum proteins and NPs, D-enantiomer (D-PAV or D-PAV-b-PHEMA) coated NPs were incubated in a culture medium containing various concentrations of FBS (0.1–50% (v/v)). As shown in Fig. 4b, the amount of serum proteins adsorbed on D-PAV–AuNPs and D-PAV-b-PHEMA–AuNPs increased along with the increase of the FBS concentration, and leveled off at about 20% FBS (Fig. 4b). More importantly, the binding affinity of serum proteins is strongly reduced on D-PAV-b-PHEMA–AuNPs, which is represented by the gentle initial slope of the binding isotherm.
Taking all the results into consideration, the most probable scenario that governs the different cellular uptake behaviors of PAV–AuNPs and PAV-b-PHEMA–AuNPs in a medium containing concentrated FBS should be the different protein adsorption behaviors (Fig. 5). A large amount of adsorbed proteins on PAV–AuNPs fully cover the PAV molecules, which screen the accessibility of the chiral units to the cancer cell surface (Fig. 5, left). By contrast, the very hydrophilic PHEMA block can significantly reduce non-specific adsorption of serum proteins, enabling the efficient interaction of the upper PAV block on PAV-b-PHEMA–AuNPs with cells and thereby the maintenance of chiral-selective cellular uptake (Fig. 5, right).
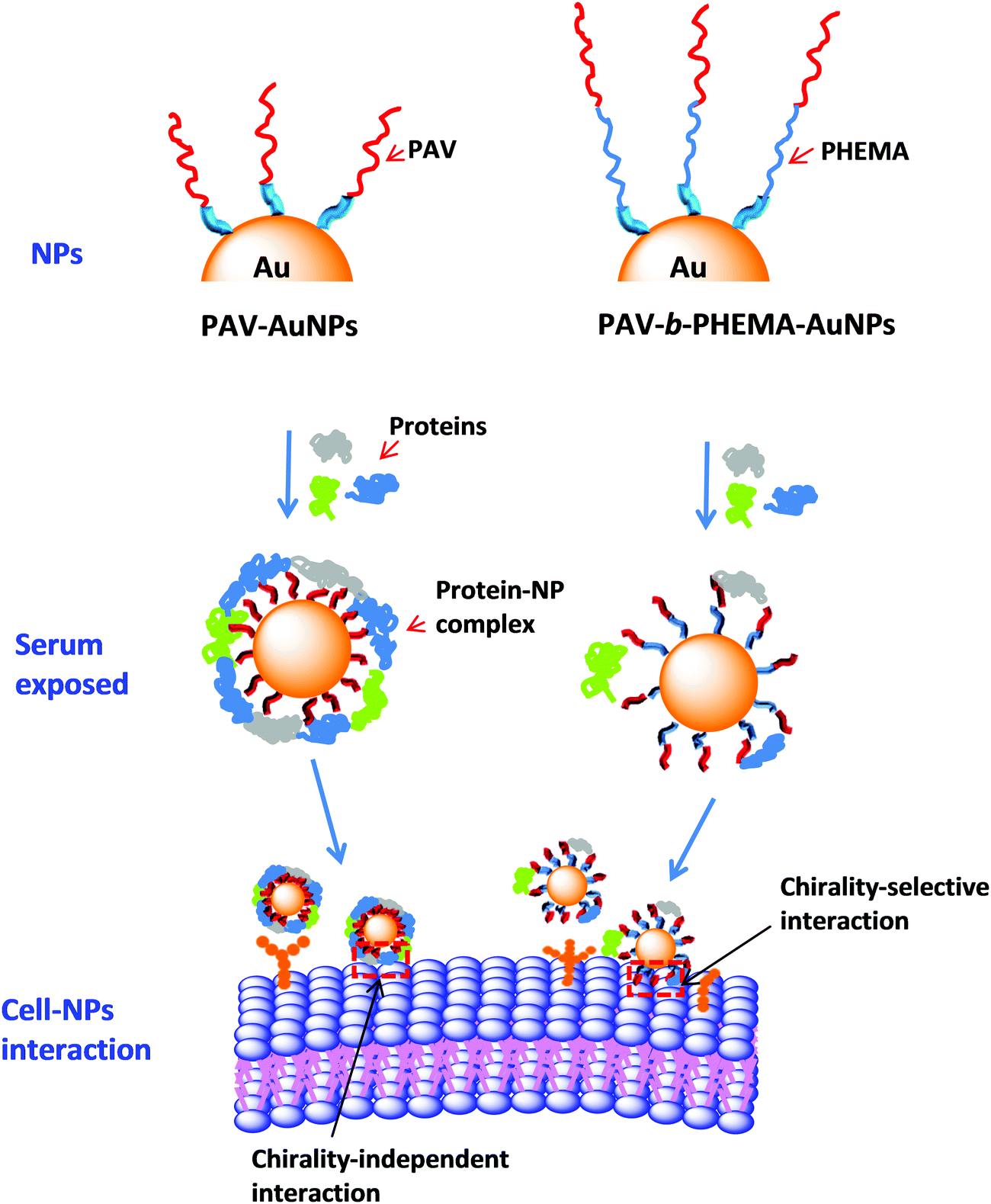 | ||
| Fig. 5 Schematic illustration showing the influences of surface chirality on serum protein adsorption and thereby uptake of the AuNPs by HepG2 and A549 cells. When incubated in a serum-rich medium, PAV–AuNPs lose their chirality-selective cellular uptake due to the screening effect of abundant protein corona. By significantly reducing the adsorption of serum proteins, PAV-b-PHEMA–AuNPs can still exhibit chirality-selective cell uptake in a medium with concentrated FBS. | ||
Our results display that even though chirality recognition is successfully achieved in a model system such as DMEM with a low concentration of FBS in vitro, this effect can be fully screened under realistic physiological conditions (e.g. blood and serum-rich medium) in vivo. Besides the cellular uptake, the other critical issue is the capability of the NPs to discriminate between different targets, particularly in the complex biological milieu in which they are applied. Success in similar systems by no means implies success in vivo. However, if specific discrimination or targeting is not observed in relevant fluids, the apparent effect in vivo may be a consequence of other poorly understood phenomena.
4. Conclusions
In this study, two groups of AuNPs modified with PAV and PAV-b-PHEMA of different surface chiralities were successfully prepared. These two groups of NPs had a diameter about 15 nm and a narrow size distribution in the dry state. The density of PAV and PAV-b-PHEMA grafted on AuNPs was 0.26 molecules per nm2 and 0.15 molecules per nm2, respectively. Compared to PAV, PAV-b-PHEMA grafting endowed AuNPs with decreased optical activity due to a lower density of chiral units. L-PAV–AuNPs and D-PAV–AuNPs or L-PAV-b-PHEMA–AuNPs and D-PAV-b-PHEMA–AuNPs possessed good colloidal stability in culture medium, and showed similar physicochemical properties except for reversed ellipticity.Compared with PAV, PAV-b-PHEMA grafting reduced the interaction with serum proteins significantly. The high adsorption of serum proteins on the NP surface could screen the PAV molecules from being recognized by the cells after incubation in FBS with a high concentration (50%), and thereby L- and D-PAV–AuNPs did not show a significant difference in terms of the uptake amount by A549 cells and HepG2 cells. By contrast, the chiral polymer-capped PAV-b-PHEMA–AuNPs with much lower protein adsorption did show chirality-dependent uptake behaviors, and D-PAV-b-PHEMA–AuNPs were internalized with a significantly larger amount than L-PAV-b-PHEMA–AuNPs in the same medium containing concentrated FBS. Taking into account that rather simple chiral molecules such as PAV can be selectively recognized by cancer cells, the combination of stealth nanocarriers with chiral-based targeting moieties is very attractive to overcome the loss of chiral-selectivity in terms of cellular uptake by cancer cells.
Acknowledgements
This study was financially supported by the Key Science Technology Innovation Team of Zhejiang Province (2013TD02), and the Natural Science Foundation of China (21374097 and 51120135001).References
- M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- S. Schöttler, K. Landfester and V. Mailänder, Angew. Chem., Int. Ed., 2016, 55, 8806–8815 CrossRef PubMed.
- S. Nie, Y. Xing, G. J. Kim and J. W. Simons, Annu. Rev. Biomed. Eng., 2007, 9, 257–288 CrossRef CAS PubMed.
- S. Y. Qin, M. Y. Peng, L. Rong, B. Li, S. B. Wang, S. X. Cheng, R. X. Zhuo and X. Z. Zhang, Regener. Biomater., 2015, 2, 159–166 CrossRef PubMed.
- F. M. Gong, Z. Q. Zhang, X. D. Chen, L. Zhang, X. S. Yu, Q. H. Yang, X. T. Shuai, B. L. Liang and C. Du, Chin. J. Polym. Sci., 2014, 32, 321–332 CrossRef CAS.
- Y. Zhang, S. Liu, X. Wang, Z. Y. Zhang, X. B. Jing, P. Zhang and Z. G. Xie, Chin. J. Polym. Sci., 2014, 32, 1111–1118 CrossRef CAS.
- X. Q. Zhang, X. Y. Zhang, B. Yang and Y. Wei, Chin. J. Polym. Sci., 2014, 32, 871–879 CrossRef CAS.
- X. Q. Zhang, X. Y. Zhang, B. Yang and Y. Wei, Chin. J. Polym. Sci., 2014, 32, 1479–1488 CrossRef CAS.
- C. Z. Geng, X. Hu, G. H. Yang, Q. Zhang, F. Chen and Q. Fu, Chin. J. Polym. Sci., 2015, 33, 61–69 CrossRef CAS.
- C. Nelson, Y. Khan and C. T. Laurencin, Regener. Biomater., 2014, 1, 3–9 CrossRef PubMed.
- R. Govindan, N. Engl. J. Med., 2015, 372, 1760–1761 CrossRef PubMed.
- J. Li, Y. Tian and A. Wu, Regener. Biomater., 2015, 2, 215–219 CrossRef PubMed.
- Z. Zhang, J. Jia, Y. Lai, Y. Ma, J. Weng and L. Sun, Bioorg. Med. Chem., 2010, 18, 5528–5534 CrossRef CAS PubMed.
- L. E. van Vlerken and M. M. Amiji, Expert Opin. Drug Delivery, 2006, 3, 205–216 CrossRef CAS PubMed.
- A. Bicho, I. N. Peça, A. C. A. Roque and M. M. Cardoso, Int. J. Pharm., 2010, 399, 80–86 CrossRef CAS PubMed.
- B. Kang, P. Okwieka, S. Schöttler, S. Winzen, D. C. J. Langhanki, K. Mohr, T. Opatz, V. M. Nder, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 7436–7440 CrossRef CAS PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee and M. Mahmoudi, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- J. Deng, M. Sun, J. Zhu and C. Gao, Nanoscale, 2013, 5, 8130–8137 RSC.
- J. Deng, H. Zheng, S. Wu, P. Zhang and C. Gao, RSC Adv., 2015, 5, 22792–22801 RSC.
- J. Deng, D. H. Yu and C. Y. Gao, Sci. China: Chem., 2013, 56, 1533–1541 CrossRef CAS.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- A. Lesniak, A. Campbell, M. P. Monopoli, I. Lynch, A. Salvati and K. A. Dawson, Biomaterials, 2010, 31, 9511–9518 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- J. Deng, T. Ren, J. Zhu, Z. Mao and C. Gao, Regener. Biomater., 2014, 1, 17–25 CrossRef PubMed.
- M. Horie, K. Nishio, K. Fujita, S. Endoh, A. Miyauchi, Y. Saito, H. Iwahashi, K. Yamamoto, H. Murayama and H. Nakano, Chem. Res. Toxicol., 2009, 22, 543–553 CrossRef CAS PubMed.
- J. Llop, I. Estrela-Lopis, R. F. Ziolo, A. González, J. Fleddermann, M. Dorn, V. G. Vallejo, R. Simon-Vazquez, E. Donath and Z. Mao, Part. Part. Syst. Charact., 2014, 31, 24–35 CrossRef CAS.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- Z. Li, A. Köwitsch, G. Zhou, T. Groth, B. Fuhrmann, M. Niepel, E. Amado and J. Kressler, Adv. Healthcare Mater., 2013, 2, 32–44 CrossRef.
- J. Deng, L. Zheng, M. Yao and C. Gao, Langmuir, 2016, 32, 5608–5616 CrossRef CAS PubMed.
- X. Wang, H. Gan and T. Sun, Adv. Funct. Mater., 2011, 21, 3276–3281 CrossRef CAS.
- B. Hu, J. Deng, H. Zheng, S. Yu and C. Gao, Macromol. Rapid Commun., 2016, 37, 1331–1336 CrossRef CAS PubMed.
- T. Jian, P. Rong and D. Jiandong, Biomaterials, 2010, 31, 2470–2476 CrossRef PubMed.
- W. Liu, Z. Zhu, K. Deng, Z. Li, Y. Zhou, H. Qiu, Y. Gao, S. Che and Z. Tang, J. Am. Chem. Soc., 2013, 135, 9659–9664 CrossRef CAS PubMed.
- Y. Zhou, M. Yang, K. Sun, Z. Tang and N. A. Kotov, J. Am. Chem. Soc., 2010, 132, 6006–6013 CrossRef CAS PubMed.
- Y. Zhou, Z. Zhu, W. Huang, W. Liu, S. Wu, X. Liu, Y. Gao, W. Zhang and Z. Tang, Angew. Chem., Int. Ed., 2011, 50, 11456–11459 CrossRef CAS PubMed.
- Y. Li, Y. Zhou, H. Y. Wang, S. Perrett, Y. Zhao, Z. Tang and G. Nie, Angew. Chem., Int. Ed., 2011, 123, 5982–5986 CrossRef.
- J. Deng, S. Wu, M. Y. Yao and C. Y. Gao, Sci. Rep., 2016, 6, 31595 CrossRef CAS PubMed.
- B. Mrabet, M. N. Nguyen, A. Majbri, S. Mahouche, M. Turmine, A. Bakhrouf and M. M. Chehimi, Surf. Sci., 2009, 603, 2422–2429 CrossRef CAS.
- C. Yoshikawa, Y. Hashimoto, S. Hattori, T. Honda, K. Zhang, D. Terada, A. Kishida, Y. Tsujii and H. Kobayashi, Chem. Lett., 2010, 39, 142–143 CrossRef CAS.
- C. Yoshikawa, S. Hattori, T. Honda, C. F. Huang and H. Kobayashi, Mater. Lett., 2012, 83, 140–143 CrossRef CAS.
- J. E. Gagner, M. D. Lopez, J. S. Dordick and R. W. Siegel, Biomaterials, 2011, 32, 7241–7252 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- J. Song, E. Saiz and C. R. Bertozzi, J. Am. Chem. Soc., 2003, 125, 1236–1243 CrossRef CAS PubMed.
- F. J. Xu, J. P. Zhao, E. T. Kang, K. G. Neoh and J. Li, Langmuir, 2007, 23, 8585–8592 CrossRef CAS PubMed.
- T. Ren, Z. Mao, J. Guo and C. Gao, Langmuir, 2013, 29, 6386–6395 CrossRef CAS PubMed.
- J. O. You and D. T. Auguste, Biomaterials, 2008, 29, 1950–1957 CrossRef CAS PubMed.
- N. Shibuya-Fujiwara, F. Hirayama, Y. Ogata, H. Ikeda and K. Ikebuchi, Life Sci., 2001, 70, 291–300 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi and C. Reinhardt, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- S. Dominguezmedina, L. Kisley, L. J. Tauzin, A. Hoggard, B. Shuang, S. Chen, L. Y. Wang, P. J. Derry, A. Liopo and E. R. Zubarev, ACS Nano, 2016, 10, 2103–2112 CrossRef CAS PubMed.
- N. A. Monteiro-Riviere, M. E. Samberg, S. J. Oldenburg and J. E. Riviere, Toxicol. Lett., 2013, 220, 286–293 CrossRef CAS PubMed.
- C. D. Walkey and W. C. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00163g |
| This journal is © the Partner Organisations 2017 |