
Exploring the potential of linear polymer structures for the synthesis of fluorescent gold nanoclusters†
Nik Nik M.
Adnan
a,
Syafiq
Ahmad
a,
Rhiannon P.
Kuchel
b and
Cyrille
Boyer
*ac
aAustralian Centre for Nanomedicine (ACN), School of Chemical Engineering, University of New South Wales, Sydney, Australia 2052. E-mail: cboyer@unsw.edu.au
bElectron Microscope Unit (EMU), Mark Wainwright Analytical Centre, University of New South Wales, Sydney, Australia 2052
cCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, University of New South Wales, Sydney, Australia 2052. E-mail: cboyer@unsw.edu.au
First published on 17th August 2016
Abstract
In this study, fluorescent gold nanoclusters (AuNCs) were synthesized via the one-pot reduction of gold ions (Au3+) in the presence of thiolated copolymers. Well-defined copolymers, which are comprised of oligo(ethylene glycol) methyl ether methacrylate (OEGMA) and 2-(acetylthio)ethyl methacrylate (AcSEMA) monomers in either a block or random structure, were prepared via reversible addition–fragmentation chain transfer (RAFT) polymerization. Following deprotection of the AcSEMA thioester pendant group to yield a thiol, the formation of gold nanoclusters was performed at various relative molar concentrations of thiol to gold ions in order to investigate the effect on the fluorescent properties. Random copolymer stabilized gold nanoclusters (R@AuNCs) displayed higher emission intensity in comparison to block copolymer stabilized gold nanoclusters (B@AuNCs). In aqueous media, the hydrodynamic diameter of B@AuNCs (10.4–13.4 nm) were relatively larger compared to the R@AuNCs (5.9–6.6 nm) as determined by dynamic light scattering (DLS). However, the AuNCs cores were estimated to be similar in size according to the Jellium model (≅0.49 nm, ∼Au25), indicating the negligible effect of different polymer structure on the size of the fluorescent AuNCs core. Interestingly, these nanoclusters displayed linear temperature-dependent fluorescence emission intensity (≅0.7% °C−1) which may be important in biosensing applications.
Introduction
Fluorescent gold nanoclusters (AuNCs) are an emerging class of nanomaterials since they offer significant advantages over small molecule organic dyes1 and quantum dots1–3 for biomedical applications. For example, these metal nanoclusters are very appealing for intracellular fluorescence imaging and sensing due their low photobleaching,4 long fluorescence lifetime5,6 and well-known inert properties associated with elemental gold.7 Furthermore, AuNCs display interesting biological properties including effective renal clearance, long circulation times and effective tumor accumulation.8,9 These metal clusters are comprised of a few to several hundreds of gold atoms with a typical diameter less than 2 nm, a size comparable to the Fermi wavelength of an electron and small enough for the quantum confinement effect to take place. As a result, AuNCs exhibit molecule-like and distinct photo-physical behavior. Unlike gold nanoparticles (>2 nm),10–12 AuNCs do not possess the localized surface plasmon resonance property. Instead, they possess discrete size-dependent electronic states and size-dependent fluorescence behaviour.13–16Currently, the origin of the fluorescence is not fully understood. Theories have been proposed that indicate that the fluorescence of AuNCs could be related to the quantization of the metal core (quantum confinement effect) as well as ligand-to-metal charge transfer (surface ligand effect).17,18 In a study performed by Dickson et al., AuNCs with different sizes yielded fluorescence excitation and emission at different wavelengths, i.e. the larger size AuNCs core displays emission at a lower energy (longer wavelength).17 Jin et al. also discovered that electron-rich ligands play a key role in improving the fluorescence properties.18 Furthermore, Xie et al. highlighted the effect of AuNCs degree of aggregation on the luminescence properties via an aggregation-induced emission (AIE) mechanism.19,20 In recent years, great effort has been devoted to the synthesis of fluorescent AuNCs that exert excellent photoluminescence properties using a variety of ligands including thiol-bearing molecules and oligomers,5,21,22 dendrimers,17,23 peptides,19 proteins,24,25 DNA26 and synthetic polymers.27–32 Among these highly fluorescent AuNCs, long lifetime fluorescence within the near-infrared (NIR) region is particularly attractive for deep tissue imaging simply due to the superior NIR light penetration through biological tissue and blood as well as reduced background fluorescence.5,21,24,32,33
However, there are a limited number of studies exploring the potential of synthetic polymers for the synthesis of AuNCs.27–32 Tan and co-workers reported blue fluorescent AuNCs stabilized by multidentate thioether homopolymers (poly(vinyl acetate) (PVAc), poly(methyl methacrylate) (PMMA), poly(n-butyl methacrylate) (PnBMA) and poly(tert-butyl methacrylate (PtBMA)).28,31 Among these hybrid polymer nanoclusters, the AuNC stabilized by the PtBMA recorded the highest quantum yield in THF (20.1%). However, the AuNCs could not be dispersed in water, thus limiting its translation into biomedical application. The same group also studied the synthesis of AuNCs using multidentate thioether poly(methacrylic acid), PMAA.29,30,32 The resulting AuNCs were dispersible in water and were found to have a red fluorescence with quantum yield of up to 5.3%. Although the multidentate thioether polymeric ligands have been successfully employed to grow fluorescent AuNCs in the blue and red spectrum, the AuNCs exhibited weak fluorescence dependence towards temperature.28–32 In these seminal works, the effect of the polymer architecture was not extensively investigated.
Inspired by the study performed by Jin et al.,18 the fluorescent AuNCs were prepared by employing thiolated copolymers with different polymer architectures (block and random copolymer) synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization.34–38 The different monomer arrangement in the polymer backbone can be correlated with the polymer electron donating capability to the metal core via the Au–S bond, resulting in different fluorescence intensities of the AuNCs. The thiol moieties were introduced into the polymer by copolymerization of 2-(acetylthio)ethyl methacrylate (AcSEMA), a monomer bearing the protected thiol and oligo(ethylene glycol) methyl ether methacrylate (OEGMA), a monomer having poly(ethylene glycol) (PEG) group. The PEGylated polymer ligand confers remarkable properties to AuNCs, including improved colloidal stability in a biological system through steric stabilization. Suitable colloidal stability can be difficult to achieve when using thiolated small molecule ligands as stabilizers for AuNCs, particularly in the presence of high salt concentrations and varying pH. More importantly, the AuNCs prepared by our approach showed photoluminescence in the red region of the visible spectrum (λEm = 640–710 nm) which is advantageous for its higher depth penetration in biological systems. In previous studies, polymer stabilized AuNCs showed an independent emission intensity change versus temperature.28 In accordance with other studies,6,39,40 we observed a strong linear dependence of AuNCs emission intensity on temperature for each polymer architecture (0.73 and 0.71% °C−1 for the random and block copolymer respectively).
Materials and method
Materials
4-Cyanopentanoic acid dithiobenzoate (279.4 g mol−1, CPADB) was prepared according to the approach of Mitsukami et al.41 2,2′-Azobisisobutyronitrile (AIBN, Wako Chemicals) was crystallized twice from methanol before use. 2-Bromoethanol (Sigma, 95%), methacryloyl chloride (Sigma 97%), oligo(ethylene glycol) methyl ether methacrylate (OEGMA, Sigma, 300 g mol−1), potassium thioacetate (Sigma, 98%), gold(III) chloride trihydrate, (Sigma, HAuCl4·3H2O, ≥99.9%), acetonitrile (Sigma, HPLC), N,N-dimethylacetamide (Sigma, HPLC), dichloromethane (Merck) and triethylamine (Scharlau, 99.5%) were used without further purification. Diethyl ether, acetone, n-hexane, petroleum spirit, ammonia, dimethylformamide (DMF), magnesium sulphate (MgSO4) and sodium bicarbonate (NaHCO3) were obtained from Chem-Supply Australia and used without further purification. Deuterated dimethylsulfoxide (DMSO-d6) and chloroform (CDCl3) were obtained from Cambridge Isotope Laboratories, Inc. Milli-Q water (17.8 mΩ cm) was obtained using the Milli-Q purification system.Instrumental analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1. Polymer solutions at 2–3 mg mL−1 were prepared in the eluent and filtered through 0.45 µm filters prior to injection.
000000 g mol−1. Polymer solutions at 2–3 mg mL−1 were prepared in the eluent and filtered through 0.45 µm filters prior to injection.
Experimental procedures
Step 1: In a 200 mL round bottom flask equipped with a stirrer bar, 2-bromoethanol (10.0 g, 0.08 mol) was dissolved in 40 mL of dichoromethane. Triethylamine (13.4 mL, 0.1 mol) in 10 mL dichloromethane was added gradually into the 2-bromoethanol solution. The mixture was stirred for 30 min then cooled to 0 °C in an ice bath. A solution of methacryloyl chloride (12.5 g, 0.12 mol) in 10 mL dichloromethane was added dropwise into the mixture in 15 min at 0 °C and quickly formed a slurry reaction mixture. The reaction mixture was stirred overnight at room temperature and filtered. The filtrate was washed thoroughly by 3 × 100 mL of 1 M hydrochloric (HCl) acid and 3 × 100 mL of sodium bicarbonate (NaHCO3) solution and dried by anhydrous magnesium sulphate (MgSO4). Solvent was removed under vacuum by rotary evaporator at room temperature to yield 2-bromoethyl methacrylate in the form of yellow oil (total yield 67%). 1H-NMR (300 MHz, CDCl3): δ (ppm) 6.17 (s, 1H, CHH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)–), 5.62 (s, 1H, CHHC(CH3)–), 4.45 (t, 2H, CH2–O–), 3.56 (t, 2H, CH2–Br), 1.96 (s, 3H CH2C(CH3)–).
C(CH3)–), 5.62 (s, 1H, CHHC(CH3)–), 4.45 (t, 2H, CH2–O–), 3.56 (t, 2H, CH2–Br), 1.96 (s, 3H CH2C(CH3)–).
Step 2: Potassium thioacetate (5.2 g, 0.05 mol) was dissolved in 100 mL of acetone. 2-Bromoethyl methacrylate (8.0 g, 0.04 mol) in 10 mL acetone was then added dropwise while stirring at room temperature. The reaction mixture was stirred for 24 h at room temperature and filtered. Acetone was then removed from the crude product by rotary evaporator at room temperature and dichloromethane was added to re-dissolve the crude product. The crude product was washed with 5 × 100 mL NaHCO3 solution and 100 mL water. The product was dried with MgSO4 and solvent was removed by rotary evaporator to yield a brown oil product. The brown oil product was further purified through a silica flash column chromatography using mixture of hexane and ethyl acetate (19:1 v/v), which resulted in 4.5 g of the product, 2-(acetylthio)ethyl methacrylate in a form of brown oil (total yield 58%). 1H-NMR (300 MHz, CDCl3): δ (ppm) 1.95 (s, 3H, CH3C(CH2)), 2.25 (s, 3H, CH3CO), 3.18 (t, 2H, J = 7.5 Hz, CH2S), 4.22 (t, 2H, J = 7.5 Hz, CH2OCO), 5.48 (m, 1H, CHHC), 6.04 (m, 1H, CHHC) (Fig. S1, ESI†).
:[CPADB]:[AIBN] = 50.0:1.0:0.2. The reaction mixture was degassed with nitrogen for 30 min. The degassed solution was then immersed in a pre-heated oil bath at 70 °C for 6 h. The reaction was then placed in an ice bath for about 15 min to terminate the polymerization and the crude aliquot was sampled for 1H-NMR analysis to determine OEGMA monomer conversion (61%). OEGMA monomer conversion was determined via1H-NMR spectroscopy by the following equation: where ∫ is the peak integral of monomer (vinyl proton at 5.6 ppm, 1H) and the polymer (ester proton at 4.1 ppm, 2H).
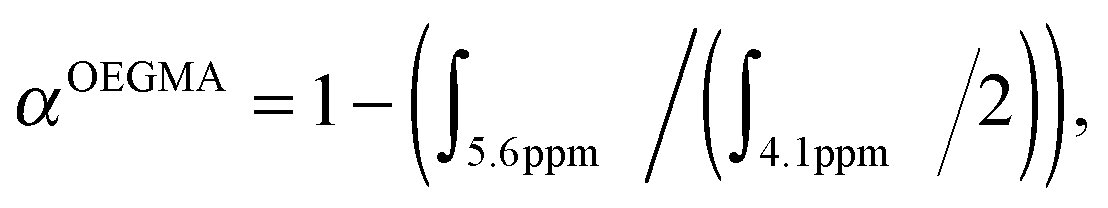
The polymer product was precipitated in petroleum spirit (boiling range 40–60 °C) and centrifuged (7000 rpm for 5 min). The precipitation and centrifugation steps were repeated three times to remove any traces of unreacted monomer and then the reaction medium was dried in a vacuum oven (40 °C). The purified P(OEGMA) was finally analyzed by 1H-NMR and SEC. Mn,NMR was calculated to be 9400 g mol−1 and SEC results yielded Mn,SEC of 12600 g mol−1 with polydispersity index (Đ) of 1.11.
:[AcSEMA]:[AIBN] = 1:25:0.2. The reaction mixture was then degassed with nitrogen for 20 min. The degassed solution was immersed in a pre-heated oil bath at 70 °C for 6 h. The reaction was then placed in an ice bath for about 15 min to terminate the polymerization. An aliquot was collected after polymerization for 1H-NMR analysis to determine AcSEMA monomer conversion (70%). AcSEMA monomer conversion was determined via1H-NMR spectroscopy by comparing the peak integral of monomer at t = 0 and t = 6 h using the following equation: where ∫ is the peak integral of monomer (vinyl proton at 5.4 ppm, 1H) and the polymer (CH2–S proton at 3.1 ppm, 2H).
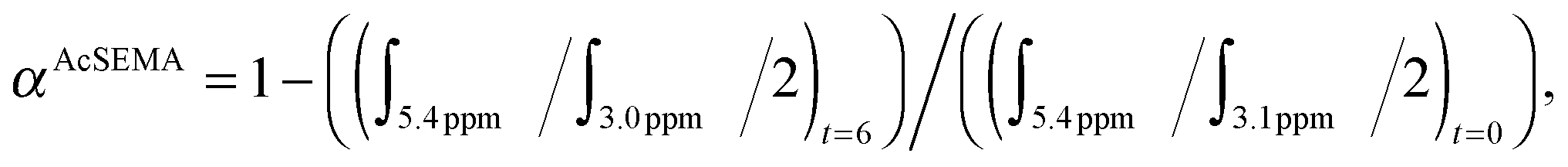
The product was precipitated in a mixture of diethyl ether and petroleum spirit (3:7 v/v) and centrifuged (7000 rpm for 5 min). The purification process was repeated three times and the reaction medium was dried in vacuum oven (40 °C). The purified copolymer product was analyzed using SEC (Mn,SEC = 16100 g mol−1, Đ = 1.12) and 1H-NMR (Mn,NMR = 12700 g mol−1).
:[AcSEMA]:[CPADB]:[AIBN] = 50.0:25:1.0:0.175. The reaction mixture was degassed with nitrogen for 30 min. The degassed solution was then immersed in a pre-heated oil bath at 70 °C for 6 h. The reaction was then placed in an ice bath for about 15 min to terminate the polymerization and the crude aliquot was sampled for 1H-NMR analysis to determine OEGMA and AcSEMA total monomer conversion (57%). Total monomer conversion was determined via1H-NMR spectroscopy by the following equation: where ∫ is the peak integral of monomer (vinyl proton at 5.6 ppm, 1H) and the polymer (ester proton at 4.1 ppm, 2H).
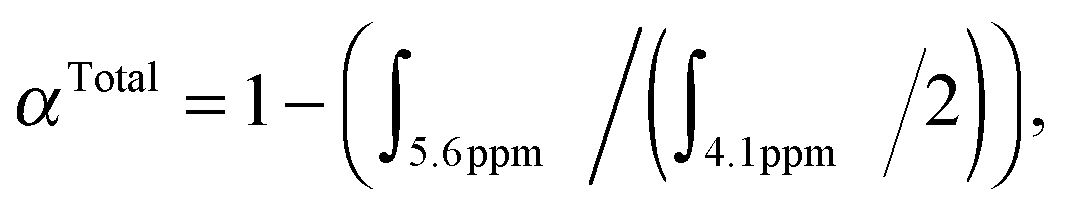
The polymer product was precipitated in petroleum spirit (boiling range 40–60 °C) and centrifuged (7000 rpm for 5 min). The precipitation and centrifugation steps were repeated three times to remove any traces of unreacted monomer and then the reaction medium was dried in a vacuum oven (40 °C). The purified P(OEGMA-co-AcSEMA) was finally analyzed by 1H-NMR and SEC to determine monomer repeating unit and Đ. Mn,NMR was calculated to be 10800 g mol−1 and SEC results yielded Mn,SEC of 13100 g mol−1 with Đ of 1.18.
:1 v/v). Then, an aqueous solution of HAuCl4 (0.50 mL, 20 mM) was added into the polymer solution at 25 °C and the reaction mixture was heated to 60 °C for 24 h under gentle stirring (500 rpm). The reaction mixture was then subjected to centrifugation (14800 rpm, 5 min) to isolate aggregates and the yellow-colored supernatant was collected. The supernatant was further purified by dialysis against Milli-Q water in a dialysis tubing cellulose membrane (≈14000 MWCO) to remove free, unreacted gold ions and DMF. The gold nanoclusters were finally dried, re-dissolved in 3 mL of Milli-Q water and subjected to extensive characterizations by ICP-OES, UV-Vis, DLS, ATR-FTIR, fluorescence and XPS.
Results and discussion
Synthesis and characterization of copolymer
First, 2-(acetylthio)ethyl methacrylate (AcSEMA), a methacrylic monomer with an acetyl-protected thiol (thioester) pendant group was synthesized (ESI,† Fig. S1).42,43 In this work, reversible addition–fragmentation chain transfer (RAFT) polymerization technique was employed to synthesize the well-defined linear copolymers, using CPADB and AIBN as RAFT agent and thermal initiator respectively (Fig. 1). OEGMA was chosen as the co-monomer as it offers several advantages for biological application including enhance biocompatibility, antifouling properties and solubility in aqueous medium.44,45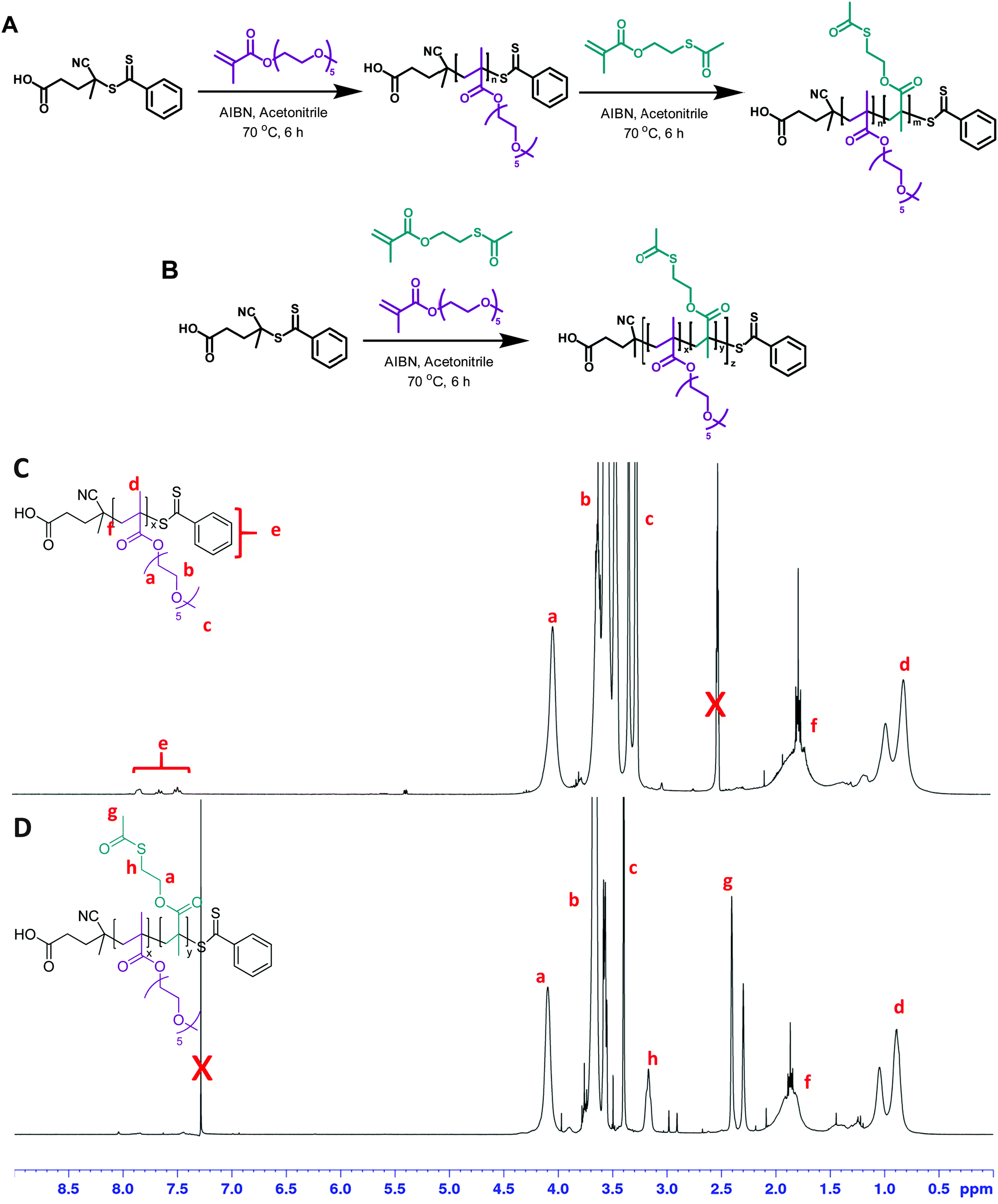 | ||
| Fig. 1 Synthetic procedures to the (A) block copolymer P(OEGMA)-b-P(AcSEMA) and (B) random copolymer P(OEGMA-co-AcSEMA) via conventional RAFT polymerization employing CPADB as the RAFT agent and AIBN as the thermal initiator at 70 °C. 1H-NMR analysis of (C) P(OEGMA) in DMSO-d6 and (D) P(OEGMA)-b-P(AcSEMA) in CDCl3 confirmed the successful polymerization of OEGMA and AcSEMA. | ||
Two copolymers, a random and a block copolymer, were prepared. The block P(OEGMA)-b-P(AcSEMA) copolymer was prepared by sequential polymerization, where the P(OEGMA) homopolymer was first prepared and then chain extended with AcSEMA. The random P(OEGMA-co-AcSEMA) copolymer was synthesized by copolymerization of OEGMA and AcSEMA in one pot. In both cases, the polymerizations proceeded in controlled fashion as indicated by SEC analysis (Đ < 1.4) (Table 1). Furthermore, 1H-NMR analysis indicated the successful polymerization of the monomers by the appearance of signals corresponding to the ester (4.1 ppm), methyl of thioester (2.4 ppm) and thioester (3.1 ppm) proton (Fig. 1). The copolymer composition was determined by NMR to be 30/18 and 27/13 repeating units of OEGMA/AcSEMA for the block and random copolymer, respectively. ATR-FTIR analyses also confirmed the presence of signals attributed to the ether (1100 cm−1) and ester (1740 cm−1) functionalities in the P(OEGMA) homopolymer as well as methyl thioester (610 cm−1) and thioester (1690 cm−1) in the copolymer of OEGMA and AcSEMA, which are in agreement with the data published in literature (Fig. 2(B)).43,46,47
| Polymer | [M1]:[M2]:[RAFT]:[I]a |
α (%) | M n,NMR (g mol−1) | M n,SEC (g mol−1) | Đ | F AcSEMA |
|---|---|---|---|---|---|---|
| a M1, M2 and I correspond to OEGMA, AcSEMA and initiator (AIBN). b Total monomer conversion (α) was calculated by 1H-NMR of the reaction mixture by the ratio of vinyl signal at 5.6 ppm to the ester signal at 4.1 ppm for P(OEGMA), P(OEGMA)-b-P(AcSEMA) and P(OEGMA-co-AcSEMA). c Molecular weights were calculated from the equation Mn,NMR = ([M]o/[RAFT]o) × α × Fmonomer × Mw,monomer + Mw,RAFT, where [M]o, [RAFT]o, α, F, Mw,monomer, and Mw,RAFT represent monomer and RAFT agent molar ratio, monomer conversion, ratio of the monomer in the polymer, molecular weight of monomers and RAFT agent, respectively. Depending on the polymerization, the RAFT agent can be the CPADB or the P(OEGMA) macroRAFT. d M n,SEC and polydispersity (Đ) was measured by SEC using N,N′-dimethyl acetamide (DMAc) as eluent and poly(methyl methacrylate) standards. e AcSEMA monomer unit was determined by 1H-NMR of the purified polymer samples by comparing the ester signal at 4.1 ppm and thioester signal at 3.1 ppm. | ||||||
| P(OEGMA) | 50:0:1:0.2 |
61 | 9400 | 12600 |
1.11 | — |
| P(OEGMA)-b-P(AcSEMA) | 0:25:1:0.2 |
70 | 12700 |
16100 |
1.12 | 0.34 |
| P(OEGMA-co-AcSEMA) | 50:25:1:0.175 |
57 | 10800 |
13100 |
1.18 | 0.32 |
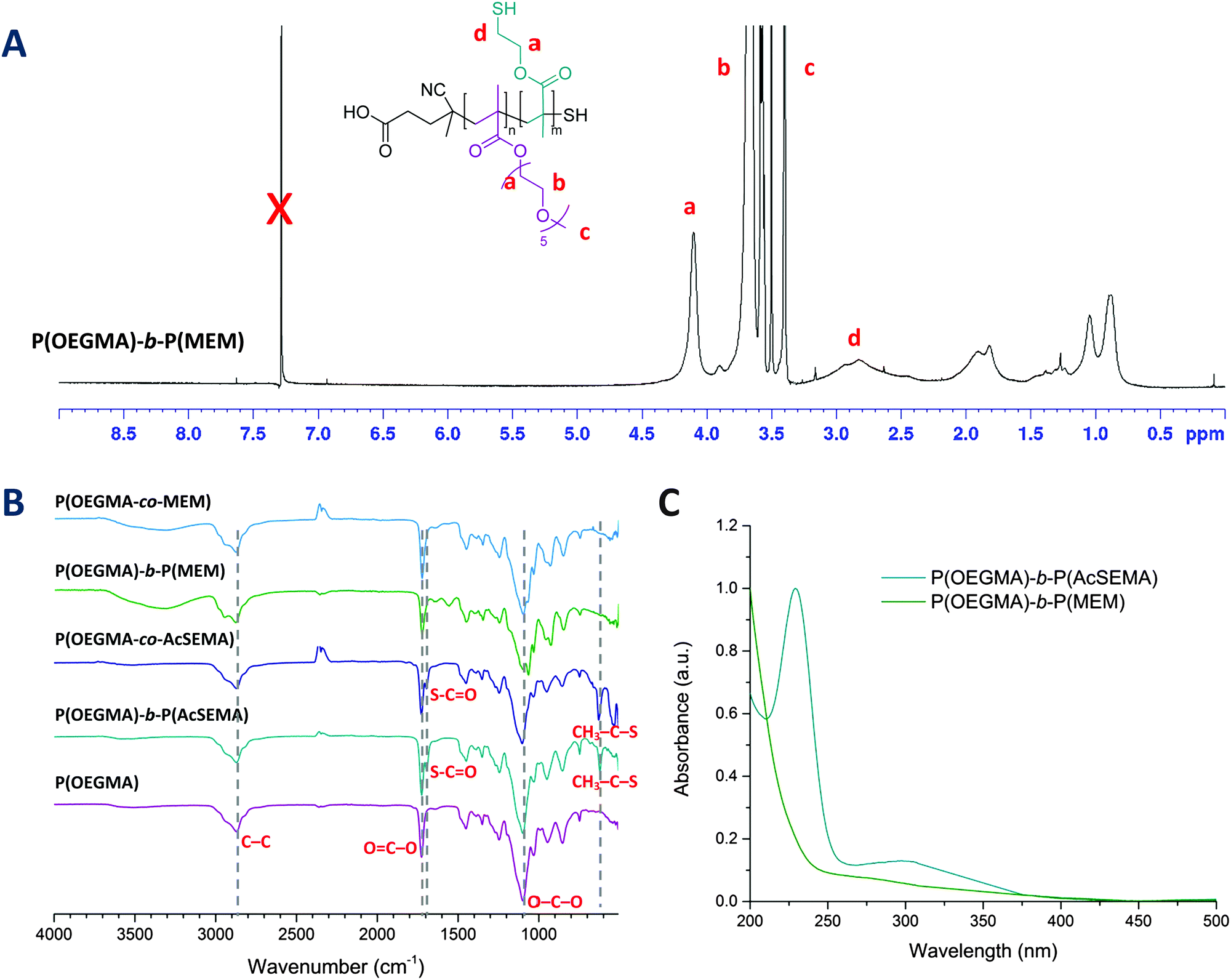 | ||
| Fig. 2 (A) 1H-NMR of the block copolymer after deprotection, P(OEGMA)-b-P(MEM) in CDCl3 confirming the disappearance of proton signals from thioester (3.1 ppm) and methyl thioester (2.4 ppm). (B) ATR-FTIR spectra of the polymers. (C) UV-Vis spectra of the block copolymer before and after deprotection showing the disappearance of RAFT chain end absorbance. | ||
The acetyl-protected thiols in the copolymers were then subjected to hydrolysis by using excess ammonia, yielding thiol pendant groups identified as 2-mercaptoethyl methacrylate (MEM) in the copolymer backbone.46 Deprotection was performed under a nitrogen atmosphere in order to limit the spontaneous oxidation of thiols to form disulphide bonds in the presence of oxygen.48 After purification by dialysis, the copolymers were sampled for 1H-NMR and FTIR analyses. 1H-NMR analysis of the copolymers confirmed the disappearance of the signal from the thioester methyl protons (2.4 ppm) and the broadening of the signal from the thioester proton adjacent to the thiol (3.1 ppm), indicating successful removal of thioester protecting group (Fig. 2(A)).47 Similar results were obtained from ATR-FTIR analysis, in which both the methyl thioester and thioester signals disappeared (Fig. 2(B)). Additionally, the degradation of RAFT end group was also noted by UV-Vis analysis after ammonia treatment due to the loss of the characteristic dithiobenzoate absorption at 300 nm (Fig. 2(C)). This was attributed to aminolysis of the RAFT chain end in basic aqueous conditions.49
Formation and photophysical characterization of fluorescent gold nanoclusters (AuNCs)
The AuNCs were synthesized utilizing the deprotected copolymers as both reducing and stabilizing agents. In a typical reaction, freshly prepared copolymer samples were dissolved in mixture of Milli-Q water and N,N-dimethylformamide (DMF) (8:1 v/v) and HAuCl4 solution was added rapidly. Upon mixing, the yellow-colored HAuCl4 solution immediately turned colorless and then the reaction mixture was heated to 60 °C for 24 h to form the light yellowish fluorescent AuNCs.
The formation of AuNCs was first optimized by varying the relative molar concentration of thiol moiety in the copolymer to HAuCl4 salt. In general, an excess of thiol relative to the HAuCl4 was necessary for the formation of AuNCs, which is consistent with the synthesis of AuNCs with small ligands.21 At thiol:HAuCl4 = 1:1, the formation of gold nanoparticles was observed along with the AuNCs resulting in a red to purple solution mixture. The formation of non-fluorescent gold nanoparticles for thiol:HAuCl4 = 1:1 was also confirmed by UV-Vis analysis by the appearance of the characteristic surface plasmon resonance absorbance at 500–600 nm (ESI,† Fig. S4). By increasing the relative molar concentration of thiol group, we observed a reduction in the yield of gold nanoparticles. At the highest relative concentration studied, the formation of gold nanoparticles and aggregates was completely suppressed to yield exclusively AuNCs as indicated by the light yellowish color (ESI,† Fig. S5). Furthermore, after purification, inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis of the AuNCs revealed up to 40% and 26% reaction yield for the B@AuNCs and R@AuNCs respectively (Table 2).
| Thiol:HAuCl4a |
d DLS (nm) | Yieldc (%) | λ Ex/λEm (nm) | Stoke shiftd (nm) | d core (nm) | Aunf | |
|---|---|---|---|---|---|---|---|
| a The relative concentration of sulphur moiety in the copolymer to HAuCl4. Number of sulphur repeating units was determined by 1H-NMR. b The hydrodynamic diameter, d is the number-average size determined from DLS. c The yield was calculated by the relative concentration of starting concentration of gold reactant and the gold concentration in the AuNCs determined by ICP-OES. d Stoke shift values were measured from the differences in the maxima of fluorescence excitation and emission of the AuNCs. e AuNCs core size, dcore was determined by Jellium model. f Number of Au atoms in the AuNCs core, Aun was determine by Jellium model. | |||||||
| B@AuNCs | 3:1 |
13.3 ± 0.3 | 15.4 | 390/660 | 270 | 0.50 | 26 |
| 5:1 |
13.4 ± 0.4 | 19.0 | 390/660 | 270 | 0.50 | 26 | |
| 7:1 |
10.4 ± 0.1 | 40.0 | 390/650 | 260 | 0.49 | 25 | |
| R@AuNC | 3:1 |
6.6 ± 0.1 | 10.5 | 390/710 | 320 | 0.54 | 33 |
| 5:1 |
5.9 ± 0.2 | 26.3 | 400/640 | 240 | 0.49 | 24 | |
| 7:1 |
5.9 ± 1.4 | 25.8 | 400/640 | 240 | 0.49 | 24 | |
Fluorescence emission and excitation of the AuNCs were studied by fluorescence spectroscopy. As the polymers alone were found to be non-fluorescence (ESI,† Fig. S7), the observed fluorescence was found to be originating from the formed AuNCs. The AuNCs displayed fluorescence excitation between 200–500 nm and strong emission from 540 nm to 870 nm for both block and random copolymers. Interestingly, the fluorescence maxima of these AuNCs were slightly different according to the polymer structure and the ratio of thiol:HAuCl4 (Table 2). Furthermore, at a similar gold concentration of 0.2 mg mL−1 (as determined by ICP-OES), the emission intensity of AuNCs prepared using block copolymer, B@AuNCs, were lower than the ones made using random copolymer, R@AuNCs. As the relative molar concentration of thiol group was increased, the B@AuNCs displayed an increase in emission intensity (Fig. 3(A)). In contrast, the emission intensity of R@AuNCs increased up to thiol:HAuCl4 = 5:1, and then was reduced at higher ratio (Fig. 3(B)). Taking into account the results for R@AuNCs, we believe the optimum conditions were achieved by the random copolymer at relative molar concentration of five.
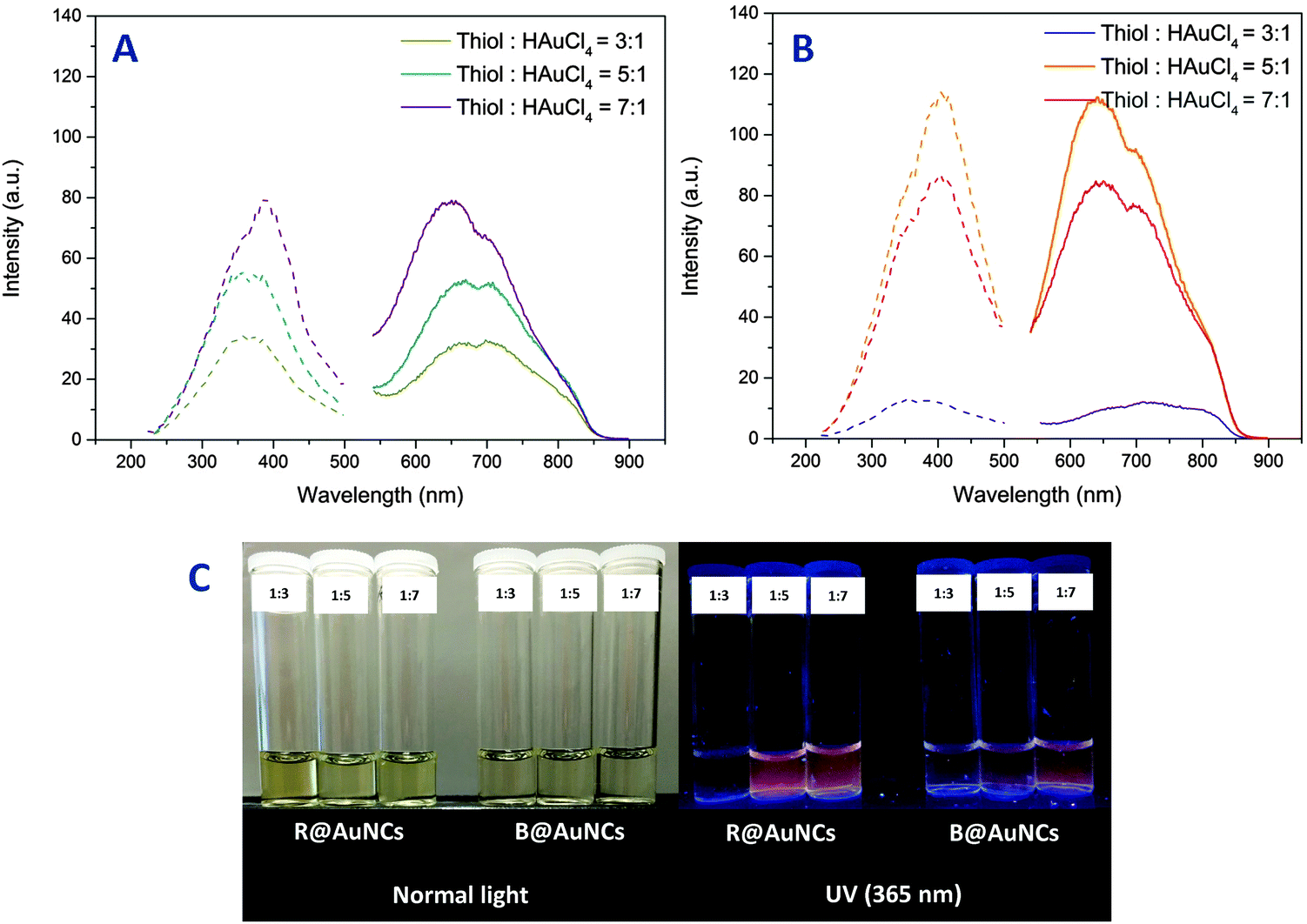 | ||
| Fig. 3 (A) Fluorescence spectra of the B@AuNCs and (B) R@AuNCs. (C) The AuNCs solution under normal and UV light (365 nm) irradiation. Note: the AuNCs were analyzed and imaged at gold concentration of 0.2 mg mL−1 as determined by ICP-OES analyses. | ||
At the optimized ratio of thiol:HAuCl4, the AuNCs were only weakly fluorescent as confirmed by quantum yield calculations using coumarin 153 (QY: 53%)50 as a standard. The random and block copolymer stabilized AuNCs recorded quantum yield values of 0.24% and 0.17% respectively. The values are 107 higher compared to the native fluorescence of bulk gold (1 × 10−8%)51 and comparable to Au25SG18 clusters reported in literature.52 In contrast to the non-fluorescent gold nanoparticles, the fluorescent gold nanoclusters contain a substantial amount of Au(I) oxidation state in the form of a Au(I) ligand complex.14 The presence of the Au(I) oxidation state was observed in various fluorescent AuNCs experimentally. The Au(I) oxidation state is stabilized by thiolate ligands and has been shown to play a critical role in the fluorescence generation.19,28,53 The AuNCs were further analyzed by X-ray photoelectron spectroscopy (XPS) to determine the composition in Au(I) and Au(0) of these AuNCs. The deconvolution of the Au 4f7 binding energy revealed the presence of Au(I) and Au(0) due to the presence of two signals at 84.0–84.2 and 84.6–84.8 eV corresponding to Au(I) and Au(0) respectively. Furthermore, the composition of Au(I) was higher for R@AuNCs (thiol:HAuCl4 = 5:1, Au(I) = 24%) compared to B@AuNCs (thiol:HAuCl4 = 7:1, Au(I) = 11%) (ESI,† Fig. S8). Interestingly, the lower Au(I) content of B@AuNCs was correlated to the lower emission intensity of B@AuNCs (thiol:HAuCl4 = 7:1) when compared to R@AuNCs (thiol:HAuCl4 = 5:1), confirming the effect of Au(I) content in the AuNCs on the intensity of fluorescence emission.
Transmission electron microscopy showed the size of the as-synthesized AuNCs was below 2 nm (ESI,† Fig. S9). Dynamic light scattering (DLS) measurements of the AuNCs in Milli-Q water revealed a relatively smaller number average size of R@AuNCs (5.9 ± 1.4 nm) compared to B@AuNCs (10.4 ± 0.1 nm) at the highest relative thiol molar ratio investigated (Table 2). This observation can be attributed to the difference in the monomer arrangement, which resulted in a significant difference in the size of the polymer shell surrounding the AuNCs core. In addition, further analysis of the R@AuNCs and B@AuNCs with SEC also supported the size determined by DLS. The copolymer stabilized gold nanoclusters were significantly larger compared to their respective single chain copolymers, Mn of ≅48000 and ≅109000 g mol−1 for the R@AuNCs and B@AuNCs respectively (ESI,† Fig. S10). Together, these observations suggest that the structure of the stabilizing copolymer is an important factor in the formation of AuNCs with different sizes in aqueous medium.
According to the Jellium model (ESI,† eqn (S1)), the emission frequency of an AuNCs is inversely proportional to the size (radius) of the metal clusters core.16,54,55 Dickson et al. showed that the model can accurately predict the core size of AuNCs with emission maxima in the range of 400–530 nm, however, a variation to the model has to be introduced into the equation for emission maxima of lower energy (λEm > 530 nm).16 Apart from the emission maxima of R@AuNCs (thiol:HAuCl4 = 3:1, λEm = 710 nm), the gold nanoclusters displayed an emission maxima at ≅640 ± 10 nm, corresponding to an estimated core size of ≅0.49 ± 0.01 nm (Au24 ∼ Au26). Thus based on the Jellium model, we hypothesized that the polymer structure has a negligible effect on the size of the fluorescent gold core, as indicated by the approximately similar emission maxima of the R@AuNCs and B@AuNCs. Our results are consistent with previous data reported in the literature (λEm ≅ 640–660 nm, ∼Au25),16,17,24,56,57 which identified that Au25 is the most stable form.58,59
We also explored the potential of the AuNCs as a temperature sensor by monitoring changes in the fluorescence intensity. It should be noted that the copolymers used in this study displayed lower critical solution temperature (LCST) behavior in water ranging from 40 to 85 °C (ESI,† Fig. S11). It is the point where polymer become more hydrophobic, decreasing the overall solubility of the polymers in solution. Interestingly however, the copolymer stabilized AuNCs did not exhibit these LCST-like properties in contrast to previous studies,60,61 which could be due to the presence of thiol group in the copolymer. Instead, the AuNCs exhibited a slow decrease in the fluorescence versus temperature without a shift in emission wavelength (Fig. 4 and 5). The fluorescence response with temperature recorded by the R@AuNCs (≈0.73% °C−1) was relatively similar to B@AuNCs (≈0.71% °C−1). In particular, the intensity of R@AuNCs decreased by 26% as the temperature increased from 20 to 50 °C; a range that is particularly relevant for biological applications. To the best of our knowledge, this is the first time such a property has been observed for AuNCs prepared using synthetic polymers. Others have reported AuNCs stabilized with BSA39 (≈1.02% °C−1) and DHLA6 (≈1.91% °C−1) which possess a similar temperature response. More importantly, the emission intensity of the AuNCs was highly reversible, indicative of the AuNCs excellent stability at elevated temperature, after multiple heating and cooling cycles (Fig. 5).
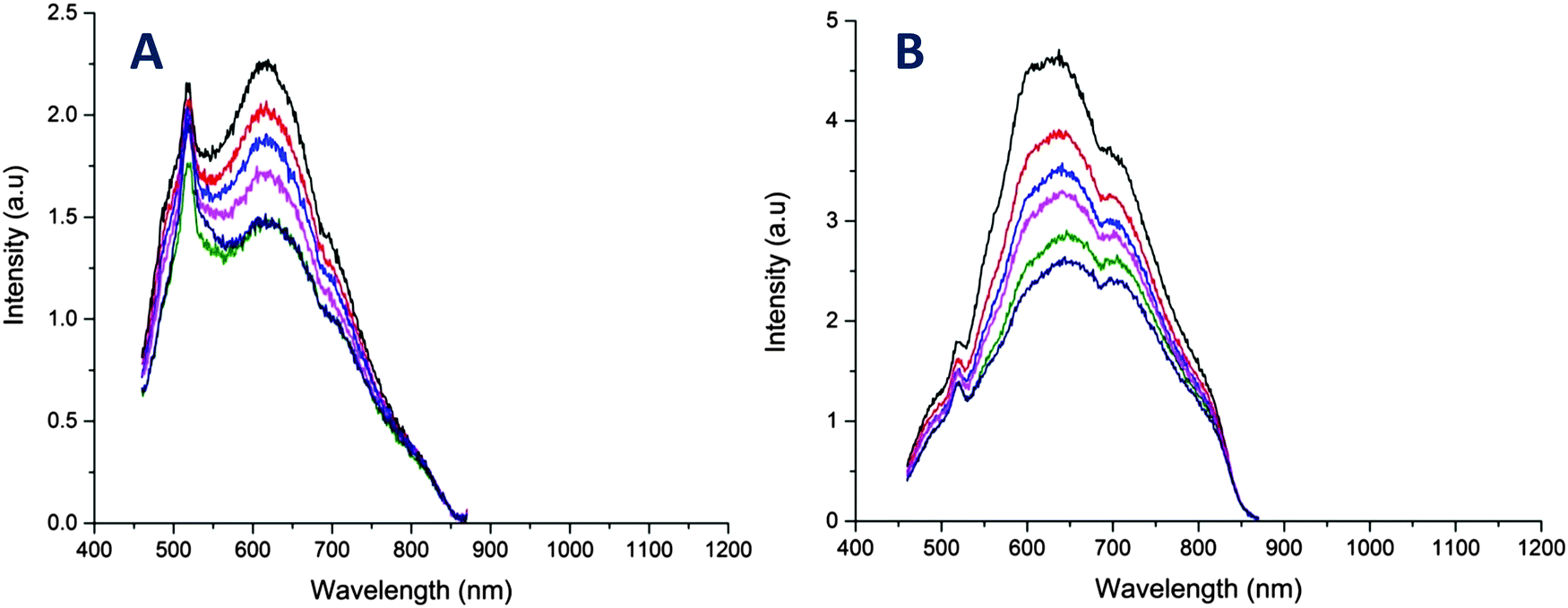 | ||
| Fig. 4 Temperature dependence of the fluorescence emission of AuNCs in Milli-Q water. (A) Fluorescence emission spectra of B@AuNCs for different temperature in the range of 30 to 80 °C. (B) Fluorescence emission spectra of R@AuNCs for different temperature in the range of 20 to 80 °C. Note: the spike centred at 525 nm is an artefact from the quartz cuvette, which was described by Z. Wu and R. Jin, for [Au25(SR)18] clusters.18 | ||
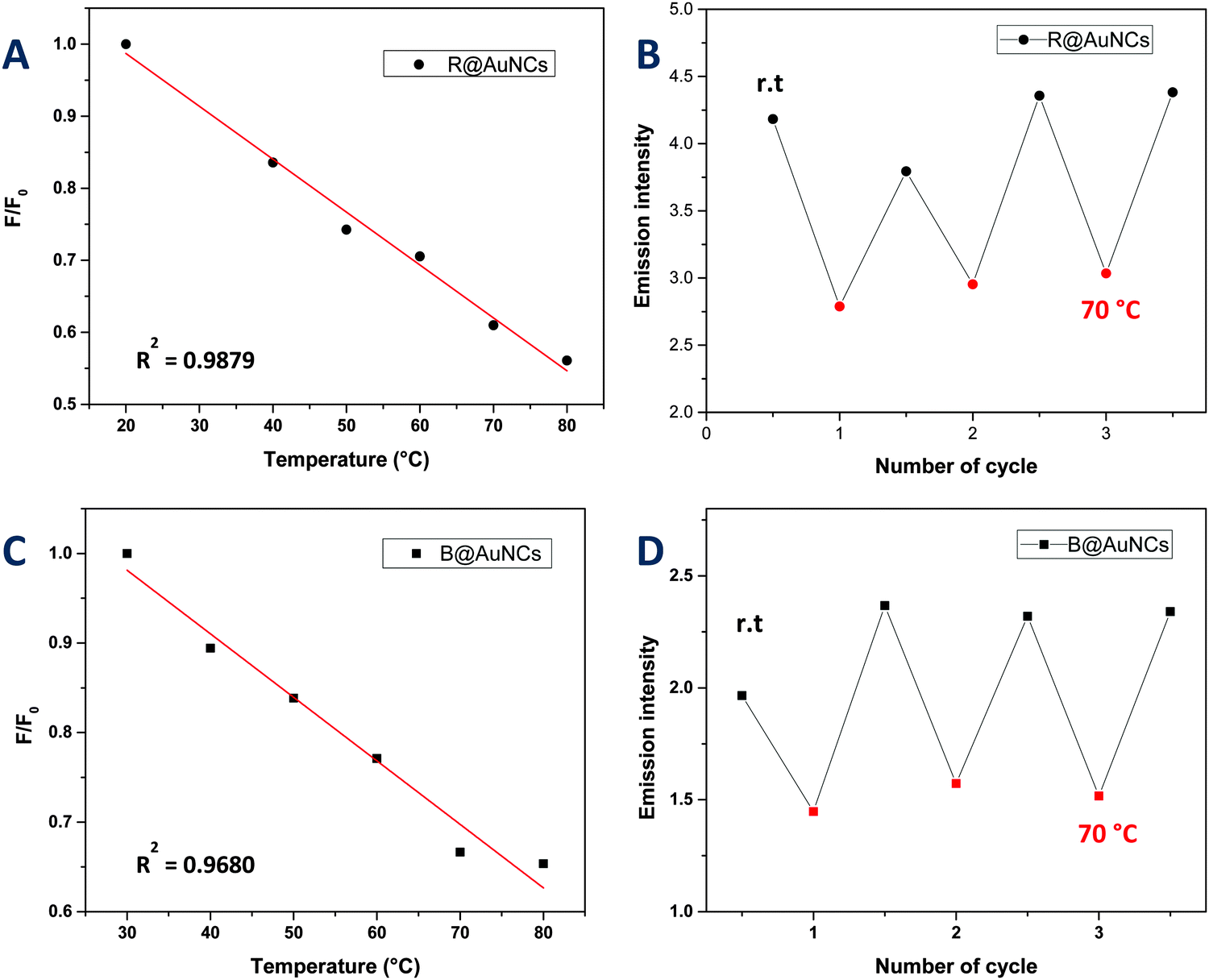 | ||
| Fig. 5 Temperature dependence of the fluorescence emission of the AuNCs in Milli-Q water. (A) Intensity of fluorescence emission of R@AuNCs at 640 nm was plotted against temperature range between 20–80 °C. (B) Reversible fluorescence emission of R@AuNCs at 640 nm upon cycling the temperature three times between room temperature and 70 °C. (C) Intensity of fluorescence emission of B@AuNCs at 650 nm was plotted against temperature range between 30–80 °C. (D) Reversible fluorescence emission of B@AuNCs at 650 nm upon cycling the temperature three times between room temperature and 70 °C. | ||
Conclusions
In this study, we have successfully fabricated AuNCs by employing copolymers synthesized with RAFT polymerization that act as both reducing and stabilizing agent. The random copolymer stabilized AuNCs exhibited superior fluorescence emission over block copolymer stabilized AuNCs. Although the fluorescence emissions were relatively weak, further enhancement on the quantum yield of the AuNCs can be achieved by incorporation of electron rich groups into the copolymer. Furthermore, the significant difference in the hydrodynamic size of the gold nanocluster coated with different polymer structures allow for specific size tuning for biological applications, especially to achieve long blood circulation and excellent tumor penetration in vivo. Although the hydrodynamic diameter is different for the random and block copolymer stabilized gold nanoclusters, the Jellium model suggested a relatively similar gold core size, indicated by the similar fluorescence emission maxima. Importantly, the AuNCs displayed fluorescence emission centered in the red region of the visible spectrum which may be important for deep tissue imaging and sensing. In addition to this, a marked emission intensity response to changes in temperature was noted and it is proposed that AuNCs could be used as a spatially resolved fluorescence thermometer in biological experiments.Acknowledgements
CB acknowledges the Australian Research Council (ARC) for his Future Fellowship and UNSW for funding (SPF01).References
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614–2620 CrossRef CAS PubMed.
- L. Shang, F. Stockmar, N. Azadfar and G. U. Nienhaus, Angew. Chem., Int. Ed., 2013, 52, 11154–11157 CrossRef CAS PubMed.
- Z. Luo, K. Zheng and J. Xie, Chem. Commun., 2014, 50, 5143–5155 RSC.
- X.-D. Zhang, J. Chen, Z. Luo, D. Wu, X. Shen, S.-S. Song, Y.-M. Sun, P.-X. Liu, J. Zhao, S. Huo, S. Fan, F. Fan, X.-J. Liang and J. Xie, Adv. Healthcare Mater., 2014, 3, 133–141 CrossRef CAS PubMed.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978–4981 CrossRef CAS PubMed.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 1699–1709 CrossRef CAS PubMed.
- W. Yan, L. Xu, C. Xu, W. Ma, H. Kuang, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 15114–15121 CrossRef CAS PubMed.
- L. Xu, Y. Zhu, W. Ma, W. Chen, L. Liu, H. Kuang, L. Wang and C. Xu, J. Phys. Chem. C, 2011, 115, 3243–3249 CAS.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073–4083 RSC.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509–2521 CrossRef CAS PubMed.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS PubMed.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- P. L. Xavier, K. Chaudhari, P. K. Verma, S. K. Pal and T. Pradeep, Nanoscale, 2010, 2, 2769–2776 RSC.
- T. A. C. Kennedy, J. L. MacLean and J. Liu, Chem. Commun., 2012, 48, 6845–6847 RSC.
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 2412–2413 CrossRef CAS PubMed.
- L. Li, Z. Li, H. Zhang, S. Zhang, I. Majeed and B. Tan, Nanoscale, 2013, 5, 1986–1992 RSC.
- N. Schaeffer, B. Tan, C. Dickinson, M. J. Rosseinsky, A. Laromaine, D. W. McComb, M. M. Stevens, Y. Wang, L. Petit, C. Barentin, D. G. Spiller, A. I. Cooper and R. Levy, Chem. Commun., 2008, 3986–3988, 10.1039/B809876J.
- H. Zhang, X. Huang, L. Li, G. Zhang, I. Hussain, Z. Li and B. Tan, Chem. Commun., 2012, 48, 567–569 RSC.
- X. Huang, B. Li, L. Li, H. Zhang, I. Majeed, I. Hussain and B. Tan, J. Phys. Chem. C, 2012, 116, 448–455 CAS.
- X. Huang, Y. Luo, Z. Li, B. Li, H. Zhang, L. Li, I. Majeed, P. Zou and B. Tan, J. Phys. Chem. C, 2011, 115, 16753–16763 CAS.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- X. Chen, J. B. Essner and G. A. Baker, Nanoscale, 2014, 6, 9594–9598 RSC.
- P. Yu, X. Wen, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 6567–6571 CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- M. Liras, I. Quijada-Garrido, M. Palacios-Cuesta, S. Munoz-Durieux and O. Garcia, Polym. Chem., 2014, 5, 433–442 RSC.
- M. P. Montero-Rama, M. Liras, O. García and I. Quijada-Garrido, Eur. Polym. J., 2015, 63, 37–44 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- M. Liras, O. Garcia, N. Guarrotxena, M. Palacios-Cuesta and I. Quijada-Garrido, Polym. Chem., 2013, 4, 5751–5759 RSC.
- M. T. Cook, S. A. Schmidt, E. Lee, W. Samprasit, P. Opanasopit and V. V. Khutoryanskiy, J. Mater. Chem. B, 2015, 3, 6599–6604 RSC.
- P. Nagy, Antioxid. Redox Signaling, 2013, 18, 1623–1641 CrossRef CAS PubMed.
- D. B. Thomas, A. J. Convertine, R. D. Hester, A. B. Lowe and C. L. McCormick, Macromolecules, 2004, 37, 1735–1741 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- A. Mooradian, Phys. Rev. Lett., 1969, 22, 185–187 CrossRef CAS.
- M. A. H. Muhammed, A. K. Shaw, S. K. Pal and T. Pradeep, J. Phys. Chem. C, 2008, 112, 14324–14330 CAS.
- C. Zhou, C. Sun, M. Yu, Y. Qin, J. Wang, M. Kim and J. Zheng, J. Phys. Chem. C, 2010, 114, 7727–7732 CAS.
- S. Chattoraj, A. Amin, B. Jana, S. Mohapatra, S. Ghosh and K. Bhattacharyya, ChemPhysChem, 2016, 17, 253–259 CrossRef CAS PubMed.
- S. Chattoraj and K. Bhattacharyya, J. Phys. Chem. C, 2014, 118, 22339–22346 CAS.
- K. Chaudhari, P. L. Xavier and T. Pradeep, ACS Nano, 2011, 5, 8816–8827 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC.
- M. Luzon, C. Boyer, C. Peinado, T. Corrales, M. Whittaker, L. Tao and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2783–2792 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, UV-Vis spectra, SEC traces, XPS data, fluorescence spectra (Fig. S1–S11). See DOI: 10.1039/c6qm00109b |
| This journal is © the Partner Organisations 2017 |