
Spin transitions in a series of [Fe(pybox)2]2+ complexes modulated by ligand structures, counter anions, and solvents†
Yuan-Yuan
Zhu
 *a,
Hong-Qing
Li
a,
Zhong-Yu
Ding
a,
Xiao-Jin
Lü
b,
Liang
Zhao
b,
Yin-Shan
Meng
c,
Tao
Liu
*b and
Song
Gao
c
*a,
Hong-Qing
Li
a,
Zhong-Yu
Ding
a,
Xiao-Jin
Lü
b,
Liang
Zhao
b,
Yin-Shan
Meng
c,
Tao
Liu
*b and
Song
Gao
c
aSchool of Chemistry and Chemical Engineering, Hefei University of Technology and Anhui Key Laboratory of Advanced Functional Materials and Devices, Hefei 230009, China. E-mail: yyzhu@hfut.edu.cn
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Road, Dalian 116024, China. E-mail: liutao@dlut.edu.cn
cBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 28th October 2016
Abstract
Herein, we investigate the spin transition properties in a series of mononuclear Fe(II) complexes constructed by two pybox ligands and four counter anions. 1(BF4) undergoes an abrupt spin-transition at around 150 K, which proceeds to 50% completeness and is accompanied by a structural phase transition. 1(BPh4)·MeCN, 2(ClO4), and 2(BF4) all display gradual and complete SCO behaviours around or above ambient temperature. The desolvated compound 1(BPh4) exhibits a gradual but incomplete SCO. 2(PF6)·MeCN, 2(PF6) and 2(BPh4) show a two-step SCO. The well-defined two-step SCO of 2(BPh4) takes place at 100–200 K (T1/2 = 150 K) and 400–500 K (T1/2 = 446 K). In contrast, compounds 1(BF4)·MeCN, 1(PF6)·MeCN·Et2O and 1(PF6) remain high-spin at the measured temperatures. The LIESST (light induced excited spin state trapping) effect of several SCO compounds are also investigated, which display photo-conversion during light irradiation at low temperature at a relatively high level. Our results demonstrate that the [Fe(pybox)2]2+ series of complexes will become a new versatile system in spin-crossover research.
Introduction
Recently, there have been big breakthroughs in switching materials at the molecular level in the field of chemistry, physics, and materials science. Among them, spin-crossover (SCO) complexes represent one of the most attractive examples of switching materials. In these complexes, the electronic states of metal ions can be reversibly switched between high-spin (HS) and low-spin (LS) states via external perturbations such as heat, pressure, light, solvent, and magnetic field.1,2 The spin-transition (ST) process is often accompanied by changes in several physical properties including magnetic moment, color, dielectric constant, structural phase transition, and electrical resistance.3–6 These novel properties have been widely investigated over the past several decades due to their potential applications in information storage, molecular switches, molecular electronics, and optical devices.7–11 In order to achieve practical or potential applications in molecular devices, the spin-transition should be abrupt with a 30–50 K thermal hysteresis and spanned ambient temperature.12–14 However, only a few SCO compounds with desired SCO properties have been reported because the prediction and regulation of the occurrence of SCO properties is still difficult in most cases.15–21In SCO research, a well-designed ligand is considered to be the most important requirement. Normally, octahedral six-coordinated Fe(II) complexes belong to the well-studied SCO systems. The nitrogen-donor ligands with a moderate ligand-field strength can produce the expected spin-transition behaviour. However, a ligand-field with strong or weak strength leaves the compound in the LS or HS state, respectively.22 In addition, counter anions, solvents, and intermolecular/intramolecular cooperative interactions (such as hydrogen-bonding, π–π stacking, and van der Waals interaction) can affect the molecular packing in the solid state and thus change the coordination sphere of metal ions, which also exerts strong influence on the ligand-field and thus affects the occurrence of SCO.23–31 According to the literature results, Halcrow correlated the presence or absence of a thermal induced spin-transition in several complex systems with their ligand structures.32,33 His results disclose that the molecular structures in the HS form are often seriously distorted in their coordination geometry or ligand conformation. Compared with the LS form, the HS form usually exhibits greater structural flexibility and structural distortions. Complexes in the solid state whose HS structures differ too strongly from that expected for their LS forms may not undergo SCO on cooling because the greater structural change from the HS state to the LS state is difficult to occur in a rigid solid lattice, thus they tend to remain their original HS states.
To date, hundreds of SCO compounds, including mononuclear and multinuclear complexes as well as coordination polymers, have been reported.34–38 Among the ocean of SCO compounds, the ligand structures are practically composed of various heterocyclic ring composites. Most importantly, the mononuclear complexes [Fe(1-bpp)2]2+ and [Fe(3-bpp)2]2+ and their derivatives constructed by bpp type tridentate nitrogen ligands (1-bpp = 2,6-di[pyrazol-1-yl]pyridine; 3-bpp = 2,6-di[1H-pyrazol-3-yl]pyridine) are some of the most widely investigated complexes in SCO research.39–42 Due to their structural similarity, pyridine-2,6-bis(oxazoline) (pybox) type ligands are also a family of tridentate heterocyclic ligands. These type ligands have been extensively employed in asymmetric organic synthesis as privileged chiral ligands.43 This type of ligand moieties has also been used to construct luminescent and magnetic mononuclear complexes as well as CPs.44–51 On the basis of our long research interests towards molecular magnetism, we started to explore the possibility to assemble SCO complexes containing pybox type ligands. Very recently, we found that a mononuclear SCO Fe(II) complex constructed by the pybox type ligand L1 (2,6-bis{4,4-dimethyl-4,5-dihydrooxazol-2-yl}pyridine) could exhibit multi-induced spin-crossover behaviour mediated by heat, light, pressure, and solvent. This compound also undergoes a structural phase transition, which accompanies the incomplete and abrupt spin-transition process.52 In this contribution, we continue to study the SCO behaviour in a series of [Fe(pybox)2]2+ compounds with structural modification. It is discovered that variations in the ligand structures, counter anions, and solvents may greatly influence the coordination geometry of the central Fe2+ ion and further significantly affect the SCO properties.
Experimental
Materials and synthesis
All solvents and reagents used for synthesis were of analytical grade and used without further purification. Solvents were dried and stored over molecular sieves, and the syntheses of complexes were carried out under the protection of an inert atmosphere of N2. The ligands L1 and L2 were synthesized according to the modified method established by our group (see ESI† for details of the synthesis).49,50,52 Desolvated samples of 1(BF4)′, 1(PF6), 1(BPh4), and 2(PF6) were obtained by heating at 200 °C under an N2 atmosphere for 12 h, and completion was monitored trough TG analysis and elemental analysis.Physical measurements
X-ray data collection and structure determinations
Crystals suitable for single crystal X-ray diffraction were covered in a thin layer of hydrocarbon oil, mounted on a glass fiber attached to a copper pin, and placed under an N2 cold stream. Data for 1(BF4)·MeCN at 153 K, 2(ClO4) at 173 K, and 2(PF6)·MeCN at 153 K were obtained on a Rigaku Saturn724+ CCD diffractometer with Confocal-monochromator Mo-Kα radiation (λ = 0.71073 Å). Intensities were processed using the CrystalClear (Rigaku Inc., 2008) software and absorption corrections were applied using the ‘multi-scan’ method. Data for 1(BF4) at 180 K and 2(BPh4) at 100 K and 300 K were collected on a Rigaku SuperNova Atlas Dual system with a (Mo) microfocus source and focusing multilayer mirror optics (λ = 0.71073 Å). Intensities were processed using CrysAlisPro (Rigaku, Version 1.171.36.32) and absorption corrections were applied using the ‘multi-scan’ method. Data for 1(BF4) at 120 K, 1(BPh4)·MeCN at 180 K, and 2(BPh4) at 180 K were collected on a Rigaku SuperNova Atlas Dual system with a Cu-Kα microfocus source and focusing multilayer mirror optics (λ = 1.5406 Å). Intensities were processed using CrysAlisPro (Rigaku, Version 1.171.36.32) and absorption corrections were applied using the ‘multi-scan’ method. Data for 1(PF6)·MeCN·Et2O at 296 K, 1(BPh4)·MeCN at 296 K, 2(BF4) at 296 K, and 2(PF6)·MeCN at 296 K were collected on a Bruker CCD area detector using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 298 K. Intensity data were collected in the variable ω-scan mode. Crystal structures were all solved by the direct methods and refined on F2 by full-matrix least squares using SHELXTL-97 and Olex 2 (version 1.2.6).56,57 All non-hydrogen atoms were refined with anisotropic thermal parameters, whereas all hydrogen atoms were placed at calculated positions and refined using a riding model. Full crystallographic data for thirteen structures are presented in Table S1.†Synthetic procedures
Results and discussion
Syntheses
The pybox ligands L1 and L2 were synthesized according to our modified method (see ESI† for details). Subsequently, eight Fe(II) complexes were prepared via two different approaches. 1(BF4) was crystallized from a cold methanol solution, and the others were all obtained from the slow gas evaporation of ether into their diluted acetonitrile solution. The structure and purity of all the complexes were characterized by single crystal X-ray diffraction, powdered XRD, FT-IR, thermogravimetric (TG) analysis, and elemental analysis (for the PXRD patterns, see Fig. S7–S14;† and for the TGA traces, see Fig. S15–S22†). Compounds 1(BF4)·MeCN, 1(PF6)·MeCN·Et2O, 1(BPh4)·MeCN, and 2(PF6)·MeCN contained the corresponding solvent molecules during the crystallization process. Therefore, desolvated samples of 1(BF4)′, 1(PF6), 1(BPh4), and 2(PF6) were obtained by heating under the protection of an N2 atmosphere (Scheme 1).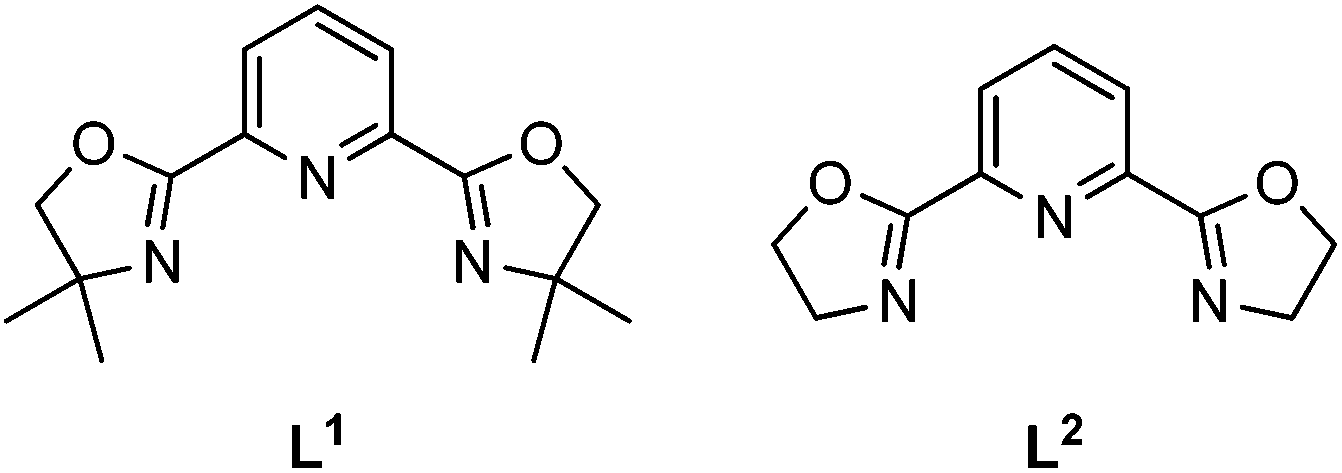 | ||
| Scheme 1 Structures of the two pybox type ligands used in this work. | ||
Solid-state structure study
Crystal structures of the eight compounds were determined by X-ray crystallography. The detailed crystallography data and selected molecular geometries are presented in Table S1† and Table 1, respectively. In general, the basic molecular moiety in this family of compounds is the compressed octahedral Fe(II) core coordinated by two tridentate pybox ligands in an approximately perpendicular fashion. Different types of counter anions (ClO4−, BF4−, PF6−, and BPh4−) are located in their periphery to keep the charge balance. Although these compounds possess similar coordination geometries, their respective coordination polyhedron shows considerably different distortion degrees from the idealized octahedron. It is well known that the distortion of the coordination sphere from an idealized octahedron is related to the ligand-field strength on the iron centre, which is important for the occurrence of SCO behaviour. The degree of distortion can be calculated quantitatively in terms of several structural parameters. Therefore, in the following structural discussion, several structural distortion parameters, such as Σ,58–60θ and ϕ,61–63 and S,64 are introduced. The calculation methods are defined and the values are listed in Table 2.| 1(BF4) | 1(BF4)·MeCN | 1(PF6)·MeCN·Et2O | 1(BPh4)·MeCN | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 180 K | 120 K | 153 K | 296 K | 296 K | 180 K | |||||||||
| Fe1–N1 | 2.249(2) | Fe1–N1 | 2.219(7) | Fe2–N7 | 1.903(7) | Fe1–N1 | 2.274(2) | Fe1–N1 | 2.208(3) | Fe2–N7 | 2.229(3) | Fe1–N1 | 2.204(3) | 2.013(2) |
| Fe1–N2 | 2.097(2) | Fe1–N2 | 2.111(7) | Fe2–N8 | 2.031(7) | Fe1–N2 | 2.124(2) | Fe1–N2 | 2.122(3) | Fe2–N8 | 2.109(3) | Fe1–N2 | 2.089(3) | 1.900(2) |
| Fe1–N3 | 2.184(2) | Fe1–N3 | 2.253(7) | Fe2–N9 | 1.916(7) | Fe1–N3 | 2.208(2) | Fe1–N3 | 2.281(3) | Fe2–N9 | 2.250(3) | Fe1–N3 | 2.219(3) | 2.024(2) |
| Fe1–N4 | 2.184(2) | Fe1–N4 | 2.200(7) | Fe2–N10 | 2.010(7) | Fe1–N4 | 2.221(2) | Fe1–N4 | 2.207(3) | Fe2–N10 | 2.199(3) | Fe1–N4 | 2.199(3) | 2.022(2) |
| Fe1–N5 | 2.093(2) | Fe1–N5 | 2.126(6) | Fe2–N11 | 2.001(6) | Fe1–N5 | 2.116(2) | Fe1–N5 | 2.114(3) | Fe2–N11 | 2.111(3) | Fe1–N5 | 2.093(3) | 1.906(2) |
| Fe1–N6 | 2.217(2) | Fe1–N6 | 2.234(7) | Fe2–N12 | 2.035(7) | Fe1–N6 | 2.223(2) | Fe1–N6 | 2.279(3) | Fe2–N12 | 2.257(3) | Fe1–N6 | 2.195(3) | 2.017(2) |
| 2(ClO4) | 2(BF4) | 2(PF6)·MeCN | 2(BPh4) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 173 K | 296 K | 296 K | 153 K | T/K | 100 K | 180 K | 300 K | |||
| Fe1–N1 | 1.983(3) | Fe1–N1 | 1.975(3) | Fe1–N1 | 2.185(3) | 2.011(4) | Fe1–N1 | 1.959(1) | 1.965(7) | 1.964(2) |
| Fe1–N2 | 1.898(2) | Fe1–N2 | 1.905(3) | Fe1–N2 | 2.117(3) | 1.939(4) | Fe1–N2 | 1.892(2) | 1.893(8) | 1.889(2) |
| Fe1–N3 | 1.974(3) | Fe1–N3 | 1.981(3) | Fe1–N3 | 2.188(3) | 2.022(4) | Fe1–N3 | 1.988(1) | 2.120(6) | 2.170(2) |
| Fe1–N4 | 1.971(2) | Fe1–N4 | 1.993(3) | Fe1–N4 | 2.199(3) | 2.008(4) | Fe1–N4 | 1.933(2) | 2.076(9) | 2.138(2) |
| Fe1–N5 | 1.899(2) | Fe1–N5 | 1.907(3) | Fe1–N5 | 2.108(3) | 1.936(4) | ||||
| Fe1–N6 | 1.971(2) | Fe1–N6 | 1.985(3) | Fe1–N6 | 2.189(3) | 1.994(4) | ||||
| Entry | T/K | Σ (°) | θ (°) | ϕ (°) | Fe–Nav (Å) | S(Oh) | Spin state |
|---|---|---|---|---|---|---|---|
| 1(BF4) | 180 | 134.9 | 89.7 | 171.1 | 2.171 | 4.75 | HS |
| 1(BF4) , Fe1 | 120 | 143.0 | 87.2 | 170.2 | 2.190 | 5.10 | HS |
| 1(BF4) , Fe2 | 120 | 91.8 | 88.0 | 176.6 | 1.983 | 2.35 | LS |
| 1(BF4)·MeCN | 153 | 140.9 | 89.7 | 170.0 | 2.194 | 5.15 | HS |
| 1(PF6)·MeCN·Et2O , Fe1 | 296 | 140.2 | 88.9 | 168.8 | 2.202 | 5.16 | HS |
| 1(PF6)·MeCN·Et2O , Fe2 | 296 | 140.0 | 89.2 | 172.7 | 2.192 | 5.02 | HS |
| 1(BPh4)·MeCN | 296 | 135.27 | 89.3 | 176.7 | 2.166 | 4.65 | HS |
| 1(BPh4)·MeCN | 180 | 90.07 | 90.0 | 179.2 | 1.980 | 2.28 | LS |
| 2(ClO4) | 173 | 90.13 | 89.0 | 179.1 | 1.949 | 2.22 | LS |
| 2(BF4) | 296 | 92.00 | 88.8 | 178.7 | 1.958 | 2.30 | LS |
| 2(PF6)·MeCN | 296 | 143.41 | 88.6 | 174.8 | 2.164 | 5.17 | HS → LS |
| 2(PF6)·MeCN | 153 | 96.38 | 89.5 | 177.5 | 1.985 | 2.53 | LS |
| 8 (100 K) Fe1 | 100 | 85.228 | 90 | 180 | 1.937 | 1.99 | LS |
| 8 (100 K), Fe2 | 100 | 99.392 | 90 | 180 | 1.970 | 2.62 | LS |
| 8 (180 K), Fe1 | 180 | 87.76 | 90 | 180 | 1.941 | 2.09 | LS |
| 8 (180 K), Fe2 | 180 | 138.88 | 90 | 180 | 2.105 | 4.78 | HS |
| 8 (300 K), Fe1 | 300 | 84.848 | 90 | 180 | 1.939 | 1.98 | LS |
| 8 (300 K), Fe2 | 300 | 149.60 | 90 | 180 | 2.149 | 5.39 | HS |
Compounds 1(BF4) and 1(BF4)·MeCN crystallize in the orthorhombic space group Pbca at 180 K and 153 K, respectively. For these two compounds, there is only one independent Fe(II) centre in their lattice. The average Fe–N bond lengths of 2.171 Å for 1(BF4) and 2.194 Å for 1(BF4)·MeCN show that the two complexes are in the HS state at the measured temperature. The co-crystallized acetonitrile molecules in 1(BF4)·MeCN result in serious deviation of the FeN6 coordination sphere from idealized octahedron in the solid state with the values of Σ and S as 140.86° and 5.146, respectively. It is notable that the conformation of one ligand is obviously distorted (Fig. 1). The pyridine ring in one of the L1 ligands deviates from the ligand plane (defined through three coordinated nitrogen atoms), which results in a bending angle of 9.34° (see Fig. S23†).
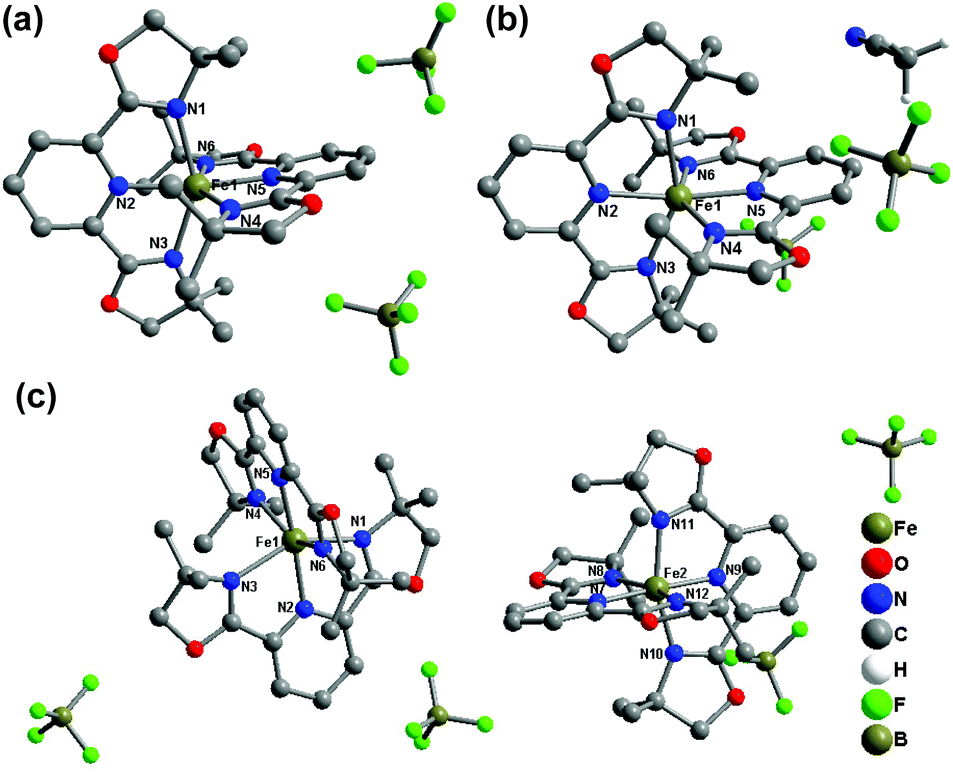 | ||
| Fig. 1 Molecular structures of (a) 1(BF4) at 180 K, (b) 1(BF4)·MeCN at 153 K, and (c) 1(BF4) at 120 K. The hydrogen atoms in the complex cations are omitted for clarity. | ||
As the temperature decreases to 120 K, the space group of 1(BF4) undergoes a significant change from Pbca to P21/c, which indicates that a phase transition process takes place (see Fig. 2). The color of the crystals also changes from dark red to red during this process (see Fig. S24†). The single crystal X-ray structure at 120 K shows that there are two crystallographically independent Fe(II) centres in the crystal lattice at the low-temperature phase (LTP). As shown in Table 1, the average Fe–N bond lengths of the two Fe(II) centres are 2.190 Å for Fe1 and 1.983 Å for Fe2. On the basis of the Fe–N distances and structural parameters (see Table 2), it can be determined that the Fe1 centre is in the HS state, whereas the Fe2 centre is in the LS state, which reveals that 1(BF4) adopts a mixed 1HS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1LS state population at LTP.
:1LS state population at LTP.
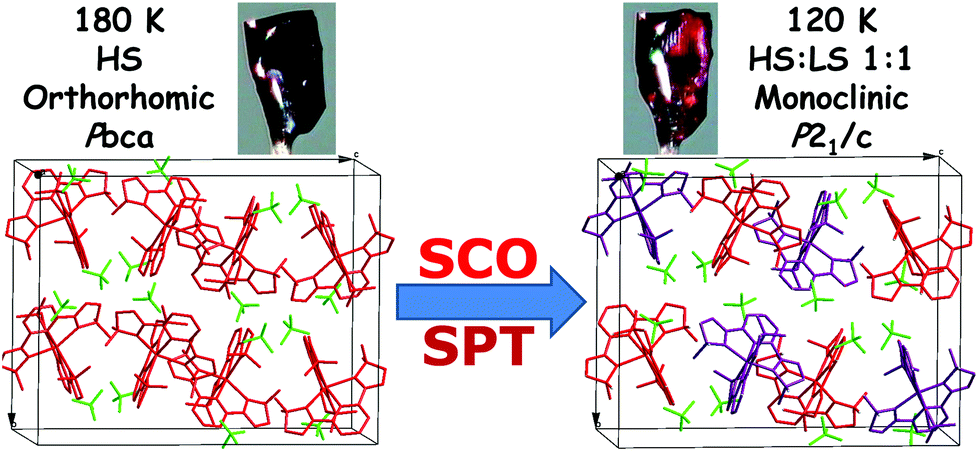 | ||
| Fig. 2 Illustration of the spin-crossover and structural phase transition of 1(BF4) in one unit cell. | ||
In order to accurately determine the phase transition temperature of 1(BF4), differential scanning calorimetry (DSC) measurement was carried out over the temperature range of 100–250 K with a heating/cooling rate of 5 K min−1 (see Fig. 3). We observed an endothermic peak at 151.1 K (ΔH = 2.67 kJ mol−1 and ΔS = 17.7 J mol−1 K−1) upon heating and an exothermic peak at 138.8 K (ΔH = 2.40 kJ mol−1 and ΔS = 17.3 J mol−1 K−1) in the cooling process, which indicate that the structural phase transition (SPT) of 1(BF4) takes place in this narrow temperature range within 15 K.
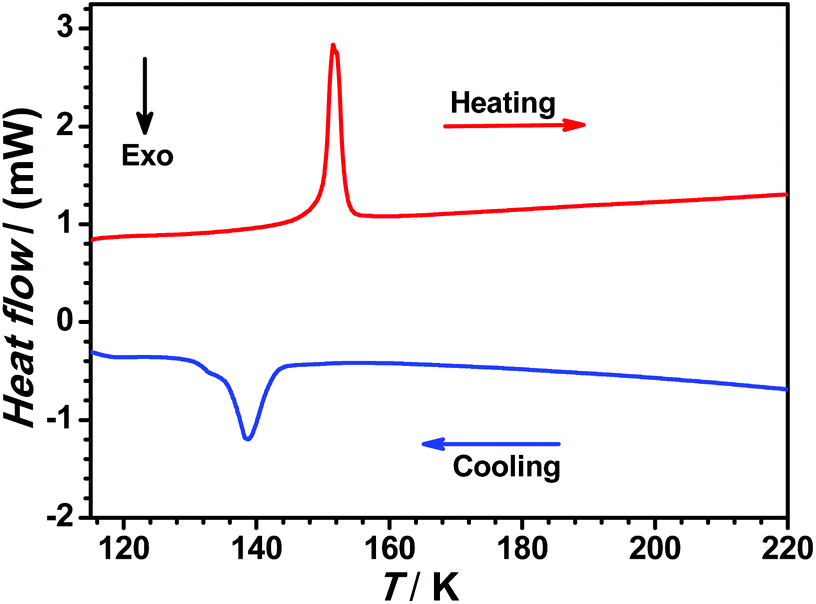 | ||
| Fig. 3 Differential scanning calorimetry (DSC) profile of 1(BF4) in heating (red line) and cooling (blue line) modes. | ||
The crystal structure of 1(PF6)·MeCN·Et2O and 1(BPh4)·MeCN crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the monoclinic space group P2/c, respectively. 1(PF6)·MeCN·Et2O contains two crystallographically independent Fe(II) centres in its asymmetric unit (see Fig. 4(a)). The average Fe–N bond lengths of the Fe1 and Fe2 spheres are 2.202(3) and 2.192(3) Å, respectively, which reveal that both Fe(II) centres are in the HS state at 296 K. The subsequent decrease in temperature shows no significant crystallographic change, which indicates that the HS state is still maintained at low temperature. The average Fe–N bond length of the only Fe(II) sphere in 1(BPh4)·MeCN is 2.166 Å at 296 K, which suggests an incomplete HS state. As the temperature decreases to 180 K, although the space group is maintained, the Fe–N bond length undergoes a noticeable contraction (1.980 Å on average), which shows that the Fe(II) centre has transferred to the LS state. The crystallographic measurements preliminarily determine that 1(BPh4)·MeCN is an SCO compound and the spin-transition process occurs above 180 K.
and the monoclinic space group P2/c, respectively. 1(PF6)·MeCN·Et2O contains two crystallographically independent Fe(II) centres in its asymmetric unit (see Fig. 4(a)). The average Fe–N bond lengths of the Fe1 and Fe2 spheres are 2.202(3) and 2.192(3) Å, respectively, which reveal that both Fe(II) centres are in the HS state at 296 K. The subsequent decrease in temperature shows no significant crystallographic change, which indicates that the HS state is still maintained at low temperature. The average Fe–N bond length of the only Fe(II) sphere in 1(BPh4)·MeCN is 2.166 Å at 296 K, which suggests an incomplete HS state. As the temperature decreases to 180 K, although the space group is maintained, the Fe–N bond length undergoes a noticeable contraction (1.980 Å on average), which shows that the Fe(II) centre has transferred to the LS state. The crystallographic measurements preliminarily determine that 1(BPh4)·MeCN is an SCO compound and the spin-transition process occurs above 180 K.
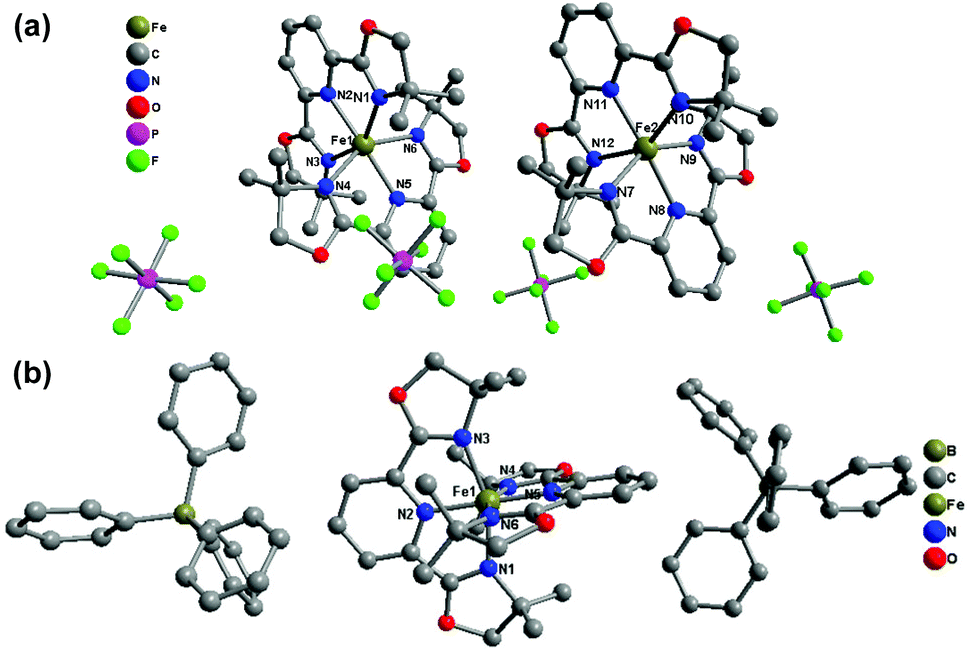 | ||
| Fig. 4 Molecular structures of (a) 1(PF6)·MeCN·Et2O at 296 K and (b) 1(BPh4)·MeCN at 153 K. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
Compounds 2(ClO4), 2(BF4), 2(PF6)·MeCN, and 2(BPh4) were all constructed from the ligand L2 (2,6-bis(4,5-dihydrooxazol-2-yl)pyridine) and Fe(II) salts with different counter anions. Overall, the different counter anions (ClO4−, BF4−, PF6−, and BPh4−) exert a strong impact on the deviation of the FeN6 coordination sphere from idealized D2d symmetry, and the molecular packing in the solid state. 2(ClO4) (at 173 K) and 2(BF4) (at 296 K) both crystallize in the monoclinic space group P21/n with the average Fe–N bond lengths of 1.949 Å and 1.958 Å, respectively, which indicate that both complexes are in the LS state at the measured temperatures. The single crystal X-ray structure of 2(PF6)·MeCN was determined at 296 K and 153 K. One acetonitrile molecule per Fe2+ complex cation co-crystallizes in the crystal lattice. At 296 K, it crystallizes in the monoclinic space group P21/c with the average Fe–N bond length of 2.164(3) Å, which shows that the Fe(II) centre has not achieved a complete HS state. As temperature decreases to 153 K, the original space group is maintained but the average Fe–N bond length undergoes a sizeable decrease to 1.985(4) Å, which indicates it is involved in a spin-transition process upon cooling (Fig. 5).
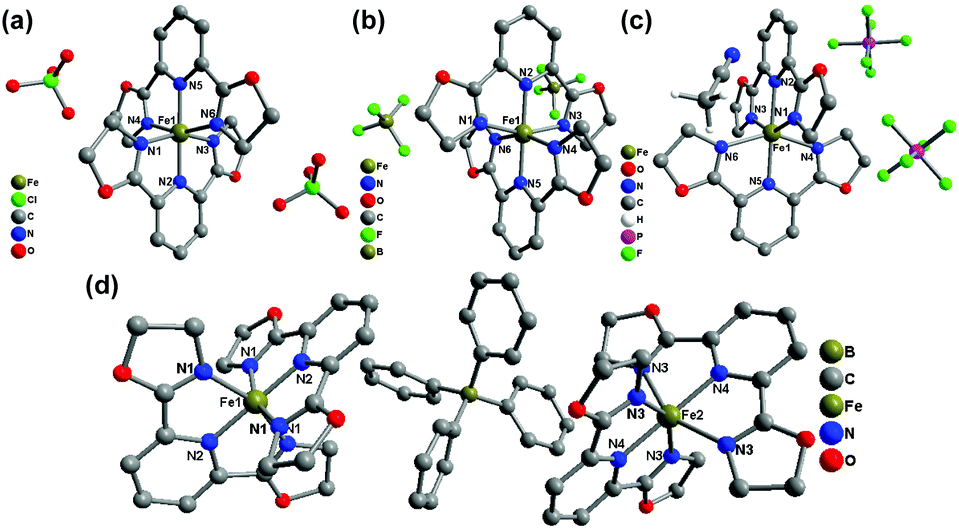 | ||
| Fig. 5 Molecular structures of (a) 2(ClO4), (b) 2(BF4), (c) 2(PF6)·MeCN, and (d) 2(BPh4). Hydrogen atoms and solvent molecules are omitted for clarity. | ||
Structural analyses of 2(BPh4) were carried out at 300 K, 180 K, and 100 K. At 300 K, it crystallizes in the tetragonal space group I41/a with Z = 16. There are two crystallographically independent Fe(II) centres in the crystal lattice. Both Fe(II) sites possess relatively high symmetry and belong to the point group D2d with θ = 90° and ϕ = 180°. Two Fe(II) complex cations are well-isolated by the bulky BPh4− counter anions, and the shortest Fe1–Fe2 distance is 12.382 Å. Upon cooling, the space group is unchanged, whereas the cell dimension decreases slightly by 2.9% from 11707.2 Å3 at 300 K to 11369.6 Å3 at 100 K, which indicates that a thermal induced spin-transition takes place. At 300 K, the average Fe–N bond lengths are 1.939(2) Å for the Fe1 site and 2.149(2) Å for the Fe2 site, which imply that the Fe1 site is in the LS state while the Fe2 site is in the HS state. At 180 K, the average Fe–N bond lengths are 1.941(8) Å for the Fe1 site and 2.105(8) Å for the Fe2 site, which show that the Fe1 site is still in the LS state but the Fe2 site begins to undergo SCO. At 100 K, the average Fe–N bond lengths are 1.937(2) Å for the Fe1 site and 1.970(2) Å for the Fe2 site, which are typical for the complete LS state of Fe2+ ions. This detailed structural investigation demonstrates that 50% of the Fe2+ ions has a complete spin transition from the HS to the LS state in the temperature range of 300–100 K.
Magnetic studies
In order to evaluate their SCO behaviour, magnetic measurements for these compounds, including the desolvated samples, were performed on polycrystalline samples using Quantum-Design MPMS and PPMS magnetometers under a dc field of 5000 Oe and the results are presented in the form of χMT vs. T curves, where χM is the molar magnetic susceptibility and T is the absolute temperature. To our delight, we observed different spin-state properties modulated by molecular structure including ligands, counter anions, and solvents. These structural factors display a strong impact on the occurrence of SCO in this family of compounds.The χMT vs. T plot of 1(BF4) in the range of 300–2 K is shown in Fig. 6. At 300 K, the χMT value of 1(BF4) is 3.72 cm3 K mol−1 and it remains roughly constant down to 150 K, whereas the χMT value decreases abruptly from 3.73 cm3 K mol−1 at 150 K to 1.96 cm3 K mol−1 at 140 K with the T1/2 temperature of 149 K. This variation in χMT value demonstrates that about 50% of the HS Fe2+ ions have transformed to LS Fe2+ ions statistically. This result is well consistent with the above single crystal structural analysis. Further cooling below 10 K leads to a second decrement due to the zero-field splitting effect of the rest of HS Fe2+ ions with the spin state S = 2. The cooling and heating cycle measurement shows a small thermal hysteresis with the width of about 5 K (see Fig. S25†). We consider that the hysteresis loop is closely related to the crystallographic structural phase transition.
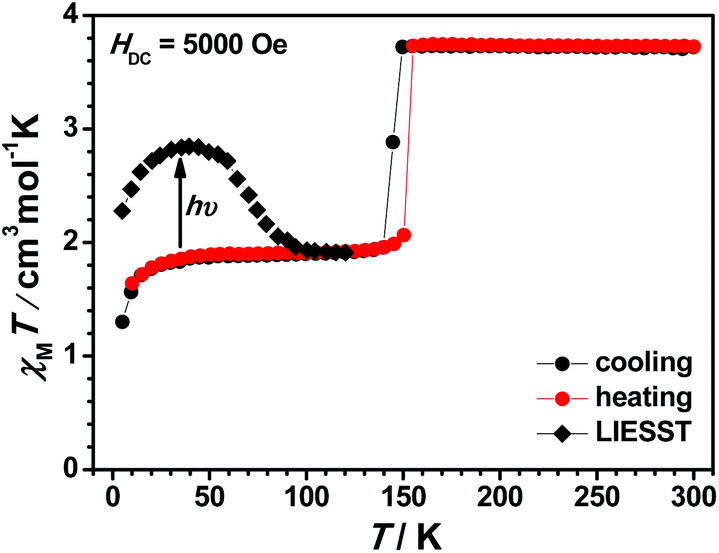 | ||
| Fig. 6 Plot of χMT vs. T for 1(BF4). | ||
In contrast, 1(BF4)·MeCN remains the HS state in the temperature range of 300–10 K (see Fig. S26†). The desolvated sample of 1(BF4)·MeCN (named 1(BF4)′) was subjected to VT-magnetic measurements as well. An abrupt and 50% complete ST and narrow hysteresis loop with a 5 K width were observed, which reveal identical SCO behaviour compared to 1(BF4) (see Fig. S27†). A similar situation was also observed in our reported compound 1(ClO4)·MeCN.52 These investigations suggest that the co-crystallized solvent molecules impose a strong effect on the occurrence of SCO.
1(PF6)·MeCN·Et2O remains totally the HS state in the temperature range of 300–10 K (see Fig. S28(a)†). The structural analysis reveals that the two symmetry independent Fe(II) centres are both in the HS state at 298 K. Thus, the HS state of the two Fe(II) centres is always maintained during the cooling process. The desolvated sample 1(PF6) is still a high spin compound (see Fig. S28(b)†). However, its spin-state behaviour is different when the counter anion is changed to the bulky BPh4− (see Fig. 7). The χMT value of 1(BPh4)·MeCN is 3.50 cm3 K mol−1 at 300 K, which indicates that the majority of Fe2+ ions approach the HS state. As the temperature decreases, the χMT value drops gradually to 0.02 cm3 K mol−1 at 150 K, thus all the Fe2+ ions are in the complete LS state below this temperature. The magnetic data is consistent with the structural analyses at different temperatures. The subsequent heating mode reveals no thermal hysteresis loop and the T1/2 temperature is 235 K. Further investigation of the desolvated sample 1(BPh4) shows a gradual but incomplete SCO. At 400 K, the χMT value is 3.85 cm3 K mol−1, which indicates the achievement of a complete HS state. Upon cooling, the χMT value gradual decreases to 2.74 cm3 K mol−1 at 170 K. It can be estimated that about 30% of the Fe2+ ions underwent SCO. However, further spin transition does not occur significantly below this temperature, and no thermal hysteresis loop is observed in the subsequent heating process.
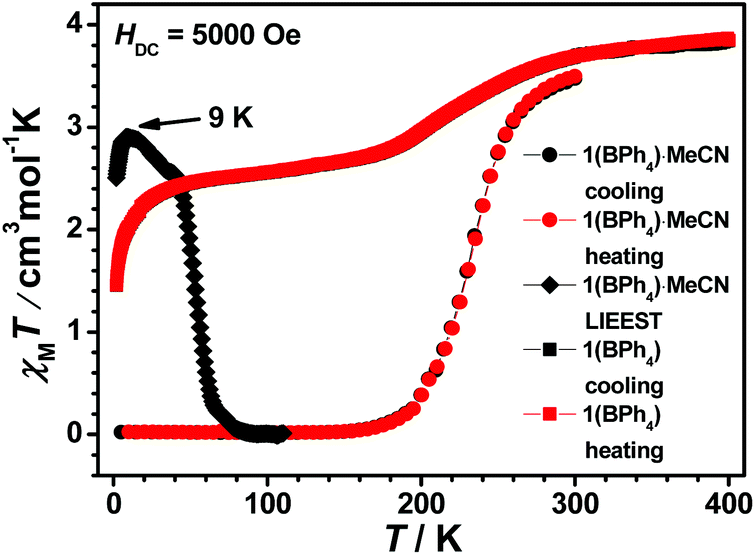 | ||
| Fig. 7 Plot of χMT vs. T for 1(BPh4)·MeCN and 1(BPh4). | ||
The four compounds 2(ClO4), 2(BF4), 2(PF6)·MeCN, and 2(BPh4) constructed by ligand L2 with different counter anions all display SCO behaviour and spin-transition temperatures around or above the ambient temperature (see Fig. 8). At 380 K, the χMT values of 2(ClO4) and 2(BF4) are 2.52 and 2.14 cm3 K mol−1, respectively, which suggest that the Fe2+ ions are still involved in the spin transition process. TG analyses show that the decomposition of 2(ClO4) and 2(BF4) starts at 488 K and 540 K, respectively (see Fig. S19 and S20†), thus magnetic measurements in a higher temperature region can be carried out safely. The χMT values of 2(ClO4) at 450 K and 2(BF4) at 500 K are 3.39 cm3 K mol−1 and 3.40 cm3 K mol−1, respectively, which indicate that most of the Fe2+ ions approach a complete HS state. Upon cooling, a gradual and complete spin transition is observed and the process into the LS state is complete at approximately 230 K for these two compounds. The subsequent heating mode reveals that there is no thermal hysteresis loop and the T1/2 temperature is 345 K for 2(ClO4) and 360 K for 2(BF4).
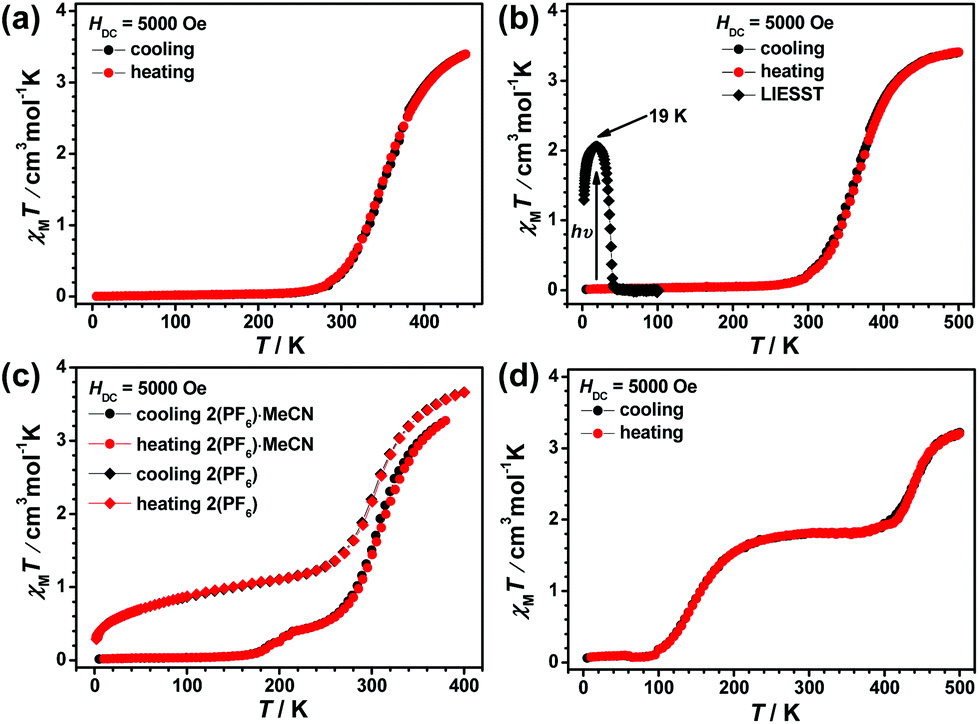 | ||
| Fig. 8 Plot of χMT vs. T for (a) 2(ClO4), (b) 2(BF4), (c) 2(PF6)·MeCN and 2(PF6), and (d) 2(BPh4). | ||
TG analysis of 2(PF6)·MeCN reveals that the loss of solvent starts above 380 K (see Fig. S23†), therefore the magnetic measurement was started from this temperature. The χMT value of 2(PF6)·MeCN is 3.27 cm3 K mol−1 at 380 K, which suggests it approaches the complete HS state. During the cooling process, the χMT value first decreases gradually to 0.47 cm3 K mol−1 at 240 K and then descends slowly to 0.07 cm3 K mol−1 at 160 K. Further decrease in the χMT value ceases below 160 K, which suggests that all the Fe2+ ions are close to the complete LS state. The first derivative of the χMT product relative to temperature, dχMT/dT vs. T, clearly shows two peaks at 305 K and 196 K (see Fig. S29†), which confirm the SCO feature of an overlapped two-step process. However, the desolvated sample 2(PF6) displays different SCO behaviour. Its χMT value at 400 K is 3.66 cm3 K mol−1, which is typical of a complete HS state for Fe2+ ions. Upon cooling, the fast decrease terminates at about 150 K with a χMT value of 1.44 cm3 K mol−1 and T1/2 in the first step is about 300 K (see Fig. S30†). However, further cooling to 2 K only causes a slow and gradual decrement, thus indicating that the second SCO step is significantly weakened after the desolvation process.
TG analysis demonstrates that compound 2(BPh4) is stable below 500 K (see Fig. S21†), thus VT-magnetic measurement began at 500 K (see Fig. 8(d)). This compound unexpectedly shows a well defined two-step SCO behaviour. Its χMT value is 3.21 cm3 K mol−1 at 500 K, which indicates the approach to the HS state. Upon cooling, the χMT value decreases in two distinguishable and well-isolated platforms, to 1.83 cm3 K mol−1 at 360 K for the first step, which proceeds to 50% completeness, and then to a minimum value below 90 K of approximately 0.1 cm3 K mol−1 for the second step, thus completing SCO to reach the LS state. No thermal hysteresis is observed in the subsequent heating mode. The two T1/2 values for the low- and high-temperature steps are 145 K and 440 K, respectively (see Fig. S24†) and the large plateau between the two steps is approximately 160 K.
Photomagnetic study
In the previous work, we found that 1(ClO4) displays a significant LIESST effect at low temperature.52 In order to test the universality of the photomagnetic effect in this family of compounds and discover new LIESST phenomena, 1(BF4), 1(BPh4)·MeCN, and 2(BF4) were selected as examples to perform photomagnetic measurements. After irradiation, a significant increment in the χMT value for these three compounds can be observed, which is attributed to the LS to HS spin transition of the Fe2+ ions (see the ◆ dots in Fig. 6, 7, and 8(b)). The photo-induced χMT value of 1(BF4) increases steeply and reaches a maximum value of 2.84 cm3 K mol−1 at 39 K, which corresponds to about 80% Fe2+ ions in the HS state after irradiation. When the temperature is above 100 K, the metastable 30% HS Fe2+ ions recover to the LS state again. The TLIESST temperature of 1(BF4) is equal to 74.5 K (the TLIESST temperature is determined as the minimum of the d(χMT)/dT vs. T curve, see Fig. S32(a)†).651(BPh4)·MeCN displays the most significant LIESST effect in this series of compounds. Irradiation at 2 K results in a pronounced increase in χMT value from 0.02 to 2.51 cm3 K mol−1. The photo-induced curve reaches a maximum value of 2.90 cm3 K mol−1 at 9 K with a TLIESST temperature of 53.3 K (see Fig. S32(b)†), which suggests about 80% photo-induced conversion of Fe2+ ions. Then it returns to the complete LS state when the temperature warms up to 90 K. The irradiation of 2(BF4) at 2 K yields a considerable increase in χMT value from 0.01 to 1.29 cm3 K mol−1. It increases steeply and reaches a sharp maximum value of 2.06 cm3 K mol−1 at 19 K with a TLIESST temperature of 38.0 K (see Fig. S32(c)†), which implies that about 60% of the Fe2+ ions undergo LS to HS conversion. However, the metastable HS state of 2(BF4) is less stable, and it relaxes back to the complete LS state quickly by warming to 43 K. We compared the data in the L1 series (1(ClO4), 1(BF4), and 1(BPh4)·MeCN) to disclose the relation between TLIESST and thermal spin transition (T1/2) temperatures and obtained a good linear correlation equation: TLIESST = T0 − 0.25T1/2, T0 = 111.5 (see Fig. S32(d)†), which is similar to the results in the [Fe(bpp)2]2+ series of compounds.65Variable-temperature Raman spectra
Since changes in molecular vibrations may drive SCO through the entropic effect, variable-temperature Fourier-transform Raman spectroscopy has become a new and powerful tool to monitor the spin-transition details of metal complexes.66–68 In this series of compounds, Raman measurements were performed on 1(BF4), 1(BPh4)·MeCN, 2(ClO4), and 2(BF4) at selected temperatures. The peaks between 1400 to 1700 cm−1 can be ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch of the oxazole and pyridine ring, which are very sensitive to the spin state of the coordinated Fe2+ ions. In this wavenumber region, distinguishable changes in the four compounds before and after spin-transition can be observed (see Fig. 9). For 1(BF4), considerable decrease in the peak at 1576 cm−1 and increase in the peak at 1644 cm−1 (relative to the peak intensity at 1458 cm−1) occur when the temperature is above 130 K (see Fig. S33†), which is consistent with the observation of abrupt SCO behaviour around 150 K from the magnetic results. The change trends in 1(BPh4)·MeCN, 2(ClO4), and 2(BF4) are very similar. The intensity of the peak at 1633 cm−1 (LS form) gradually decreases, however the peak at 1642 cm−1 (HS form) gradually increases with a continuous increase in temperature. Overall, the SCO properties of this family of compounds derived from variable-temperature Raman spectra agree quite well with that from other characterizations, such as crystal structure analysis and magnetic measurements.
N stretch of the oxazole and pyridine ring, which are very sensitive to the spin state of the coordinated Fe2+ ions. In this wavenumber region, distinguishable changes in the four compounds before and after spin-transition can be observed (see Fig. 9). For 1(BF4), considerable decrease in the peak at 1576 cm−1 and increase in the peak at 1644 cm−1 (relative to the peak intensity at 1458 cm−1) occur when the temperature is above 130 K (see Fig. S33†), which is consistent with the observation of abrupt SCO behaviour around 150 K from the magnetic results. The change trends in 1(BPh4)·MeCN, 2(ClO4), and 2(BF4) are very similar. The intensity of the peak at 1633 cm−1 (LS form) gradually decreases, however the peak at 1642 cm−1 (HS form) gradually increases with a continuous increase in temperature. Overall, the SCO properties of this family of compounds derived from variable-temperature Raman spectra agree quite well with that from other characterizations, such as crystal structure analysis and magnetic measurements.
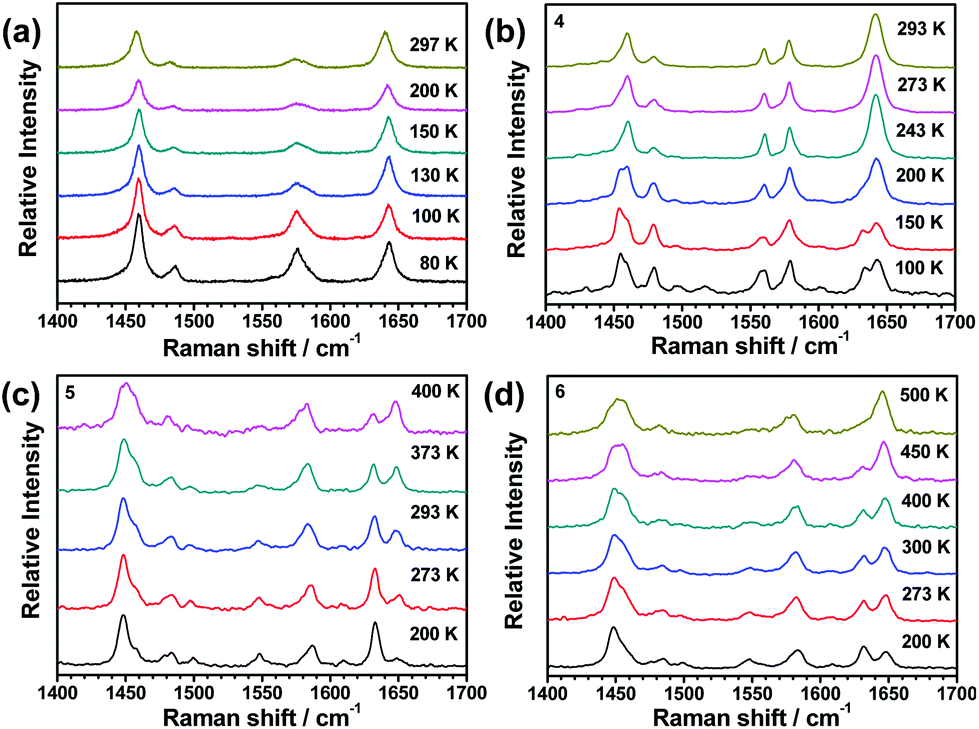 | ||
| Fig. 9 Raman spectra of 1(BF4), 1(BPh4)·MeCN, 2(ClO4), and 2(BF4) at selected temperatures. | ||
Discussion
Ligand effect
As a new type of tridentate ligand in the construction of SCO complexes, pybox ligands show a relatively moderate ligand-field strength to coordinate with Fe2+ ions. In our laboratory, effective synthetic methods have been established to introduce the substituted group at specific positions of the ligand skeleton. In this work, we only focus on the complexes constructed from two representative ligands, L1 and L2. Structural and magnetic investigations show that the L2 series of compounds are all SCO compounds and most SCO processes all take place at a relatively high temperature region with T1/2 values above 300 K.Four methyl groups are introduced into the oxazole ring in ligand L1, which we anticipate will weaken the relative ligand-field strength compared with L2 through the steric effect but still maintain the symmetry of the ligand. As expected, such modification generally reduces the spin-transition temperatures of 1(ClO4), 1(BF4), and 1(BPh4)·MeCN. The other compounds, such as 1(ClO4)·MeCN, 1(BF4)·MeCN, 1(PF6), and 1(BPh4), are all HS compounds. The spin transition of 1(ClO4) and 1(BF4) can only proceed to 50% completeness and is associated with a crystallographic structural phase transition, which indicate that the methyl groups introduce structural complexity in the crystal arrangement.
Counter anion effect
Similar to other SCO systems, the counter anions not only keep the charge balance but also exert an obvious influence on SCO properties. In the solid state, the tetrahedral anions of ClO4− and BF4− often fall into disorder in the crystal lattice (see 1(BF4) and 2(ClO4)). They possess similar sizes and the same electric charge, thus the SCO behaviour in this pair is very similar (see 1(ClO4)52vs. 1(BF4), 1(ClO4)·MeCN52vs. 1(BF4)·MeCN, and 2(ClO4)vs. 2(BF4)). However, the influence of other counter anions, such as PF6− and BPh4−, on the SCO properties is complicated due to their larger sizes. 1(PF6)·MeCN·Et2O and the desolvated sample 1(PF6) are high-spin at all temperatures. Different from the analogues of ClO4− and BF4−, there are two independent Fe(II) centres in 1(PF6)·MeCN·Et2O, which imply that the octahedral PF6− anions affect the packing of the [Fe(L1)2]2+ complex cations due to their larger size. Compounds 2(PF6)·MeCN, 2(PF6), and 2(BPh4) all exhibit a two-step SCO process. It is reasonable to expect that the anions with a larger size produce a delicate influence on the coordination geometry of the Fe(II) centre in the solid state. However, the relationship between the counter anion and stepwise SCO in mononuclear Fe(II) systems is still an open question. In the further studies, we will focus on a deeper understanding of the size effect of counter anions and the origin of two-step SCO in this family of compounds.Solvent effect
In this series, compounds 1(ClO4)·MeCN, 1(BF4)·MeCN, 1(PF6)·MeCN·Et2O, 1(BPh4)·MeCN, and 2(PF6)·MeCN contain the corresponding solvent molecules during the crystallization process. Compared with the solvent-free SCO analogues 1(ClO4) and 1(BF4), the solvated analogues 1(ClO4)·MeCN and 1(BF4)·MeCN are both HS complexes. According to the structural analysis, it can be found that co-crystallization of solvent molecules will cause serious deviation from the idealized coordination geometry or distorted ligand conformation (see 1(ClO4)·MeCN52 and 1(BF4)·MeCN). On the one hand, the distorted coordination sphere affords a weaker ligand-field strength, on the other hand, the inserted solvent makes the crystal lattice more rigid and thus impedes the possible structural change from the HS to LS state in the solid state. Therefore, such solvated compounds tend to maintain the HS state. When the solvents in 1(BF4)·MeCN are completely removed, the crystals transform back to the solvent-free phase 1(BF4), thus the SCO behaviour is recovered. 1(PF6)·MeCN·Et2O and the desolvated sample 1(PF6) are both HS complexes, which imply that the solvents do not affect the spin state strongly in this compound. In contrast, solvent molecules impose a strong influence on the SCO behaviours of 1(BPh4)·MeCN and 2(PF6)·MeCN. When the solvents are removed completely, the SCO of 1(BPh4)·MeCN becomes incomplete and the second step of SCO of 2(PF6)·MeCN significantly slows down. Indeed the removal of solvent molecules results in a rearrangement of complexes in the crystals. Single crystal X-ray structures of the desolvated samples were not obtained, therefore we cannot compare the detailed structural change after the desolvation process. As pointed out by Bürgi et al., the engineering of lattice solvation in SCO research is still a big challenge, thus the control and prediction of the solvent factor are very limited at this stage.69Magneto-structural relationship
Several structural parameters are beneficial for the comparison of the structural characteristics in this series of compounds. The distortion parameter, ∑, is defined as the sum of the deviations from 90° of the 12 cis angles in the coordination sphere.58–60 The angle θ is the dihedral angle between the last-squares planes of the tridentate ligands and the angle ϕ refers to the trans-N{pyridine}–Fe–N{pyridine} angle.61–63 The distance Fe–Nav is the average value of the six Fe–N bond lengths in one FeN6 coordination sphere. S(Oh) is the CShMs calculation result, which is the deviation value of ideal Oh symmetry.64 This family of compounds exhibit variable spin-state behaviours in the solid state, depending on the ligand structures, counter anions, and solvents. In this series compounds, the HS forms all adapt a twisted hexa-coordinated geometry, which is caused by the angular Jahn–Teller distortion of the 5T high-spin manifold. Halcrow et al. successfully used two independent structural parameters ϕ and θ to measure and analyze the deviation of the [Fe(bpp)2]2+ series of compounds from their idealized D2d symmetry.33 In [Fe(pybox)2]2+ derivatives, the values observed in practice for these two parameters were also measured, which are 168.4° ≤ ϕ ≤ 180° and 87.0° ≤ θ ≤ 90° (see Fig. 10(a)). The range of structural distortions for this family of compounds with the plot of ϕ vs. θ is shown in Fig. 10(b). It shows that the HS forms exhibit significant deviations from the idealized D2d symmetry. Normally, the HS forms are able to undergo SCO, and their deviation degree is smaller and located near the LS region of the graph. However, in the HS forms, their distortion of the coordination geometry or ligand conformation is larger, which means that the HS structures differ very strongly from their LS forms, which cannot undergo SCO on cooling.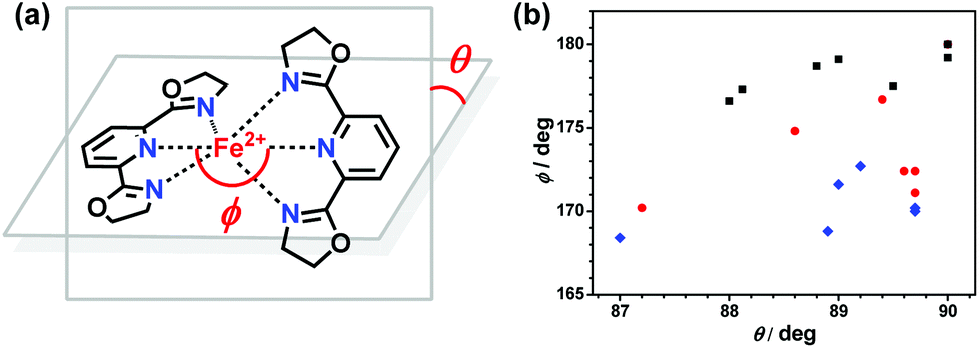 | ||
| Fig. 10 (a) Definition of the angular Jahn–Teller distortion parameters θ and ϕ in [Fe(pybox)2]2+ complexes (θ ≤ 90°, ϕ ≤ 180°). (b) Plot of θ vs. ϕ for high-spin complexes that remain high-spin (black), SCO complexes in their HS state (red), and LS state plus LS complexes (blue) from the [Fe(pybox)2]2+ series. There is one SCO compound with θ = 90° and ϕ = 180° in its high-spin state at two measured temperatures (300 K and 180 K). | ||
Compared with the LS forms, there are obvious deviations in the coordination geometries of the HS forms in this family of compounds. The structural distortion can be expressed quantitatively through the structural parameters ∑ and S as well. The parameter ∑ was introduced by Hendrickson et al. to measure the small differences in the octahedral coordination geometries and to confirm the spin state of the Fe2+ ions in the crystal structure.58 The continuous symmetry measures (CShMs) method has also been proven to be a conceptually simple but powerful technique to display structural diversities and to identify the relationship between molecular symmetry and properties. Alvarez successfully used this method to establish the relationships between the symmetry of the metal coordination sphere and the spin-crossover transition in several complexes.64 In this [Fe(pybox)2]2+ series, the S(Oh) values of each FeN6 core based on Oh symmetry were calculated by the SHAPE 2.0 program.70 The relationship between structural distortions and spin state with ∑ and S(Oh) vs. average Fe–N length is shown in Fig. 11. A perfect octahedral coordination sphere gives ∑ = S(Oh) = 0. We found that two structural parameters are sensitive indicators to determine the spin state of this family of compounds. The ∑ and S(Oh) values of the HS forms are more than 130° and 4.5, whereas those of the LS forms are below 100° and 2.0, respectively. The HS to LS transition forms are located in the intermediate region. The HS forms with larger values of ∑ and S(Oh) cannot undergo spin crossover on cooling. Similar to other SCO systems, these two structural parameters will help to identify the spin state of new complexes in this [Fe(pybox)2]2+ series.71
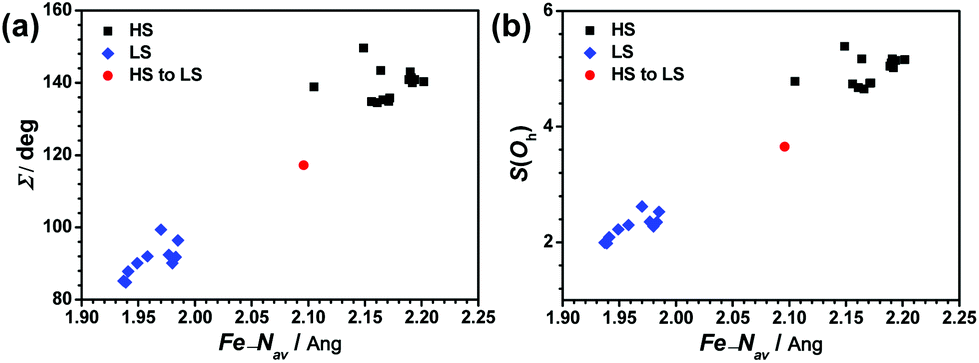 | ||
| Fig. 11 Plots of (a) Σ vs. Fe–Nav and (b) S(Oh) vs. Fe–Nav for HS forms, HS to LS transition forms, and LS forms in the [Fe(pybox)2]2+ series. | ||
Comparison with 1-bpp and 3-bpp series complexes
The complexes iron(II) of 2,6-di(pyrazol-1-yl) (1-bpp), 2,6-di(pyrazol-3-yl) (1-bpp) and their derivatives have proven to be very flexible compounds in the research of SCO. Chemists have developed efficient synthetic routes for the functionalization of almost every site of the bpp skeleton. Our work reveals that the pybox series of compounds is a new tridentate ligand for the construction of SCO compounds. A list of SCO behaviours in several representative 1-bpp and 3-bpp series complexes with pybox series complexes is provided in Table 3. It is found that the spin transition behaviours of the 1-bpp and 3-bpp series complexes are also highly dependent on ligand structures, counter anions, and solvent. Especially, the 1-bpp series is very sensitive to counter anions. For instance, the BF4− ClO4−, and [Co(C2B8H11)]− based complexes are SCO complexes with transition temperatures around ambient temperature. However, the PF6−, ClO4−, and SbF6− complexes are HS complexes. Similar situations are also observed in our L1 series of compounds. The complicated SCO behaviours and structural phase transition occur in the 3-bpp series due to the introduction of intermolecular hydrogen bonding and methyl groups. In our L1 series, some interesting phenomena, such as hysteresis loops and structural phase transitions, are observed as well. These results demonstrate that the modification of the prototype skeleton not only tunes SCO behaviour but also may produce several synergetic effects. Among them, L2 is a versatile ligand for the construction of SCO compounds. Complexes from the L2 ligand all display SCO behaviour, regardless of the counter anion and solvent, thus exhibiting potential applications in the functional materials field, similar to the famous complex Fe(1-bpp)2[BF4]2.| Compounds | Spin state | T 1/2 | Hysteresis loop |
|---|---|---|---|
| Fe(1-bpp)2[BF4]261,62,72,73 |
SCO (abrupt) | 261 K | 3 K |
| [Fe(1-bpp)2][BF4]2·2.9CH3NO2·0.25H2O72 | SCO (irreversible) | 260–265 K | — |
| [Fe(1-bpp)2][Co(C2B9H11)2]273 |
SCO (gradual, 50%) | 318 K | No |
| Fe(1-bpp)2[PF6]262,72 |
HS | — | — |
| Fe(1-bpp)2[ClO4]2·(CH3)2CO73 | SCO | 175 ± 3 K | No |
| Fe(1-bpp)2[SbF6]262 |
HS | — | — |
| Fe(3-Me-1-bpp)2[BF4]2·xH2O (x = 0–1/3)74 | SCO (two step) | 1st: 150 K (↑), 144 K (↓) | 1st: 6 K |
| 2nd: 114 K (↑), 96 K (↓) | 2nd: 18 K | ||
| Fe(3-bpp)2[BF4]2·2H2O75,76 | SCO (gradual) | 300 K | No |
| Fe(3-bpp)2[BF4]2·2H2O (dehydrate)75,76 | SCO (abrupt) | 170 K (↓), 180 K (↑) | 10 K |
| Fe(3-bpp)2[BF4]2·3H2O77 | LS | — | — |
| [Fe(3-bpp)2]I2·4H2O77 | LS | — | — |
| [Fe(3-bpp)2]I2·4H2O (dehydrate)77 | SCO (abrupt) | 203 K (↓), 205 K (↑) | 2 K |
| [Fe(3-bpp)2](SCN)2·2H2O78 | SCO (two-step, abrupt) | 1st: 256 K (↑), 247 K (↓) | 9 K (1st) |
| 2nd: 219 K (↑), 193 K (↓) | 24 K (2nd) | ||
| [Fe(3-bpp)2](SeCN)278 |
SCO (abrupt) | 231 K | ≤1 K |
| [Fe(3-bpp)2](CF3SO3)2·H2O79,80 | SCO abrupt, one-step (↓) | 140 K (↓), 147 K (↑) (1st) | Asymmetric and wide |
| Two-step (↑) | 285 K (↑) (2nd) | ||
| [Fe(5-Me-3-bpp)2](BF4)281 |
SCO | 205 K | 65 K to 37 K |
| [Fe(5-Me-3-bpp)2](ClO4)282 |
SCO | <200 K | 65 K |
| [Fe(L1)2](ClO4)2 | SCO | 170 K | No |
| [Fe(L1)2](ClO4)2·MeCN | HS | — | — |
| [Fe(L1)2](BF4)2 | SCO | 145 K | 5 K |
| [Fe(L1)2](BF4)2·MeCN | HS | — | — |
| [Fe(L1)2](PF6)2·0.5MeCN·0.5Et2O | HS | — | — |
| [Fe(L1)2](PF6)2 | HS | — | — |
| [Fe(L1)]2(BPh4)2·MeCN | SCO | 235 K | No |
| [Fe(L1)]2(BPh4)2 | SCO (incomplete) | 240 K | No |
| [Fe(L2)]2(ClO4)2 | SCO | 355 K | No |
| [Fe(L2)2](BF4)2 | SCO | 365 K | No |
| [Fe(L2)2](PF6)2·MeCN | SCO (two step) | 196 K, 305 K | No |
| [Fe(L2)2](PF6)2 | SCO | 300 K | No |
| [Fe(L2)2](BPh4)2 | SCO (two step) | 150 K, 446 K | No |
Conclusions
In summary, a family of [Fe(pybox)2]2+ type mononuclear Fe(II) complexes has been synthesized with variation in the ligand structures, counter anions, and solvents in order to explore the possible SCO behaviour and elucidate the potential relationship between structure and magnetic properties. In this series, 1(BF4) exhibits abrupt SCO at around 150 K and proceeds to the LS state with 50% completeness, which is accompanied by a synergetic structural phase transition. Compounds 1(BPh4)·MeCN, 2(ClO4), and 2(BF4) all exhibit one step SCO with T1/2 near or above ambient temperature. Desolvated compounds 1(BPh4) and 2(PF6) display a gradual but incomplete SCO. The SCO behaviours of 2(PF6)·MeCN and 2(BPh4) occur in a two-step fashion. These results show that the iron(II) complexes constructed from pybox ligands belong to a new family of FeN6 SCO compounds. Furthermore, we have discussed the influence of ligand structures, counter anions, and solvents on SCO properties, the magneto-structural correlation, and compare well-known bpp series compounds in detail. Several structural parameters have been proven to be sensitive indices to determine the occurrence of SCO. In the next step, we will focus on the synthesis of new mononuclear complexes, multinuclear Fe(II) clusters and coordination polymers based on the pybox moiety and aim to introduce several new functions in this SCO system.83–87 The combination and synergy of SCO with other physical properties, such as redox, fluorescence, ferroelectricity, and conductivity, in this system will also be attempted.Acknowledgements
This work was partly supported by NSFC (21302035, 21371043, 21421005, 21322103, and 91422302) and the Fundamental Research Funds for the Central Universities. We thank Prof. Ming-Liang Tong and Mr Yan-Cong Chen (Sun Yat-Sen University) for kindly help in magnetic measurements at high temperature range and Prof. Wei-Xiong Zhang (Sun Yat-Sen University) and Ms Lin Wang (Beijing Institute of Technology) for kindly help and discussion in crystal structure refinement.Notes and references
- P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds I–III, Top. Curr. Chem., 2004, pp. 233–235 Search PubMed.
- M. A. Halcrow, Spin-Crossover Materials - Properties and Applications, John Wiley & Sons, Chichester, 2013, p. 568 Search PubMed.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068–2093 CrossRef.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed.
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- O. Sato, J. Tao and Y.-Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed.
- T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CAS.
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Angew. Chem., Int. Ed., 2015, 54, 823–827 CrossRef CAS PubMed.
- C.-F. Wang, R.-F. Li, X.-Y. Chen, R.-J. Wei, L.-S. Zheng and J. Tao, Angew. Chem., Int. Ed., 2015, 54, 1574–1577 CrossRef CAS PubMed.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97–128 CrossRef CAS.
- H. A. Goodwin, Top. Curr. Chem., 2004, 234, 23–47 CrossRef CAS.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- J.-F. Létard, P. Guionneau, E. Codjovi, O. Lavastre, G. Bravic, D. Chasseau and O. Kahn, J. Am. Chem. Soc., 1997, 119, 10861–10862 CrossRef.
- S. Hayami, Z.-Z. Gu, H. Yoshiki, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2001, 123, 11644–11650 CrossRef CAS PubMed.
- V. Niel, J. M. Martinez-Agudo, M. C. Munoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2001, 40, 3838–3839 CrossRef CAS PubMed.
- B. Weber, W. Bauer and O. Jaroslava, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed.
- N. F. Sciortino, S. M. Neville, J. F. Letard, B. Moubaraki, K. S. Murray and C. J. Kepert, Inorg. Chem., 2014, 53, 7886–7893 CrossRef CAS PubMed.
- L. Pineiro-Lopez, M. Seredyuk, M. C. Munoz and J. A. Real, Chem. Commun., 2014, 50, 1833–1835 RSC.
- W. Liu, L. Wang, Y.-J. Su, Y.-C. Chen, J. Tucek, R. Zboril, Z.-P. Ni and M.-L. Tong, Inorg. Chem., 2015, 54, 8711–8716 CrossRef CAS PubMed.
- A. Hauser, Top. Curr. Chem., 2004, 233, 49–58 CrossRef CAS.
- G. Agusti, A. B. Gaspar, M. C. Munoz and J. A. Real, Inorg. Chem., 2007, 46, 9646–9654 CrossRef CAS PubMed.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, P. D. Southon, J. D. Cashion, J. F. Létard, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 2869–2876 CrossRef CAS PubMed.
- M. Arai, W. Kosaka, T. Matsuda and S. Ohkoshi, Angew. Chem., Int. Ed., 2008, 47, 6885–6887 CrossRef CAS PubMed.
- P. Nielsen, H. Toftlund, A. D. Bond, J. F. Boas, J. R. Pilbrow, G. R. Hanson, C. Noble, M. J. Riley, S. M. Neville, B. Moubaraki and K. Murray, Inorg. Chem., 2009, 48, 7033–7047 CrossRef CAS PubMed.
- Z.-P. Ni and M. P. Shores, Inorg. Chem., 2010, 49, 10727–10735 CrossRef CAS PubMed.
- O. Roubeau, Chem. – Eur. J., 2012, 18, 15230–15244 CrossRef CAS PubMed.
- F. Setifi, E. Milin, C. Charles, F. Thetiot, S. Triki and C. J. Gomez-Garcia, Inorg. Chem., 2014, 53, 97–104 CrossRef CAS PubMed.
- J.-Y. Li, Y.-C. Chen, Z.-M. Zhang, W. Liu, Z.-P. Ni and M.-L. Tong, Chem. – Eur. J., 2015, 21, 1645–1651 CrossRef CAS PubMed.
- X.-H. Zhao, S.-L. Zhang, D. Shao and X.-Y. Wang, Inorg. Chem., 2015, 54, 7857–7867 CrossRef CAS PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2008, 37, 278–289 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1–47 CrossRef.
- H. A. Goodwin, Top. Curr. Chem., 2004, 233, 59–90 CrossRef CAS.
- G. J. Long, F. Grandjean and D. L. Reger, Top. Curr. Chem., 2004, 233, 91–122 CrossRef CAS.
- P. J. van Konngsbruggen, Top. Curr. Chem., 2004, 233, 123–149 CrossRef.
- M. C. Muñoza and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068–2093 CrossRef.
- M. A. Halcrow, Coord. Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS.
- J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203–240 CrossRef.
- L. J. K. Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef.
- G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119–3154 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, S. Viswanathan and A. Rollett, J. Am. Chem. Soc., 2007, 129, 15436–15437 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, P. S. Barber, S. Viswanathan, D. T. de Lill, A. Rollett, G. Ling and S. Altun, Inorg. Chem., 2010, 49, 8848–8861 CrossRef CAS PubMed.
- J. Yuasa, T. Ohno, K. Miyata, H. Tsumatori, Y. Hasegawa and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9892–9902 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987–6994 CrossRef CAS PubMed.
- S. Chorazy, K. Nakabayashi, K. Imoto, J. Mlynarski, B. Sieklucka and S.-I. Ohkoshi, J. Am. Chem. Soc., 2012, 134, 16151–16154 CrossRef CAS PubMed.
- Y.-Y. Zhu, C. Cui, N. Li, B.-W. Wang, Z.-M. Wang and S. Gao, Eur. J. Inorg. Chem., 2013, 3101–3111 CrossRef CAS.
- N. Liu, L.-H. Jia, Z.-Q. Wu, Y.-Y. Zhu, B.-W. Wang and S. Gao, Chin. J. Inorg. Chem., 2014, 30, 1660–1666 CAS.
- A. Duerrbeck, S. Gorelik, J. Hobley, J. Wu, A. Hor and N. Long, Chem. Commun., 2015, 51, 8656–8659 RSC.
- Y.-Y. Zhu, C.-W. Liu, J. Yin, Z.-S. Meng, Q. Yang, J. Wang, T. Liu and S. Gao, Dalton Trans., 2015, 44, 20906–20912 RSC.
- R. L. Carlin, Magnetochemistry, Springer-Verlag Press, Berlin, Heidelberg, 1996 Search PubMed.
- J. F. Letard, P. Guionneau and L. Goux-Capes, Top. Curr. Chem., 2004, 225, 221–249 CrossRef.
- J. F. Létard, L. Capes, G. Chastanet, N. Moliner, S. Létard, J. A. Real and O. Kahn, Chem. Phys. Lett., 1999, 313, 115–120 CrossRef.
- G. M. Sheldrick, SHELXS-97, University of Göttingen, Göttingen, Germany, 1990 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. K. McCusker, A. L. Rheingold and D. N. Hendrickson, Inorg. Chem., 1996, 35, 2100–2112 CrossRef CAS.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97–128 CrossRef CAS.
- M. Marchivie, P. Guionneau, J.-F. Létard and D. Chasseau, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 25–28 CrossRef PubMed.
- J. M. Holland, J. A. McAllister, Z. Lu, C. A. Kilner, M. Thornton-Pett and M. A. Halcrow, Chem. Commun., 2001, 577–578 RSC.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2005, 1693–1700 Search PubMed.
- K. Takahashi, H.-B. Cui, Y. Okano, H. Kobayashi, H. Mori, H. Tajima, Y. Einaga and O. Sato, J. Am. Chem. Soc., 2008, 130, 6688–6689 CrossRef CAS PubMed.
- S. Alvarez, J. Am. Chem. Soc., 2003, 125, 6795–6802 CrossRef CAS PubMed.
- J.-F. Létard, J. Mater. Chem., 2006, 16, 2550–2559 RSC.
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1–47 CrossRef.
- X. Bao, P.-H. Guo, W. Liu, J. Tucek, W.-X. Zhang, J.-D. Leng, X.-M. Chen, I. Gural′skiy, L. Salmon, A. Bousseksou and M.-L. Tong, Chem. Sci., 2012, 3, 1629–1633 RSC.
- H. L. C. Feltham, C. Johnson, A. B. S. Elliott, K. C. Gordon, M. Albrecht and S. Brooker, Inorg. Chem., 2015, 54, 2902–2909 CrossRef CAS PubMed.
- M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Angew. Chem., Int. Ed., 2004, 43, 4589–4593 CrossRef CAS PubMed.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE, version 2.0, Universitat de Barcelona, Barcelona, Spain, 2010 Search PubMed.
- M. Marchivie, P. Guionneau, J.-F. Létard and D. Chasseau, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 25–28 CrossRef PubMed.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 548–554 RSC.
- J. Elhaïk, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2006, 823–830 RSC.
- V. A. Money, C. Carbonera, J. Elhaïk, M. A. Halcrow, J. A. K. Howard and J.-F. Létard, Chem. – Eur. J., 2007, 13, 5503–5514 CrossRef CAS PubMed.
- K. H. Sugiyarto and H. A. Goodwin, Aust. J. Chem., 1988, 41, 1645–1663 CrossRef CAS.
- T. Buchen, P. Gütlich and H. A. Goodwin, Inorg. Chem., 1994, 33, 4573–4576 CrossRef CAS.
- K. H. Sugiyarto, D. C. Craig, A. D. Rae and H. A. Goodwin, Aust. J. Chem., 1994, 47, 869–890 CrossRef CAS.
- K. H. Sugiyarto, M. L. Scudder, D. C. Craig and H. A. Goodwin, Aust. J. Chem., 2000, 53, 755–765 CrossRef CAS.
- K. H. Sugiyarto, K. Weitzner, D. C. Craig and H. A. Goodwin, Aust. J. Chem., 1997, 50, 869–874 CrossRef CAS.
- T. Buchen, P. Giitlich, K. H. Sugiyarto and H. A. Goodwin, Chem. – Eur. J., 1996, 2, 1134–1138 CrossRef CAS.
- T. D. Roberts, F. Tuna, T. L. Malkin, C. A. Kilner and M. A. Halcrow, Chem. Sci., 2012, 3, 349–354 RSC.
- T. D. Roberts, M. A. Little, F. Tuna, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2013, 49, 6280–6282 RSC.
- L. J. K. Cook, R. Kulmaczewski, S. A. Barrett and M. A. Halcrow, Inorg. Chem. Front., 2015, 2, 662–670 RSC.
- M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Dalton Trans., 2011, 40, 2154–2156 RSC.
- M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Inorg. Chem., 2016, 55, 2263–2273 CrossRef PubMed.
- R. Ohtani, S. Egawa, M. Nakaya, H. Ohmagari, M. Nakamura, L. F. Lindoy and S. Hayami, Inorg. Chem., 2016, 55, 3332–3337 CrossRef CAS PubMed.
- H.-Q. Li, Z.-Y. Ding, Y. Pan, C.-H. Liu and Y.-Y. Zhu, Inorg. Chem. Front., 2016, 3, 1363–1375 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic route of ligands L1–2, NMR spectra of intermediates and ligands (Fig. S1–S6), structural details (Fig. S7–S24), full crystallographic data (Table S1), magnetic and Raman characterization (Fig. S25–S33). CCDC 1468980–1468991 and 1507077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00417b |
| This journal is © the Partner Organisations 2016 |