
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ stabilizer formation from methacrylic acid macromonomers in emulsion polymerization†
Ingeborg
Schreur-Piet
 and
Johan P. A.
Heuts
*
and
Johan P. A.
Heuts
*
Department of Chemical Engineering & Chemistry, Eindhoven University of Technology, P O Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: j.p.a.heuts@tue.nl
First published on 17th October 2017
Abstract
Oligomers of methacrylic acid containing a propenyl ω-endgroup (i.e. MAA-macromonomers) were synthesized by cobalt-mediated catalytic chain transfer polymerization and used as precursors to stabilizers in emulsion polymerization. It was found that only in those polymerizations in which these precursors were sufficiently quickly converted into amphiphilic molecules, via a type of polymerization induced self-assembly (PISA) mechanism, stable emulsion polymerization could be carried out. This process was too slow in the emulsion polymerization of methyl methacrylate (MMA) and in order to obtain stable latexes, the addition of a conventional surfactant (sodium dodecyl sulphate, SDS) was necessary. In the emulsion polymerization of butyl acrylate, however, reactions with the macromonomers were faster and because of the more hydrophobic nature of BA (as compared to MMA), stabilizers were sufficiently quickly formed in situ and stable latexes were produced without the need for additional SDS. Also the emulsion polymerization of butyl methacrylate (BMA), which reacts via the same “sulfur-free RAFT” mechanism as MMA, could be carried out in the absence of SDS because of the greater hydrophobicity of the monomer. Copolymerizations of MMA with >30% of BA or 85% BMA also resulted in stable latexes without the addition of SDS. The synthesized macromonomers and in situ formed copolymers were characterized by means of size exclusion chromatography (SEC), 1H NMR spectroscopy and MALDI-ToF MS.
Introduction
Polymer latexes prepared by emulsion polymerization have found and are continuously finding applications in many fields, ranging from coatings and adhesives to biomedical applications.1–3 In emulsion polymerization surfactants play an important role in controlling the particle diameter, the stability and the surface functionality of the formed latex particles. During latex film formation, surfactants can migrate from the surface of the particles towards the film interface and may have a negative effect on final film properties such as water sensitivity, gloss, adhesion and blocking.4–7 A generally employed strategy used to circumvent these problems is the use of reactive surfactants which are chemically bound to the polymer particles.8–11 On the one hand an ideal reactive surfactant should not be too reactive during emulsion polymerization at low monomer conversion to avoid burying of the surfactant groups inside the latex particles and/or the formation of water-soluble polymer chains that cause bridging flocculation.12 On the other hand, all of the surfactants should have reacted by the end of the polymerization so that a stable latex with bound surfactants is obtained.13 Surfactants containing a propenyl end-group would be promising candidates displaying just the right reactivity. Oligomers containing these end-groups (called macromonomers in the remainder of this paper) are readily prepared via Co-mediated catalytic chain transfer (CCT)14–16 (see Scheme 1) and their subsequent copolymerization behavior has been described previsously.17–19 In earlier studies also, amphiphilic macromonomers were synthesized and subsequently used as stabilizers in emulsion polymerization.20,21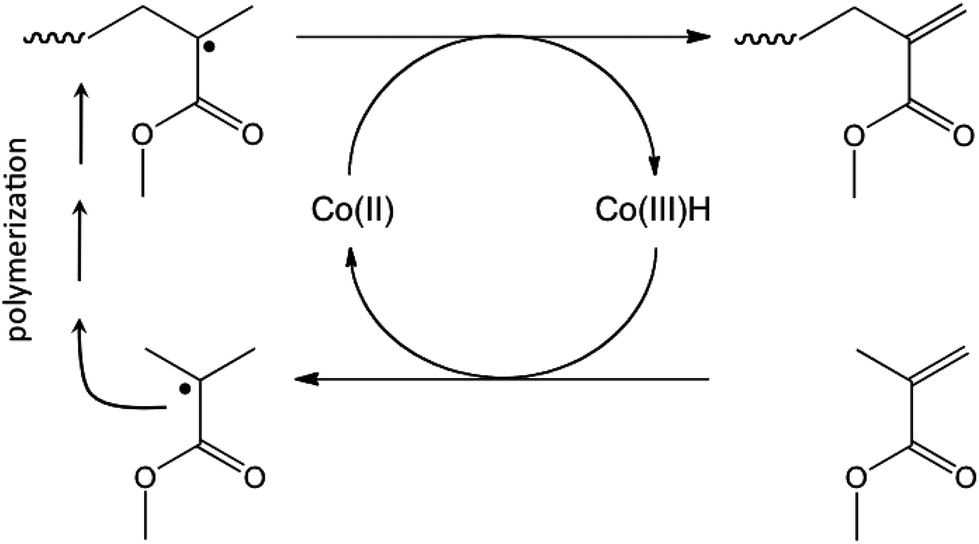 | ||
| Scheme 1 Schematic representation of the Co-mediated CCT polymerization of methacrylates. | ||
In the current study, methacrylic acid macromonomers (synthesized via the Co-mediated CCT polymerization of t-butyl methacrylate followed by acidolysis of the t-butyl groups) are used as precursors to stabilizing agents in the emulsion polymerization of methacrylates and acrylates. Both acrylates and methacrylates were used because it is known that methacrylic macromonomers react differently with these two classes of monomers14,22 and will yield reactive surfactants with different architectures. The macromonomers will react via an addition–fragmentation chain transfer mechanism with methacrylates and the process will result in surface-active block copolymers. In fact it is exactly this mechanism (in combination with low monomer concentrations) that has recently been coined “sulfur-free RAFT” by Haddleton and co-workers,23,24 and was recently used by Zetterlund and co-workers25 in a “non-living version” of polymerization induced self-assembly (PISA).26 With acrylates the macromonomers will ultimately yield graft copolymers.14 The influence of the length and the added amount of macromonomers on the particle size of the latex particles and on the stability of the final latex will be examined. Also the influence of the architecture of the in situ formed copolymer on these properties will be discussed.
Experimental
Materials
All monomers, methyl methacrylate (MMA), n-butyl acrylate (BA), n-butyl methacrylate (BMA), ethyl acrylate (EA) and tert-butyl methacrylate (t-BMA), were obtained from Sigma-Aldrich (99%). The monomers were passed over a column of an inhibitor remover (Aldrich) to remove the inhibitor. Azobis(isobutyronitrile) (AIBN, Merck) was recrystallized from methanol. The bis-methanol complex of cobaloxime boron fluoride (COBF) was prepared as described previously27 (measured CT for MMA in bulk = 34 × 103 at 60 °C). Toluene (AR, Biosolve), dichloromethane (DCM, AR, Biosolve) and trifluoro acetic acid (TFA, 95%, Aldrich) were all used as received. Sodium dodecyl sulfate (SDS, 99%), potassium persulfate (KPS, p.a.) and sodium carbonate (dehydrated, p.a.) were purchased from Merck and used as received.Synthesis of the t-butyl methacrylate macromonomer
For the t-butyl methacrylate macromonomer (t-BMA-MM) synthesis, t-BMA was polymerized with AIBN in toluene in a round-bottom flask at 60 °C.28 The initially added amount of the COBF catalyst was varied to obtain macromonomers with different molecular weights. In a typical experiment the procedure is as follows: 141 g t-BMA (1.0 mol) and 200 g toluene (2.2 mol) were deoxygenated by purging for 30 minutes with nitrogen at 0 °C in a flask (A) sealed airtight with a septum. To a separate flask (B), fitted with a magnetic stirring bar, 600 mg AIBN (3.7 × 10−3 mol) and 10 mg COBF (2.6 × 10−5 mol) were added; the flask was consecutively evacuated and purged with nitrogen three times.Then the monomer solution A was added to flask B via a cannula and the solution was heated to 60 °C. The reaction mixture was left with continuous stirring for 24 h. The t-BMA-MM was isolated by evaporation of toluene and residual monomer under reduced pressure and subsequently dried in a vacuum oven at 60 °C for 24 h (120 g yield, 85%). The molar mass distributions and the number-average degrees of polymerization (DPn) were determined by SEC and 1H NMR, and the results are summarized in Table 1 (see ESI† for more details).
| # | [COBF]/[t-BMA] | t-BMA-MM | MAA-MM | Sample name | ||
|---|---|---|---|---|---|---|
DP
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Đ |
DP
nb |
DP
nb |
|||
| a Determined via SEC using appropriate Mark-Houwink constants to convert the polystyrene calibration curve;29 for samples 1 and 2 DPn = Mw/(2 × 142),30 for sample 3 DPn = Mn/142. b From 1H NMR. | ||||||
| 1 | 3.0 × 10−6 | 14 ± 5 | 1.9 | 17 ± 1 | 17 ± 3 | MAA16 |
| 2 | 2.4 × 10−6 | 80 ± 12 | 2.3 | 70 ± 4 | 80 ± 8 | MAA80 |
| 3 | 0.9 × 10−6 | 350 ± 24 | 2.0 | 330 ± 25 | 335 ± 25 | MAA350 |
Synthesis of the methacrylic acid macromonomer
For the synthesis of the MAA macromonomer (MAA-MM), t-BMA-MM was acidolized using trifluoroacetic acid (TFA) in dichloromethane (DCM). A typical procedure is as follows: a round-bottom flask fitted with a magnetic stirring bar was charged with t-BMA-MM (100 g, MSECn = 2300 g mol−1, Đ = 1.9) and DCM (100 mL). The mixture was stirred until the polymer was dissolved after which TFA (100 mL, 1.3 mol) was added. Subsequently, the mixture was allowed to stir at room temperature for 48 h. DCM and excess TFA were removed by evaporation under reduced pressure and the resulting polymer was dried in a vacuum oven at 60 °C for 2 days to yield the MAA-MM (60 g yield, 100%). The number-average degrees of polymerization were determined via1H NMR, and the results are summarized in Table 1.Emulsion polymerization
Emulsion polymerizations were carried out in both batch and semi-batch operation. All experiments were carried out under argon in a jacketed and baffled glass reactor (250 mL), thermostated at 60 °C and equipped with a mechanical four-bladed turbine stirrer. The monomer conversions during and after the reaction were determined gravimetrically.For the batch emulsion polymerizations all ingredients except the initiator solution were charged into the reactor, stirred at 350 rpm, purged with argon for 30 minutes and subsequently heated to 60 °C. Five minutes after reaching a constant temperature, an aqueous KPS solution (10 mL containing 0.08 g of KPS) was injected to initiate the polymerization.
For the semi-batch emulsion polymerizations the reactor was charged with water, buffer, stabilizer(s) and 3 g of the monomer (i.e. 10% of the overall monomer content), stirred at 350 rpm, purged with argon for 30 minutes and subsequently heated to 60 °C. Five minutes after reaching a constant temperature, an aqueous KPS solution (10 mL containing 0.08 g of KPS) was injected to initiate the polymerization. Starting 1 h after initiation, the remaining monomer (27 g) was added at a constant feeding rate of 5 mL h−1.
Particle size distributions were determined by dynamic light scattering (DLS) and some of the latexes were examined using scanning (SEM) and transmission electron microscopy (TEM).
Analysis
SEC for high molecular weight macromonomers and polymers was carried out using a Waters Alliance system equipped with a Waters 2695 separation module, a Waters 2414 refractive index detector (40 °C), a Waters 2487 dual UV absorbance detector, a PSS SDV 5 μm bead size guard column along with two PSS SDV 5 μm bead size linear XL columns in series (300 × 8 mm) at 40 °C. Tetrahydrofuran (THF stabilized with BHT, Biosolve) with 1 v/v% acetic acid was used as the eluent at a flow rate of 1.0 mL min−1. The system was calibrated with polystyrene standards (Polymer Laboratories, Mn = 580 g mol−1 − Mn = 7.1 × 106 g mol−1), after which correction was applied using the appropriate Mark–Houwink parameters (polystyrene: KpS = 1.14 × 10−4 dL g−1, apS = 0.716 and poly(tert-butyl methacrylate) Kpt-BMA = 5.84 × 10−5 dL g−1, apt-BMA = 0.76).29
:1:4 volume ratio. Subsequently, 0.30 μL of this mixture was spotted on the sample plate, and the spots were dried at room temperature.
Results and discussion
Emulsion polymerization of methyl methacrylate
First, the emulsion polymerization of MMA in the presence of MAA macromonomers was investigated. In the initial experiments we used SDS to ensure sufficient stabilization of the latex particles and investigated the co-stabilizing effect of the MAA macromonomers. The effects of the amount and the chain length of the MAA macromonomers on the polymerization rate, the particle formation and the particle size distribution in the emulsion polymerization were studied; the macromonomer amount was varied between 1 and 4 wt% and chain lengths (DPn) of 16, 80 and 350 were used. In all cases SDS was added at a concentration of 10 mM (= 1.5 wt%), which is just above its critical micelle concentration (CMC = 9.5 mM at 60 °C).33 A comparative experiment without the macromonomer was also performed. The conversion versus time curves were measured and the results are shown in Fig. 1. All polymerizations proceeded to high monomer conversion and resulted in stable latexes without significant amounts of coagulum.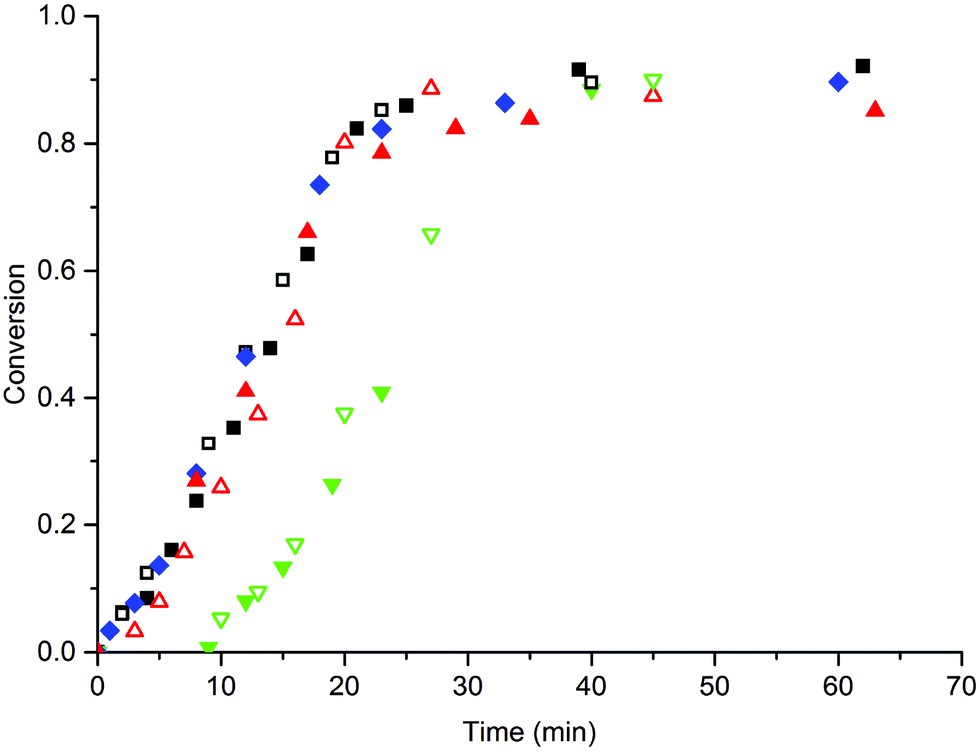 | ||
Fig. 1 Conversion-time curve of the emulsion polymerization of MMA in the presence of 1.5 wt% (= 10 mM) SDS. The polymerizations were performed in the absence of a macromonomer ( ), 1 wt% (open symbols) and 4 wt% (closed symbols) MAA16 (■), MAA80 ( ), 1 wt% (open symbols) and 4 wt% (closed symbols) MAA16 (■), MAA80 ( ) and MAA350 ( ) and MAA350 ( ). Standard polymerization conditions as listed in Table 2. ). Standard polymerization conditions as listed in Table 2. | ||
From the results shown in Fig. 1, it is immediately clear that the addition of MAA16 or MAA80 to the system does not significantly affect the polymerization rate as compared to the system that is only stabilized by SDS; only the addition of MAA350 results in a significantly delay in the onset of polymerization, but then proceeds with a polymerization rate similar to the other systems (the conversion rate ≈0.040 ± 0.005 min−1 during interval II in all cases). These similar rates imply that the particle numbers are also similar in all reactions (Np ≈ (1.3 ± 0.3) × 1017 L−1), determined from Dn, which in turn suggests that SDS is the main stabilizer in all these reactions, without any significant contribution from the macromonomers. The initial delay in the MAA350 systems is conceivably explained by the fact that these longer hydrophilic chains require the addition of more hydrophobic MMA units before they become surface-active and as such initially act as a kind of “propagating radical sink”. This is supported by the observation that during the experiments with MAA350 the viscosity of the latex increased because of flocculation, which is indicative of the presence of a water-soluble polymer.34
Particle size distributions (PSD) of the latexes were measured as a function of the conversion. The polydispersity index (PdI) starts for all reactions at around 0.1 and increases significantly at higher conversions (not shown). A summary of the PSD characteristics of the final latexes is given in Table 3. The DLS results in this table suggest an increasing Dn with an increasing macromonomer content (which is at odds with similar polymerization rates observed in Fig. 1), but this increase is clearly not observed in our SEM images (see Fig. 2).
 | ||
| Fig. 2 SEM images of the final latex made using 1.5 wt% of SDS and (a) 1 wt% and (b) 4 wt% of MAA80. | ||
| Ingredient | Amount |
|---|---|
| a Batch: all monomer was added at the start; semi-batch: 10% of the monomer was added initially, the remaining monomer was added at a rate of 5 mL h−1, starting 1 h after initiation. b wt% = weight percentage relative to the monomer (= g per 100 g of monomer). | |
| Water | 120 g |
| Na2CO3 | 0.4 g (0.02 M) |
| Macromonomer | 1.5 g (5 wt%b, varied between 0–11 wt%) |
| SDS | 0.3 g (1.5 wt%b, varied between 0–1.5 wt%) |
| Monomera | 30 g (solids content 20%) |
| KPS | 0.08 g (0.25 wt%b; 2.5 × 10−3 M) |
In Fig. 2, the SEM images of the final latexes produced using 1 wt% and 4 wt% MAA80 are shown. These images show monomodal distributions suggesting that the high PdI values observed in the DLS measurements are caused by the (reversible) aggregation of particles. The number mean particle diameter of both latexes obtained by SEM is around 85 nm (particle count >100).
From the results presented thus far it is safe to conclude that the presence of the MAA macromonomers in the emulsion polymerization does not significantly affect the polymerization rate and the particle size, and that SDS dominates the particle stabilization. In order to study the stabilizing properties of the macromonomers explicitly, we performed emulsion polymerizations using decreasing amounts of SDS and a constant amount of macromonomer (4 wt%). In Fig. 3 conversion versus time curves of the emulsion polymerization of MMA with 4 wt% MAA80 and variable amounts of SDS are shown (for MAA16 and MAA350, see ESI†). It is clear from this figure that the polymerizations using 1.0 and 1.5 wt% SDS result in high monomer conversions and stable latexes, but in the absence of SDS it was impossible to reach a conversion of higher than 40%. In the latter polymerization and that carried out using 0.5 wt% of SDS, a strong increase in the viscosity was observed above 40% conversion. This viscosity increase was caused by heavy flocculation and therefore trapping of water inside the flocs.34 In both cases the flocs eventually collapsed and the polymer particles sedimented.
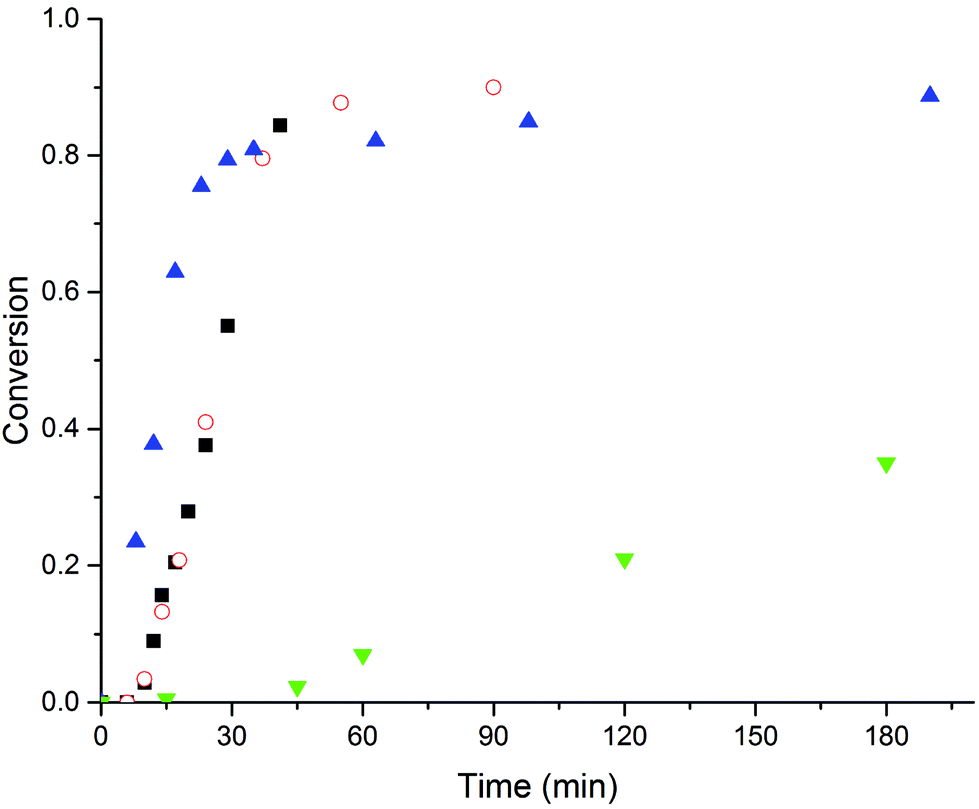 | ||
Fig. 3 Batch emulsion polymerization of MMA with 4 wt% MAA80 and variable amounts of SDS. Concentration SDS: ( ) 0 wt%, (■) 0.5 wt%, ( ) 0 wt%, (■) 0.5 wt%, ( ) 1 wt% SDS and ( ) 1 wt% SDS and ( ) 1.5 wt%. Standard polymerization conditions as listed in Table 2. ) 1.5 wt%. Standard polymerization conditions as listed in Table 2. | ||
From Fig. 3 it can also be seen that the polymerizations using SDS concentrations below the CMC (= 9.5 mM ≈ 1.4 wt%) show a significant delay in the onset of polymerization. This result can be explained by the fact that in these reactions initially no micellar nucleation takes place and that the MAA macromonomers are converted into surfactants by the addition of a monomer unit via a (reversible) addition fragmentation chain transfer19 (or “sulfur-free RAFT”)23,24 mechanism; hence particle formation takes place by a “PISA-like” mechanism25,26 aided by SDS when present. The process, however, is not fast/efficient enough to provide enough stabilization for the particles when no SDS is present. With the aim of reducing the monomer concentration and slowing down the polymerization we changed from a batch process to a semi-batch process (and changing the amounts of the macromonomer); in the case of SDS-free polymerizations, no stable latexes could be obtained.
In summary, it can be concluded that in the case of the emulsion homopolymerization of MMA the synthesized MAA macromonomers are too slowly converted into surfactants to provide sufficient stabilization and that some SDS is always needed. The use of insufficient SDS gives rise to heavy flocculation of the system, ultimately resulting in complete sedimentation of the polymer particles.
Emulsion polymerization of butyl acrylate
The emulsion polymerization of n-butyl acrylate (BA) was also investigated as this monomer is not only more reactive than MMA, but it also reacts via a different mechanism with the MAA-macromonomer as already mentioned in the introduction.14 Initial experiments showed that in these BA polymerizations the MAA macromonomers provided sufficient stabilization and that no SDS was required; hence no experiments were performed using these combinations. In Fig. 4 the overall conversion of BA is given as a function of time for batch and semi-batch reactions with various amounts of the MAA-macromonomer MAA80 in the absence of SDS.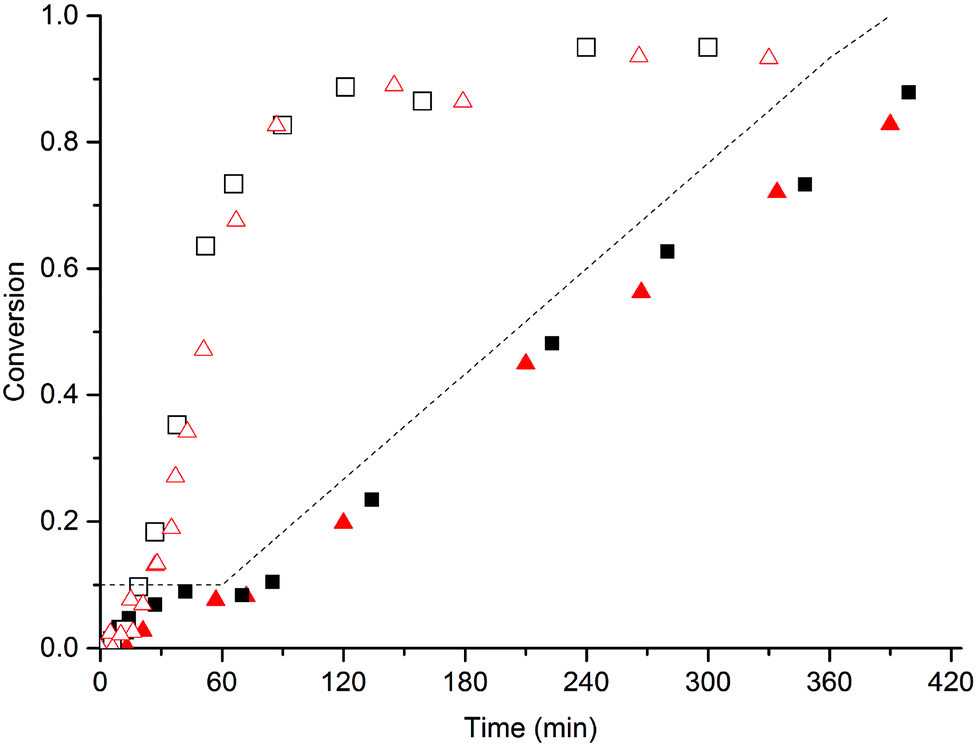 | ||
Fig. 4 Batch (open symbols) and semi-batch (closed symbols) emulsion polymerization of BA in the presence of MAA80, without SDS. Used concentrations of MAA80: 5 (■) and 10 ( ) wt%, respectively. Standard polymerization conditions as listed in Table 2. The dotted line indicates the addition profile of BA in the semi-batch reaction. ) wt%, respectively. Standard polymerization conditions as listed in Table 2. The dotted line indicates the addition profile of BA in the semi-batch reaction. | ||
First, two batch reactions were performed using 5 and 10 wt% of MAA80, respectively. It is clear from Fig. 4 that the amount of the macromonomer has no large influence on the conversion rate. Furthermore, monomer droplets were visible in samples taken up to monomer conversions of about 30% during these batch polymerizations. In order to prevent these potentially complicating monomer droplets we also performed semi-batch reactions. As is clear from the results in Fig. 4, the conversion of the BA in the semi-batch reaction closely follows the addition profile, indicating a near-full instantaneous conversion. Although in all cases minor coagulation is visible just after reaction (mostly around the stirrer and on the reactor wall), the latexes remained stable for periods of over 12 months. These results all suggest that efficient stabilizers are formed in these polymerizations.
The characteristics of the final latexes are summarized in Table 4 and two things are immediately clear from this table: (i) the particles obtained in the batch process are much larger than those obtained in the semi-batch process and (ii) increasing the amount of the macromonomer leads to a decrease in particle size. The first observation is consistent with a particle growth that is relatively fast as compared to stabilizer formation in the batch process, so after nucleation has started the newly formed stabilizers are used to stabilize the fast growing particles rather than form new particles. In the semi-batch process the initial batch period is mainly used for stabilizer and particle formation; the low amount of monomer precludes fast particle growth and additionally increases the probability of radicals to react with the macromonomers. Hence more surfactants are available for particle nucleation in the batch than in the semi-batch process. The second observation is more easily explained; increasing the amounts of the MAA macromonomer lead to increasing stabilizer concentrations, which in turn lead to increasing particle numbers (and concomitant decreasing particle diameters).
| MAA80 (wt%) | DLS | SEM/TEMa | Fraction polymer coagulatedb | ||
|---|---|---|---|---|---|
| D n (nm) | PdI | D n (nm) | |||
| a Number mean particle diameter; error 10%. b All obtained latexes from the semi-batch reactions showed good stability (>12 months), latexes obtained from the batch reactions coagulated over time. | |||||
| Batch | 5 | 1200 | 0.09 | 970 | 0.01 |
| 10 | 750 | 0.10 | 800 | 0.03 | |
| Semi-batch | 5 | 375 | 0.17 | 310 | 0.05 |
| 10 | 175 | 0.11 | 130 | 0.14 | |
The particle diameters in the batch reactions are high compared to that of a reference latex stabilized with 1.5 wt% SDS (Dn = 60 nm, PdI = 0.1), because fewer micelles are formed initially by the (reacted) macromonomer. Although almost no coagulation took place immediately during or after the reaction, the latexes obtained from the batch reactions coagulated over time.
In summary, it can be concluded that in the case of the emulsion polymerization of BA the synthesized MAA macromonomers are sufficiently and fast converted into efficient stabilizers. This significantly different behavior as compared to that observed in the MMA polymerization is conceivably explained by one or more of the following reasons: BA reacts faster than MMA, the resulting surfactant in the BA polymerization is different (graft copolymer) as compared to that in MMA polymerization (block copolymer),14 and finally, BA is more hydrophobic than MMA, so fewer monomer units are required to convert the MAA macromonomer into a surfactant. In what follows, we will investigate this in more detail.
Emulsion copolymerization of BA and MMA
Considering the fact that adequate stabilization was observed in the BA system, we first investigated whether the addition of BA to MMA could also lead to stable latexes. We therefore carried out several different SDS-free emulsion copolymerizations of the two monomers with varying amounts of MAA80 and in order to avoid the presence of monomer droplets (see the previous section) these polymerizations were carried out in a semi-batch. The conversion-time curves of these polymerizations using 10 wt% of MAA80 and varying monomer feed compositions are shown in Fig. 5 (for the 5 and 15 wt% data, see ESI†).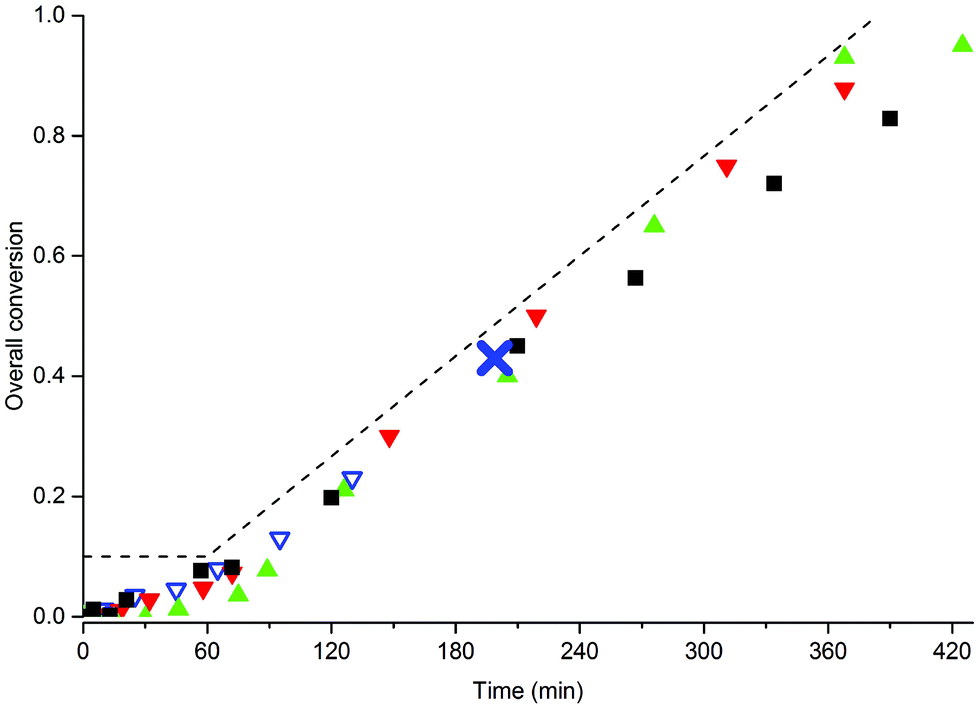 | ||
Fig. 5 Semi-batch copolymerization of BA and MMA with 10 wt% MAA80, no SDS. Monomer feed compositions (weight fractions of BA): wBA = 1 (■), wBA = 0.5 ( ), wBA = 0.3 ( ), wBA = 0.3 ( ) and wBA = 0.05 ( ) and wBA = 0.05 ( ). Standard polymerization conditions as listed in Table 2. The dotted line indicates the addition profile of the semi-batch reaction. The cross in the data set for wBA = 0.05 indicates major coagulation. ). Standard polymerization conditions as listed in Table 2. The dotted line indicates the addition profile of the semi-batch reaction. The cross in the data set for wBA = 0.05 indicates major coagulation. | ||
It is immediately clear from this figure that polymerizations with monomer feed compositions of wBA ≥ 0.3 all lead to stable latexes, and that only the system with 5% BA resulted in severe coagulation. A summary of the properties of the final latexes is given in Table 5, and comparison with the data in Table 4 shows that particle sizes are very similar and that increasing macromonomer concentrations lead to decreasing particle sizes. TEM and SEM images of the final latexes are shown in the ESI.†
| W BA | MAA80 (wt%) | D n (nm) | PdI | Fraction polymer coagulateda |
|---|---|---|---|---|
| a All obtained latexes showed good long term stability (>12 months). | ||||
| 1.0 | 5 | 375 | 0.10 | 0.05 |
| 10 | 175 | 0.10 | 0.10 | |
| 15 | 175 | 0.17 | 0.01 | |
| 0.7 | 15 | 120 | 0.05 | 0.01 |
| 0.5 | 5 | 500 | 0.08 | 0.05 |
| 10 | 300 | 0.12 | 0.10 | |
| 15 | — | — | — | |
| 0.3 | 5 | — | — | — |
| 10 | 400 | 0.12 | 0.02 | |
| 15 | 145 | 0.16 | 0.03 | |
| 0.05 | 5 | Coagulated | ||
| 10 | Coagulated | |||
| 15 | Coagulated | |||
In summary, it can be concluded that the addition of ≥30 wt% of BA to MMA results in the formation of efficient stabilizers for the SDS-free emulsion polymerization and that stable latexes are obtained. This result is of practical importance for coating applications, where these two monomers are often copolymerized.
Effects of monomer reactivity and hydrophobicity on emulsion polymerization
In order to investigate in more detail whether the higher reactivity and/or higher hydrophobicity are the cause of the better stabilization in the BA system, we studied the emulsion polymerizations of ethyl acrylate (EA) and n-butyl methacrylate (BMA). The former monomer has a similar reactivity to BA, but a hydrophobicity similar to MMA, and the latter monomer has a similar reactivity to MMA, but a hydrophobicity similar to BA. Both batch and semi-batch reactions were performed using these monomers in the presence of 5 wt% MAA80 and the absence of SDS. Conversion-time curves and the particle size distributions in the final latexes of both batch and semi-batch emulsion polymerizations of EA and BMA with MAA80 are shown in the ESI.† Only the BMA polymerization resulted in stable latexes when performed in batch (Dn = 475 nm, PdI = 0.40), whereas the EA batch polymerization resulted in severe coagulation at around 30% monomer conversion. The semi-batch reactions resulted in stable latexes for both monomers, although all reactions showed some minor amounts of coagulum (mainly around the stirrer and on the reaction walls). After removal of the coagulum all latexes remained stable in the long term (the pEA latex produced in semi-batch coagulated after about one month). The particle diameter of the semi-batch latex of pBMA (Dn = 350 nm, PdI = 0.06) is similar to that observed in the pBA latex produced under similar conditions (Dn = 375 nm, PdI = 0.17) and is much smaller than that observed in the semi-batch pEA latex (Dn = 1000 nm, PdI = 0.19). From both the batch and the semi-batch results it can be concluded that stabilizer formation is more efficient in the BMA system than in the EA system, implying that hydrophobicity is more important than reactivity. A higher reactivity, however, is advantageous, as can be concluded from the fact that the semi-batch EA polymerization does lead to stable latexes, whereas this was not possible for MMA.Emulsion copolymerization of BMA and MMA
In order to investigate whether the addition of small amounts of BMA to MMA could lead to stable SDS-free all-methacrylate latexes, we investigated the semi-batch emulsion copolymerization of BMA and MMA with several different monomer feed compositions. Conversion-time curves for monomer feed compositions with weight fractions of BMA (wBMA) down 0.5 are shown in Fig. 6 and it is immediately clear that more than 70% BMA is required for obtaining stable latexes.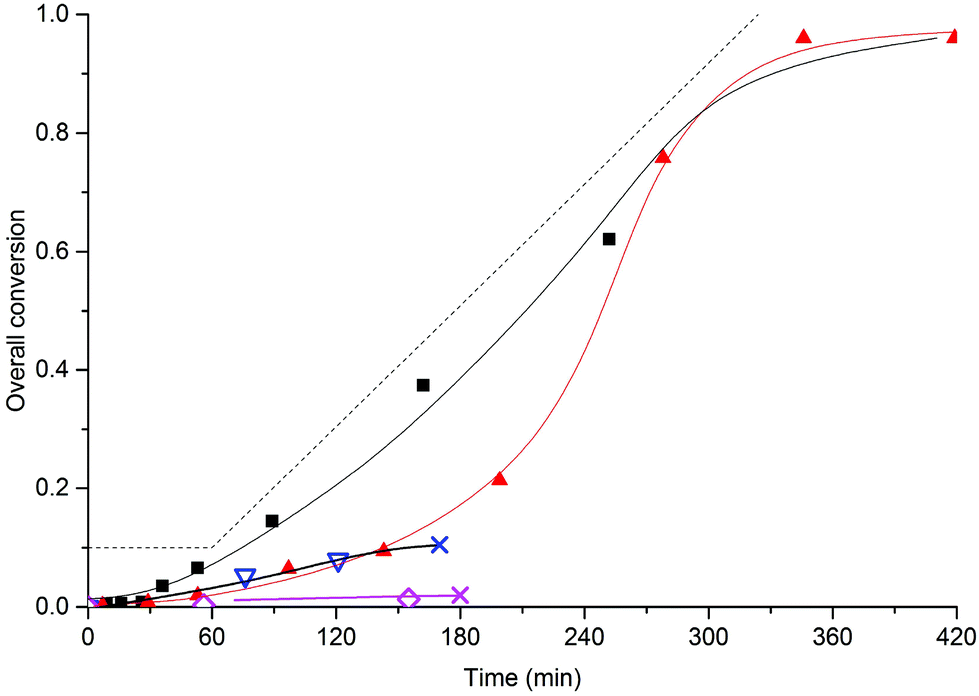 | ||
Fig. 6 Semi-batch copolymerization of BMA and MMA with 5 wt% MAA80. Monomer feed compositions (weight fractions of BMA): wBMA = 1 (■), wBMA = 0.85 ( ), wBMA = 0.7 ( ), wBMA = 0.7 ( ) and wBMA = 0.5 ( ) and wBMA = 0.5 ( ). Standard polymerization conditions as listed in Table 2. The dashed line is the addition profile for the semi-batch reactions, full curves are guides to the eye. Crosses indicate that major coagulation takes place. ). Standard polymerization conditions as listed in Table 2. The dashed line is the addition profile for the semi-batch reactions, full curves are guides to the eye. Crosses indicate that major coagulation takes place. | ||
In the cases with using wA ≤ 0.70 very low polymerization rates were observed and major coagulation occurred after 5 to 10% conversion; both of these observations are indicative of insufficient stabilization, which in turn is caused by a too slow production rate of the stabilizer. When comparing the other two systems, i.e., the BMA homopolymerization and the copolymerization with wBMA = 0.85, there is a very large difference in the polymerization rates. Although the homopolymerization proceeds roughly at the same rate as the monomer addition rate, the copolymerization is much slower in the beginning. This is indicative of a lower particle number in the latter case, which in turn is caused by a slower stabilizer production. This is also evident from the respective particle diameters (see Table 6); the particle size is much larger for the copolymerization as compared to that in the homopolymerization.
In the homopolymerization, stabilizer production is relatively fast as the addition of a few monomer units to the MAA macromonomer quickly leads to a surface-active block copolymer and combined with a slow particle growth, this leads to a larger number of particles that can be nucleated. This is in contrast to the copolymerization case. Since MMA is more water-soluble than BMA, it is likely that the chain extension of the MAA macromonomer will involve a significant number of MMA units and therefore a longer block (≈ more addition steps) is required to obtain a surface-active block copolymer. Hence stabilizer production will now be slowed down as compared to particle growth and the newly formed stabilizer during the polymerization is used to stabilize the growing particles, rather than to form new particles. This reasoning is completely analogous to that used for the explanation of the differences in particle sizes observed in the BA homopolymerizations in batch and semi-batch processes (see above, Fig. 4). Finally on comparing these BMA-MMA copolymerizations with the BA-MMA copolymerizations, it is clear that not only the hydrophobicity of the comonomer is important, but also the rate at which the stabilizer is formed.
Structure analysis of the in situ formed copolymers
It is clear from the results so far that the MAA macromonomers were converted into efficient stabilizers most effectively in the emulsion polymerization of BA and in order to obtain more detailed structural information on these stabilizers we examined a sample of the initially formed product at a conversion of 5% by MALDI-ToF MS. The obtained spectrum is shown in Fig. 7 and it is immediately clear that this spectrum is quite complex and does not originate from a single population of polymer chains only differing in degree of polymerization.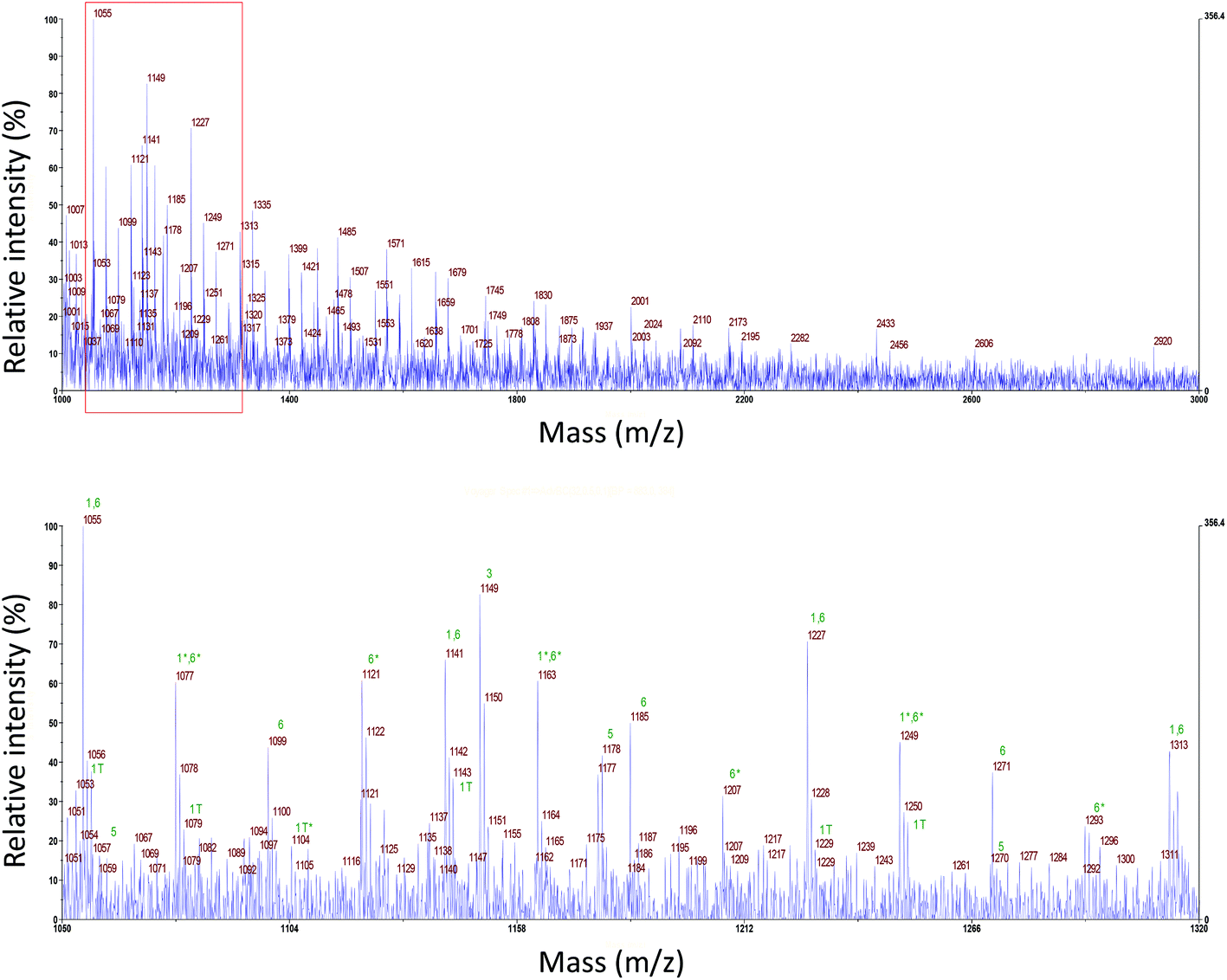 | ||
| Fig. 7 MALDI-ToF Mass spectrum of the initially formed oligomers in the emulsion polymerization of BA with 15 wt% MAA80, overview and enlarged from m/z 1050 to 1320. Each number indicates a reaction product shown in Scheme 2; * exchange of one or more H+ with Na+ in MAA chain; 1 T terminated PMAA chain by recombination. | ||
In order to simplify the discussion of this spectrum it is useful to consider the copolymerization of the MAA macromonomers with BA and so identify the potential structures in the MALDI spectrum. This process is schematically shown in Scheme 2.14 Propagating BA radicals (initially formed by the addition of SO4˙− radicals to BA and later in the process this could be any propagating radical) will undergo an addition fragmentation chain transfer (AFCT) reaction with the MAA macromonomer (1).22 This results in a MAA radical (which can propagate with BA) and a new macromonomer now containing a BA penultimate unit (2). This new macromonomer now will not undergo an AFCT reaction with a BA propagating radical, but will copolymerize and form a graft upon further polymerization.22 It should be noted here that many different grafts are possible as macromonomer 2 may not only contain a homo pBA block, but it could easily contain a pMAA block followed by a pBA block or even contain one or more grafts as any propagating BA radical can react with the initial MAA macromonomer 1. As an additional complicating factor it should be noted that the degrees of polymerization of all these blocks can vary.
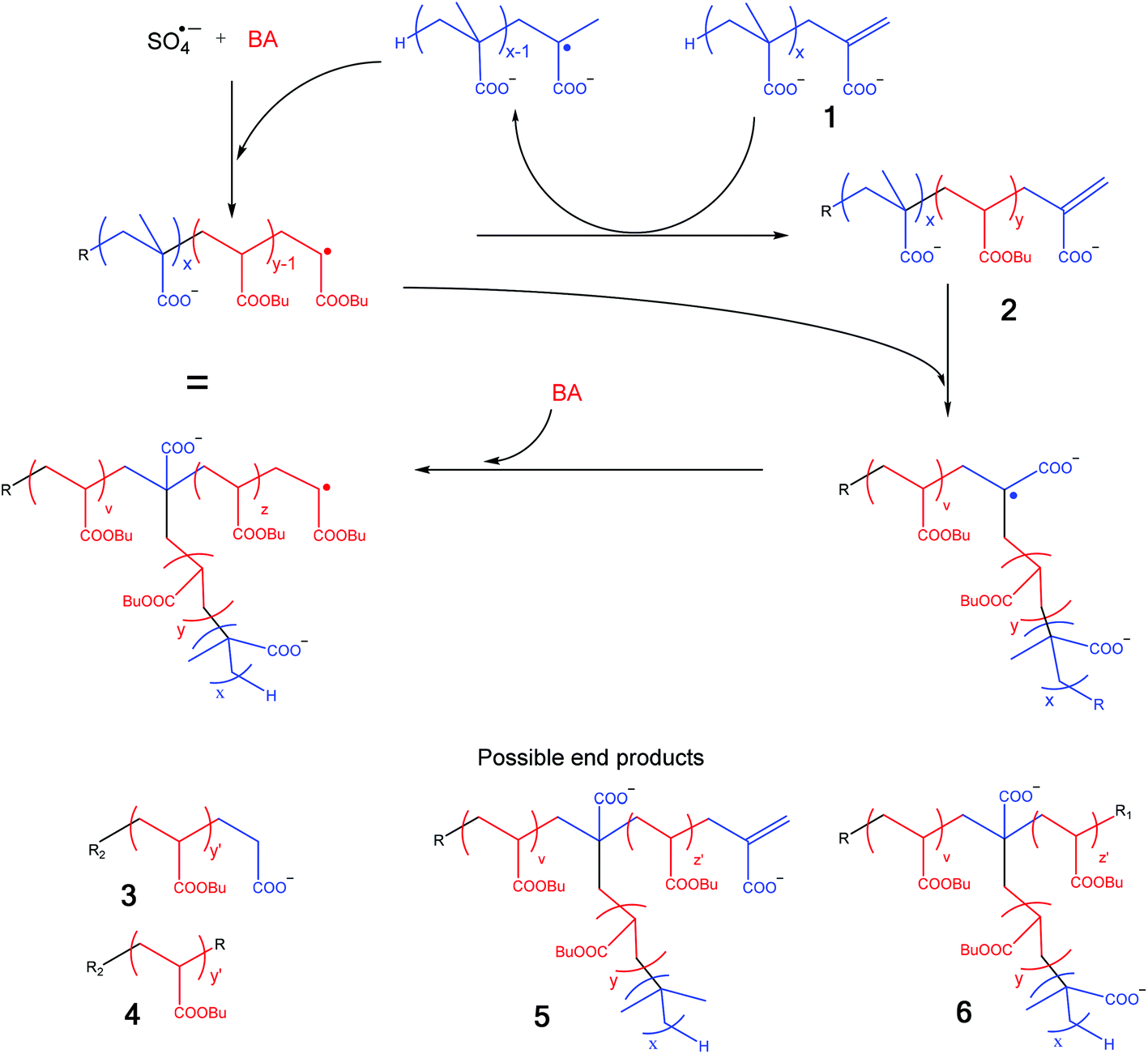 | ||
Scheme 2 Possible pathways for in situ formation of amphiphilic copolymers, R = H, OH or SO4−; R1 = H, OH, SO4−, MAAC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, R2 = OH or SO4−; y ≥ 1. C, R2 = OH or SO4−; y ≥ 1. | ||
The potential final products (resulting from AFCT and/or termination by recombination and disproportionation) of this process are also shown in Scheme 2 and with this scheme in hand we tried to identify the peaks in Fig. 7.
From Fig. 7, it can be seen that most of the measured oligomers are graft (or block) copolymers (6) of MAA and BA; in rare cases also macromonomers (thus terminated with H and vinyl group) (5) were formed. Still some of the unreacted macromonomer (1) seems to be present, although it should be noted that the m/z of this structure is the same as that of copolymer structure 6. We also observe a small number of peaks from the recombination peaks of two smaller pMAA radicals (1T). The intermediate reaction product of a growing pBA chain with one MAA macromonomer (3) was observed with an OH end group only. The expected endgroup formed with KPS is SO4−, but depending on the type of the monomer and the reaction conditions also other end groups like OH have been observed.35,36 No intermediate macromonomers (2) or oligomers of BA (4) were observed. Larger polymers of pBA, immediately formed during the emulsion polymerization, are above the measuring range of the MALDI-ToF MS. A more elaborate discussion of the spectrum is given in the ESI.†
In summary, it can be concluded that from the start of the emulsion polymerization the MAA macromonomers were copolymerized with BA to form stabilizers in situ.
Conclusions
Methacrylic acid macromonomers prepared via catalytic chain transfer polymerization were shown to be suitable precursors for stabilizers in (surfactant-free) emulsion polymerization. It was found that a prerequisite for efficient in situ stabilizer formation is that the conversion of the macromonomer into amphiphilic stabilizer molecules should be sufficiently fast. This is not the case in the homopolymerization of MMA, which neither polymerizes quickly with the macromonomer, nor is it very hydrophobic; in all MMA homopolymerizations, the addition of SDS was necessary. Hydrophobic monomers such as BA and BMA, however, convert the macromonomer more quickly into a stabilizer upon the addition of a few monomer units and the polymerization can be carried out without the addition of SDS. Furthermore, BA reacts more quickly with the MAA macromonomers than MMA (and BMA), which results in a quicker incorporation of hydrophobic units into the precursor/stabilizer. The faster reaction of acrylates also resulted in stable (surfactant-free) emulsion polymerization in the case of ethyl acrylate (with a similar hydrophobicity to MMA), but only in the case of a semi-batch process and not in the batch process; this latter observation can be explained by the fact that particle growth is much slower in the case of a semi-batch process and that there was sufficient time for in situ stabilizer formation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the “Stichting Emulsion Polymerisation” (SEP) for their financial support. Electron microscopy was performed at the Center for Multiscale Electron Microscopy (CMEM) at the Eindhoven University of Technology. The support from Rinske Knoop and Anne Spoelstra with SEM and TEM imaging is greatly acknowledged.References
- Polymer dispersions and their industrial applications, ed. D. Urban and K. Takamura, Wiley-VCH, Weinheim, 2002 Search PubMed.
- A. Elaissari, Colloidal polymers: synthesis and characterization, Marcel Dekker, New York, 2003, p. 115 Search PubMed.
- Chemistry and technology of emulsion polymerisation, ed. A. M. v. Herk, Wiley, Chichester, 2nd edn, 2013 Search PubMed.
- E. Aramendia, J. Mallégol, C. Jeynes, M. J. Barandiaran, J. L. Keddie and J. M. Asua, Distribution of surfactants near Acrylic Latex Film Surfaces: A Comparison of Conventional and reactive surfactants (Surfmers), Langmuir, 2003, 19, 3212–3221 CrossRef CAS.
- K. W. Evanson, T. A. Thorstenson and M. W. Urban, Surface and interfacial FTIR spectroscopic studies of latexes. II. Surfactant–copolymer compatibility and mobility of surfactants, J. Appl. Polym. Sci., 1991, 42, 2297–2307 CrossRef CAS.
- P. A. Steward, J. Hearn and M. C. Wilkinson, An overview of polymer latex film formation and properties, Adv. Colloid Interface Sci., 2000, 86, 195–267 CrossRef CAS PubMed.
- M. A. Winnik, Latex film formation, Curr. Opin. Colloid Interface Sci., 1997, 2, 192–199 CrossRef CAS.
- A. Guyot and K. Tauer, Reactive surfactants in emulsion polymerization, Springer, Berlin, Heidelberg, 1994, vol. 111, 43–65 Search PubMed.
- A. Guyot, Advances in reactive surfactants, Adv. Colloid Interface Sci., 2004, 108–109, 3–22 CrossRef CAS PubMed.
- F. Vidal, J. Guillot and A. Guyot, Surfactants with transfer agent properties (transurfs) in styrene emulsion polymerization, Colloid Polym. Sci., 1995, 273, 999–1007 CAS.
- K. Tauer, Polymer Nanoparticles with Surface Active Initiators and Polymer Initiators, Adv. Polym. Nanopart., 2010, 3299–3214 Search PubMed.
- J. Gregory, Polymer adsorption and flocculation in sheared suspensions, Colloids Surf., 1988, 31, 231–253 CrossRef CAS.
- J. M. Asua and H. A. S. Schoonbrood, Reactive surfactants in heterophase polymerization, Acta Polym., 1998, 49, 671–686 CrossRef CAS.
- J. P. A. Heuts and N. M. B. Smeets, Catalytic chain transfer and its derived macromonomers, Polym. Chem., 2011, 2, 2407 RSC.
- A. A. Gridnev and S. D. Ittel, Catalytic chain transfer in free-radical polymerizations, Chem. Rev., 2001, 101, 3611–3660 CrossRef CAS PubMed.
- S. Slavin, K. McEwan and D. M. Haddleton, Cobalt-Catalyzed Chain Transfer Polymerization: A Review, in Polym. Sci. Compr. Rev, 2012, vol. 3, ch. 9 Search PubMed.
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby, A. Clarke and S. N. Richards, Radical-addition-fragmentation and co-polymerization of methyl methacrylate macromonomers from catalytic chain transfer polymerization (CCTP), Polymer, 1997, 38, 6207–6217 CrossRef CAS.
- K. G. Suddaby, D. M. Haddleton, J. J. Hastings, S. N. Richards and J. P. O'Donnell, Catalytic Chain transfer for molecular weight control in the emulsion polymerization of methyl methacrylate and methyl methacrylate-styrene, Macromolecules, 1996, 29, 8083–8091 CrossRef CAS.
- J. Krstina, C. L. Moad, G. Moad, E. Rizzardo, C. T. Berge and M. Fryd, A new form of controlled growth free radical polymerization, Macromol. Symp., 1996, 111, 13–23 CrossRef CAS , Wiley Online Library.
- J. Huybrechts, P. Bruylants, K. Kirshenbaum, J. Vrana and J. Snuparek, New applications of catalytic chain transfer polymerization to waterborne binders for automotive paint systems, Prog. Org. Coat., 2002, 45, 173–183 CrossRef CAS.
- L. Chen, L. Yan, Q. Li, C. Wang and S. Chen, Controllable synthesis of new polymerizable macrosurfactants via CCTP and RAFT techniques and investigation of their performance in emulsion polymerization, Langmuir, 2010, 26, 1724–1733 CrossRef CAS PubMed.
- B. Yamada, F. Oku and T. Harada, Substituted propenyl end groups as reactive intermediates in radical polymerization, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 645–654 CrossRef CAS.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Sequence-controlled methacrylic multiblock copolymers via sulfur-free RAFT emulsion polymerization, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- G. Nurumbetov, N. Enelis, J. Godfrey, R. Hand, A. Anastasaki, A. Simula, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2017, 8, 1084–1094 RSC.
- D. Zhou, R. P. Kuchel and P. B. Zetterlund, Polym. Chem., 2017, 8, 4177–4181 RSC.
- (a) C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS; (b) B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS; (c) P. B. Zetterlund, S. C. Thicket, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed; (d) S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- A. Bakac, M. E. Brynildson and J. H. Espenson, Characterization of the structure, properties, and reactivity of a cobalt(II) macrocyclic complex, Inorg. Chem., 1986, 25, 4108–4114 CrossRef CAS.
- L. M. Muratore, J. P. A. Heuts and T. P. Davis, Synthesis of 3-tris (trimethylsilyloxy) silyl propyl methacrylate macromers using catalytic chain transfer polymerization: a kinetic and mechanistic study, Macromol. Chem. Phys., 2000, 201, 985–994 CrossRef CAS.
- G. E. Roberts, T. P. Davis, J. P. A. Heuts and G. E. Ball, Monomer substituent effects in catalytic chain transfer polymerization: tert-butyl methacrylate and dimethyl itaconate, Macromolecules, 2002, 35, 9954–9963 CrossRef CAS.
- J. P. A. Heuts, D. Kukulj, D. J. Forster and T. P. Davis, Copolymerization of styrene and methyl methacrylate in the presence of a catalytic transfer agent, Macromolecules, 1998, 31, 2894–2905 CrossRef CAS.
- International Standard ISO22412, Particle size analysis - dynamic light scattering, International Organisation for Standardisation (ISO); 2008.
- International Standard ISO13321, Methods for determination of particle size distribution part 8: photon correlation spectroscopy, International Organisation for Standardisation (ISO), 1996.
- A. Berthod and C. Garcia-Alvarez-Coque, Micellar liquid chromatography, Marcel Dekker Inc., New York, 2000, vol. 83, 33–35 Search PubMed.
- P. C. Hiemenz and R. Rajagoplan, Principles of colloid and surface chemistry, Marcel Dekker Inc., New York, 3rd edn, 1997, ch. 4 and 13 Search PubMed.
- P. Daswani, PhD Thesis: Entry in emulsion copolymerization, Technische Universiteit Eindhoven, Eindhoven, 2012 Search PubMed.
- C. Liang and H. Su, Identification of sulfate and hydroxyl radicals in thermally activated persulfate, Ind. Eng. Chem. Res., 2009, 48, 5558–5562 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01583f |
| This journal is © The Royal Society of Chemistry 2017 |