
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen and carbon dioxide dual gas-responsive homopolymers and diblock copolymers synthesized via RAFT polymerization†
Xue
Jiang
a,
Feng
Chun
*ab,
Guolin
Lu
a and
Huang
Xiaoyu
 *a
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China. E-mail: cfeng@mail.sioc.ac.cn; xyhuang@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925606 Tel: +86-21-54925310
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, 220 Handan Road, Shanghai 200433, People's Republic of China
First published on 26th December 2016
Abstract
The monomer, 2,2,2-trifluoroethyl 3-(N-(2-(diethylamino)ethyl)acrylamido)propanoate (TF-DEAE-AM), which contains both O2 and CO2-responsive functionalities, was first synthesized from commercially available N,N-diethylethylenediamine, 2,2,2-trifluoroethyl acrylate, and acryloyl chloride. Subsequently, a series of dual-gas responsive polymers, poly(2,2,2-trifluoroethyl 3-(N-(2-(diethylamino)ethyl)acrylamido)propanoate) (poly(TF-DEAE-AM)) and poly(ethylene glycol)-b-poly(2,2,2-trifluoroethyl 3-(N-(2-(diethylamino)ethyl)acrylamido)propanoate) (PEG-b-poly(TF-DEAE-AM)), were synthesized by employing reversible addition–fragmentation chain transfer (RAFT) polymerization. Due to the protonation between CO2 and DEAE groups, and the specific van der Waals interactions between O2 and C–F bonds, micelles consisting of poly(TF-DEAE-AM) or PEG-b-poly(TF-DEAE-AM) display distinct CO2 and O2 responsiveness in aqueous media. Pyrene, which is a model hydrophobic drug, is able to be effectively encapsulated in the micelles based on PEG-b-poly(TF-DEAE-AM). It is found that the release of pyrene sharply increases after bubbling CO2 or O2 compared to N2, and the release rate of the solution bubbling with CO2 is the fastest. Since both CO2 and O2 are key gases for the human body, the CO2- and O2-induced release property of poly(TF-DEAE-AM) may open opportunities for the preparation of functional materials with special CO2 and O2 responsiveness for potential applications in biomedicine.
Introduction
Environmentally responsive polymers have attracted significant attention in the last decade due to their prospective applications in biomedicine, nano-therapeutics, and gene transportation.1–6 As a type of “smart” polymer, their solubility, conformation, or chemical structure, e.g. breakage or formation of covalent and non-covalent bonds, can be tuned in response to an external trigger, such as temperature,7,8 pH,9,10 light,11,12 ionic strength,13 and redox.14Recently, intensive attention has been paid to abundant and environmentally benign gas stimulation modes, including CO2, O2, and NO. The reversible and dynamic features of gas-responsive polymers make them suitable and desirable for biomedical applications, including controlled drug release, tissue culture, and peripheral vascular embolization materials.15–28 Among them, CO2-responsive polymers have gained growing interest. On one hand, they can be reversibly protonated by introducing CO2 with an increase in their solubility or deprotonated by bubbling an inert gas, such as N2 or Ar, with a decrease in their solubility in an aqueous solution. On the other hand, since CO2 is a key metabolite for life and has good biocompatibility and membrane permeability, CO2-responsive polymers have potential applications in sensors, surface adhesives, and drug delivery.15–24 For example, Yuan et al. developed the new idea of utilizing a specific amidine-containing block copolymer to fabricate CO2-responsive vesicles with a biomimetic “breathing” feature, and the membrane permeability of the vesicles can be simply tuned by regulating the CO2 stimulation time.17 By utilization of the controlled semi-permeability of CO2-responsive polymers, hierarchical cargo release and selective separation of guest molecules could be easily realized.18 In addition to the amidine group, some polymers containing tertiary amine groups, such as poly((N,N-dimethylamino)ethyl methacrylate) (PDMAEMA) and poly((N,N-diethylamino)ethyl methacrylate) (PDEAEMA), were proven to possess CO2 responsiveness.20–24 For instance, Zhao et al. designed a series of triblock copolymers composed of an outer hydrophilic poly(ethylene oxide) segment, middle hydrophobic polystyrene bridging block, and CO2-responsive interior PDEAEMA flank, which could mimic gas-controllable diversiform deformations of organelles.24 Similar to CO2, O2 is another important gas trigger. Both aromatic and aliphatic fluoro-substituted polymers have unique O2 responsiveness because of the special van der Waals interactions between O2 molecules and C–F bonds.25–29 Zhu et al. reported the synthesis of a new type of fluorinated copolymer via the atom transfer radical polymerization (ATRP) of commercially available 2,2,2-trifluoroethyl methacrylate (TFMA) and N,N-dimethylaminoethyl methacrylate (DMAEMA).26 The obtained copolymer showed a prominent increase in lower critical solution temperature (LCST) from 24.5 °C to 50 °C by the bubbling of O2, which probably results from its improved solubility due to the interactions between O2 molecules and 2,2,2-trifluoroethyls.
In contrast to a single-stimulus system, a multi-stimuli system may show a synergistic effect, thereby enhancing the recognition efficiency for distinct drug delivery and release behaviors.30–34 Since both CO2 and O2 are important to life, the design of dual gas-responsive polymers will significantly expand the scope of stimuli-responsive materials. Although dual CO2- and O2-responsive polymers can be prepared by block or random copolymerization of CO2- and O2-responsive monomers, the time-consuming steps in the preparation and purification of block copolymers and ill-defined monomer distribution of random copolymer are inherent drawbacks of the block and random copolymer systems, respectively.27,28 In order to avoid these drawbacks, installation of CO2- and O2-responsive functionalities into the same repeated unit on the basis of the maturity of organic synthesis, that is, the preparation of CO2- and O2-responsive homopolymers, has been considered as an effective and promising alternative. Therefore, it is not surprising that homopolymers with multi-stimuli responsiveness have drawn growing interest in polymer science.35–39 However, to the best of our knowledge, the currently available multi-stimuli responsive homopolymers are mainly based on temperature and pH stimuli, and reports on the synthesis of homopolymers with CO2 and O2 responsiveness are rare.29
In this study, we develop a new strategy to prepare homopolymers with CO2 and O2 responsiveness by the installation of CO2- and O2-responsive functional groups in the same repeated unit. Commercially available N,N-diethylethylenediamine (DEAE) and 2,2,2-trifluoroethyl acrylate are first connected by the aza-Michael addition reaction (Scheme 1) to give 2,2,2-trifluoroethyl 3-(2-(diethylamino)ethylamino)-propanoate (TF-DEAE). Next, the new monomer, 2,2,2-trifluoroethyl 3-(N-(2-(diethylamino)ethyl)acrylamido)propanoate (TF-DEAE-AM), was obtained by the reaction between TF-DEAE and acryloyl chloride. Subsequently, the target poly(TF-DEAE-AM) homopolymers are prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization using 4-cyano-4-(dodecylsulfanylthiocarbonylsulfanyl)pentanoic acid (CDSTSP) as the chain transfer agent with controlled molecular weights and relatively narrow molecular weight distributions. The CO2 and O2 responsiveness of the obtained homopolymers are examined via TEM, 1H NMR, 19F NMR, DLS, and UV-vis spectroscopy. In addition, PEG-b-poly(TF-DEAE-AM) diblock polymers are prepared by the RAFT polymerization of TF-DEAE-AM using a PEG-based macromolecular chain transfer agent instead of CDSTSP. It is observed that a variation in both CO2 and O2 not only induces a change in the morphology of the aggregates in the aqueous media, but also results in difference in release profiles for hydrophobic dye (pyrene) loading in micelles based on PEG-b-poly(TF-DEAE-AM). The CO2- and O2-induced release property of poly(TF-DEAE-AM) may open opportunities for the preparation of functional materials with special CO2 and O2 responsiveness for potential applications in biomedicine.
 | ||
| Scheme 1 Synthetic route for poly(TF-DEAE-AM) and PEG-b-poly(TF-DEAE-AM). | ||
Experimental
Materials
2,2′-Azobis(isobutyronitrile) (AIBN, Aldrich, 98%) was recrystallized from anhydrous ethanol. Tetrahydrofuran (THF, Aldrich, 99%) and dichloromethane (CH2Cl2, Aldrich, 99%) were dried over CaH2 and distilled from sodium and benzophenone under N2, prior to use. Triethylamine (Et3N, Aldrich, 99.5%) was dried over KOH and distilled over CaH2 under N2, prior to use. N-Phenyl-1-naphthylamine (PNA, Alfa Aesar, 97%) was purified by recrystallization in ethanol three times. 4-Dimethylaminopyridine (DMAP, Aldrich, 98%) was recrystallized three times from toluene. 2,2,2-Trifluoroethyl acrylate (TCI, 98%), N,N-diethylethylenediamine (DEAE, J&K, 99%), N,N′-dicyclohexylcarbodiimide (DCC, Aldrich, 99%), acryloyl chloride (Aladdin, 96%), and poly(ethylene glycol) monomethyl ether (PEG-OH, Mn = 2000 g mol−1, Aldrich, 99%) were used as received. 4-Cyano-4-(dodecylsulfanyl-thiocarbonylsulfanyl)pentanoic acid (CDSTSP) was synthesized according to previous literature.42Measurements
All 1H, 13C, and 19F NMR analyses were performed on a JEOL ECZ 400S spectrometer (400 MHz) in CDCl3 and D2O; tetramethylsilicone (1H) and CDCl3 (13C) were used as internal standards, and CF3CO2H was used as the external standard for 19F NMR. FT-IR spectra were recorded on a Nicolet AVATAR-360 spectrophotometer with a 4 cm−1 resolution. Elemental analysis was carried out on a Carlo-Erba 1106 system. Relative molecular weights and molecular weight distributions were measured using a conventional gel permeation chromatography (GPC) system equipped with a Waters 515 Isocratic HPLC pump, Waters 2414 refractive index detector, and a set of Waters Styragel columns (HR3 (500–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), HR4 (5000–600000), and HR5 (50000–4000000), 7.8 × 300 mm, particle size: 5 μm). GPC measurements were carried out at 35 °C using THF as the eluent with a flow rate of 1.0 mL min−1. The system was calibrated with linear poly(methyl methacrylate) (PMMA) standards. The transmittance of polymer solutions and UV-vis spectra were measured using a Hitachi U-2910 spectrophotometer. Steady-state fluorescence spectra were measured at 20 °C on a Hitachi F-2700 fluorescence spectrophotometer with the band width of 5 nm for excitation and emission, and the emission intensity at 418 nm was recorded to determine the critical micelle concentration (cmc), where the excitation wavelength (λex) was 340 nm. Hydrodynamic diameter (Dh) was measured by dynamic light scattering (DLS) with a Malvern Nano ZS90 Zetasizer after equilibrating for 2 min at 20 °C. The zeta-potential of the solutions containing polymer aggregates was determined in KCl aqueous media (5.0 mM) at 25 °C on a Malvern Nano ZS90 Zetasizer. Transmission electron spectroscopy (TEM) images were obtained on a JEOL JEM-1230 instrument operated at 80 kV.
000), HR4 (5000–600000), and HR5 (50000–4000000), 7.8 × 300 mm, particle size: 5 μm). GPC measurements were carried out at 35 °C using THF as the eluent with a flow rate of 1.0 mL min−1. The system was calibrated with linear poly(methyl methacrylate) (PMMA) standards. The transmittance of polymer solutions and UV-vis spectra were measured using a Hitachi U-2910 spectrophotometer. Steady-state fluorescence spectra were measured at 20 °C on a Hitachi F-2700 fluorescence spectrophotometer with the band width of 5 nm for excitation and emission, and the emission intensity at 418 nm was recorded to determine the critical micelle concentration (cmc), where the excitation wavelength (λex) was 340 nm. Hydrodynamic diameter (Dh) was measured by dynamic light scattering (DLS) with a Malvern Nano ZS90 Zetasizer after equilibrating for 2 min at 20 °C. The zeta-potential of the solutions containing polymer aggregates was determined in KCl aqueous media (5.0 mM) at 25 °C on a Malvern Nano ZS90 Zetasizer. Transmission electron spectroscopy (TEM) images were obtained on a JEOL JEM-1230 instrument operated at 80 kV.
Synthesis of 2,2,2-trifluoroethyl 3-(2-(diethylamino)ethylamino)propanoate
N,N-Diethylethylenediamine (8.4 g, 72.0 mmol) was added dropwise to 2,2,2-trifluoroethyl acrylate (9.3 mL, 60.0 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min and then at room temperature for additional 12 h. The mixture was purified via flash chromatography (silica gel column, eluent: ethyl acetate/hexane (v:v = 1:4 with Et3N)) to give 8.5 g (52.4% yield) of amine 1, 2,2,2-trifluoroethyl 3-(2-(diethylamino)ethylamino)propanoate (TF-DEAE). FT-IR: ν (cm−1): 3313, 2971, 2935, 2816, 1760, 1453, 1412, 1384, 1283, 1168, 1068, 976. 1H NMR (400 MHz, CDCl3): δ (ppm): 0.97 (6H, CH2N(CH2CH3)2), 1.78 (1H, O2CCH2CH2NHCH2), 2.49 (6H, CH2N(CH2CH3)2), 2.62 (4H, O2CCH2CH2NHCH2), 2.91 (2H, O2CCH2CH2NHCH2), 4.45 (2H, CO2CH2CF3). 13C NMR (101 MHz, CDCl3): δ (ppm): 11.9 (CH2N(CH2CH3)2), 34.5 (O2CCH2CH2NHCH2), 45.0 (O2CCH2CH2NHCH2), 47.2 (CH2N(CH2CH3)2), 47.5 (O2CCH2CH2NHCH2), 52.7 (CH2N(CH2CH3)2), 60.3 (CO2CH2CF3), 123.1 (CO2CH2CF3), 171.2 (CO2CH2CF3). 19F NMR (376 MHz, CDCl3): δ (ppm): −73.9 (CO2CH2CF3). Anal. Calcd for C11H21N2O2F3: C, 48.88%; H, 7.83%; N, 10.36%. Found: C, 48.46%; H, 7.83%; N, 10.16%.
Synthesis of 2,2,2-trifluoroethyl 3-(N-(2-(diethylamino)ethyl)acrylamido)-propanoate
2,2,2-Trifluoroethyl 3-(2-(diethylamino)ethylamino)propanoate (8.1 g, 30.0 mmol) and Et3N (10.0 mL, 72.0 mmol) were dissolved in 100 mL of anhydrous CH2Cl2. The solution was cooled to 0 °C, followed by the addition of acryloyl chloride (3.3 g, 36.0 mmol) dropwise within 15 min. The mixture was slowly warmed to room temperature and stirred for 12 h. The volatiles were evaporated under reduced pressure and the residue was purified using flash chromatography (silica gel column, eluent: ethyl acetate/hexane (v:v = 1:4 with Et3N)) to give 3.0 g (30.9% yield) of the corresponding acrylamide monomer 2, 2,2,2-trifluoroethyl 3-(N-(2-(diethylamino)-ethyl)acrylamido)propanoate (TF-DEAE-AM). FT-IR: ν (cm−1): 2970, 2937, 2810, 1758, 1652, 1615, 1448, 1425, 1374, 1281, 1169, 1071, 977, 785. 1H NMR (400 MHz, CDCl3): δ (ppm): 0.99 (6H, CH2N(CH2CH3)2), 2.51 (6H, CH2N(CH2CH3)2), 2.77 (2H, O2CCH2CH2NCH2), 3.43 (2H, O2CCH2CH2NCH2), 3.67 (2H, O2CCH2CH2NCH2), 4.44 (2H, CO2CH2CF3), 5.68, 6.32 (2H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO), 6.54 (1H, CH2CHCO). 13C NMR (101 MHz, CDCl3): δ (ppm): 12.0 (CH2N(CH2CH3)2), 32.4 (O2CCH2CH2NCH2), 43.8 (CH2N(CH2CH3)2), 47.6 (CH2N(CH2CH3)2), 48.1 (O2CCH2CH2NCH2), 52.7 (O2CCH2CH2NCH2), 60.5 (CO2CH2CF3), 121.6 (CH2CHCO), 124.4 (CH2CHCO), 128.0 (CO2CH2CF3), 166.6 (CH2CHCO), 170.7 (CO2CH2CF3). 19F NMR (376 MHz, CDCl3): δ (ppm): −73.8 (CO2CH2CF3). MS-DART: calculated C14H24N2O3F3 for 325.1734; found 325.1732 [M + H]+. Anal. Calcd for C14H23N2O3F3: C, 51.84%; H, 7.15%; N, 8.64%. Found: C, 51.86%; H, 7.15%; N, 8.67%.
CHCO), 6.54 (1H, CH2CHCO). 13C NMR (101 MHz, CDCl3): δ (ppm): 12.0 (CH2N(CH2CH3)2), 32.4 (O2CCH2CH2NCH2), 43.8 (CH2N(CH2CH3)2), 47.6 (CH2N(CH2CH3)2), 48.1 (O2CCH2CH2NCH2), 52.7 (O2CCH2CH2NCH2), 60.5 (CO2CH2CF3), 121.6 (CH2CHCO), 124.4 (CH2CHCO), 128.0 (CO2CH2CF3), 166.6 (CH2CHCO), 170.7 (CO2CH2CF3). 19F NMR (376 MHz, CDCl3): δ (ppm): −73.8 (CO2CH2CF3). MS-DART: calculated C14H24N2O3F3 for 325.1734; found 325.1732 [M + H]+. Anal. Calcd for C14H23N2O3F3: C, 51.84%; H, 7.15%; N, 8.64%. Found: C, 51.86%; H, 7.15%; N, 8.67%.
RAFT homopolymerization of TF-DEAE-AM
In a typical procedure, AIBN (1.6 mg, 0.01 mmol), CDSTSP (12.4 mg, 0.031 mmol), and TF-DEAE-AM (0.50 g, 1.54 mmol) were first added to a 25 mL Schlenk flask (flame-dried under vacuum prior to use) sealed with a rubber septum for degassing and kept under N2. Next, dry THF (0.5 mL) was charged via a gastight syringe. The flask was degassed by three cycles of freezing–pumping–thawing, followed by immersing the flask into an oil bath set at 65 °C. The polymerization lasted for 15 h and it was terminated by placing the flask into liquid N2. The reaction mixture was precipitated in cold n-hexane. The crude product was purified by repeated dissolution in THF and precipitation in n-hexane, followed by drying in vacuo overnight to give 0.18 g of light yellow solid, poly(TF-DEAE-AM) 3b. GPC: Mn = 10500 g mol−1, Mw/Mn = 1.36. FT-IR: ν (cm−1): 2969, 2928, 2812, 1758, 1642, 1452, 1422, 1379, 1280, 1169, 1070, 976, 842. 1H NMR (400 MHz, CDCl3): δ (ppm): 1.02 (6H, CH2N(CH2CH3)2), 1.25 (18H, SCH2CH2(CH2)9CH3), 1.79 (2H, CH2CHCON), 2.58 (1H, CH2CHCON; 6H, CH2N(CH2CH3)2; 2H, O2CCH2CH2NCH2), 3.58 (4H, O2CCH2CH2NCH2), 4.46 (2H, CO2CH2CF3).
Preparation of PEG-CTA
PEG-OH (Mn = 2000 g mol−1, 25.0 g, 12.5 mmol –OH), DMAP (0.3 g, 2.5 mmol), CDSTSP (10.1 g, 25.0 mmol), and dry CH2Cl2 (300.0 mL) were first added to a 500 mL flask (flame-dried under vacuum prior to use) sealed with a rubber septum under N2. The solution was cooled to 0 °C, followed by the addition of 25 mL of CH2Cl2 solution containing DCC (5.2 g, 25.0 mmol) dropwise within 15 min. The mixture was slowly warmed to room temperature and stirred for 12 h. The solid was filtered and the filtrate was concentrated, followed by precipitation in cold diethyl ether. After repeated purification by dissolving in CH2Cl2 and precipitation in cold diethyl ether, 16.7 g (56.0% yield) of yellow solid, PEG-CTA, was obtained after drying in vacuo overnight. GPC: Mn = 3800 g mol−1, Mw/Mn = 1.02. 1H NMR (400 MHz, CDCl3): δ (ppm): 0.87 (3H, SCH2CH2(CH2)9CH3), 1.25 (18H, SCH2CH2(CH2)9CH3), 1.68 (2H, SCH2CH2(CH2)9CH3), 1.87 (3H, O2CCH2CH2C(CN)CH3), 2.36–2.52 (2H, O2CCH2CH2C(CN)CH3), 2.65 (2H, O2CCH2CH2C(CN)CH3), 3.31 (2H, SCH2CH2(CH2)9CH3), 3.37 (3H, OCH3), 3.45–3.63 (4H, OCH2CH2), 3.81 (2H, CO2CH2CH2O), 4.25 (2H, CO2CH2CH2O).RAFT block copolymerization of TF-DEAE-AM mediated by PEG-CTA
In a typical procedure, AIBN (1.6 mg, 0.01 mmol), PEG-CTA (72.3 mg, 0.030 mmol trithiocarbonate group), and TF-DEAE-AM (0.6 g, 1.85 mmol) were first added to a 25 mL Schlenk flask (flame-dried under vacuum prior to use) sealed with a rubber septum for degassing and kept under N2. Next, dry THF (0.6 mL) was charged via a gastight syringe. The flask was degassed by three cycles of freezing–pumping–thawing, followed by immersion of the flask into an oil bath set at 65 °C. The polymerization lasted 48 h and it was terminated by placing the flask into liquid N2. The reaction mixture was precipitated in diethyl ether. The collected supernatant was then purified by repeated dissolution in THF and precipitation in diethyl ether followed by drying in vacuo overnight to give 0.26 g of light yellow solid, PEG-b-poly(TF-DEAE-AM) 4c. GPC: Mn,GPC = 8700 g mol−1 (Mn,NMR = 11600 g mol−1), Mw/Mn = 1.24. FT-IR: ν (cm−1): 2970, 2931, 2872, 1757, 1642, 1452, 1422, 1350, 1281, 1147, 976, 845. 1H NMR (400 MHz, CDCl3): δ (ppm): 0.88 (3H, SCH2CH2(CH2)9CH3), 1.00 (6H, CH2N(CH2CH3)2), 1.26 (18H, SCH2CH2(CH2)9CH3), 1.84 (3H, O2CCH2CH2C(CN)CH3), 1.94 (2H, CH2CHCON), 2.51 (1H, CH2CHCON and 6H, CH2N(CH2CH3)2), 2.69 (2H, O2CCH2CH2NCH2), 3.30 (4H, O2CCH2CH2NCH2), 3.38 (3H, OCH3), 3.64 (OCH2CH2), 4.21 (2H, CO2CH2CH2O), 4.46 (2H, CO2CH2CF3).
Micellar morphology
A THF solution of poly(TF-DEAE-AM) 3b homopolymer or PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer (10.0 mg mL−1) was added dropwise to distilled water under vigorous stirring and bubbling with N2 until the concentration of the polymer reached 0.5 mg mL−1. THF was evaporated by moderate stirring overnight at room temperature. For TEM studies, 50 μL of micelle solution was deposited onto an electron microscopy copper grid coated with carbon film, and the water was evaporated at room temperature.Titration experiment
A 50 mL three-neck round-bottom flask equipped with a Thermo Scientific™ Orion™ ROSS Ultra™ Refillable pH/ATC Triode™ combination electrode and a Lei-Ci DJS-1C conductance electrode was used to conduct potentiometric titrations. 20 mL of poly(TF-DEAE-AM) 3b homopolymer (1.0 mg mL−1) in aqueous salt solution ([NaCl] = 10 mM, pH = 3.0) was titrated using a standard NaOH solution (1.0 M) under constant stirring and bubbling of N2.Transmittance experiment
Poly(TF-DEAE-AM) 3b homopolymer was uniformly dispersed in distilled water by ultrasonication until the concentration of the homopolymer reached 0.5 mg mL−1. The gas-based transmittance changes of the aqueous solution were measured using a Hitachi U-2910 spectrophotometer at a constant wavelength of 500 nm.Determination of critical micelle concentration
PNA was used as a fluorescence probe to measure the cmc of the PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer in the aqueous media. An acetone solution of PNA ([PNA] = 2 mM) was added to a large amount of water until the concentration of PNA reached 0.002 mM. The solutions for fluorescence measurement were obtained by adding different amounts of THF solutions of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer (1, 0.1, 0.01, 0.001, or 0.0001 mg mL−1) to water containing PNA ([PNA] = 0.002 mM). THF was evaporated by constant stirring and bubbling N2 overnight at room temperature. The solution was sealed in vials prior to measurement to avoid the influence of CO2 or O2 present in air.Results and discussion
Design and synthesis of TF-DEAE-AM trifunctional acrylamide monomer
Given the well-studied CO2-responsive PDEAEMA24 and O2-responsive PTFMA,26,27 we attempted to incorporate DEAE and 2,2,2-trifluoroethyl (TF) functional groups into a single monomer, and utilize RAFT polymerization to construct dual-gas-stimulus responsive polymers. The key in the preparation of the target monomer is to incorporate a polymerizable group, DEAE group, and TF group within a monomer. Our group designed a two-step strategy to connect two different temperature- and pH-responsive groups and acrylamide group together.40,41 This strategy possesses several advantages, such as facile sample preparation and possibility to accomplish multifunctional materials, and great opportunities to enlarge the applications of this type of polymer. Inspired by the study, we employed a similar strategy in the current study by applying aza-Michael reaction as a key approach to provide the target secondary amine (TF-DEAE) intermediate in a single step, which could be utilized in a further reaction to afford a polymerizable acrylamide monomer (TF-DEAE-AM).The key intermediate of 2,2,2-trifluoroethyl 3-(2-(diethylamino)ethylamino)-propanoate 1 secondary amine was first prepared through aza-Michael addition reaction using 2,2,2-trifluoroethyl acrylate and N,N-diethylethylenediamine. 1H, 13C, and 19F NMR spectra, shown in Fig. S1,† display the expected resonance signals of the target compound, which confirm the chemical structure of the target intermediate, i.e. TF-DEAE 1. Subsequently, TF-DEAE 1 was treated with acryloyl chloride to afford the desired TF-DEAE-AM 2 acrylamide monomer. The chemical structure of TF-DEAE-AM 2 monomer was characterized via1H NMR, 13C NMR, 19F NMR, HR-MS, FT-IR, and EA in detail. Fig. 1A shows 1H NMR spectrum of the monomer, which exhibits typical proton resonance signals of a double bond at 6.54, 6.32, and 5.68 ppm (peaks “a” and “b”), 2,2,2-trifluoroethyl unit at 4.44 ppm (peak “c”), and DEAE moiety at 0.99 ppm (peak “i”). The 13C NMR spectrum (Fig. 1B) displays the expected resonance signals of carbonyls at 166.6 and 170.7 ppm (peaks “c” and “f”) and double bond at 121.6 and 124.4 ppm (peaks “a” and “b”), respectively. The only resonance signal located at −73.8 ppm in the 19F NMR spectrum (Fig. 1C) was attributed to fluorine atoms in –CF3, this verified the existence of TF unit. Furthermore, HR-MS result (325.1732) was also consistent well with the theoretical value (C14H24N2O3F3, 325.1734). All these results clearly confirmed the structure of the acrylamide monomer TF-DEAE-AM 2.
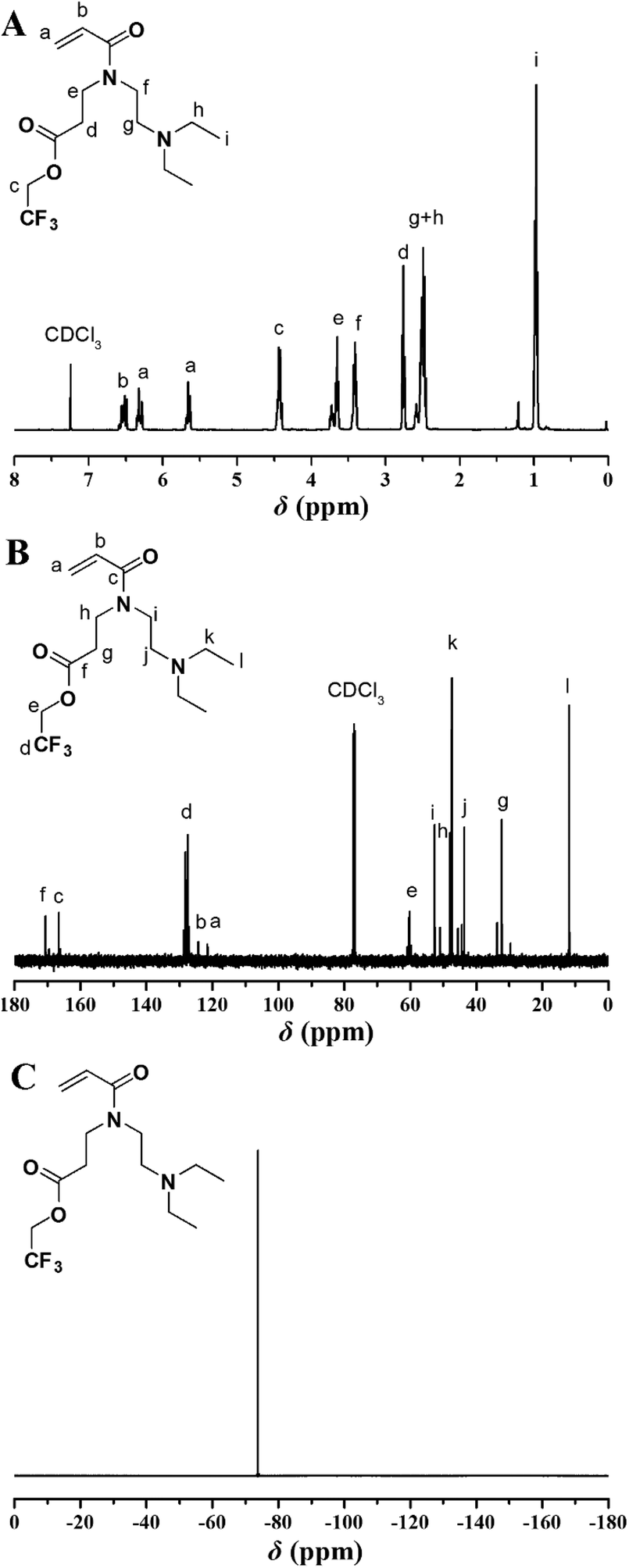 | ||
| Fig. 1 1H (A), 13C (B), and 19F (C) NMR spectra of TF-DEAE-AM 2 in CDCl3. | ||
Synthesis of poly(TF-DEAE-AM) homopolymer
In the present study, RAFT polymerization43–47 was chosen for the synthesis of poly(TF-DEAE-AM) 3 homopolymer since RAFT polymerization is one of the most powerful and versatile reversible degenerative radical polymerization (RDRP) processes that ensures precise control over molecular weight and molecular weight distribution in mild reaction conditions. Two poly(TF-DEAE-AM) 3 homopolymers were prepared by varying the feeding ratio of TF-DEAE-AM 2 monomer to CTA of CDSTSP (30:1 and 50:1) and reaction time, as listed in Table 1.
| Sample | [2]:[CTA] |
Time (h) | Conv.b (%) |
M
n,Theo
(g mol−1) |
M
nd (g mol−1) |
M
w/Mnd |
|---|---|---|---|---|---|---|
|
a [AIBN]:[CTA] = 1:3, solvent: THF, temperature: 65 °C.
b Measured by 1H NMR.
c
M
n,Theo = Mn,2 × ([2]:[CTA]) × conversion + Mn,CTA.
d Measured by GPC in THF at 35 °C.
|
||||||
| 3a | 30:1 |
12 | 80.5 | 8236 | 5500 | 1.29 |
| 3b | 50:1 |
15 | 78.3 | 13101 |
10500 |
1.36 |
Both homopolymers show symmetric and unimodal eluent peaks in their GPC curves (Fig. 2A) with relatively narrow molecular weight distributions (Mw/Mn < 1.40), which are indicative of the good choice of CDSTSP as CTA for mediating the polymerization and well-defined structure of the obtained poly(TF-DEAE-AM) 3 homopolymer. The molecular weights of the homopolymers measured by GPC are smaller than theoretical values due to the difference in the structure of poly(TF-DEAE-AM) 3 homopolymer and PMMA standards. A typical 1H NMR spectrum of poly(TF-DEAE-AM) 3 homopolymer is shown in Fig. 2B, which shows the disappearance of the proton resonance signals of the double bond and the retention of the peaks of TF moiety (peak “a”) and DEAE unit (peak “g”). These observations affirmed the chemical structure of poly(TF-DEAE-AM) 3 homopolymer.
 | ||
| Fig. 2 (A) GPC traces of poly(TF-DEAE-AM) 3 homopolymer in THF; and (B) 1H NMR spectrum of poly(TF-DEAE-AM) 3 homopolymer in CDCl3. | ||
Solution behavior of poly(TF-DEAE-AM) homopolymer
As a type of weak polybase, PDEAEMA homopolymer is soluble in water as a weak cationic polyelectrolyte in acidic surroundings due to the protonation of its tertiary amine group; however, it becomes insoluble at room temperature in a neutral or basic environment. The pKa of its conjugated acid is about 7.3.48–51 Although it is expected that poly(TF-DEAE-AM) 3 homopolymer should have similar pH stimulus responsiveness as PDEAEMA, the difference in their chemical structure might result in a change in pKa of the conjugated acid of DEAE group. In order to measure the degree of protonation of DEAE group on pH of aqueous solution, pH value and conductivity of an aqueous solution of poly(TF-DEAE-AM) 3b homopolymer were measured at the same time by adding NaOH solution (1.0 M) to the acidic poly(TF-DEAE-AM) 3b solution according to the previous report.52As shown in Fig. 3A, three regions can be divided on the basis of the changes in pH and conductivity, in which the second region between the two transition points (point a, pH ∼ 4.7 and point b, pH ∼ 8.4) corresponds to the deprotonation process of NH+(C2H5)2 group from the fully protonated state to the unionized state. We define that the degree of protonation of DEAE group is 100% when the pH of the solution is 4.7, and the degree of protonation of DEAE group is 0% when pH of the solution is 8.4. Based on this information, we obtained the profile of the dependence of degree of protonation of DEAE group with pH (Fig. 3B). The pH value of a saturated CO2 aqueous solution is about 3.9 at room temperature and atmosphere pressure so that 100% of tertiary amino of poly(TF-DEAE-AM) 3 homopolymer would be protonated by carbonic acid under this condition.20–24 As a result, it can be expected that poly(TF-DEAE-AM) 3 homopolymer will exhibit CO2-responsive behavior in an aqueous solution.
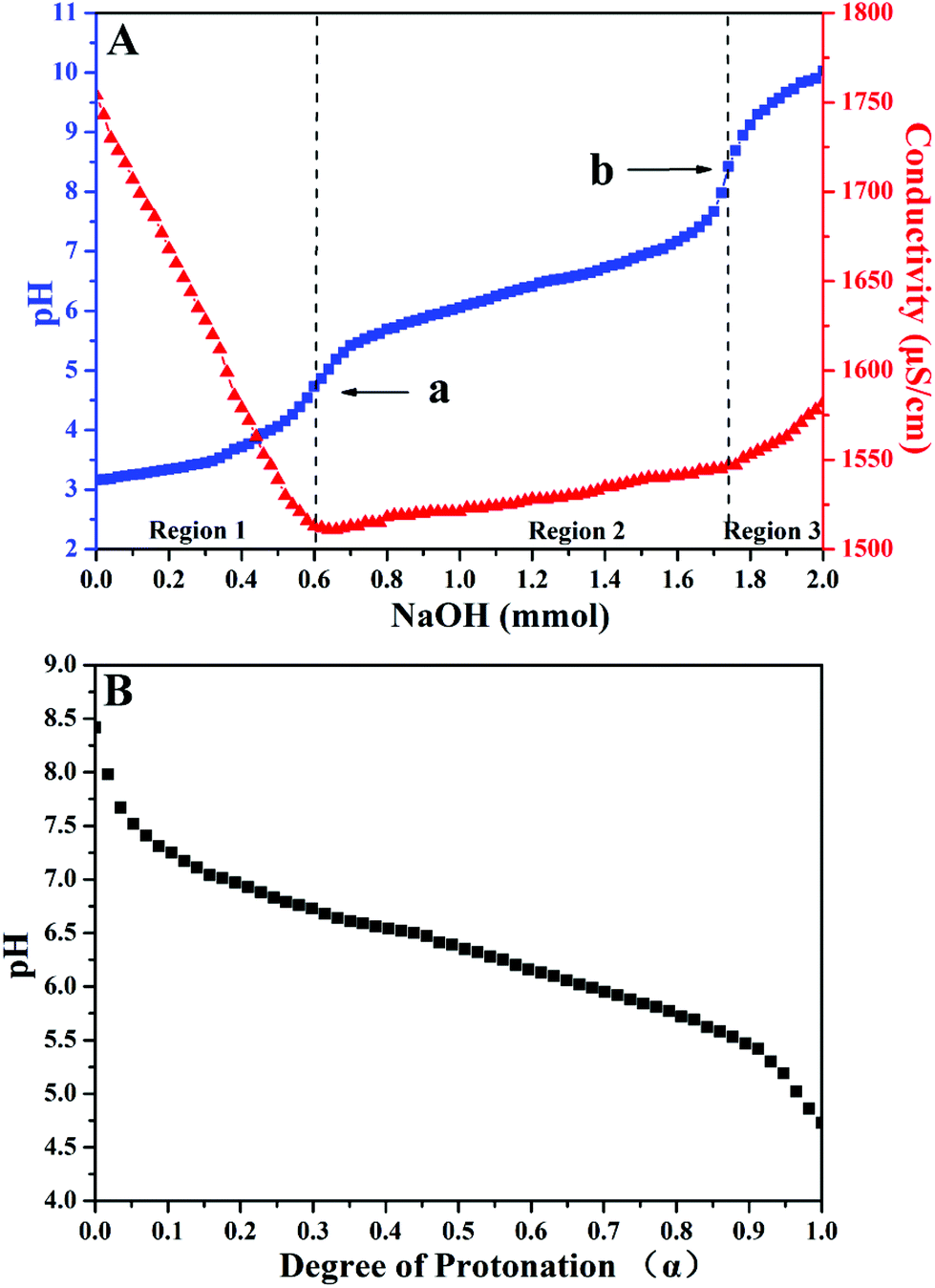 | ||
| Fig. 3 (A) Titration curves of poly(TF-DEAE-AM) 3b homopolymer (1.0 mg mL−1) at 25 °C in 10 mM NaCl; and (B) profile for the dependence of protonation on pH. | ||
The transmittance results of poly(TF-DEAE-AM) 3b aqueous solution at room temperature in response to CO2 are shown in Fig. 4 and it was found that a turbid solution was well observed at 25 °C (Fig. 4A) before treatment with CO2 (10 mL min−1). The solution became entirely transparent after the injection of CO2, while pH decreased from 8.0 to 4.7 due to the complete protonation of the tertiary amine group. The zeta-potential of the solution containing poly(TF-DEAE-AM) 3b aggregates greatly increased from 15.5 mV to 47.5 mV after the injection of CO2, which indicates that the protonated DEAE+ chains indeed formed a new corona with positive charges.
 | ||
| Fig. 4 Optical images (A) and transmittance changes (B) of poly(TF-DEAE-AM) 3b aqueous solution (0.5 mg mL−1) with CO2/N2 gas triggers; and 1H (C) and 19F (D) NMR spectra of poly(TF-DEAE-AM) 3b in D2O before (green) and after CO2 (red) or CO2/N2 (blue) treatment. | ||
Subsequently, after passing N2 (10 mL min−1) through the solution to remove CO2, a sharp drop in transmittance was observed (Fig. 4B) because of the deprotonation of DEAE group. Repeatable cycles under alternating CO2/N2 stimulation (Fig. 4B) indicated the CO2-responsive reversibility of poly(TF-DEAE-AM) 3 homopolymer. Since the removal of CO2 by N2 in aqueous solution represents a physical process, it needs enough time to completely recover. However, considering that a rather long time purging would lead to an evident loss of water, we herein set a consistent time of 1 h to bubble the gas, and thus, an apparent difference in turbidity was observed during the process of deprotonation.
In order to gain deep insight into the solubility of poly(TF-DEAE-AM) 3 homopolymer in aqueous solution upon bubbling CO2, 1H and 19F NMR spectra of poly(TF-DEAE-AM) 3b in D2O before and after bubbling gas were recorded. Before bubbling CO2, no signal was found in both 1H and 19F NMR spectra (green line in Fig. 4C and D). In CO2-saturated D2O, however, the characteristic signals of DEAE group attributed to the protons of “a”, “b” and “c” appear at 1.12 and 3.02 ppm, respectively, which is indicative of the good solubility of tertiary amine groups (red line in Fig. 4C). On the other hand, 19F NMR spectrum exhibits a weak and broad signal centered at −74.1 ppm after treatment with CO2 (red line in Fig. 4D), which suggests that TF groups are also partly soluble due to the increase in solubility of the close protonated DEAE groups after bubbling CO2. After the removal of CO2 by N2, the two typical signals of DEAE group in 1H NMR spectrum and characteristic peak originating from TF moiety in 19F NMR spectrum disappear again because of the deprotonation of DEAE group (blue line in Fig. 4C and D). Moreover, the protonation/deprotonation process could be reproduced over several cycles. It is believed that the interactions between CO2 and DEAE groups increased the solubility of poly(TF-DEAE-AM). In comparison with the chemical process of protonation by CO2, van der Waals interactions between O2 and C–F bonds appeared to be too weak, and there was no significant change in both transmittance and NMR spectrum after O2 treatment (10 mL min−1).
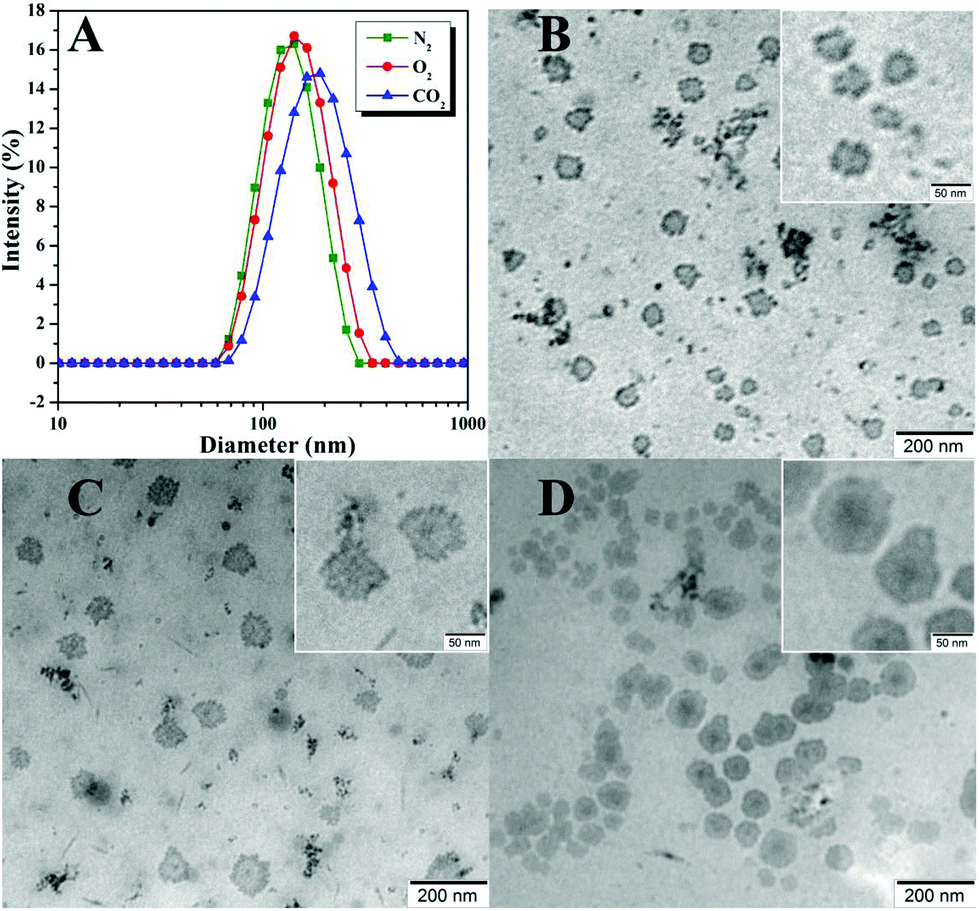
We then utilized DLS and TEM to examine the size and morphology of the aggregates formed by poly(TF-DEAE-AM) 3b homopolymer upon the treatment of N2, O2, and CO2, respectively. The micellar solution of poly(TF-DEAE-AM) 3b homopolymer was prepared by adding 0.5 mL of THF solution of poly(TF-DEAE-AM) 3b homopolymer (10 mg mL−1) in 10 mL of water, followed by removing THF by stirring and bubbling of N2 at room temperature over 24 h. Two additional aliquots (3.0 mL) taken from the micellar solution were treated with CO2 and O2, respectively.
As shown in Fig. 5A, spindle-shaped aggregates with an average hydrodynamic diameter of 379 nm (green line in Fig. 5D) were formed in aqueous media with the bubbling of N2. After the solution was bubbled with O2 for 1 h, interestingly, only spherical aggregates (Fig. 5B) with an average hydrodynamic diameter of 234 nm (red line in Fig. 5D), clearly smaller than that without the treatment with O2, were observed. This phenomenon might indicate that the large spindle-shaped aggregates fell apart and formed spherical aggregates. Although we cannot observe noticeable changes in the solubility, and 1H and 19F NMR spectra of poly(TF-DEAE-AM) 3b homopolymer upon bubbling O2, this evident change in morphology of aggregates clearly demonstrates the O2-stimulus responsiveness of poly(TF-DEAE-AM) 3 homopolymer. Similarly, the spindle-shaped aggregates also changed into ill-defined spherical micelles (Fig. 5C) with an average hydrodynamic diameter of 327 nm (blue line in Fig. 5D) after bubbling CO2 for 1 h. Different from the spherical micelles observed after the treatment with O2, there were several isolated darker “dots” in the matrix of the aggregates. This observation suggests that the aggregates composed of these darker dots are formed by poly(TF-DEAE-AM) 3 homopolymer.
 | ||
| Fig. 5 TEM images of poly(TF-DEAE-AM) 3b after N2 (A), O2 (B), and CO2 (C) treatment and the corresponding hydrodynamic diameter distributions (D). | ||
Synthesis of PEG-b-poly(TF-DEAE-AM) diblock copolymer
Although poly(TF-DEAE-AM) 3 homopolymer has both CO2 and O2-stimuli responsiveness, the homopolymer is completely insoluble in physiological environment and as a result, its applications in the biomedical field would be limited. It is necessary to connect poly(TF-DEAE-AM) with a hydrophilic and biocompatible segment for the construction of potential intelligent drug delivery systems, in which the release of hydrophobic drugs loaded in the core of aggregates formed by poly(TF-DEAE-AM) segment could be tuned as a response to CO2 and O2. In the current study, we selected PEG as the hydrophilic segment due to its unique biocompatibility and broad application in biomedicine.53–56A PEG-based macro-CTA (PEG-CTA) was first prepared via the attachment of the acidic functional trithiocarbonate of CDSTSP to PEG-OH (Mn = 2000 g mol−1) following a commonly-used procedure.57–601H NMR spectrum of PEG-CTA (Fig. S2†) exhibits the attachment of CDSTSP onto the chain end of PEG. Then, PEG-b-poly(TF-DEAE-AM) 4 diblock copolymers were obtained by RAFT polymerization of TF-DEAE-AM 2 using PEG-CTA as the chain transfer agent. By varying the polymerization time and feeding ratio of monomer 2 to PEG-CTA, three PEG-b-poly(TF-DEAE-AM) 4 diblock copolymers with different chain lengths of poly(TF-DEAE-AM) were prepared, as summarized in Table 2.
| Sample | [2]:[CTA] |
Time (h) | Conv.b (%) |
M
n,NMRc (g mol−1) |
M
n,Theod (g mol−1) |
M
n,GPCe (g mol−1) |
M
w/Mne |
f | cmcg (g mL−1) |
|---|---|---|---|---|---|---|---|---|---|
|
a [AIBN]:[CTA] = 1:3, solvent: THF, temperature: 65 °C.
b Measured by 1H NMR.
c Obtained from 1H NMR.
d
M
n,Theo = Mn,2 × ([2]:[CTA]) × conv. + Mn,CTA.
e Measured by GPC in THF at 35 °C.
f Weight fraction of hydrophobic poly(TF-DEAE-AM) segment in copolymer 4.
g Determined by fluorescence spectroscopy using PNA as the probe.
|
|||||||||
| 4a | 30:1 |
12 | 67.9 | 4200 | 8991 | 5800 | 1.13 | 0.43 | 6.8 × 10−5 |
| 4b | 30:1 |
60 | 98.6 | 8900 | 11978 |
6700 | 1.11 | 0.73 | 1.8 × 10−5 |
| 4c | 60:1 |
48 | 95.0 | 11600 |
20872 |
8700 | 1.24 | 0.79 | 8.3 × 10−6 |
Fig. 6A shows the GPC traces of the PEG-CTA and PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer. Compared to PEG-CTA, the elution time of the PEG-b-poly(TF-DEAE-AM) 4 diblock polymers decreased with an increase in polymerization time and feeding ratio of monomer 2 to PEG-CTA, indicating the extension of the polymer chain of poly(TF-DEAE-AM) from PEG-CTA. In addition, unimodal and symmetrical elution peaks with narrow molecular weight distributions (Mw/Mn ≤ 1.24) were observed for PEG-b-poly(TF-DEAE-AM) 4 diblock polymers. The chemical structure of PEG-b-poly(TF-DEAE-AM) 4 diblock polymer was characterized via1H NMR observations (Fig. 6B). The typical proton resonance signals originating from PEG block (3.64 ppm) still appear in 1H NMR spectrum. The peak “a” located at 4.46 ppm is attributed to two protons of CO2CH2CF3 moiety in TF-DEAE-AM repeated unit, whereas the peak “f” located at 4.21 ppm corresponds to two protons of CO2CH2CH2O linkage between poly(TF-DEAE-AM) and PEG in the main chain. On the basis of these results and chain length of PEG, the “absolute” molecular weight of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer could be calculated by 1H NMR observations according to eqn (1) (Sa and Sf are integral areas of peak “a” and “f” in Fig. 6B, respectively; and 325 and 2385 are the molecular weights of monomer 2 and PEG-CTA, respectively). Since the chemical structure of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer is different from PMMA standards, the molecular weights obtained from 1H NMR analysis were employed in the current study, which are indeed very different from those obtained from GPC. As listed in Table 2, three different PEG-b-poly(TF-DEAE-AM) 4 diblock copolymers with 6, 20, and 29 TF-DEAE-AM repeated units were prepared in the current case.
| Mn = 325(Sa/Sf) + 2385 | (1) |
 | ||
| Fig. 6 (A) GPC traces of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer in THF (Mn was obtained from 1H NMR in CDCl3); and (B) 1H NMR spectrum of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer in CDCl3. | ||
Solution behavior of PEG-b-poly(TF-DEAE-AM) diblock copolymer
The critical micelle concentration (cmc) of the amphiphilic PEG-b-poly(TF-DEAE-AM) 4 diblock polymer in aqueous solution without external stimuli was firstly determined via fluorescence spectroscopy using PNA as the fluorescence probe.61 The fluorescence intensity of PNA is sensitive to the environment and the polarity of its surroundings and it could be easily quenched by polar solvents such as water.61 Once micelles are formed in aqueous solution, the hydrophobic PNA could be encapsulated into their hydrophobic core, and thus, the value of I/I0 would dramatically increase (I and I0 refer to the fluorescence intensities of PNA at 418 nm with and without PEG-b-poly(TF-DEAE-AM) in aqueous solution, respectively).61 The relationships of I/I0 of PNA as a function of the concentration of PEG-b-poly(TF-DEAE-AM) 4 diblock polymer are plotted in Fig. 7. It is clear that when the concentration of copolymer 4 exceeds a certain value, I/I0 increases sharply due to the transferring of PNA probe into the hydrophobic core region of the micelles. Thus, the cmc of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer was determined to be the intersection of two straight lines and the results are summarized in Table 2. The cmc of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer increased from 8.3 × 10−6 g mL−1 to 6.8 × 10−5 g mL−1 with a decrease in the chain length of poly(TF-DEAE-AM) block, which is consistent with a previous report.62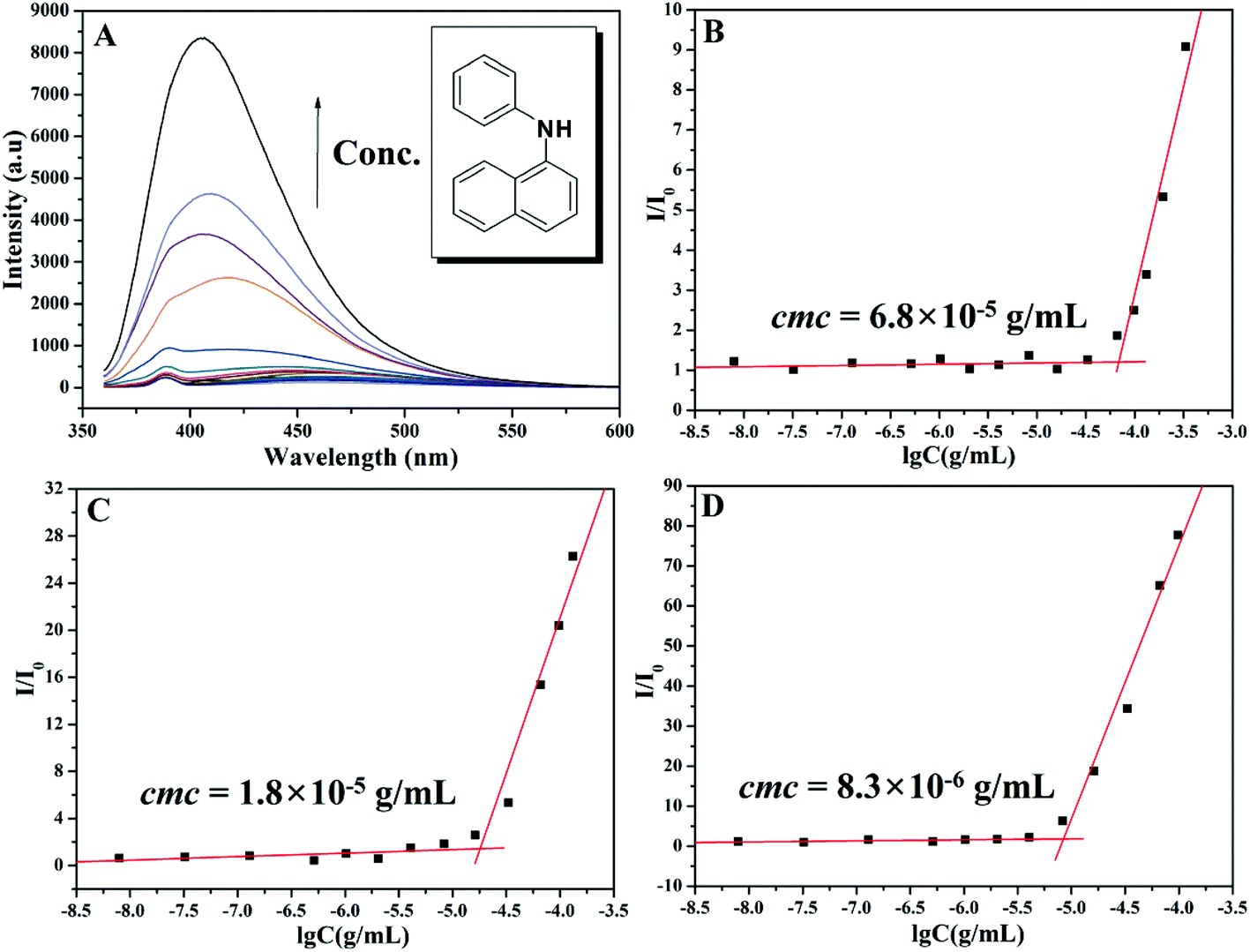 | ||
| Fig. 7 (A) Fluorescence emission spectra of PNA (0.002 mM) in an aqueous solution of PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer. Dependence of the fluorescence intensity (I/I0) of the PNA emission band at 418 nm on the concentration of PEG-b-poly(TF-DEAE-AM) 4a (B), 4b (C), and 4c (D) diblock polymers. | ||
The potentiometric and conductometric titration of PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer (Fig. S3†) illustrates that the degree of protonation of DEAE groups was 100% when the pH of the solution was 4.7, whereas the degree of protonation was 0% when the pH of the solution was 8.7. We then examined the CO2 and O2-stimuli response behaviors of the micelles formed by PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer. 0.5 mL of THF solution of PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer (10 mg mL−1) was added to 10 mL of water with stirring and bubbling of N2 (10 mL min−1). THF was then removed via evaporation with stirring and bubbling of N2 at room temperature over 24 h. Two additional aliquots (3.0 mL) taken from the micellar solution were treated with CO2 and O2, separately. PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer formed spherical aggregates (Fig. 8B) with a Dh of 140 nm (green line in Fig. 8A). Upon bubbling CO2 (10 mg mL−1) for 1 h, the aggregates were still spherical (Fig. 8D), although the size of the aggregates increased from 140 nm to 191 nm (blue line in Fig. 8A). The surface of the aggregates became more smooth (Fig. 8D) compared to the aggregates without bubbling CO2 (Fig. 8B). The zeta potential of the solution containing PEG-b-poly(TF-DEAE-AM) 4b aggregates greatly increased from 9.8 mV to 20.9 mV after CO2 was injected, which demonstrates that DEAE groups were protonated to DEAE+ in order to be extruded out from the core to form a new corona. Fig. 9 shows 1H and 19F NMR spectra of PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer in D2O treated with N2 and CO2, respectively. The typical peak at 3.64 ppm corresponding to four protons of the PEG segment could be observed upon the treatment of N2, whereas characteristic signals originating from poly(TF-DEAE-AM) block could not be detected both in 1H (blue line in Fig. 9A) and 19F (blue line in Fig. 9B) NMR spectra. However, after bubbling CO2, the proton resonance signals attributed to DEAE moiety appeared at 1.12 and 3.05 ppm in 1H NMR spectrum (red line in Fig. 9A), along with the appearance of a signal (−74.2 ppm) coming from TF group in 19F NMR spectrum (red line in Fig. 9B). This observation clearly indicates that DEAE moieties in poly(TF-DEAE-AM) block became soluble, which also improved the solubility of TF units. Thus, it can be concluded that the increase in size of the aggregates upon bubbling CO2 resulted from the hydration of both DEAE and TF functionalities on the basis of the aforementioned evidence.
 | ||
| Fig. 8 Hydrodynamic diameter distributions (A) and TEM images of PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer in aqueous solution after treatment with N2 (B), O2 (C), and CO2 (D). | ||
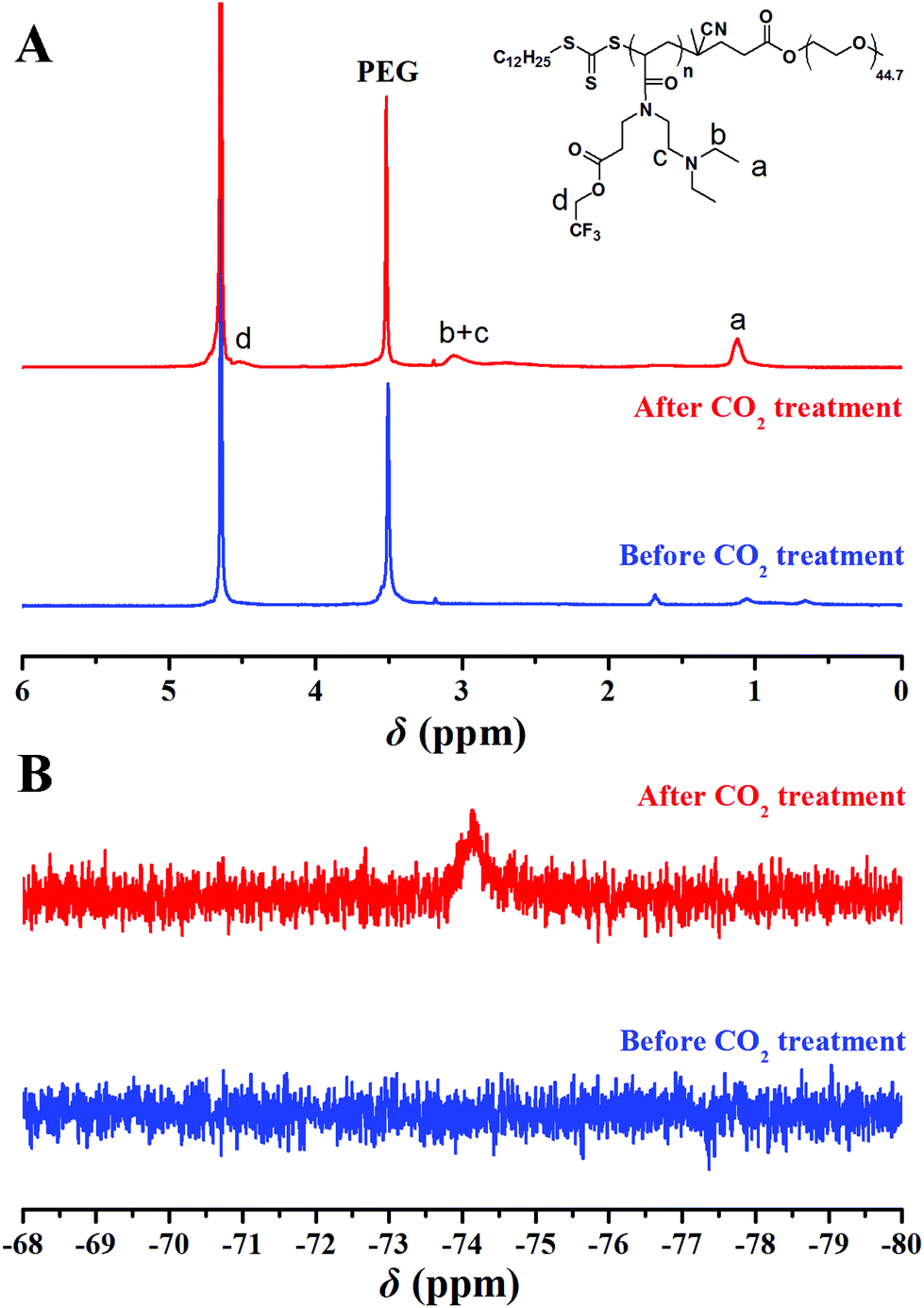 | ||
| Fig. 9 1H (A) and 19F (B) NMR spectra of PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer in D2O before (blue) and after (red) CO2 treatment. | ||
Upon bubbling O2 (10 mg mL−1), although the size of the aggregates only increased from 140 nm to 153 nm (red line in Fig. 8A), an evident change in the outmost layer of aggregates can be noticed (Fig. 8C), where the surface of the aggregates became rougher after bubbling O2. We speculate that the weak interaction between TF domains with O2 improved the solubility of TF domains to some extent, and thus, the size of the aggregates increased because of the partial hydration of TF domains. In addition, the increase in solubility of TF domains might result in micro-phase separation within the core of the aggregates, which leads to a much more rough surface of the aggregates and vogue boundary between the aggregates and supporting membrane of TEM grid, although the mobility (solubility) of TF moieties was still not good enough to be detected in 19F NMR spectrum.
In order to further prove the CO2- and O2-stimuli response behaviors of the aggregates formed by PEG-b-poly(TF-DEAE-AM) 4 diblock copolymer and potential application of these aggregates in CO2- and O2-responsive drug delivery, we examined the release behavior of hydrophobic pyrene, as a model hydrophobic drug loaded in the core of the aggregates upon treatment, with CO2 and O2. The typical UV absorption at around 335 nm attributed to pyrene was observed in an aqueous micellar solution of PEG-b-poly(TF-DEAE-AM) 4b diblock copolymer (blue line in Fig. 10A), which was prepared by adding THF solution containing both pyrene and copolymer 4b into water, followed by filtration to remove free pyrene and evaporation to remove THF, according to previous literature.63 Since the aqueous solution containing either pyrene (green line in Fig. 10A) or copolymer 4b (red line in Fig. 10A) prepared via a similar procedure showed almost no UV absorption originating from pyrene, the evident UV absorption in the aqueous solution of copolymer 4b prepared with the addition of pyrene indicates that pyrene was loaded within the core of aggregates.
 | ||
| Fig. 10 (A) UV/vis absorption spectra of pyrene/water, PEG-b-poly(TF-DEAE-AM) 4b/water, and pyrene/PEG-b-poly(TF-DEAE-AM) 4b/water; (B) cumulative release profiles of pyrene from the aggregates of PEG-b-poly(TF-DEAE-AM) 4b with the treatment of N2, O2, and CO2. | ||
Three aliquots of aqueous micellar solution of copolymer 4b containing pyrene (5 mL) were bubbled with N2, O2, and CO2, separately, and the release of pyrene from the aggregates was traced by monitoring UV absorption of these aqueous solutions varying with time. Fig. 10B shows the cumulative release of pyrene with the stimuli of N2, O2, and CO2. Upon bubbling with O2, the release rate increased with an initial burst of about 20% within 5 min and about 55% of pyrene was released from the aggregates after 1 h (red line in Fig. 10B). For the solution treated with CO2, almost 40% of the initial pyrene was released within 5 min and the total amount of released pyrene reached about 60% after 1 h (blue line in Fig. 10B). Since the bubbling of gas might also enhance the release of pyrene from the aggregates even for inert gas, we also treated the solution with N2 as a control experiment. As expected, 7% of the initial pyrene was released from the aggregates after 5 min and about 38% of pyrene escaped from the aggregates after 1 h (green line in Fig. 10B). The release rate of pyrene in both O2- and CO2-bubbling solutions was much faster than that of the solution bubbling with N2, which demonstrates that O2 and CO2 make the core of the aggregates more hydrophilic. Furthermore, compared to the solution treated with O2, the release rate of the solution bubbling with CO2 was much faster. This result demonstrates that the core of the aggregates treated with CO2 was looser than that treated with O2, which is consistent with the phenomena observed in DLS and TEM observations (Fig. 11).
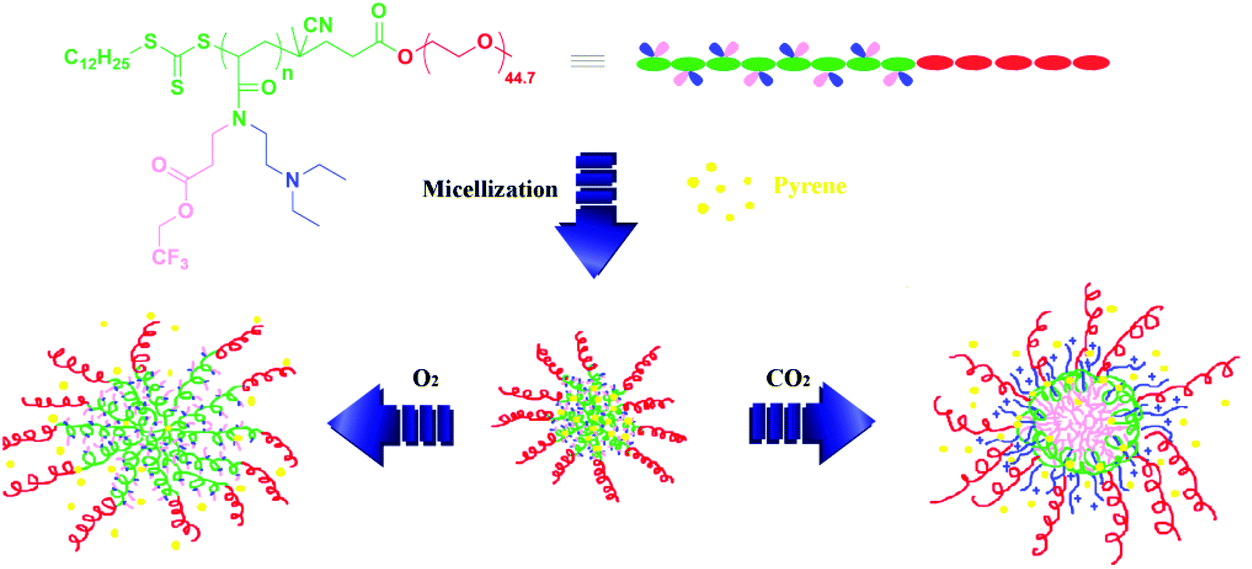 | ||
| Fig. 11 Fabrication of O2- and CO2-responsive drug-loaded micelles for the controlled delivery of pyrene. | ||
Conclusion
In this study, we synthesized a new type of CO2/O2 dual-gas-responsive acrylamide monomer by combining CO2-responsive DEAE moiety, O2-responsive TF group, and polymerizable double bond into a single monomer via aza-Michael addition and amidation reactions. RAFT polymerization was utilized to obtain the target poly(TF-DEAE-AM) homopolymer and PEG-b-poly(TF-DEAE-AM) diblock copolymer using CDSTSP and PEG-CTA as the chain transfer agents, respectively. In aqueous solution, the aggregates formed by poly(TF-DEAE-AM) homopolymer and PEG-b-poly(TF-DEAE-AM) diblock copolymer show CO2 and O2 responsiveness, as demonstrated by the changes in the size and morphologies of the aggregates on bubbling with CO2 and O2. In addition, in vitro release measurement indicates that pyrene was remarkably blocked within the micelles in the absence of gas stimuli and the release of pyrene was dramatically increased after bubbling with CO2 or O2 compared to N2. This study not only explores dual-gas-responsive drug delivery systems, but also provides promising potential applications of poly(TF-DEAE-AM)-containing materials in the biomedical area.Acknowledgements
The authors thank the financial support from the National Basic Research Program of China (2015CB931900), National Natural Science Foundation of China (21474127, 51373196, and 21504102), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), Youth Innovation Promotion Association of the Chinese Academy of Sciences (2016233), and Shanghai Scientific and Technological Innovation Project (16JC1402500, 16520710300, and 14520720100).Notes and references
- T. Tanaka, Collapse of Gels and the Critical Endpoint, Phys. Rev. Lett., 1978, 40, 820–823 CrossRef CAS.
- E. S. Matsuo and T. Tanaka, Patterns in Shrinking Gels, Nature, 1992, 358, 482–485 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Stimuli-Reponsive Polymers and Their Bioconjugates, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- A. Topete, S. Barbosa and P. Taboada, Intelligent Micellar Polymeric Nanocarriers for Therapeutics and Diagnosis, J. Appl. Polym. Sci., 2015, 132, 42650 CrossRef.
- T. Thambi, J. H. Park and D. S. Lee, Stimuli-Responsive Polymersomes for Cancer Therapy, Biomater. Sci., 2016, 4, 55–69 RSC.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Stimuli-Responsive Polymers and Their Applications, Polym. Chem., 2017, 8, 127–143 RSC.
- X. N. An, Q. Q. Tang, W. Zhu, K. Zhang and Y. L. Zhao, Synthesis, Thermal Properties, and Thermo-responsive Behaviors of Cyclic Poly(2-(dimethylamino) ethyl Methacrylate)s, Macromol. Rapid Commun., 2016, 37, 980–986 CrossRef CAS PubMed.
- J. He, X. Tong, L. Tremblay and Y. Zhao, Corona-Cross-Linked Polymer Vesicles Displaying a Large and Reversible Temperature-Responsive Volume Transition, Macromolecules, 2009, 42, 7267–7270 CrossRef CAS.
- G. Kocak, C. Tuncer and V. Butun, pH-Responsive Polymers, Polym. Chem., 2017, 8, 144–176 RSC.
- J. J. Li, Y. N. Zhou and Z. H. Luo, Smart Fiber Membrane for pH-Induced Oil/Water Separation, ACS Appl. Mater. Interfaces, 2015, 7, 19643–79650 CAS.
- O. Bertrand and J. F. Gohy, Photo-Responsive Polymers: Synthesis and Applications, Polym. Chem., 2017, 8, 52–73 RSC.
- J. M. Hu and S. Y. Liu, Responsive Polymers for Detection and Sensing Applications: Current Status and Future Developments, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- P. Kaewsaiha, K. Matsumoto and H. Matsuoka, Salt Effect on the Nanostructure of Strong Polyelectrolyte Brushes in Amphiphilic Diblock Copolymer Monolayers on the Water Surface, Langmuir, 2007, 23, 7065–7071 CrossRef CAS PubMed.
- A. Napoli, M. J. Boerakker, N. Tirelli, R. J. M. Nolte, N. A. J. M. Sommerdijk and J. A. Hubbell, Glucose-oxidase Based Self-Destructing Polymeric Vesicles, Langmuir, 2004, 20, 3487–3491 CrossRef CAS PubMed.
- H. Liu, S. Lin, Y. Feng and P. Theato, CO2-Responsive Polymer Materials, Polym. Chem., 2017, 8, 12–23 RSC.
- A. C. Feng and J. Y. Yuan, Smart Nanocontainers: Progress on Novel Stimuli- Responsive Polymer Vesicles, Macromol. Rapid Commun., 2014, 35, 767–779 CrossRef CAS PubMed.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Y. Yuan, CO2-Responsive Polymeric Vesicles that Breathe, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS PubMed.
- Q. Yan, J. Wang, Y. Yin and J. Y. Yuan, Breathing Polymersomes: CO2-Tuning Membrane Permeability for Size-Selective Release, Separation, and Reaction, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- Q. Yan and Y. Zhao, Polymeric Microtubules That Breathe: CO2-Driven Polymer Controlled-Self-Assembly and Shape Transformation, Angew. Chem., Int. Ed., 2013, 52, 9948–9951 CrossRef CAS PubMed.
- P. G. Jessop, L. Kozycz, Z. G. Rahami, D. Schoenmakers, A. R. Boyd, D. Wechsler and A. M. Holland, Tertiary Amine Solvents Having Switchable Hydrophilicity, Green Chem., 2011, 13, 619–623 RSC.
- D. Han, X. Tong, O. Boissiere and Y. Zhao, General Strategy for Making CO2-Switchable Polymers, ACS Macro Lett., 2012, 1, 57–61 CrossRef CAS.
- D. Han, O. Boissiere, S. Kumar, X. Tong, L. Tremblay and Y. Zhao, Two-Way CO2-Switchable Triblock Copolymer Hydrogels, Macromolecules, 2012, 45, 7440–7445 CrossRef CAS.
- B. Yan, D. Han, O. Boissiere, P. Ayotte and Y. Zhao, Manipulation of Block Copolymer Vesicles Using CO2: Dissociation or “Breathing”, Soft Matter, 2013, 9, 2011–2016 RSC.
- Q. Yan and Y. Zhao, CO2-Stimulated Diversiform Deformations of Polymer Assemblies, J. Am. Chem. Soc., 2013, 135, 16300–16303 CrossRef CAS PubMed.
- J. Y. Choi, J. Y. Kim, H. J. Moon, M. H. Park and B. Jeong, CO2- and O2-Sensitive Fluorophenyl End-Capped Poly(ethylene glycol), Macromol. Rapid Commun., 2014, 35, 66–70 CrossRef CAS PubMed.
- Q. Zhang and S. P. Zhu, Oxygen-Nitrogen Switchable Copolymers of 2,2,2-Trifluoroethyl Methacrylate and N,N-Dimethylaminoethyl Methacrylate, Macromol. Rapid Commun., 2014, 35, 1692–1696 CrossRef CAS PubMed.
- Q. Zhang and S. P. Zhu, Oxygen and Carbon Dioxide Dual Responsive Nanoaggregates of Fluoro- and Amino-Containing Copolymer, ACS Macro Lett., 2014, 3, 743–746 CrossRef CAS.
- L. Lei, Q. Zhang, S. X. Shi and S. P. Zhu, Oxygen and Carbon Dioxide Dual Gas- Responsive and Switchable Microgels Prepared from Emulsion Copolymerization of Fluoro- and Amino-Containing Monomers, Langmuir, 2015, 31, 2196–2201 CrossRef CAS PubMed.
- L. Lei, Q. Zhang, S. X. Shi and S. P. Zhu, Oxygen and Carbon Dioxide Dual Gas- Switchable Thermoresponsive Homopolymers, ACS Macro Lett., 2016, 5, 828–832 CrossRef CAS.
- E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Stimuli- Responsive Polymers and Their Applications in Nanomedicine, Biointerphases, 2012, 7, 9 CrossRef CAS PubMed.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Multi-stimuli Responsive Macromolecules and Their Assemblies, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- P. Schattling, F. D. Jochum and P. Theato, Multi-stimuli Responsive Polymers - the all-in-one Talents, Polym. Chem., 2014, 5, 25–36 RSC.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Multi- Stimuli-Responsive Polymeric Materials, Chem. – Eur. J., 2015, 21, 13164–13174 CrossRef CAS PubMed.
- H. G. Schild, Poly(N-isopropylacrylamide): Experiment, Theory and Application, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- S. H. Jung, H. Y. Song, Y. Lee, H. M. Jeong and H. I. Lee, Novel Thermoresponsive Polymers Tunable by pH, Macromolecules, 2011, 44, 1628–1634 CrossRef CAS.
- X. Cai, L. Zhong, Y. Su, S. L. Lin and X. H. He, Novel pH-Tunable Thermoresponsive Polymers Displaying Lower and Upper Critical Solution Temperatures, Polym. Chem., 2015, 6, 3875–3884 RSC.
- Z. Song, K. Wang, C. Gao, S. Wang and W. Q. Zhang, A New Thermo-, pH-, and CO2-Responsive Homopolymer of Poly[N,-[2-(diethylamino)ethyl]acrylamide]: Is the Diethylamino Group Underestimated?, Macromolecules, 2016, 49, 162–171 CrossRef CAS.
- C. Luo, Y. Liu and Z. B. Li, Thermo- and pH-Responsive Polymer Derived from Methacrylamide and Aspartic Acid, Macromolecules, 2010, 43, 8101–8108 CrossRef CAS.
- T. Maji, S. Banerjee, Y. Biswas and T. K. Mandal, Dual-Stimuli-Responsive L-Serine-Based Zwitterionic UCST-Type Polymer with Tunable Thermosensitivity, Macromolecules, 2015, 48, 4957–4966 CrossRef CAS.
- X. Jiang, C. Feng, G. L. Lu and X. Y. Huang, Thermoresponsive Homopolymer Tunable by pH and CO2, ACS Macro Lett., 2014, 3, 1121–1125 CrossRef CAS.
- X. Jiang, C. Feng, G. L. Lu and X. Y. Huang, Synthesis of Temperature and pH/CO2 Responsive Homopolymer Bearing Oligo(ethylene glycol) Unit and N,N-diethylamino Ethyl Group and Its Solution Property, Polymer, 2015, 64, 268–276 CrossRef CAS.
- K. Ponnusamy, R. P. Babu and R. Dhamodharan, Synthesis of Block and Graft Copolymers of Styrene by RAFT Polymerization, Using Dodecyl-Based Trithiocarbonates as Initiators and Chain Transfer Agents, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1066–1078 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, P. T. Le Tam, R. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. L. McCormick and A. B. Lowe, Aqueous RAFT Polymerization: Recent Developments in Synthesis of Functional Water-Soluble (Co)polymers with Controlled Structures, Acc. Chem. Res., 2004, 37, 312–325 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Living Radical Polymerization by the RAFT Process, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Living Radical Polymerization by the RAFT Process-A First Update, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Living Radical Polymerization by the RAFT Process-A Second Update, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- A. S. Lee, A. P. Gast, V. Butun and S. P. Armes, Characterizing the Structure of pH Dependent Polyelectrolyte Block Copolymer Micelles, Macromolecules, 1999, 32, 4302–4310 CrossRef CAS.
- S. Y. Liu, N. C. Billingham and S. P. Armes, A Schizophrenic Water-Soluble Diblock Copolymer, Angew. Chem., Int. Ed., 2001, 40, 2328–2331 CrossRef CAS PubMed.
- E. J. Lobb, I. Ma, N. C. Billingham and S. P. Armes, Facile Synthesis of Well- Defined, Biocompatible Phosphorylcholine-Based Methacrylate Copolymers via Atom Transfer Radical Polymerization at 20 °C, J. Am. Chem. Soc., 2001, 123, 7913–7914 CrossRef CAS PubMed.
- X. Chen, D. P. Randall, C. Perruchot, J. F. Watts, T. E. Patten, T. Werne and S. P. Armes, Synthesis and Aqueous Solution Properties of Polyelectrolyte- Grafted Silica Particles Prepared by Surface-Initiated Atom Transfer Radical Polymerization, J. Colloid Interface Sci., 2003, 257, 56–64 CrossRef CAS PubMed.
- E. He, P. Ravi and K. C. Tam, Synthesis and Self-Assembly Behavior of Four-Arm Poly(ethylene oxide)-b-poly(2-(diethylamino)ethyl methacrylate) Star Block Copolymer in Salt Solutions, Langmuir, 2007, 23, 2382–2388 CrossRef CAS PubMed.
- M. Szwarc, M. Levy and R. Milkovich, Polymerization Initiated by Electron Transfer to Monomer. A New Method of Formation of Block Polymers, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- M. Szwarc, ‘Living’ Polymers, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- D. Uhrig and J. W. Mays, Experimental Techniques in High-vacuum Anionic Polymerization, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6179–6222 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Controlled/living Radical Polymerization: Features, Developments, and Perspectives, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, Stability and Utility of Pyridyl Disulfide Functionality in RAFT and Conventional Radical Polymerizations, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef CAS.
- M. Achilleos, T. M. Legge, S. Perrier and C. S. Patrickios, Poly(ethylene glycol)- Based Amphiphilic Model Conetworks: Synthesis by RAFT Polymerization and Characterization, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7556–7565 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, A More Versatile Route to Block Copolymers and Other Polymers of Complex Architecture by Living Radical Polymerization: The RAFT Process, Macromolecules, 1999, 32, 2071–2074 CrossRef CAS.
- S. Kumar, R. Acharya, U. Chatterji and P. De, Side-Chain Amino-Acid-Based pH-Responsive Self-Assembled Block Copolymers for Drug Delivery and Gene Transfer, Langmuir, 2013, 29, 15375–15385 CrossRef CAS PubMed.
- P. S. Xu, H. D. Tang, S. Y. Li, J. Ren, E. Van Kirk, W. J. Murdoch, M. Radosz and Y. Q. Shen, Enhanced Stability of Core-Surface Cross-Linked Micelles Fabricated from Amphiphilic Brush Copolymers, Biomacromolecules, 2004, 5, 1736–1744 CrossRef CAS PubMed.
- L. L. Li, F. Q. Tang, H. Y. Liu, N. J. Hao, D. Chen, X. Teng and J. Q. He, In Vivo Delivery of Silica Nanorattle Encapsulated Docetaxel for Liver Cancer Therapy with Low Toxicity and High Efficacy, ACS Nano, 2010, 4, 6874–6882 CrossRef CAS PubMed.
- B. B. Xu, G. X. Gu, C. Feng, X. Jiang, J. H. Hu, G. L. Lu, S. Zhang and X. Y. Huang, (PAA-g-PS)-co-PPEGMEMA Asymmetric Polymer Brushes: Synthesis, Self-assembly, and Encapsulating Capacity for Both Hydrophobic and Hydrophilic Agents, Polym. Chem., 2016, 7, 613–624 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py02004f |
| This journal is © The Royal Society of Chemistry 2017 |