
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A self-assembled peptide mimetic of a tubular host and a supramolecular polymer†
Arpita
Paikar
,
Apurba
Pramanik
,
Tanmay
Das
and
Debasish
Haldar
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, West Bengal, India. E-mail: deba_h76@iiserkol.ac.in; deba_h76@yahoo.com
First published on 21st November 2016
Abstract
Simple and efficient strategies for the generation of a supramolecular nanotube by self-assembly of acyclic modular building blocks and guest encapsulation remain an essentially unmet challenge. A tripeptide containing leucine, Aib and serine forms a rigid type-III′ β-turn structure stabilized by multiple intramolecular N–H⋯O and O–H⋯O hydrogen bonds. The modular building blocks self-assemble in a helical manner to develop a supramolecular nanotube. The formation of a self-assembled peptide mimetic of the nanotubular host has been proven with SEM, X-ray crystallography and other spectroscopic techniques. This supramolecular nanotube with a hydrophobic core has been used as the host for modular inclusion of designer hydrophobic guests to fabricate a supramolecular polymer. The interactions between the host and the guest were analyzed by using DLS and different spectroscopic techniques. The variable temperature NMR experiments show that with the increasing temperature the hydrophobic interactions between the host and the guest decrease, which indicates the sliding of the tubular host along the guest axle. Comparison of a coumarin based guest with a naphthalenediimide based guest shows that the latter have connected two tubular hosts and thus formed a supramolecular polymer. The formation of the supramolecular polymer has been proven with DLS, SEM and AFM and other spectroscopic techniques.
Introduction
Molecular recognition, assembly and complex formation are not only important for biological processes but also for materials science.1 By precisely organizing the simple molecular building blocks by noncovalent interactions, nature has achieved complex materials with diverse structures and functions. Inspired by nature, chemists have been able to design and fabricate new synthetic materials that are efficient to recognize a variety of targets. Highly sophisticated structures and functions have been achieved by the formation of supramolecular polymers from the interactions of host and guest molecules.2 The directional and predictable supramolecular assembly mostly depends on the information coded in the complementary designer building blocks. The encapsulation of the guests by hosts is mainly attributed to the complementary nature like the size and shape of the building blocks and combination of several elemental supramolecular interactions.3 These non-covalent interactions cover hydrogen bonding, π-stacking interactions, van der Waals interactions, ionic and polar interactions and hydrophobic collapse.3 A large number of supramolecular oligomers and polymers encapsulated by a tubular host have been reported.4,5 These host–guest complexes are important as supramolecular catalysts,6 enzymes,7 advance optical materials8 and find several applications.9 The tubular host may rotate around the guest or it may slide along the guest axle. The movement of the tubular host may be manipulated by the external stimuli.10 A large number of foldamers that form helices and tubular structures and recognise small guests such as ions and small molecules have been reported.11 But fabrication of a cylindrical host or tube by assembly of acyclic modular building blocks is relatively rare.12We are looking for a supramolecular assembly of peptides into hollow helices that can encapsulate various guests.13 For this we have synthesized many Xaa-Aib-Xaa tripeptides, most have a β-turn structure, and self-assemble to form a supramolecular helix, but none form nanotubes. Herein, we have synthesized a tripeptide containing L-Leu, α-aminoisobutyric acid (Aib) and L-Ser with an assumption that the helicogenic Aib14 core leads to the hydrogen bonded folded structure and the L-Ser side chain may incorporate additional hydrogen bonding (Scheme 1). For the inclusion study guest molecules, the coumarin derivative 2 and naphthalene diimide 3, with long hydrophobic chains have been synthesized. The supramolecular assembly of peptides may provide hollow helices (Scheme 1a), which will behave like a modular host depending on the structure of the guest molecule. The host peptide has no chromophore. So, a chromophore containing guest will be helpful to study the interactions by various spectroscopic techniques. We incorporate coumarin and naphthaline diimide as the core units in the guest molecules. We propose that the coumarin derivative 2 having one hydrophobic tail will interact with the nanotube by incorporating its hydrophobic tail inside the nanotube (Scheme 1b) and finally develop a supramolecular rod-like structure (Scheme 1c). However, one naphthalene diimide 3 unit having two hydrophobic tails will connect two host nanotubes and thus develops a supramolecular polymer (Scheme 1d).
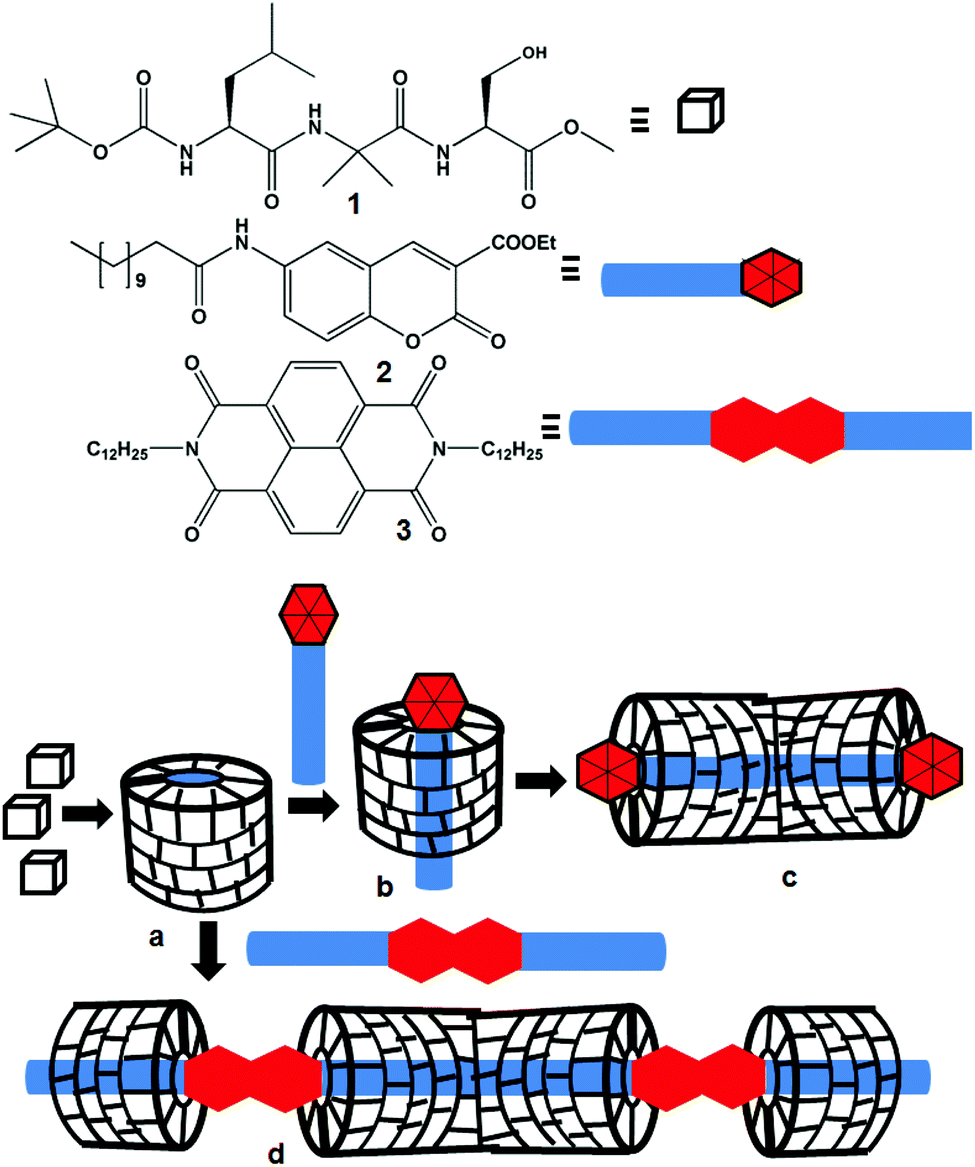 | ||
| Scheme 1 Compounds 1, 2 and 3 and the schematic presentation of (a) the supramolecular helical nanotube from peptide building blocks; (b) inclusion of the hydrophobic compound in the capsid; (c) the supramolecular columnar structure; (d) the supramolecular inclusion polymer. | ||
Experimental
Materials
All L-amino acids were purchased from Sigma chemicals. HOBt (1-hydroxybenzotriazole) and DCC (dicyclohexylcarbodiimide) were purchased from SRL.Peptide synthesis
The peptides were synthesized by a conventional solution-phase method using a racemization free fragment condensation strategy. The Boc group was used for N-terminal protection and the C-terminus was protected as a methyl ester. Couplings were mediated by dicyclohexylcarbodiimide/1-hydroxybenzotriazole (DCC/HOBt). Methyl ester deprotection was performed via the saponification method. All the intermediates were characterized by 500 MHz 1H NMR, 13C NMR and mass spectrometry. Final compounds were fully characterized by 500 MHz 1H NMR spectroscopy, 13C NMR spectroscopy, mass spectrometry, and IR spectroscopy. Peptide 3 was characterized by X-ray crystallography. The products were purified by column chromatography using silica (100–200 mesh size) gel as the stationary phase and the n-hexane–ethyl acetate mixture as the eluent.1H NMR (500 MHz, DMSO-d6, δ in ppm): 12.36 [b, 1H, COOH], 7.03–7.02 [d, 1H, J = 2 Hz, Leu(1) NH], 3.91–3.87 [m, 1H, Leu(1) CαH], 1.63–1.60 [m, 2H, Leu(1) CβH], 1.51–1.47 [m, 1H, Leu(1) CγH], 1.37 [s, 9H, Boc], 0.87–0.83 [m, 12H, 4 CH3]. 13C NMR (125 MHz, DMSO-d6, δ in ppm): 174.73, 155.64, 77.76, 66.41, 51.81, 28.24, 24.38, 22.29. FT-IR (KBr): 3462, 3355, 3311, 2983, 2968, 2934, 2877, 2525, 1727, 1699, 1675, 1533, 1456, 1419, 1385, 1368, 1296, 1197, 1172, 1087, 1048, 1018 cm−1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. Yield: 4.17 g (12.62 mmol, 77.8%).
:1) as an eluent. Yield: 4.17 g (12.62 mmol, 77.8%).
1H NMR (CDCl3, 500 MHz, δ in ppm): 6.60 [s, 1H, Aib(2) NH], 4.78 [b, 1H, Leu(1) NH], 3.98 [b, 1H, Leu (1)CαH], 3.66 [s, 3H, OMe], 1.62–1.55 [m, 2H, Leu(1) CβH], 1.46 [s, 6H, Aib(2) CβH], 1.41 [s, 9H, Boc], 0.88–0.85 [m, 7H, Leu(1) CδH + CγH]. 13C NMR (CDCl3, 125 MHz, δ in ppm): 174.74, 171.73, 155.83, 80.07, 56.37, 56.37, 52.95, 52.53, 40.82, 28.26, 24.74, 24.70, 24.67, 22.86, 22.06. FT-IR (KBr): 3323, 3078, 2959, 2872, 1748, 1685, 1534, 1458, 1393, 1366, 1320, 1281, 1247, 1153 cm−1.
1H NMR (DMSO-d6, 500 MHz, δ in ppm): 12.20 [s, 1H, COOH], 7.92 [s, 1H, Aib(1) NH], 6.74–6.72 [d, 1H, J = 9 Hz, Leu(1) NH], 3.98–3.34 [m, 1H, Leu(1) CαH], 1.63–1.56 [dd, 2H, Leu(1) CβH], 1.37 [s, 6H, Aib(2) CβH], 1.32 [s, 9H, Boc], 0.89–0.84 [m, 7H, Leu(1) CδH + CγH].13C NMR (DMSO-d6, 125 MHz, δ in ppm): 175.43, 171.69, 155.16, 77.91, 54.72, 52.44, 40.97, 28.14, 24.65, 24.12, 22.96, 21.60. FT-IR (KBr): 3308, 3067, 1665, 1528, 1376, 1284, 1171 cm−1.
:4) as an eluent. Yield: 4.06 g (8.5 mmol, 78.89%).
1H NMR (CDCl3, 500 MHz, δ in ppm): 7.0615 [b, 1H, Ser(3) NH], 6.7218 [s, 1H, Aib(2) NH], 5.0348–5.0176 [d, 1H, Boc NH, J = 8.6 Hz], 4.5825–4.5634 [t, 1H Ser(3) OH], 4.0463–4.0196 [d, 1H, CαH Leu(1), J = 13.35 Hz], 3.9757–3.9222 [m, 2H, CβH Ser(3)], 3.8726–3.8344 [dd, 1H, CαH of Ser(3)], 3.7524 [s, 3H, OMe], 1.6932–1.5711 [m, 2H, CβH Leu(1)], 1.5329–1.5119 [d, 6H, 2 × Me of Aib, J = 10.5 Hz], 1.4375 [s, 9H, Boc], 0.9490–0.9089 [m, 7H, Leu(1) CδH + CγH]. 13C NMR (CDCl3, 125 MHz, δ in ppm): 174.0582, 173.5599, 171.4655, 156.8697, 81.4060, 62.6647, 57.6815, 57.7098, 53.0232, 40.6373, 28.8292, 26.8865, 25.3048, 24.7776, 23.4777, 22.4016. FT-IR (KBr): 3303, 2948, 1745, 1654, 1541, 1276, 1178, 1049 cm−1. Mass spectra: m/z 440.2803, [M + Na]+; Mcalcd 417.25.
:1) as an eluent. Yield: 2.80 g (12.83 mmol, 68.97%).
1H NMR (400 MHz, CDCl3, δ in ppm): 8.52 (s, 1H lactone ring proton); 7.62 (m, 2H, phenyl ring protons); 7.36 (m, 2H, phenyl ring protons); 4.42 (m, 2H, CH2); 1.42 (t, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 162.99, 156.67, 155.08, 148.55, 134.28, 129.44, 124.78, 118.24, 117.80, 116.72, 61.92, 14.17.
:hexane (2:1) as an eluent. Yield: 2.5 g (9.5 mmol, 74.5%).
1H NMR (400 MHz, CDCl3, δ in ppm): 8.58 (s, 1H phenyl ring proton); 7.56 (s, 1H, lactone ring protons); 8.50–8.47 (d, 1H, J = 12.4, phenyl ring protons); 7.51–7.49 (d, 1H, J = 8.8, phenyl ring protons); 4.46 (m, 2H, CH2); 1.43 (t, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 162.01, 158.29, 154.93, 146.88, 144.19, 128.56, 125.18, 120.52, 118.05, 62.51, 14.14.
:hexane (4:1) as an eluent.
Yield: 1.25 g (5.36 mmol, 58.77%).
1H NMR (400 MHz, DMSO-D6, δ in ppm): 8.55 (s, 1H lactone ring proton); 7.16–7.14 (d, 1H, J = 9.2, phenyl ring proton); 6.98–6.96 (d, 1H, J = 9.8, phenyl ring proton); 6.88 (s, 1H, phenyl ring proton); 5.27 (s, 2H, NH2); 4.29 (m, 2H, CH2); 1.31 (t, 3H, CH3); 13C NMR (100 MHz, DMSO-D6): δ 163.03, 156.51, 148.62, 146.23, 145.93, 121.52, 118.11, 117.45, 116.51, 110.97, 61.09, 14.07.
:2 as an eluent. Yield: 1.462 g (3.52 mmol, 68.4%).
1H NMR (400 MHz, CDCl3, δ in ppm): 8.44 (s, 1H lactone ring proton); 8.11–8.10 (d, 1H, J = 4.2, phenyl ring proton); 7.47–7.46 (d, 1H, J = 2.4, phenyl ring proton); 7.26 (s, 1H, phenyl ring proton); 7.23 (t, 1H, NH); 4.37 (m, 2H, CH2); 2.37 (m, 2H, CH2); 1.69 (m, 2H, CH2); 1.38 (m, 3H, CH3); 1.25 (m, 16H); 0.84 (t, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 172.82, 162.79, 157.47, 151.90, 148.44, 135.05, 125.84, 119.46, 118.47, 117.98, 117.10, 62.00, 37.66, 31.88, 29.58, 29.44, 29.30, 29.23, 25.48, 22.66, 14.19, 14.08. ESI Mass m/z 416.1 (M + H)+Mcal 415.52.
1H NMR (CDCl3, 500 MHz, δ in ppm): 8.7536 [s, 4H, aromatic ring protons], 4.2040–4.1732 [t, 4H, CαH of the Dodec chain], 1.7682–1.7077 [m, 4H, CβH of the Dodec chain], 1.4405–1.3951 [m, 4H, CH2 of the Dodec chain], 1.3718–1.3435 [m, 4H, CH2 of the Dodec chain], 1.2496 [s, 32H, CH2 of the Dodec chain], 0.8866–0.8582 [t, 6H, 2 × Me]. 13C NMR (CDCl3, 125 MHz, δ in ppm): 173.447, 131.5220, 127.2686, 41.6015, 32.5026, 30.2206, 30.1773, 30.1123, 29.9245, 28.6824, 27.6787, 23.2664, 14.6947. ESI Mass m/z 603.804 [M + H]+Mcal 602.41.
NMR experiments
All NMR studies were carried out on Bruker AVANCE 500 MHz and Jeol 400 MHz spectrometers at 278 K. Compound concentrations were in the range 1–10 mM in CDCl3 and DMSO-d6.FT-IR spectroscopy
All reported solution FT-IR spectra were obtained with a Perkin Elmer Spectrum RX1 spectrophotometer.UV-Vis spectroscopy
UV-Vis absorption spectra were recorded on a UV-Vis spectrophotometer (Hitachi).Fluorescence spectroscopy
All the fluorescence spectra were recorded in DCM on a fluorescence spectrometer (HORIBA JOBIN YVON).Mass spectra
Mass spectra were recorded on a Q-Tof Micro YA263 high resolution (Waters Corporation) mass spectrometer by positive-mode electrospray ionization.Dynamic light scattering
The particle sizes were determined by using a DLS instrument (model ZETASIZER nano series nano zs) with the corresponding compound solution in DCM at different concentrations.Field emission scanning electron microscopy
Morphologies of all the reported compounds were investigated using field emission-scanning electron microscopy (FE-SEM). A small amount of solution of the corresponding compound was placed on a clean silicon wafer and then dried by slow evaporation. The material was then allowed to dry under vacuum at 30 °C for two days. The materials were gold-coated, and the micrographs were obtained using FE-SEM apparatus (Jeol Scanning Microscope-JSM-6700F).Atomic force microscopy
The morphology of the reported compound was investigated by atomic force microscopy (AFM). A drop of the sample solution in DCM was placed on a clean microscope cover glass and then dried by slow evaporation. The material was then allowed to dry under vacuum at 30 °C for two days. Images were obtained with an NTMDT instrument, model no. AP-0100 in semi-contact mode.Single crystal X-ray diffraction
Crystallographic data of peptide 1: C19H35N3O7, Mw = 417.50, hexagonal, space group P65, a = 19.7793(8), b = 19.7793(8), c = 11.2095(7) Å, V = 3797.8(4) Å3, Z = 6, dm = 1.095 Mg m−3, T = 100 K, R1 0.0591 and wR2 0.1625 for 4990 data with I > 2σ(I). Intensity data were collected with MoKα radiation using a Bruker APEX-2 CCD diffractometer. Data were processed using the Bruker SAINT package and the structure solution and refinement procedures were performed using SHELX9718 and deposited at CCDC with reference 1434362.Results and discussion
Peptide structure analysis
Target compounds were synthesized by conventional solution phase methods, and purified and characterized by 1H-NMR, 13C-NMR, FT-IR and mass spectrometry (MS) analysis. X-ray diffraction sheds some light on the structure and self-assembly of tripeptide 1 at the atomic level. Colorless crystals of tripeptide 1 were suitable for X-ray crystallography and obtained from dichloromethane solution by slow evaporation. Tripeptide 1 crystallizes with one molecule in the asymmetric unit. The solid state structure (Fig. 1a) of tripeptide 1 shows that the peptide backbone adopts a type III′ β-turn like conformation.13 There is a ten member intramolecular N–H⋯O hydrogen bond between Boc C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Ser NH. Another ten member intramolecular O–H⋯O hydrogen bond exists between Leu CO and Ser side chain OH. These intramolecular hydrogen bonds made the β-turn building block very rigid. The existence of this rigid β-turn like conformation of tripeptide 1 in the solution state was also confirmed by circular dichroism (CD) spectroscopy (Fig. S1 in the ESI†). The torsion angles around the central Aib residue appear to play a crucial role in dictating the overall structural features of tripeptide 1 (Table 1 in the ESI†).
O and Ser NH. Another ten member intramolecular O–H⋯O hydrogen bond exists between Leu CO and Ser side chain OH. These intramolecular hydrogen bonds made the β-turn building block very rigid. The existence of this rigid β-turn like conformation of tripeptide 1 in the solution state was also confirmed by circular dichroism (CD) spectroscopy (Fig. S1 in the ESI†). The torsion angles around the central Aib residue appear to play a crucial role in dictating the overall structural features of tripeptide 1 (Table 1 in the ESI†).
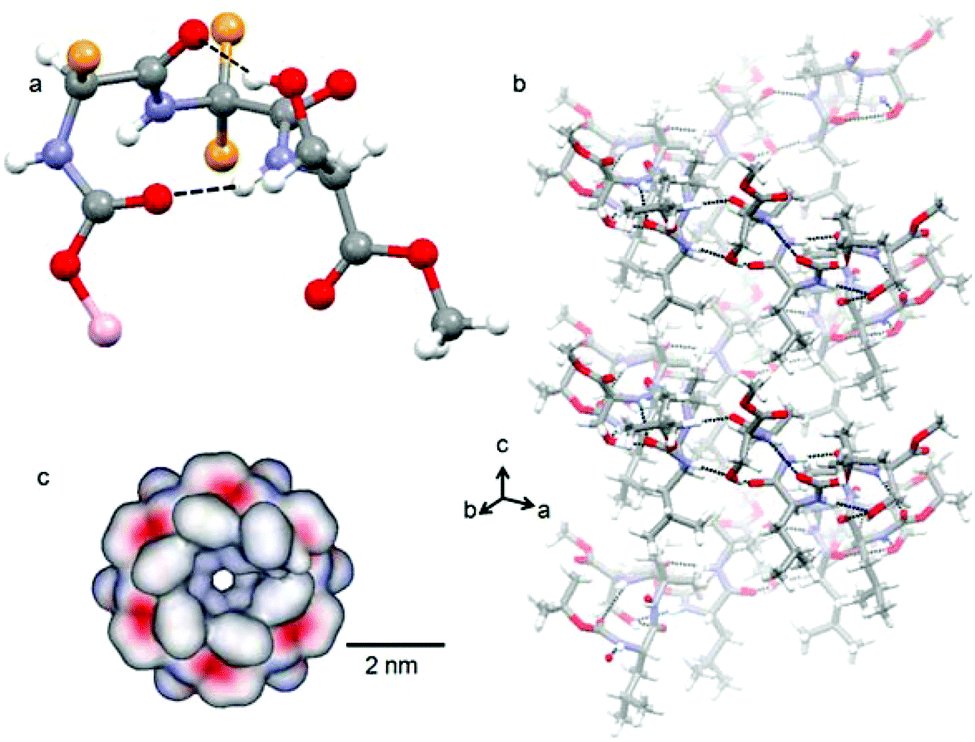 | ||
| Fig. 1 (a) The solid state structure of peptide 1. Intramolecular hydrogen bonds are shown as black dotted lines. Leu and Aib side chains here appear as orange spheres and the t-butyl group as a pink sphere. (b) Side view of the intermolecular hydrogen bonded supramolecular helical column of tripeptide 1. Intermolecular hydrogen bonds are shown as black dotted lines. (c) Top view surface diagram of the supramolecular helical column of tripeptide 1 showing a hydrophobic inner core of the tube. The red colour indicates negative charge, blue positive charge and grey uncharged areas. | ||
The crystal structure further reveals that the individual tripeptide 1 subunits are stacked by intermolecular H-bonding interaction between neighboring molecules and generate a supramolecular helical column like structure (Fig. 1b) about the crystallographic c direction.13 There are two intermolecular N–H⋯.O hydrogen bonds between Aib CO and Aib NH and Leu NH and Ser side chain OH. The hydrogen bonding parameters of peptide 1 are listed (Table 2 in the ESI†). The tripeptide 1 helix looks like a nanotube from a top view (Fig. 1c). The diameter of the tube is about 6 Å and there is no restriction on the length. The surface diagrams (Fig. 1c) show that the inner core as well as the outside of the tube is hydrophobic in nature.
DLS study of the inclusion complex
We have intended to use the nanotubes for guest inclusion. The guest molecules 2 and 3 were designed with an assumption that the hydrophobic tail of the guests will be fitted inside the hydrophobic core of the nanotubes. The assembly behaviour of tripeptide 1 and guests was studied by dynamic light scattering (DLS). From the DLS study, the mixture of peptide 1 and coumarin derivative 2 forms bigger particles (251 nm) than the pure compounds (60 nm) (Fig. 2). However, the peptide 1 and naphthalene diimide 3 mixture forms the biggest particles (1130 nm) as expected (Fig. 2).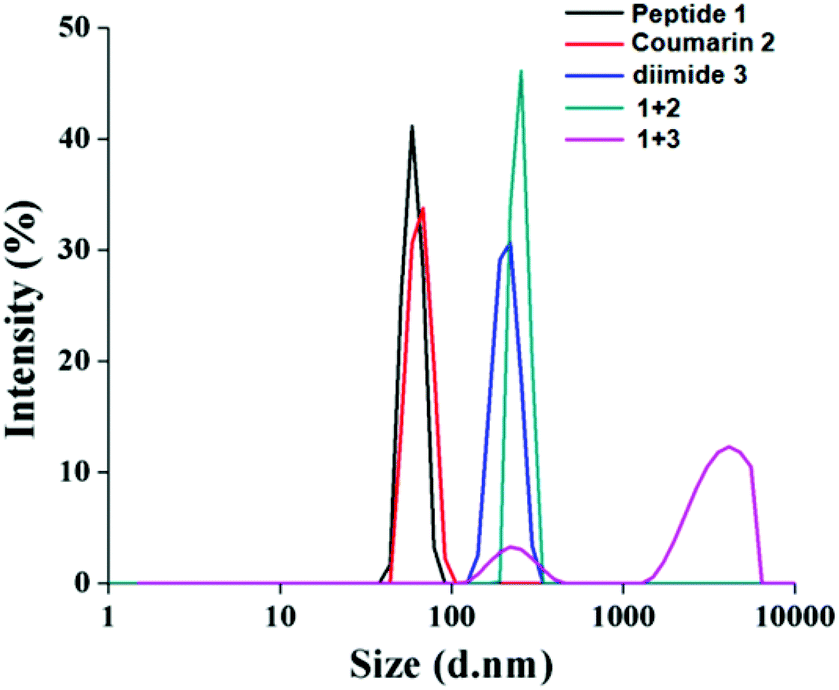 | ||
| Fig. 2 DLS study of tripeptide 1 (1.865 × 10−5 M), coumarin derivative 2 (1.1990 × 10−5 M), naphthalene diimide 3 (0.9732 × 10−5 M) and their mixtures in dichloromethane at room temperature. | ||
Spectroscopic studies of the inclusion complex
The typical UV-Vis titration experiments of the coumarin derivative 2 in dichloromethane (1.065 × 10−5 M) with tripeptide 1 show a gradual decrease of absorption bands at 250, 294 and 365 nm responsible for π to π* transition (Fig. S2 in the ESI†). The UV-Vis titration experiments of naphthalene diimide 3 in dichloromethane (2.41 × 10−5 M) with tripeptide 1 show similar results. With addition of tripeptide 1, the intensity of the peaks at 240, 356 and 386 nm responsible for π to π* transition decreases (Fig. S3 in the ESI†). The typical emission band of the coumarin derivative 2 in DCM solution at 460 nm also decreases with the addition of tripeptide 1 (Fig. S4 in the ESI†). From the emission spectra, the intensity of the peak at 401 nm for naphthalene diimide 3 also decreases with the addition of tripeptide 1 (Fig. S5 in the ESI†). The results suggest the strong interactions between the tripeptide 1 and the coumarin derivative 2 or naphthalene diimide 3. We have calculated the binding constant (0.1473 × 104 M−1) of the peptide nanotube 1 and the coumarin derivative 2 using the Bensi–Hildebrand equation (Fig. S2b in the ESI†). The binding constant for the peptide nanotube 1 and naphthalene diimide 3 is 0.1512 × 104 M−1(Fig. S3b in the ESI†). Solution FT-IR spectroscopy is an excellent method to investigate the self-assembly propensity of tripeptide 1 and the coumarin derivative 2. The FT-IR region 3500–3200 cm−1 is important for the N–H and O–H stretching and 3000–2800 cm−1 for the C–H stretching; however the range 1800–1500 cm−1 is assigned to the stretching band of amide I and the bending peak of amide II and ester groups. The FT-IR spectra of tripeptide 1 in DCM (Fig. 3a(i)) exhibits the C–H stretching frequency at 2926 cm−1.16 The coumarin derivative 2 in DCM exhibits peaks at 2888 and 2815 cm−1 corresponding to the C–H stretching frequency (Fig. 3a(ii)) which shifted to 2902 cm−1 on mixing with peptide 1 in DCM (Fig. 3a(iii)). This indicates the hydrophobic interactions between the tripeptide 1 and guest 2.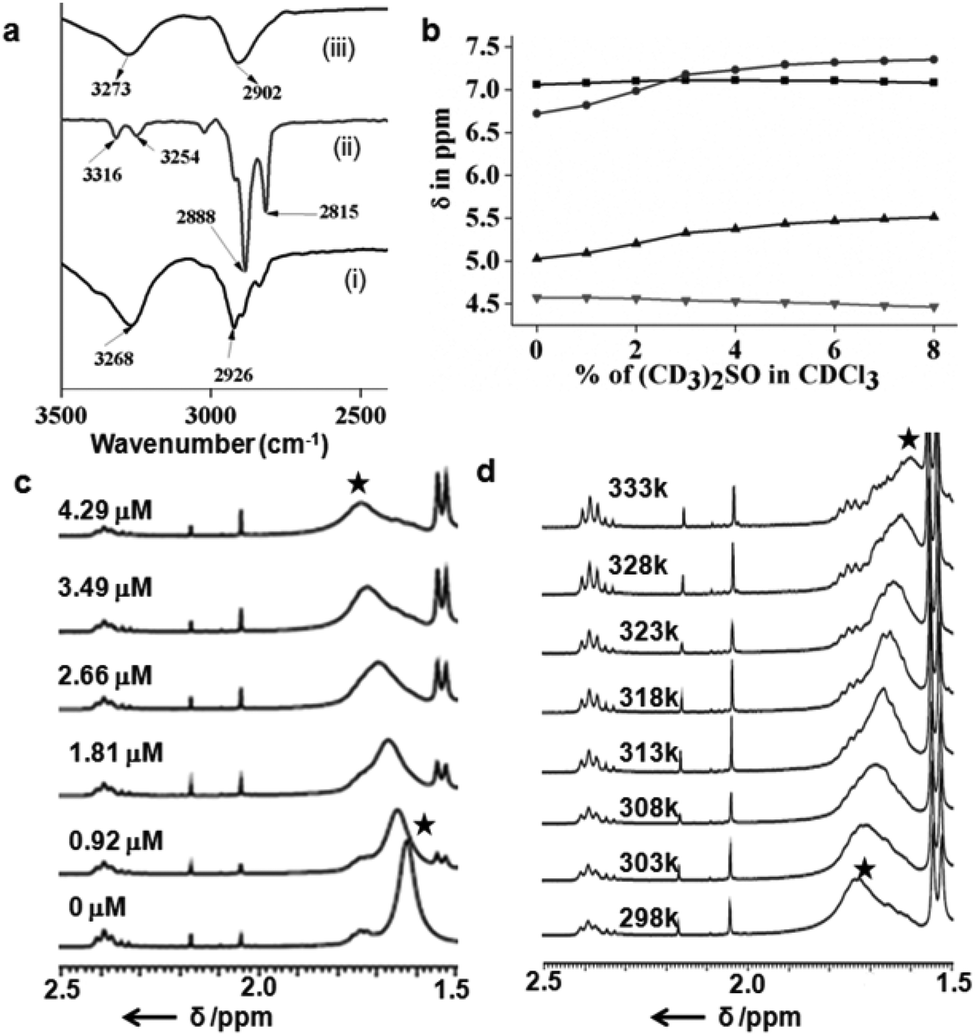 | ||
| Fig. 3 (a) The solution FT-IR spectra of (i) peptide 1, (ii) coumarin derivative 2 and (iii) tripeptide 1 and coumarin derivative 2 mixture in DCM. (b) Plot of solvent dependence of the chemical shift of peptides 1 (▼) Ser OH, (▲) Leu NH, (●) Aib NH and (■) Ser NH at varying concentrations of (CD3)2SO in CDCl3 solutions. (c) Part of 1H NMR spectra of the coumarin derivative 2 with the increasing amount of peptide 1 in CDCl3. (d) Part of the variable temperature 1H NMR spectra of the coumarin derivative 2 and peptide 1 (4.29 μM) in CDCl3. (★) indicates hydrocarbon protons of the coumarin derivative 2. | ||
In order to understand whether the amide and hydroxyl groups of peptide 1 are intramolecularly or intermolecularly hydrogen bonded, 1H NMR solvent titration experiments have been performed. Generally, addition of small amounts of (CD3)2SO in CDCl3 brings about monotonic downfield shifts of the exposed NH groups in peptides, leaving solvent-shielded NH groups largely unaffected.17Fig. 3b shows that Leu(1) and Aib(2) NHs are solvent exposed, as it is evident from their significant chemical shift (Δδ 0.48 and 0.63), upon the addition of (CD3)2SO to CDCl3 solutions. However, Ser(3) NH and OH exhibit minimum chemical shifts (Δδ 0.02 and 0.11) even at higher percentages of (CD3)2SO. Table 3 in the ESI† illustrates the chemical shift values of all NHs of peptide 1. This demonstrates that peptide 1 has an intramolecular hydrogen bonded conformation in solution like that in the solid state.15 The typical NMR titration experiments of the coumarin derivative 2 in CDCl3 (1.1359 × 10−4 M) with tripeptide 1 show the gradual down field shift and broadening of hydrocarbon protons from 1.62 to 1.76 ppm (Fig. 3c). This is due to the unique interaction between the tripeptide 1 and the coumarin derivative 2. Other non-tubular peptides including a Met analogue do not affect the spectra in a similar manner (Fig. S6–S8 in the ESI†). Further we have studied the variable temperature NMR experiments of peptides 1 and the coumarin derivative 2 in CDCl3. Fig. 3d shows the part of variable temperature 1H NMR spectra of peptides 1 and the coumarin derivative 2 in CDCl3. Upon heating (from 25 °C to 60 °C) the hydrocarbon protons at 1.76 ppm shifted to an up-field direction (Fig. 3d). With the increasing temperature the interaction between the tripeptide 1 and the coumarin derivative 2 decreases. Ser(3) NH and OH of peptide 1 show no chemical shift with the increasing temperature, suggesting no change in intramolecular hydrogen bonding. However, Leu(1) and Aib(2) NHs exhibit very little chemical shift (Δδ 0.05 and 0.08) even at higher temperatures. The supramolecular tube from tripeptide 1 encapsulates rod-like hydrophobic guests 2.
Morphological study
The freshly prepared solution of peptide 1 in DCM (0.5 mg mL−1) was drop-cast on a silicon wafer and dried under vacuum at 30 °C for two days. The FE-SEM micrographs show unbranched tube like aggregates with 100 nm average diameter and several micrometers in length (Fig. 4a). The coumarin derivative 2 from DCM solution exhibits a twisted fiber like morphology. The twisted fibers have a diameter of about 70 nm and are several micrometers in length (Fig. 4b). To examine the difference in the morphology after mixing peptide 1 and the coumarin derivative 2, FE-SEM was performed. The peptide 1 and coumarin derivative 2 mixture exhibits an unbranched polydisperse rod like morphology (Fig. 4c). The rods are hexagonal (Fig. 4c inset) in shape and rigid in nature. The average diameter of the rods is ca. 100 nm and length ca. 500 nm which may be obtained by interaction between coumarin molecules and the tubes. The existence of both components in hexagonal rods was confirmed by NMR spectroscopy (Fig. S9 in the ESI†). We have also studied the modular encapsulation of tripeptide 1 with naphthalene diimide 3 having hydrophobic dodecyl chains in both termini. The absorption as well as emission spectra exhibit the interaction between the peptide 1 and naphthalene diimide 3. We have also studied the morphology of naphthalene diimide 3 in DCM solution. Naphthalene diimide 3 exhibits a polydisperse layered sheet like morphology (Fig. 4d). However, the peptide 1 and naphthalene diimide 3 mixture exhibits a long fiber like morphology (Fig. 4e). The average diameter of the fibers is ca. 100 nm and they are several micrometers in length. The fibers also entangle like a rope. The homogeneity of the twist sense is due to chiral induction and the presence of two chiral L-amino acids in peptide 1. To investigate the topology of the fibers, atomic force microscopy (AFM) studies were performed. AFM shows a fiber like morphology (Fig. 4f). The 3D image of peptide 1 and naphthalene diimide 3 fibers indicates that the fiber surface is very rough. These microscopy studies confirm the fabrication of a long supramolecular polymer from peptide 1 and diimide 3. The docking studies also show the formation of a supramolecular polymer (ESI, Fig. S32 and S33†). So, the encapsulation is modular and one can develop different morphologies using long chain alkanes or branched alkanes instead of 2 and 3. | ||
| Fig. 4 FE-SEM images of (a) peptide 1 showing an unbranched polydisperse nanotubes morphology; (b) coumarin derivative 2 having a twisted fiber and rope like morphology; (c) unbranched polydisperse rod-like morphology of the peptide 1 and coumarin derivative 2 mixture. (Inset: Hexagonal rod-like structures); (d) naphthalene diimide 3 showing a sheet like morphology; (e) peptide 1 and naphthalene diimide 3 mixture exhibits a very long fiber of the supramolecular polymer. (Inset: The fibers entangled like a rope) (f) AFM topology of the peptide 1 and naphthalene diimide 3 showing a very rough fiber surface. | ||
Mass spectrometric analysis
Furthermore, we have performed the analysis of the hexagonal column obtained from peptide 1 and compound 2 and an unbranched fiber from peptide 1 and diimide 3 by mass spectrometry. Experimental and simulated isotopic patterns of the hexagonal column obtained from peptide 1 and compound 2 are depicted in Fig. 5.18 The mass spectrum peak at m/z 855.4801, which was assigned to the ion [(peptide 1 + compound 2) + Na]+ shows the existence of the peptide 1 and compound 2 conjugate. The mass spectrum analysis of the unbranched fiber from peptide 1 and diimide 3 exhibits a peak at m/z 1514.2728, which was assigned to the ion [(2 peptide 1 + diimide 3) + 2K]+ that shows the existence of the peptide 1 and compound 3 conjugate (Fig. S10 in the ESI†).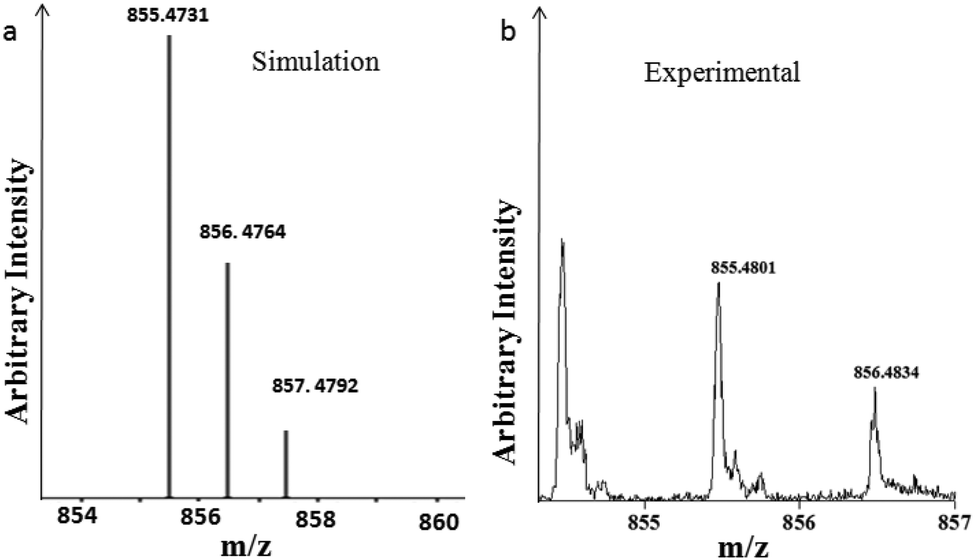 | ||
| Fig. 5 Mass spectrum of the hexagonal column obtained from peptide 1 and compound 2. (a) Simulated and (b) experimental isotopic patterns. | ||
Conclusions
We have presented a supramolecular helix from a tripeptide, which can interact with various hydrophobic guests. In organic solvents, the tripeptide 1 forms rigid type-III′ β-turn structure.In hierarchical association, the modular building blocks self-assemble in a helical manner to form supramolecular nanotubes with a hydrophobic core. The coumarin derivative 2 having a hydrophobic tail interacts with the nanotube by incorporating the hydrophobic tail inside the nanotube. However, the naphthalene diimide 3 having two hydrophobic tails interacts with the nanotube by incorporating its hydrophobic tails inside two nanotubes and thus develops a supramolecular polymer. Further the formation of supramolecular polymer has been confirmed with DLS, SEM and AFM and other spectroscopic techniques. The developed strategy could find important application in drug formulation and delivery, sensor technology and materials science.
Acknowledgements
This work is supported by CSIR, New Delhi, India (Project No. 02(0206)/14/EMR-II). This work is also supported by UGC, India (fellowship to A. Paikar) and CSIR, India (fellowship to A. Pramanik and T. Das).Notes and references
- (a) L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196 Search PubMed; (b) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 Search PubMed; (c) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 Search PubMed; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 Search PubMed; (e) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 Search PubMed; (f) C. Schmuck and H. Wennemers, in Highlights in Bioorganic Chemistry, Wiley-VCH Verlag GMBH, Weinheim, 2004 Search PubMed.
- (a) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240 Search PubMed; (b) Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521 Search PubMed; (c) W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Science, 2011, 334, 340 Search PubMed; (d) P. Cordier, F. Tournilhac, C. S. Ziakovic and L. Leibler, Nature, 2008, 451, 977 Search PubMed; (e) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 Search PubMed; (f) E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290 Search PubMed; (g) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414 Search PubMed.
- (a) S. Hecht and I. Huc, in Foldamers: structure, properties and applications, Wiley-VCH Verlag GMBH, Weinheim, 2007 Search PubMed; (b) P. Jana, S. Maity, S. K. Maity, P. K. Ghorai and D. Haldar, Soft Matter, 2012, 8, 5621 Search PubMed; (c) M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028 Search PubMed; (d) C. H. Görbitz, Chem. – Eur. J., 2007, 13, 1022 Search PubMed; (e) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- (a) A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 Search PubMed; (b) G. Crini, Chem. Rev., 2014, 114, 10940 Search PubMed; (c) G. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254 Search PubMed.
- (a) J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 Search PubMed; (b) K. Kim, Chem. Soc. Rev., 2002, 31, 96 Search PubMed.
- (a) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 Search PubMed; (b) J. Kang and J. Rebek, Nature, 1997, 385, 50 Search PubMed.
- R. Villalonga, R. Cao and A. Fragoso, Chem. Rev., 2007, 107, 3088 Search PubMed.
- (a) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 Search PubMed; (b) J. M. Haider and Z. Pikramenou, Chem. Soc. Rev., 2005, 34, 120 Search PubMed.
- (a) X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794 Search PubMed; (b) R. Haag, Angew. Chem., Int. Ed., 2004, 43, 278 Search PubMed; (c) C. J. Porter, N. L. Trevaskis and W. N. Charman, Nat. Rev. Drug Discovery, 2007, 6, 231 Search PubMed; (d) C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 235 Search PubMed; (e) F. Hirayama and K. Uekama, Adv. Drug Delivery Rev., 1999, 36, 125 Search PubMed.
- (a) D. Nishimura, T. Oshikiri, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, J. Org. Chem., 2008, 73, 2496 Search PubMed; (b) T. Oshikiri, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2005, 127, 12186 Search PubMed.
- (a) C. Bao, B. Kauffmann, Q. Gan, K. Srinivas, H. Jiang and I. Huc, Angew. Chem., Int. Ed., 2008, 47, 4153 Search PubMed; (b) H. P. Yi, C. Li, J. L. Hou, X. K. Jiang and Z. T. Li, Tetrahedron, 2005, 61, 7974 Search PubMed; (c) B. Prince, S. A. Barnes and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 2758 Search PubMed; (d) R. A. Dwek, Chem. Rev., 1996, 96, 683 Search PubMed; (e) H. Abe, N. Masuda, M. Waki and M. Inouye, J. Am. Chem. Soc., 2005, 127, 16189 Search PubMed; (f) C. Y. Tan, M. R. Pinto, M. E. Kose, I. Ghiviriga and K. S. Schanze, Adv. Mater., 2004, 16, 1208 Search PubMed.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172 Search PubMed.
- (a) P. Jana, S. Maity, S. K. Maity and D. Haldar, Chem. Commun., 2011, 47, 2092 Search PubMed; (b) A. K. Das, A. Banerjee, M. G. B. Drew, S. Ray, D. Haldar and A. Banerjee, Tetrahedron, 2005, 61, 5027 Search PubMed; (c) S. K. Maji, A. Banerjee, M. G. B. Drew, D. Haldar and A. Banerjee, Tetrahedron Lett., 2002, 43, 6759 Search PubMed.
- I. L. Karle, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 341 Search PubMed.
- C19H35N3O7, Mw = 417.50, hexagonal, space group P65, a = 19.7793(8), b = 19.7793(8), c = 11.2095(7) Å, V = 3797.8(4) Å3, Z = 6, dm = 1.095 Mg m−3, T = 100 K, R1 0.0591 and wR2 0.1625 for 4990 data with I > 2σ(I). Intensity data were collected with MoKα radiation using a Bruker APEX-2 CCD diffractometer. Data were processed using the Bruker SAINT package and the structure solution and refinement procedures were performed using SHELX9719 and deposited at CCDC with reference 1434362.
- V. Moretto, M. Crisma, G. M. Bonora, C. Toniolo, H. Balaram and P. Balaram, Macromolecules, 1989, 22, 2939 Search PubMed.
- S. K. Maji, R. Banerjee, D. Velmurugan, A. Razak, H. K. Fun and A. Banerjee, J. Org. Chem., 2002, 67, 633 Search PubMed.
- A. A. Nazarov, S. M. Meier, O. Zava, Y. N. Nosova, E. R. Milaeva, C. G. Hartinger and P. J. Dyson, Dalton Trans., 2015, 44, 3614 Search PubMed.
- G. M. Sheldrick, SHELX 97, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds 1–3, 1H NMR, 13C NMR, Fig. S1–S35. CCDC 1434362. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6py01955b |
| This journal is © The Royal Society of Chemistry 2017 |