
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resin and carbon foam production by cationic step-growth polymerization of organic carbonates†
L.
Wöckel
a,
A.
Seifert
a,
C.
Mende
b,
I.
Roth-Panke
b,
L.
Kroll
b and
S.
Spange
*a
aPolymer Chemistry, Technische Universität Chemnitz, 09107 Chemnitz, Germany. E-mail: stefan.spange@chemie.tu-chemnitz.de
bDepartment of Lightweight Structures and Polymer Technology, Technische Universität Chemnitz, 09107 Chemnitz, Germany
First published on 21st November 2016
Abstract
Acid induced step-growth polymerizations of bis(p-methoxybenzyl) carbonate (pMBC), bis(m-methoxybenzyl) carbonate (mMBC) and difurfuryl carbonate (DFC) have been performed to produce resin-foams, because controlled release of carbon dioxide takes place during polymerization of those organic carbonates. The monomers are polymerized in bulk using p-toluene sulfonic acid (pTS) as a catalyst. The volume development of the foams is assisted by use of an appropriate surfactant and the crosslinking agent 1,3,5-trioxane as co-components. A portion of carbon dioxide release is a function of the carbenium stability of the reactive intermediate derived from the monomer; DFC > pMBC ≫ mMBC. Resins derived from mMBC can be post-treated to release carbon dioxide after polymerization. The molecular structures of the resulting materials are investigated by solid state 13C-NMR spectroscopy and IR spectroscopy. Scanning electron microscopy was used to study foam morphology. The carbon dioxide release was monitored with TG-MS analysis. Finally, the polymer foams have been converted into carbon foams and investigated by means of mercury porosimetry.
Introduction
Polymer foams can be produced by physical, mechanical or chemical processing.1 Physical processes are suitable for foaming of thermoplastic polymers as polystyrene and polyethylene due to addition of blowing agents (e.g. easily volatile hydrocarbons). Mechanical foaming processes are performed by blowing the polymer with compressed air or carbon dioxide (CO2) gas. Polyurethane resins require chemical processes to develop gas (CO2) during the reaction of components. Another approach is the use of templates by emulsion due to surfactants which stabilize nitrogen gas bubbles during UV-initiated polymerization of styrene.2Isocyanate derived fire prevention foams are used for isolation in electrical systems.3,4 However, the substitution of isocyanates is of importance because of their toxicity. In this work, an alternative route for foam production by chemical processing is presented. Shape persistent polymer foams consisting of anisolic resins5,6 or polyfurfuryl alcohol7 (PFA) have a high thermal stability. Therefore, they have been chosen as suitable materials for potential application in fire prevention.8
Carbon dioxide release during formation of polyurethane foams is well established. Isocyanate groups react with traces of water and a decarboxylation reaction takes place starting from the carbamic acid intermediate (Scheme 1a).9,10Organic carbonic acid derivatives are promising candidates for cationic polymerization because they can be synthesized from benzylic alcohols and phosgene or other carbonic acid derivatives. To achieve foam formation among carbon dioxide release, the carbonic acid ester must be constructed in such a way that both, polymerization and carbon dioxide release, occur simultaneously. The idea for the structure of carbon dioxide-releasing monomers was inspired by the concept of twin polymerization of tetrafurfuryloxysilane (TFOS)11,12 (Scheme 2a). Silicon as an element was substituted by carbon. However, the 4-fold substituted orthocarbonate of furfuryl alcohol is still not reported and our attempts to synthesize this compound are not of success. Thus, the organic carbonic acid ester difurfurylcarbonate (DFC) (Scheme 2b) and additionally para- and meta-methoxybenzyl carbonates (pMBC, mMBC) (Scheme 2c) are used for this work.
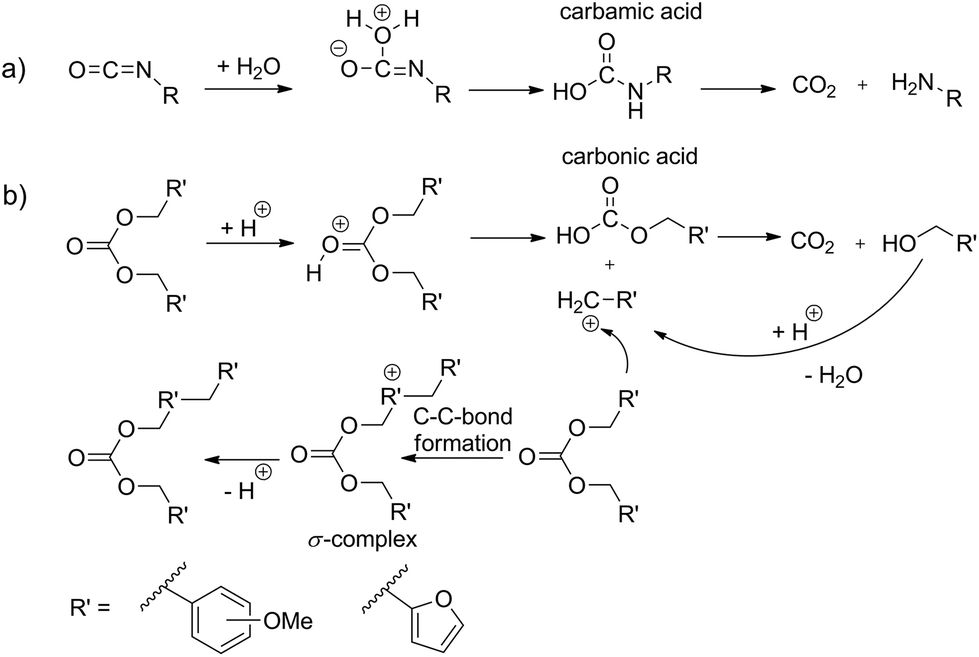 | ||
| Scheme 1 Comparison of (a) carbon dioxide release during reaction of isocyanate with water to (b) theoretically expected cationic step-growth polymerization of organic carbonates. | ||
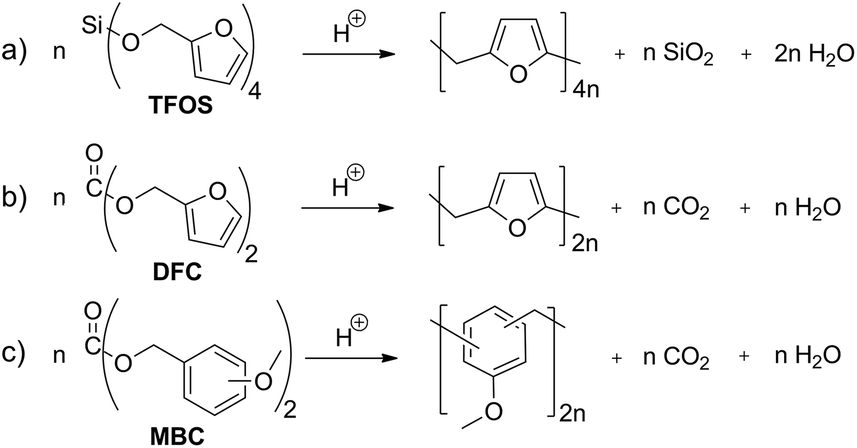 | ||
| Scheme 2 (a) Condensation twin polymerization of tetrafurfuryloxysilane (TFOS) and (b) condensation polymerization of difurfurylcarbonate (DFC) and (c) methoxybenzylcarbonate derivatives (MBC). | ||
The organic carbonates are synthesized by the transesterification reaction of diethyl carbonate with the respective alcohol (Scheme 3).
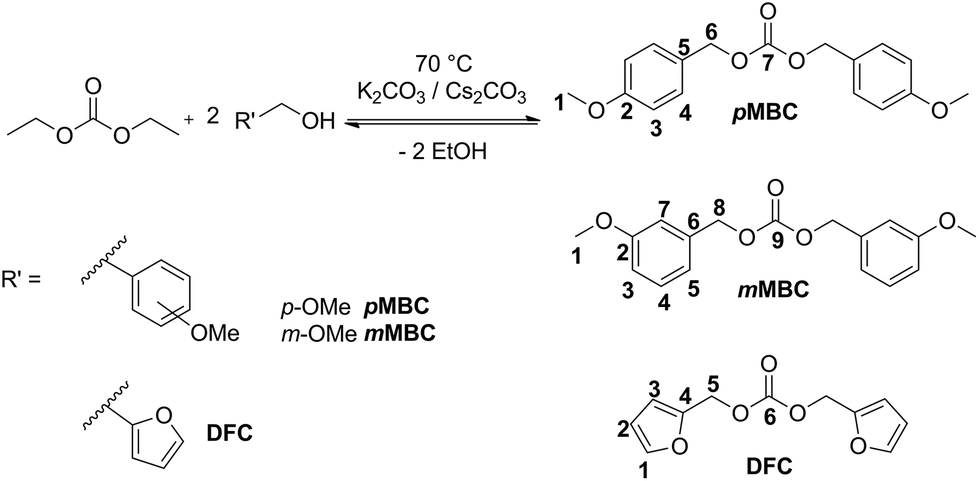 | ||
| Scheme 3 Synthesis of pMBC, mMBC and DFC from transesterification of diethyl carbonate and the respective alcohol. | ||
Basically, there are two scenarios to consider. The carbonate monomer can polymerize under the release of carbon dioxide or the carbonate moiety remains intact after polymerization. As well, both scenarios can occur simultaneously. If the carbonate moiety remains after polymerization, it is assumed that the carbon dioxide release can be triggered on demand afterwards. Theoretically, it is expected that acid catalyzed poly-condensation of carbonates proceeds via carbenium intermediates (Scheme 1b). Thus, the formation of stabilized carbenium ions should favor carbon dioxide elimination during the polymerization process (Scheme 1b). The furan ring or the methoxy substituent at the para position of the phenyl ring is well suited to stabilize benzylic carbenium ions.13,14 The increasing stabilizing effect of the substituent on carbocation stability is in accordance with the HAMMETT σ-constant which is −0.39 for 2-furanyl, −0.27 for p-OCH3 and +0.12 for m-OCH3 based on substituted benzoic acids.15–17 Therefore, it is expected that DFC and pMBC are suitable candidates as carbon dioxide sources for foam production at ambient temperature. The main objective of this study is to show the feasibility of this conception whether it is possible to deliver sufficient amount of carbon dioxide gas during resin fabrication. A crucial point is the fact that the alkylation reaction during polycondensation should also have an additional stabilizing effect on carbenium ion stability. An argument for this consideration is the significantly higher π-reactivity of the meta-substituted aromatics compared to that of the para-substituted ones. Thus the order of π-reactivity of the monomers DFC > mMBC > pMBC does not follow the corresponding carbenium stability of the intermediate derived from the same monomer.13,14
Theoretically, DFC and MBC, respectively, can produce per gram monomer 74 and 100 mL gaseous carbon dioxide if a complete conversion takes place. The objective of this study is the fabrication of foams containing anisolic5,6 and polyfurfuryl alcohol resin by acid-catalyzed polymerization and to study some basic mechanistic aspects using model reactions in solution.
Experimental
Materials and methods
Diethyl carbonate (>99%) and trifluoromethanesulfonic acid (95%) were purchased from Alfa Aesar. m-Methoxybenzyl alcohol (98%) and 1,3,5-trioxane (TO) (98%) were purchased from ABCR. p-Methoxybenzyl alcohol (98%) and caesium carbonate (Cs2CO3) (99.5%) were purchased from Acros. These reagents were used as received. Furfuryl alcohol (98%) was purchased from Merck and distilled before use. CDCl3 which was used for reaction monitoring was stored over molecular sieves. The dispersing agent was a silicone surfactant (S) named Dabco® DC 193 from Air Products that was kindly provided by BASF SE, Lemförde. The surfactant consists of a dimethylsiloxane backbone with trimethylsilyl end groups and methoxy-terminated ethylenoxide side chains as the hydrophilic part (Fig. 1).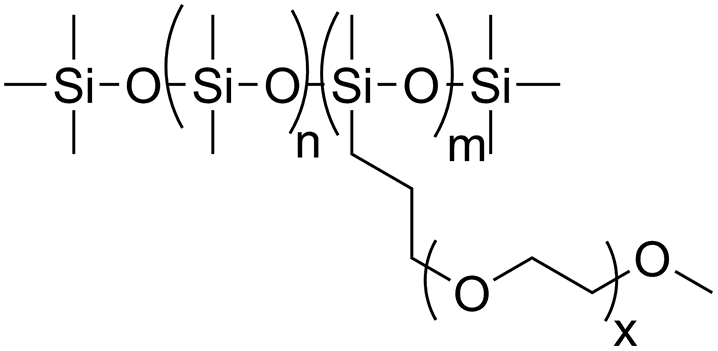 | ||
| Fig. 1 Molecular structure of the silicone polyether surfactant. | ||
Solution 1H– and 13C–{1H}-NMR experiments were performed on a Bruker DPX 250 NMR spectrometer. Deuterated chloroform (CDCl3) was used as a solvent and an internal standard (δH = 7.26 ppm; δC = 77.16 ppm).
The solid state 13C–{1H}-CP-MAS-NMR measurements were performed at 9.4 T on a Bruker Digital Avance 400 spectrometer equipped with double-tuned probes capable of MAS (magic angle spinning). A 3.2 mm standard zirconium oxide rotor was used spinning at 15 kHz. Cross polarization with a contact time of 3 ms was used to enhance sensitivity. The recycle delay was 5 s. The external standard was tetrakis(trimethylsilyl)silane (δH = 0.27 ppm; δC = 3.55 ppm) and 1H decoupling was performed with a TPPM pulse sequence.
ATR-FTIR spectra were measured using a FTS 165 spectrometer (BioRad) and a Golden Gate single ATR accessory from LOT-Oriel GmbH.
Quantitative elemental analysis was carried out with a Vario El from the company Elementar Analysensysteme GmbH.
Differential scanning calorimetry (DSC) measurements were carried out in a 40 μL aluminium crucible with a DSC 1 from Mettler Toledo under a nitrogen atmosphere (50 mL min−1). The heating rate was 10 K min−1.
Thermogravimetric (TG) analysis was performed with a thermogravimetric microbalance TG 209 F1 Iris® from NETZSCH. This device was coupled with a mass spectrometer (MS) MS 403C Aëolos®. The measurements were performed at a heating rate of 10 K min−1 under a helium atmosphere (50 mL min−1).
Size exclusion chromatography (SEC) measurements were performed with a PL-GPC 50 plus from Polymer Laboratories. An auto sampler PL-AS RT, a detector PC-RI and a column PLgel MIXED-D were used. The eluent was tetrahydrofuran (THF) at a flow rate of 1 mL min−1 at 40 °C.
Scanning electron microscopy (SEM) images were taken using a Nova NanoSEM 200 of FEI Company. The samples were sputtered with platinum before measurement.
The pores were studied at a low pressure with a Pascal 140 mercury porosimeter (Thermo Scientific). Afterwards, the pores were analysed at high pressure, utilising a Porosimeter 2000 (Fisons Instruments).
Synthetic procedures for pMBC, mMBC and DFC
DFC 18,19 and pMBC18 were prepared according to the literature, but the synthesis route was adapted for this work (Scheme 3). The target carbonates were synthesized in a transesterification reaction of diethyl carbonate with a two-fold excess of the respective alcohol with addition of potassium carbonate (K2CO3) or caesium carbonate (Cs2CO3).20–22 The reaction mixture was heated to 70 °C and ethanol was continuously removed by distillation in a vacuum. DFC is obtained as a slightly yellow liquid after distillation at 114 °C and 0.2 mbar.In the case of pMBC and mMBC Cs2CO3 was separated by washing with water. The organic products were extracted with ethyl acetate. All byproducts (methoxybenzyl alcohol and mono-substituted compound) were removed by distillation. The crude product remained in the residue. pMBC and mMBC were obtained after recrystallization with n-hexane as opaque solids with a melting point of 70 °C and 52 °C, respectively.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1609, 1512 (CC), 1456 (CH2), 789 (CH) cm−1. EA: Found: C, 67.54; H, 5.98. Calc. C17H18O5: C, 67.54; H, 6.00.
O), 1593(CC), 1456 (CH2), 791 (CH) cm−1. EA: Found: C, 67.75; H, 6.00. Calc. C17H18O5: C, 67.54; H, 6.00.
O), (CC), 1441 (CH2), 735 (CH) cm−1. EA: Found: C, 60.58; H, 4.64. Calc. C11H10O5: C, 59.46; H, 4.54.
O), 1609, 1512 (CC), 1456 (CH2), 789 (CH) cm−1. EA: Found: C, 67.54; H, 5.98. Calc. C17H18O5: C, 67.54; H, 6.00.
O), 1593(CC), 1456 (CH2), 791 (CH) cm−1. EA: Found: C, 67.75; H, 6.00. Calc. C17H18O5: C, 67.54; H, 6.00.
O), (CC), 1441 (CH2), 735 (CH) cm−1. EA: Found: C, 60.58; H, 4.64. Calc. C11H10O5: C, 59.46; H, 4.54.
Polymerization of pMBC, mMBC and DFC
The polymerization of carbonates in solution was monitored with 1H-NMR spectroscopy. Therefore, in an argon atmosphere 1 g carbonate and 0.1 g naphthalene as standards were dissolved in 1 mL CDCl3. The acid (tenth mol regarding the monomer) was mixed with 1 mL CDCl3 and added to the carbonate solution at room temperature. At different time intervals a NMR sample was taken from the reaction mixture.The polymerization in melt was performed in a Teflon vessel. 2.0 g (0.007 mol) of the monomer pMBC or mMBC was melted while heating until 70 °C and 100 °C, respectively. Afterwards, the vessel was removed from the heating bath. After five minutes 0.13 g (0.0007 mol) p-toluene sulfonic acid monohydrate (pTS) was added to the monomer melt under vigorous stirring. The Teflon vessel was removed from the heating bath after a homogenization time of two minutes.
In the case of pMBC a strong gas development at 70 °C takes place. The polymerization of pMBC was annealed at 70 °C and 150 °C each for 2 hours. For mMBC an annealing temperature of 100 °C and 150 °C was chosen each for 2 hours.
DFC is liquid at room temperature (RT). Thus, pTS can be added to the monomer at RT. For polymerization, 2.0 g (0.009 mol) DFC and 0.17 g (0.0009 mol) pTS were mixed under vigorous stirring. An immediate discoloration of slightly yellow to dark brown takes place. After two minutes, the Teflon vessel was moved to a 60 °C hot heating bath and annealed for four hours. The polymerization starts after two minutes as indicated by a strong gas evolution.
For polymerization with 1,3,5-trioxane (TO) as a crosslinking agent and/or a surfactant (S), 0.20 g (0.002 mol) TO and/or 0.02 g (1 wt%) S were mixed with the monomer. The polymerizations were also investigated at lower temperatures of 100 °C and 90 °C.
Post-condensation was performed by heating the sample up to 250 °C at a heating rate of 2 K min−1 under an argon atmosphere. The end temperature was maintained for two hours. For conversion of the polymer into a carbon material the sample was heated at 4.3 K min−1 to 800 °C and treated for 3 h at this temperature under an argon atmosphere.
The endorsements A and P of the labels indicate the performed annealing and pyrolysis process.
Results and discussion
The carbonates pMBC, mMBC and DFC were synthesized by the transesterification reaction of diethyl carbonate with the respective alcohol (Scheme 3) to avoid the use of toxic carbonyl dichloride. All organic carbonates were characterized with appropriate spectroscopic methods (see the Experimental part). DSC measurements of the carbonates with p-toluenesulfonic acid monohydrate (pTS) as an acid catalyst in a nitrogen atmosphere were performed to study the reaction behavior during polymerization (Fig. 2). DSC investigations show an exothermic peak for pMBC and mMBC at 66 °C and 89 °C, respectively indicating a higher reactivity for pMBC. DFC already reacts with pTS at 30 °C. The results were confirmed by TG-MS measurements (thermogravimetric analysis coupled with mass spectrometry) while adding pTS in a helium (He) atmosphere (Fig. 3). The TG curve of pMBC shows an onset temperature of 64 °C for polymerization and of 92 °C for mMBC. MS detects the ion current of the mass to charge ratio (m/z) of 44 and 18 which can be attributed to CO2 and water. DFC releases the carbon dioxide during polymerization over a broad temperature range (40–150 °C). A slow mass loss occurs above this temperature interval. In contrast to DFC, pMBC shows a fast CO2 displacement associated with a rapid mass loss in TG analysis. mMBC shows a slowly increasing ion current of CO2 beginning at 86 °C that reaches its maximum at 140 °C.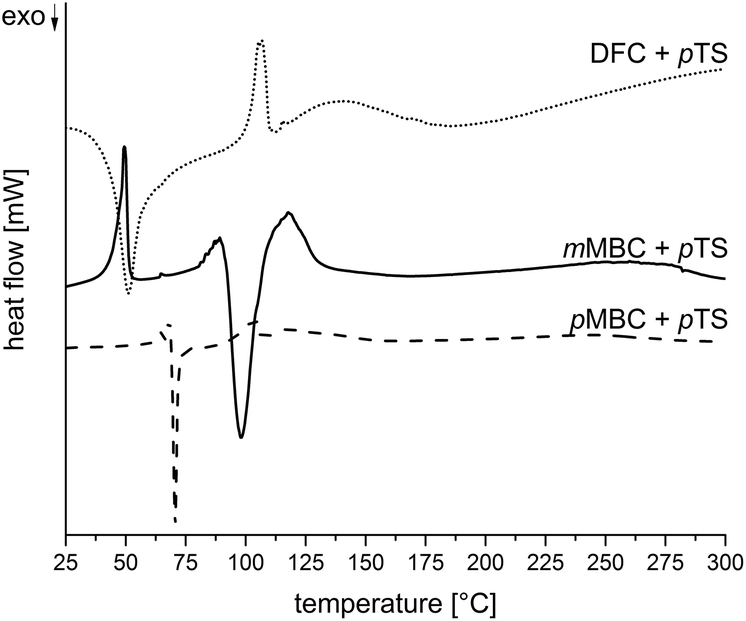 | ||
| Fig. 2 DSC curves of the carbonates pMBC, mMBC and DFC while adding pTS. | ||
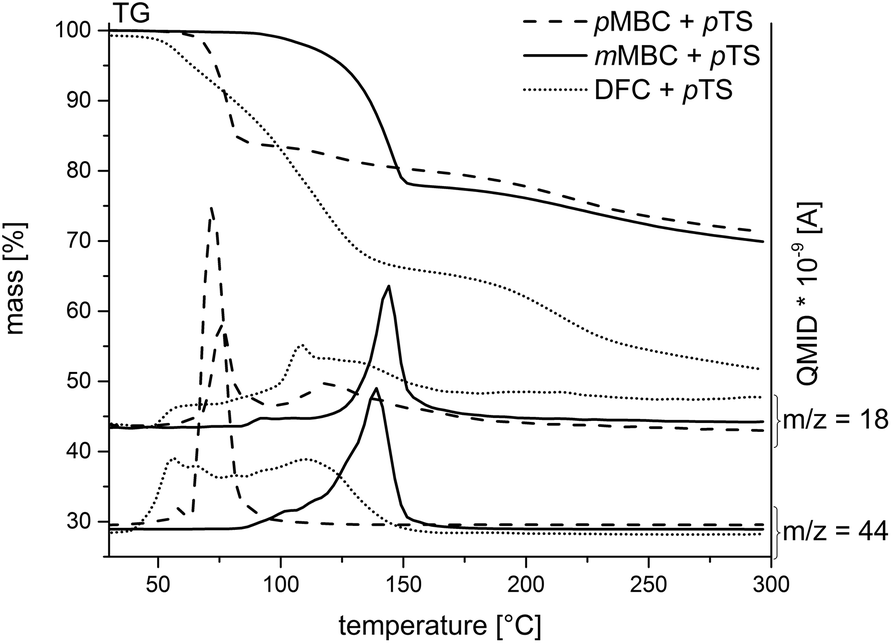 | ||
| Fig. 3 TG-MS curves of the carbonates pMBC, mMBC and DFC while adding pTS. The emission of CO2 (m/z = 44) and water (m/z = 18) was monitored with mass spectrometry. | ||
Thus, the results of DSC measurements are in accordance with the theoretically suggested stability of the carbenium intermediates. The more stable the carbenium intermediate, (DFC > pMBC ≫ mMBC), the lower the necessary trigger temperature for the polymerization and associated carbon dioxide release.
To study some basic aspects of the reaction mechanism, which was theoretically postulated in Scheme 1b, liquid 1H-NMR studies of the monomers by acid treatment in solution (CDCl3) were performed (Fig. S1, ESI†). The 1H-NMR spectra show a decrease of the carbonate concentration as a function of the reaction time while methylol signals (–CH2OH) between 4.4 and 4.7 ppm and the signals of methylene-bridges (–CH2–) (3.3 and 4.0 ppm) appear. NMR-spectra series are shown in Fig. S1, ESI.† Traces of methylether bridges (–CH2OCH2–) in the region of 4.4 and 4.9 ppm can be detected too. mMBC and pMBC conversion can be monitored using trifluoromethanesulfonic acid (TFMSA) as a catalyst (a tenth mol regarding to the amount of carbonate). DFC is too reactive to monitor polymerization induced with TFMSA by 1H-NMR spectroscopy. By dropping the acid dissolved in CDCl3 to the carbonate solution, an immediate precipitation of polyfurfurylalcohol resin takes place. Even after several days the mMBC conversion is incomplete whereas 60% of pMBC was converted after one minute. In this initial time mMBC behaves unreacted.
Water and carbon dioxide releases as by-products during the polymerization of carbonates have been evidenced (Fig. S1†). The water formed seems to have no effect on the course of the reaction, because of the good stability of these carbonates against water. Thus, p-toluenesulfonic acid monohydrate (pTS) can be promptly used as an acid catalyst.
The experimental conditions for the synthesis of the target resin materials are adjusted according to the results of DSC and TG-MS measurements. The three carbonates are polymerized in melt. Different additives such as surfactants and crosslinking agents were used to optimize the experimental parameters to achieve good quality of the foam. Experimental parameters are summarized in Table 1.
| Sample | Molar ratios [mol%] | Mass ratios [wt%] | Conditions T [°C]/t [h] | |
|---|---|---|---|---|
n
M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :npTS:nTO :npTS:nTO |
m
M:mS |
T 1 | T 2 | |
| 1TO – one additional methylene bridge per monomer. | ||||
| pMBC_150 | 1:0.1:0 |
— | 70/2 | 150/2 |
| mMBC_150 | 1:0.1:0 |
— | 100/2 | 150/2 |
| DFC_60 | 1:0.1:0 |
— | 60/4 | — |
| pMBC/1TO_150 | 1:0.1:0.3 |
— | 70/2 | 150/2 |
| mMBC/1TO_100 | 1:0.1:0.3 |
— | 100/4 | — |
| pMBC/1TO/S_150 | 1:0.1:0.3 |
1:0.01 |
70/2 | 150/2 |
| mMBC/1TO/S_150 | 1:0.1:0.3 |
1:0.01 |
100/2 | 150/2 |
| DFC/S_60 | 1:0.1:0 |
1:0.01 |
60/4 | — |
The obtained anisolic resins pMBC_150 (Table 1, Fig. 7a) and mMBC_150 (Table 1, Fig. 7d) are meltable when synthesized without the use of an additional crosslinking agent. The melting process can be seen in an endothermic peak in DSC curves in Fig. 8. Furthermore, they are soluble in polar solvents (CH2Cl2, CHCl3, tetrahydrofuran, acetone) which allows analysis by means of liquid 1H- and 13C-NMR spectroscopy and size exclusion chromatography (SEC). The results (Table 2, Fig. 4) show oligomers with a broad molecular weight distribution. The solubility can be explained by the fact that only one methylene-bridge per aromatic unit is present in the monomer. Therefore, only oligomers are formed (Table 2, Fig. 4). For this reason, 1,3,5-trioxane (TO) was added as a crosslinking agent to produce shape persistent foams. TO serves as an additional Friedel–Crafts reagent because it delivers –CH2–bridges which connect the oligomeric anisolic resin chains.
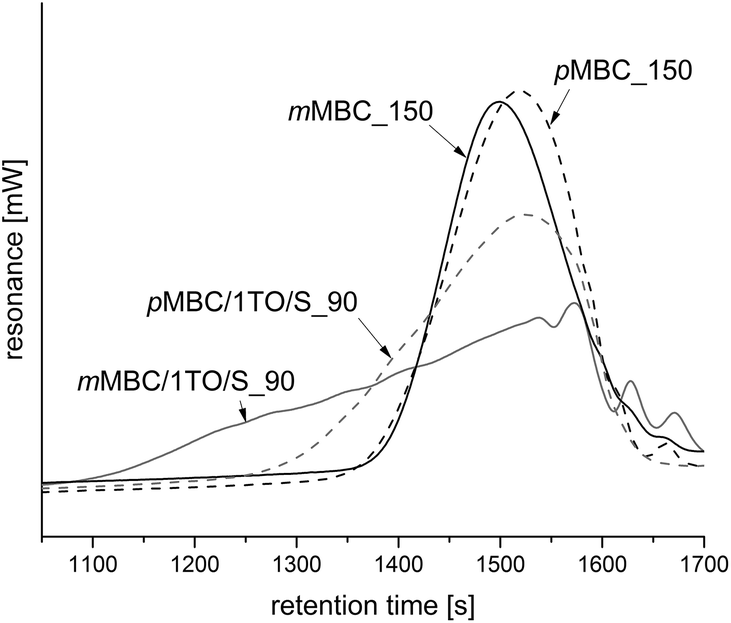 | ||
| Fig. 4 SEC profiles of the soluble anisolic resins. | ||
| Sample | M n [g mol−1] | M w [g mol−1] | PDI |
|---|---|---|---|
| pMBC_150 | 982 | 1838 | 1.87 |
| pMBC/1TO/S_90 | 1147 | 3286 | 2.86 |
| mMBC_150 | 1115 | 1874 | 1.68 |
| mMBC/1TO/S_90 | 2790 | 21205 |
7.60 |
Molecular structure formations during the crosslinking of pMBC and mMBC with TO are shown in Scheme 4. pMBC prefers the formation of carbenium ions during cationic catalysis due to the good stabilization effect of the methoxy group at the para-position. The p-methoxybenzyl carbenium ion can react in electrophilic substitution with an aromatic compound to form an anisolic resin under complete release of CO2 (Scheme 4a). In contrast, the m-methoxybenzyl carbenium ion shows low carbenium stability because the cation cannot readily be stabilized by a methoxy group at the meta-position. Thus, the formation of a carbenium ion intermediate from the carbonate moiety is inhibited in the first stage of cationic polymerization. In contrast to this fact, the mMBC shows a high reactivity for Friedel–Crafts alkylation. Thus, polymerizations of combination of mMBC with 1,3,5-trioxane yield crosslinked carbonate structures where the carbonate unit remains intact. Further annealing or acid treatment leads to the release of CO2 and the formation of methylene bridges (Scheme 4b). The subsequent release of carbon dioxide from the crosslinked mMBC resin samples is explainable by the fact that the introduced para- or ortho-alkyl substituents (Fig. 4b) improves carbenium ion stability compared to the only m-methoxy substituent previously present. This result is a strong support for the suggested mechanism and agrees with the results of the model reactions of the pure monomers when treated with an acid in solution.
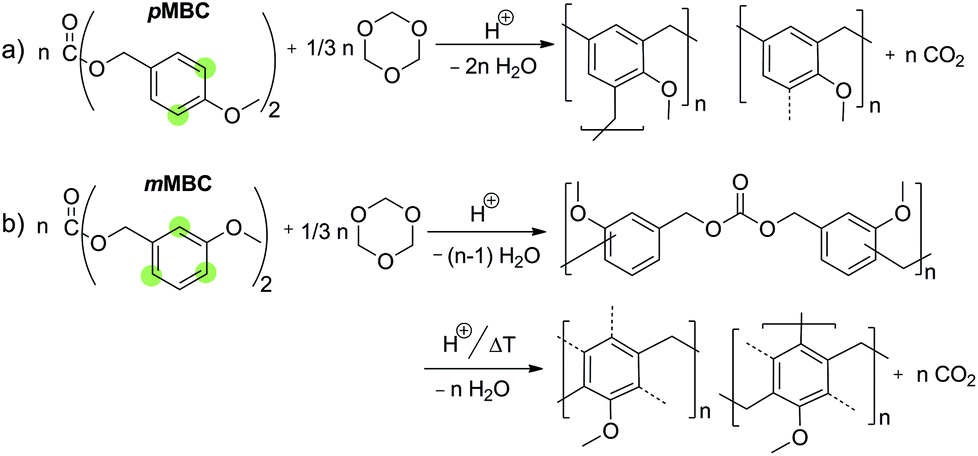 | ||
| Scheme 4 Acid induced step growth polymerization of (a) pMBC and (b) mMBC with addition of 1,3,5-trioxane as a curing agent. pMBC directly forms an anisolic resin network under the release of carbon dioxide, whereas mMBC is crosslinked while a portion of the carbonate units remains intact. Possible positions for electrophilic substitution are marked. | ||
Molecular structure investigations of the resins
Phenol formaldehyde resins (phenolic resins) have been well studied and established polymers for more than one century.23,24 Anisol derivatives can be likewise polymerized by a polycondensation reaction with formaldehyde or derivatives.5,6 Extensive investigations on molecular structure formation have not been reported in the literature, yet.The investigations of the anisolic resin molecular structures arising from cationic polymerization of pMBC and mMBC and combinations of these monomers with TO confirm the suggested reactions as postulated in Scheme 4. The molecular structures were proven by means of solid state 13C-NMR (Fig. 5c) and ATR-FTIR spectroscopy (Fig. S2, ESI†). mMBC has three positions for electrophilic substitution, two at the ortho- and an additional one at the para-position with regards to the methoxy group. The methylene group at the meta-position is predetermined from the monomer structure (Fig. 5b). Therefore, polymers from mMBC show more signals in the solid-state 13C–{1H}-CP-MAS spectra (Fig. 5c) in contrast to the spectra of pMBC polymers. Thus, two additional signals are observed. The signal C-7 (136–147 ppm) indicates a substituted aryl carbon atom at the meta-position. Signal C-6 (117–124 ppm) suggests an unsubstituted aryl carbon atom at the para-position to the methoxy group. In the case of polymers obtained from pMBC only one substitution pattern is possible (Fig. 5a), because one methylene group situated at the para-position to the methoxy group in the monomer is present.
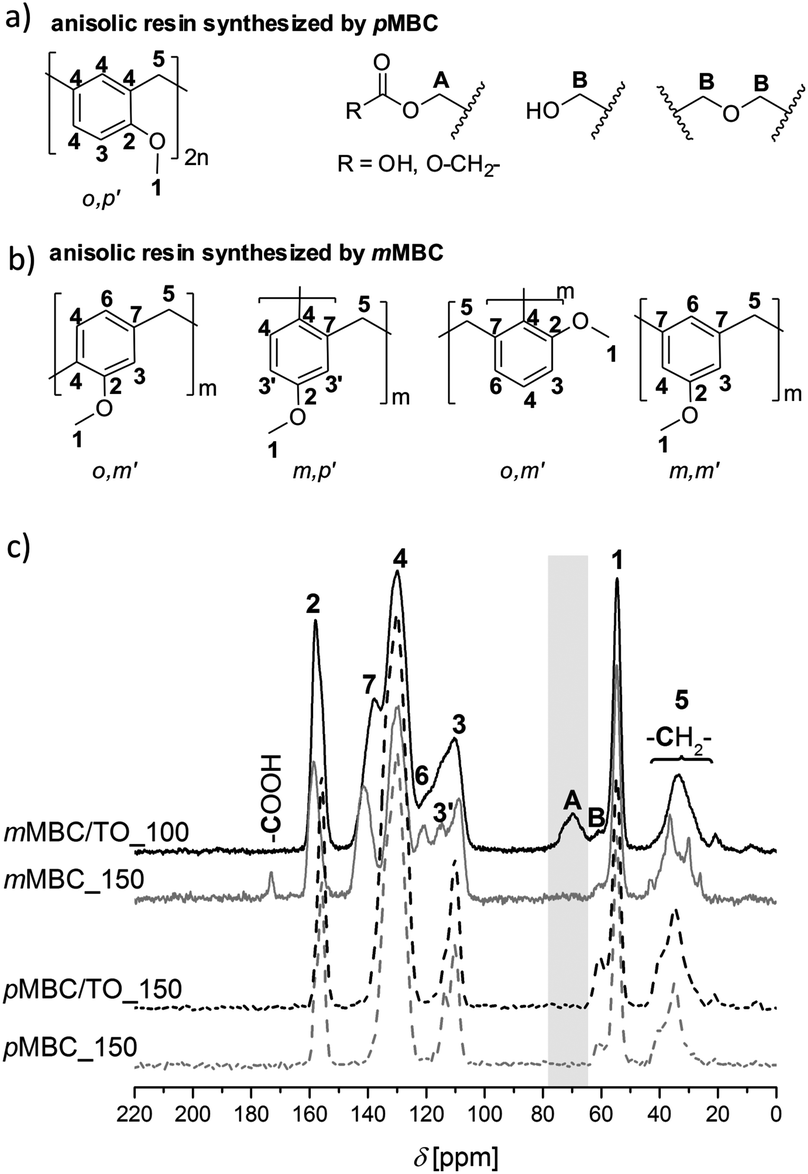 | ||
| Fig. 5 (a, b) Possible molecular structures and (c) solid-state 13C–{1H}-CP-MAS spectra of the anisolic resin obtained from pMBC (dashed lines) and mMBC (full lines) compared with the polymers crosslinked by TO. | ||
Furthermore, only one ortho connection is possible because a transformation of the o,p′-CH2- to o,o′-CH2- in phenolic resins takes place only at high temperatures under acidic conditions via quinone–methide intermediates.25 The anisolic resins (meta- and para-methoxy-substituted) show signals for the methylene (C-5), methylol (B) and methoxy group (C-1) between 29–42, 58–63 and 53–57 ppm, respectively (Fig. 5c). Because the NMR data of anisolic resins have not yet been reported in the literature, the positions of the signals were assigned by comparing with the 13C-NMR data of phenolic resins.26–28 In the solid state 13C–{1H}-CP-MAS-NMR spectrum (Fig. 5c) of sample mMBC/TO_100, a signal (A 64–77 ppm) for intact methylene bridges, which were bonded at the carbonate unit, is found. This is in accordance with the ATR-FTIR measurement (Fig. S2, ESI†) that the carbonate moiety in the polymer remains intact.
The cationic polymerizations were performed at 90 °C with trioxane (1TO – one additional methylene bridge per monomer) too. The obtained anisolic resins of mMBC and pMBC are meltable and soluble in polar organic solvents (CH2Cl2, CHCl3, tetrahydrofuran, acetone). The SEC profiles (Fig. 4) show a twice-as-high number average molecular mass (Mn = 2790 g mol−1) for mMBC/1TO_90 compared to pMBC/1TO_90 (Mn = 1147 g mol−1). The weight average molar mass (MW) of mMBC/1TO_90 exhibits a 6-fold higher value compared to pMBC/1TO_90. This resulted in a high polydispersity index (PDI) of 7.6 (Fig. 4) for mMBC/1TO_90 which shows that very different chain lengths with irregular molecular weights exist.
This result is consistent with the high possibility of mMBC to undergo cross-linking reactions due to the mesomeric structures. Furthermore, a strong carbonyl vibrational band in the ATR-FTIR spectrum (Fig. S3, ESI†) is found. In contrast, in pMBC/1TO_90 the carbonyl band (Fig. S3, ESI†) disappears completely. The liquid 13C–{1H}-NMR spectra (Fig. S6, ESI†) of the pre-polymerized organic carbonates mMBC and pMBC with TO at 90 °C exhibit the reaction behaviour more clearly. pMBC reacts completely during the formation of methylene bridges and release of CO2. The defined signals for the –CH2– carbon atoms of o,p′-, p,p′- and o,o′-linkage between aromatic units at 35.1, 40.2 and 30.2 ppm are found in the liquid 13C–{1H}-NMR spectrum (Fig. S6, ESI†), respectively. Furthermore, the signals for the –CH2– carbon atoms (93.6 ppm) of unreacted TO are found. The intact carbonyl carbon atom (C) in the 13C–{1H}-NMR spectra of mMBC/1TO_90 is overlaid with the signal of the C-2 carbon atom of the anisolic resin.
DFC shows an outstanding behavior, because it consists of reactive furfuryl moieties which additionally can undergo crosslinking at the methylene bridge (C-5-position) during polymerization in acidic media.29 This reaction is due to the fact that the furan ring stabilizes furfurylium ions in a significant manner. Thus, the carbenium ions derived from DFC prefer to abstract hydride ions from CH2-bridged furan ring chains. This is added to the –CH2 cation to form methyl-end groups. As a consequence of this behavior, conjugated polyfurfuryl alcohol structures are built. As well, the furan ring can be opened during cationic polymerization resulted in the formation of levulinic acid derivatives (Fig. S13a, ESI†).7 However, many other side reactions can occur by acid catalyzed furfuryl alcohol polymerization.7,26,29,30 The possible structures of polyfurfuryl alcohol are illustrated in Fig. 6. The polymer obtained by cationic polymerization of DFC shows the molecular structures of polyfurfuryl alcohol. Furthermore, a downfield 13C-NMR signal at 204 ppm relating to a carbonyl group and a carbonyl vibration in the ATR-FTIR spectrum (Fig. S2, ESI†) was found due to the formation of levulinic acid.7
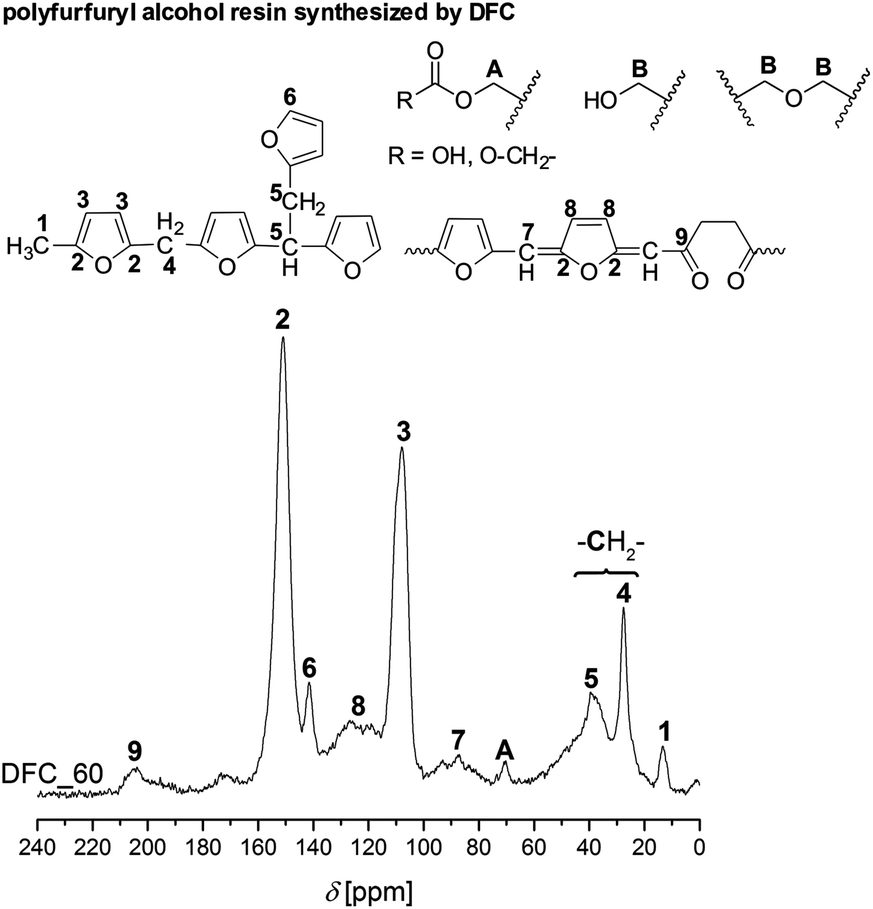 | ||
| Fig. 6 Solid-state 13C–{1H}-CP-MAS spectrum of the polyfurfuryl alcohol obtained from DFC and the assignment of the different carbon atoms to molecular structures moieties. | ||
Foam formation studies
DFC has been found to be the best carbonate monomer to achieve foams by the chemical foaming process (Fig. S7, ESI†). However, the foam collapses rapidly when DFC or the other organic carbonates are polymerized without any agent to stabilize the micro gas seeds when polymerization starts within the monomer melt. In order to achieve a high volume enlargement, the displaced carbon dioxide must be trapped as small bubbles in the reaction media. For this purpose a polydimethylsiloxane-based surfactant with methoxy-terminated ethylenoxide side chains (Fig. 1) was used as a dispersing agent while the polymerization mixture was vigorously stirred. Attempts to use other dispersing agents such as aerosil® (Ox50, 300) or polydimethyl siloxane (PDMS) give no satisfactory results (Fig. S8, ESI†). Nevertheless, the anisolic resins produced by this procedure show no high volume enlargement. Only mMBC/1TO/S_150 (Fig. 7f) has extended to 5-fold the volume of the monomer melt. A reason for this fact could be the increasing viscosity due to higher possibility for crosslinking in mMBC. Due to its reactivity and formation of side reactions, DFC shows the best results in foam formation (DFC/S_60Fig. 7h) even without using a crosslinking additive. A volume enlargement to 10-fold regarding the monomer volume can be observed. The SEM image of DFC/S_60 (Fig. 7h) shows a mixture of open- and closed-cell microstructure.1 | ||
| Fig. 7 SEM images of the polymerization products of (a) pMBC_150, (b) mMBC_150 and (c) DFC_60 in comparison to the polymers where 1,3,5-trioxane (TO) and/or a surfactant (S) was used. The inset shows the macroscopic image of the foams. The label of the picture indicates the used additive and reaction temperature. | ||
The problem during cationic polymerization of mMBC and pMBC seems to be the low viscosity of the mixture during polymerization and CO2 elimination. Therefore, the gas does escape while the polymers are not yet sufficiently solidified.
The thermal properties of the anisolic and polyfurfuryl alcohol resins were studied by DSC measurements (Fig. 8). It is conspicuous that the polymer mMBC/TO_100, which was synthesized at only 100 °C, exhibits a strong exothermic peak in DSC measurement (Fig. 8). A subsequent heating does release the chemical bonded CO2 which was proved with TG-MS measurement (Fig. S9, ESI†).
 | ||
| Fig. 8 DSC curves of anisolic and polyfurfuryl alcohol resins obtained by cationic polymerization of pMBC, mMBC and DFC. | ||
The TO quantity was varied in the ratios 1, 2, 4 and 6 regarding the methylene bridges per monomer unit (Table S2, Fig. S10, ESI†) in order to increase the viscosity of the mixture during the pre-polymerization of mMBC. However, a higher volume expansion cannot be achieved when a higher content of the crosslinking agent is used. In the ATR-FTIR spectra, increased carbonyl vibration bands from the 1 to 4 TO ratio occurred and a decrease in the extractable content with dichloromethane (DCM) (Table S2, ESI†) was observed. After extraction of soluble fractions, the carbonate structure remains in the crosslinked polymer. The extractable content and the thermal stability also show the good cross-linking ability of mMBC. During extraction with DCM the anisolic resin pMBC/4TO/S_100 loses 89.2 wt% of its mass, whereas mMBC/4TO/S_100 has a mass loss of only 12.9 wt% (Table S2, ESI†).
Additionally, the pyrolysis of pMBC/4TO/S_100 and mMBC/4TO/S_100 shows a mass loss of 37.3 wt% and 50.0 wt% (Table S2, ESI†), respectively. Moreover, a higher content of TO increases the thermal stability of mMBC polymers during pyrolysis. As well, it is found that the more intensive the carbonate vibration band (Fig. S11b†) the more carbonate structures are present in the polymer. This leads to a higher mass loss (Table S2, ESI†) during the annealing process at 250 °C (Ar) and TG analysis (Fig. S9, ESI†) at 300 °C (He). However, no additional volume enlargement was observed after annealing. In SEM images all samples show closed and open isolated spherical bubbles on the surface.1 For samples with 2 and 4 TO, areas with fine-porous structures (SEM Fig. S15, ESI†) are found. By addition of TO, the polymerization of pMBC at 100 °C (Table S2†) leads to a dense monolith (Fig. S10, ESI†) without any volume expansion and no carbonate vibration band in ATR-FTIR spectra (Fig. S11b, ESI†).
The polymerization of mMBC with 1 TO at 90 °C results in an increasing viscosity which inhibits the fast escape of the blowing gas in a thermal pre-annealing process at 250 °C. The obtained foam mMBC/1TO/S_90-A is shown in Fig. 9d. The SEM image of mMBC/1TO/S_90 (Fig. 9c) shows a flat structure. pMBC/1TO/S_90 (Fig. 9a) exhibits roundly shaped cavities in the SEM images due to CO2 elimination. After annealing of pMBC/1TO/S_90, flat structures were found, whereas mMBC/1TO/S_90-A (Fig. 9d) is foamed under formation of open- and closed-cell microstructures.1 The TG-MS measurements (Fig. 10) show for mMBC/1TO/S_90 a higher mass loss of 16.7 wt% in contrast to 9.9 wt% for pMBC/1TO/S_90. During these mass losses only mMBC/1TO/S_90 releases carbon dioxide which was monitored by MS measurement (Fig. 10, m/z = 44). The mass loss of pMBC/1TO/S_90 is due to the release of carbon monoxide and water regarding the thermal degradation of the polymer.31
 | ||
| Fig. 9 Pictures and SEM images of the anisolic resins (a) mMBC/1TO_90 and (c) pMBC/1TO_90 produced at 90 °C with 1,3,5-trioxane. The polymers were annealed (A) at 250 °C in a second step. (d) mMBC/1TO_90-A shows a volume enlargement and a change of its morphology from a flat surface to open- and closed-cell microstructures. | ||
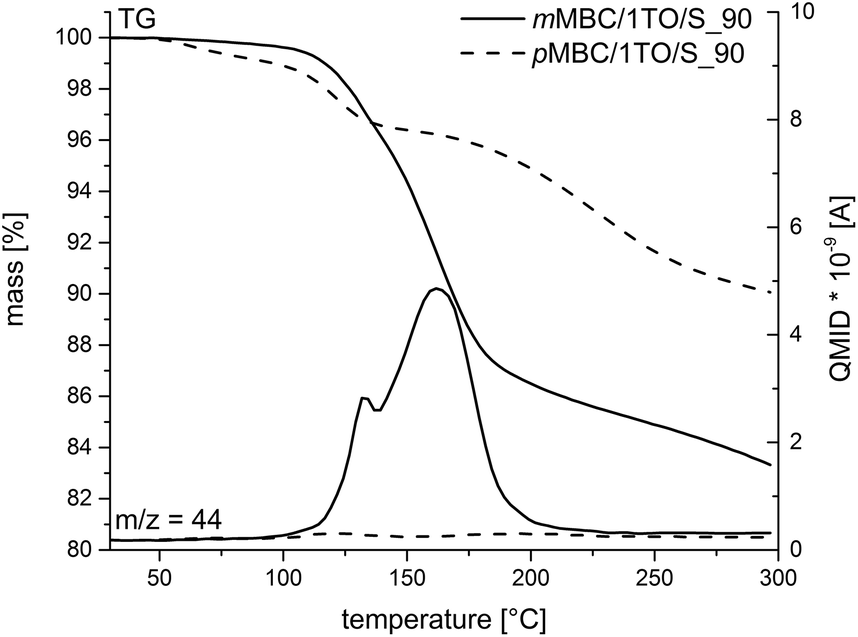 | ||
| Fig. 10 TG-MS measurements of anisolic resins obtained by cationic polymerization of pMBC and mMBC at 90 °C. The emission of CO2 (m/z = 44) was monitored with mass spectrometry (MS). | ||
Phenolic and polyfurfuryl alcohol resins are frequently used as carbon precursors.32–34 Porous carbon materials play an important role in applications such as separation, adsorption or as catalyst support materials.35–37 Thus, the polymer foams were converted into carbon foams by pyrolysis at 800 °C in an Ar atmosphere.
In order to determine the porosity and pore volume of the material, two selected carbon foams were investigated by means of mercury porosimetry (Table 3, Fig. S16, ESI†).38,39DFC/S_60-P has a high porosity of 87% and high pore volume, whereas mMBC/1TO/S_90A_P has only a porosity of 21% (Table 3). Both carbon foams have irregular pore sizes (Fig. S11, ESI†), but DFC/S_60-P shows smaller average pore diameters of 77 μm (Table 3). With nitrogen sorption measurements only very low specific surface areas could be measured.40 Thus, no micro- and meso-pores are present in the carbon foams.
| Sample | V total [cm3 g−1] | ρ bulk [g cm−3] | ρ apparent [g cm−3] | ε [%] | d pore [μm] |
|---|---|---|---|---|---|
| DFC/S_60-P | 5.1 | 0.17 | 1.4 | 87 | 77 |
| mMBC/1TO/S_90A-P | 0.2 | 1.1 | 1.4 | 21 | 117 |
Conclusions
The cationic step-growth polymerization of organic carbonates derived from furfuryl alcohol and methoxy-substituted benzyl alcohols has been presented as a suitable method to produce foamy resins by chemical processing. The gaseous carbon dioxide release in melt is the tool to obtain foams directly during polymerization. para-Toluenesulfonic acid is a suitable catalyst when dissolved in the monomer melt at a temperature which is lower as needed for the polymerization process. Both, polymerization and foam formation take place simultaneously by appropriate heating. Importantly, the use of an appropriate surfactant is essential to reproducibly produce foams of high volume expansion.It is striking that the process of carbon dioxide release is a function of the carbenium ion stability of the reactive monomer fragment. The better the carbenium ion is stabilized by the aryl substituent, the more efficient is the carbon dioxide release directly during the polymerization. Thus, the associated foam formation enhances in the order of mMBC < pMBC < DFC. DFC is found to be the best monomer for this purpose. The m-methoxy substituent is less suitable to stabilize a carbenium ion, but it improves the reactivity of the aromatic ring. mMBC readily undergoes Friedel–Crafts polymerization with 1,3,5-trioxane under maintenance of the carbonate moiety. This type of resin is suitable as a polymeric precursor to trigger foam formation by a post-thermal treatment. Future work is suggested to use these carbonate monomers in simultaneous polymerization with twin monomers to fabricate inorganic–organic hybrid foams which can be converted into micro- and meso-porous carbon foams.
Acknowledgements
Financial support by DFG Sp 392/38-1, DFG Sp 392/35-2, DFG FG 1497 and EXC 1075 is gratefully acknowledged. We thank Dr Ebling, BASF AG, Lemförde, for helpful suggestion and discussion. The surfactant was provided by BASF AG Lemförde. An additional acknowledgment goes to Dr E. Dietzsch for support during mercury porosimetry measurements and Prof. Dr Hietschold for the opportunity to measure SEM.Notes and references
- N. Mills, Polymer Foams Handbook: Engineering and Biomechanics Applications and Design Guide, Butterworth-Heinemann, 2007. ISBN: 9780080973883 Search PubMed.
- F. Schüler, D. Schamel, A. Salonen, W. Drenckhan, M. D. Gilchrist and C. Stubenrauch, Angew. Chem., Int. Ed., 2012, 124, 2256–2260 CrossRef.
- S. V. Levchik and E. D. Weil, Polym. Int., 2004, 53, 1585–1610 CrossRef CAS.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem. Int. Ed., 2013, 125, 9596–9616 CrossRef.
- K. G. Shah, D. H. Desai and B. N. Mankad, Angew. Makromol. Chem., 1973, 33, 177–190 CrossRef CAS.
- K. G. Shah and B. N. Mankad, Angew. Makromol. Chem., 1973, 32, 1–15 CrossRef CAS.
- A. M. Nathanael Guigo, Polym. Degrad. Stab., 2009, 94, 908–913 CrossRef.
- Y. Ma, J. Wang, Y. Xu, C. Wang and F. Chu, J. Therm. Anal. Calorim., 2013, 114, 1143–1151 CrossRef CAS.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- O. Bayer, Angew. Chem., 1947, 59, 257–272 CrossRef.
- S. Grund, P. Kempe, G. Baumann, A. Seifert and S. Spange, Angew. Chem., Int. Ed., 2007, 119, 636–640 CrossRef.
- T. Ebert, A. Seifert and S. Spange, Macromol. Rapid Commun., 2015, 36, 1623–1639 CrossRef CAS PubMed.
- H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remennikov and H. Schimmel, J. Am. Chem. Soc., 2001, 123, 9500–9512 CrossRef CAS PubMed.
- R. Taylor, Electrophilic Aromatic Substitution, Wiley, Chichester, 1990. ISBN 0-471-92482-2 Search PubMed.
- O. Exner and S. Böhm, J. Org. Chem., 2002, 67, 6320–6327 CrossRef CAS PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- Y. Yamazaki, K. Kakuma, Y. Du and S. Saito, Tetrahedron, 2010, 66, 9675–9680 CrossRef CAS.
- R. Griscom, DE00000244438, 1974 Search PubMed.
- J. Barry, G. Bram and A. Petit, Tetrahedron Lett., 1988, 29, 4567–4568 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- P. Kempe, Diplomarbeit, Kationische Polymerisation neuer Furan-Monomere, Technische Universität Chemnitz, 2006 Search PubMed.
- K. Hultzsch, Chemie der Phenolharze, Springer Verlag, 1950 Search PubMed.
- A. Gardziella, L. A. Pilato and A. Knop, Phenolic resins: Chemistry, Application, Standardization, Safety and Ecology, Springer, 2nd Completely Revised edn, 2000 Search PubMed.
- S. F. Wanat, J. Micro/Nanolithogr., MEMS, MOEMS, 2008, 7, 033008 CrossRef.
- I. S. Chuang and G. E. Maciel, Annu. Rep. NMR Spectrosc., 1994, 29, 169–286 CrossRef CAS.
- M.-F. Grenier-Loustalot, S. Larroque and P. Grenier, Polymer, 1996, 37, 639–650 CrossRef CAS.
- R. Rego, P. J. Adriaensens, R. A. Carleer and J. M. Gelan, Polymer, 2004, 45, 33–38 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839–3850 CrossRef CAS.
- A. Gandini, Prog. Polym. Sci., 1997, 22, 1203–1379 CrossRef CAS.
- R. T. Conley and J. F. Bieron, J. Appl. Polym. Sci., 1963, 7, 103–117 CrossRef CAS.
- Z. Yuan, Y. Zhang, Y. Zhou and J. Han, Mater. Chem. Phys., 2014, 143, 707–712 CrossRef CAS.
- Y. X. Wang, S. H. Tan, D. L. Jiang and X. Y. Zhang, Carbon, 2003, 41, 2065–2072 CrossRef CAS.
- X. Zhang and D. H. Solomon, Chem. Mater., 1999, 11, 384–391 CrossRef CAS.
- T. A. Centeno and A. B. Fuertes, J. Membr. Sci., 1999, 160, 201–211 CrossRef CAS.
- S. Farhan, R.-M. Wang, H. Jiang and N. Ul-Haq, J. Anal. Appl. Pyrolysis, 2014, 110, 229–234 CrossRef CAS.
- A. K. Roy, M. Zhong, M. G. Schwab, A. Binder, S. S. Venkataraman and Ž. Tomović, ACS Appl. Mater. Interfaces, 2016, 8, 7343–7348 CAS.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CrossRef CAS.
- H. Giesche, Part. Part. Syst. Charact., 2006, 23, 9–19 CrossRef.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01572g |
| This journal is © The Royal Society of Chemistry 2017 |