
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal syntheses of all tri-oxygenated 16-phytoprostane classes via a common precursor constructed by oxidative cyclization and alkyl–alkyl coupling reactions as the key steps†
Jakub
Smrček
,
Radek
Pohl
and
Ullrich
Jahn
 *
*
Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo náměstí 2, 16610 Prague 6, Czech Republic. E-mail: jahn@uochb.cas.cz
First published on 24th October 2017
Abstract
A unified strategy for the total synthesis of the methyl esters of all phytoprostane (PhytoP) classes bearing two ring-oxygen atoms based on an orthogonally protected common precursor is described. Racemic 16-F1t-, 16-E1-PhytoP and their C-16 epimers, which also occur as racemates in Nature, were successfully obtained. The first total synthesis of very sensitive 16-D1t-PhytoP succeeded, however, it quickly isomerized to more stable, but so far also unknown Δ13-16-D1t-PhytoP, which may serve as a more reliable biomarker for D-type PhytoP. The dioxygenated cyclopentane ring carrying the ω-chain with the oxygen functionality in the 16-position was approached by a radical oxidative cyclization mediated by ferrocenium hexafluorophosphate and TEMPO. The α-chain was introduced by a new copper-catalyzed alkyl–alkyl coupling of a 6-heptenyl Grignard reagent with a functionalized cyclopentylmethyl triflate.
Introduction
Oxidative stress is one of the important and intensely studied topics in current medicine and life sciences. It describes a situation, where the redox equilibrium of a cell and its environment is imbalanced in favor of oxidants and by which reactive oxygen species (ROS) are unspecifically generated. This excess of ROS stabilizes by reaction with biologically important primary metabolites such as nucleic acids, proteins or lipids thus abrogating their functions.1,2 Clear evidence has been gathered that increased levels of oxidative stress are related to many human diseases.Prostaglandins (PGs) are enzymatically formed by a radical reaction cascade as single enantiomers from the most prominent polyunsaturated fatty acid (PUFA) arachidonic acid regulating a wide range of biological processes (Scheme 1).3–5 However all PUFAs are essential parts of the cell membrane and react with ROS by radical reactions including peroxidation as well as fragmentation or cyclization cascades.6–9 Cyclic products of these autoxidative transformations are structural analogs of PGs. The best known and studied representatives are isoprostanes (IsoPs) produced from arachidonic (C20:4 ω-6)6,10 or eicosapentaenoic (C20:5 ω-3)11,12 acids in animals and humans. Recently metabolites of docosahexaenoic acid (C22:6 ω-3), named neuroprostanes (NeuroPs), were also discovered in the human brain.13 A number of biological activities of IsoPs and NeuroPs were reported.14
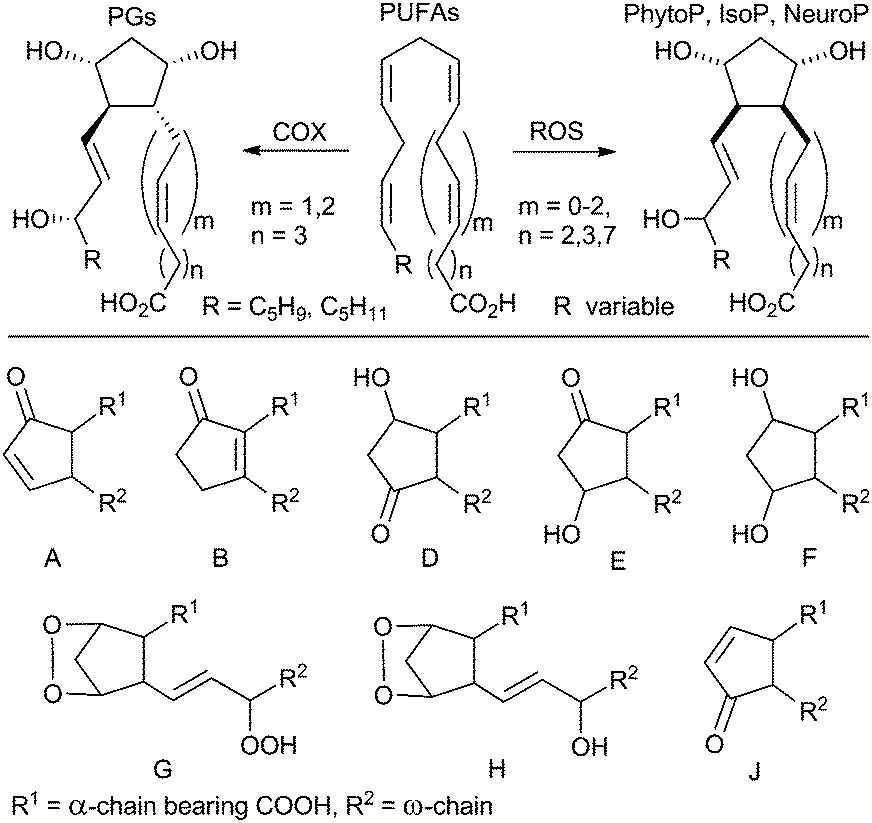 | ||
| Scheme 1 Formation of cyclic PUFA metabolites and classes based on their ring substitution. COX = cyclooxygenase for arachidonic acid, ROS = reactive oxygen species. | ||
In the plant kingdom phytoprostanes (PhytoPs) were found as metabolites of α-linolenic acid (C18:3 ω-3).8 PhytoPs are assumed to play an essential role in the protection and detoxification of plant cells and serve as a part of their oxidative injury-sensing system.15–17 A living cell is not required for their formation and the level and composition of PhytoP change during the processing of plant-originated raw materials in the food industry.18 Thus, they are recommended as suitable biomarkers of oxidative degradation in plant-derived edibles.19–26
The structural similarity of PhytoPs to PGs and IsoPs suggests possible bioactivities in humans and animals. It was demonstrated that the level of PhytoPs in the human body depends on the diet and also increases after a high consumption of α-linolenic acid.27,28 However, only a few studies have been published in this field. Traidl-Hoffmann et al. reported that E1- and F1-PhytoPs act similarly to endogenous PGs and modulate human dendritic cell function and T-cell polarization.29,30 The role of E1-PhytoP in pollen-caused allergies was subsequently studied in more detail.31,32
Because of the radical nature of their formation, PhytoPs occur in vivo as racemic mixtures of hardly separable regio- and diastereomers and the natural material is therefore not useful for a thorough study of their biological effects. The total synthesis of individual stereoisomers is the only appropriate way to provide a material for the study of their biological activity. Since the discovery of PhytoPs in 1998![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 more than ten total syntheses of individual PhytoPs have been published.6,33,34
8 more than ten total syntheses of individual PhytoPs have been published.6,33,34
Diversity oriented syntheses of PhytoPs are especially attractive for many biological investigations. So far, such approaches aimed at accessing the different diastereomers or side chain isomers for a given ring substitution pattern. For example, all eight diastereomers of 16-F1t and 9-F1t PhytoPs were published by Durand et al.,35,36 which is based on a radical 5-exo cyclization of a sugar-based precursor.37 An analogous approach was recently applied to the total syntheses of B1-PhytoPs38,39 and 9-D1t-PhytoP.40 An individual total synthesis of 16-E1-PhytoP was also published, based on the Friedel–Crafts acylation of furan, followed by a Piancatelli rearrangement and attachment of the ω-chain by 1,4-conjugate addition after enzymatic resolution.41 The total syntheses of ring-isomeric cyclopentenone PhytoP, namely 9- and 16-B1- and L1-PhytoP as well as 9-A1- and 9-J1-PhytoP, were accomplished by the Vidari–Zanoni group,42,43 whereas Riera's group developed an individual approach to 16-B1- and 9-L1-PhytoPs.44 A total synthesis of 16-D1t-PhytoP was never accomplished. The only evidence for its occurrence stems from the transformation of a mixture of all possible F1-PhytoP isomers based on a procedure originally developed for an access to PGD2.45 A 3:1 mixture of D1- to E1-PhytoP isomers was obtained, which was not further separated and identified.46 This further highlights the need for selective approaches to these natural products for meaningful biological investigations. Considering that the diversity of biological activities of cyclic PUFA metabolites rather rests on the differential substitution patterns at the cyclopentane ring than on the side chain structure, we hypothesized that a unified approach allowing a simple simultaneous access to most possible ring oxygenation patterns of PhytoP may constitute a more promising and affordable approach than designing individual strategies to them (vide supra). Another benefit of such a unified approach is the possibility of attaching different types of α-chains to the same ω-chain-containing cyclic unit,47,48 thus forming PhytoPs, IsoPs or NeuroPs with minimal effort.
We report here the total syntheses of 16-F1t-, 16-E1- and 16-D1t-PhytoP and provide evidence that 16-D1t-PhytoP will probably hardly be detectable in biological samples because of its facile isomerization to the so far unknown Δ13-16-D1t-PhytoP, which represents therefore a potentially better observable metabolite in biological materials.
Results and discussion
The retrosynthesis of 16-F1t-, 16-E1- and 16-D1t-PhytoPs 1–3 is based on common precursor 4, which can be easily transformed to all target compounds by simple deprotection, protection and oxidation reactions (Scheme 2). This orthogonally protected PhytoP structure 4 was envisaged to be obtained by copper(I)-catalyzed C(sp3)–C(sp3) coupling49 of a suitable α-chain precursor and the substituted cyclopentane core 5. An oxidative radical anion cyclization using ferrocenium hexafluorophosphate 6 and the stable radical 2,2,6,6-tetramethylpiperidine N-oxyl (TEMPO) 7 paves the way for construction of the central cyclic structure 5. Cyclization substrate 8 should be easily prepared from commercially available (E,E)-2,4-heptadienal (9) and methyl acetoacetate (10).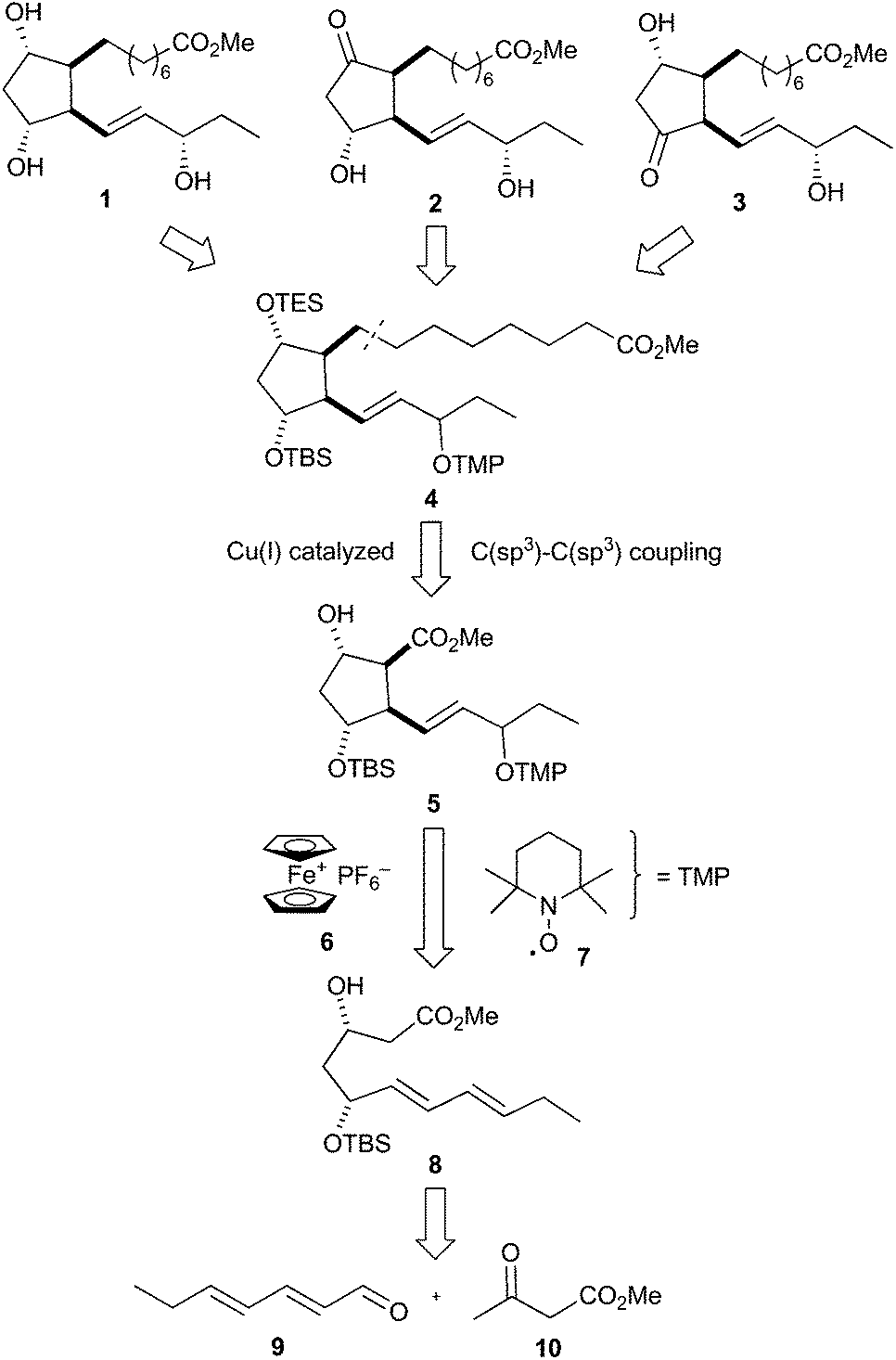 | ||
| Scheme 2 Retrosynthetic analysis of F1t-, E1- and D1t-PhytoP. | ||
The total syntheses commenced with the preparation of cyclization precursor 8 in analogy to published procedures in three steps (Scheme 3).47,48 The vinylogous aldol addition of heptadienal 9 and acetoacetate 10 had to be performed with a somewhat larger excess of bases (1.3 equiv. NaH, 1.2 equiv. BuLi) than previously. Reduction of the carbonyl group at C-3 of hydroxy keto ester 11 proceeded with good yield and exclusive syn selectivity providing diol 12. The formation of monoprotected diol 8 was accomplished with equimolar amounts of tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) in the presence of 2,6-lutidine at low temperature.
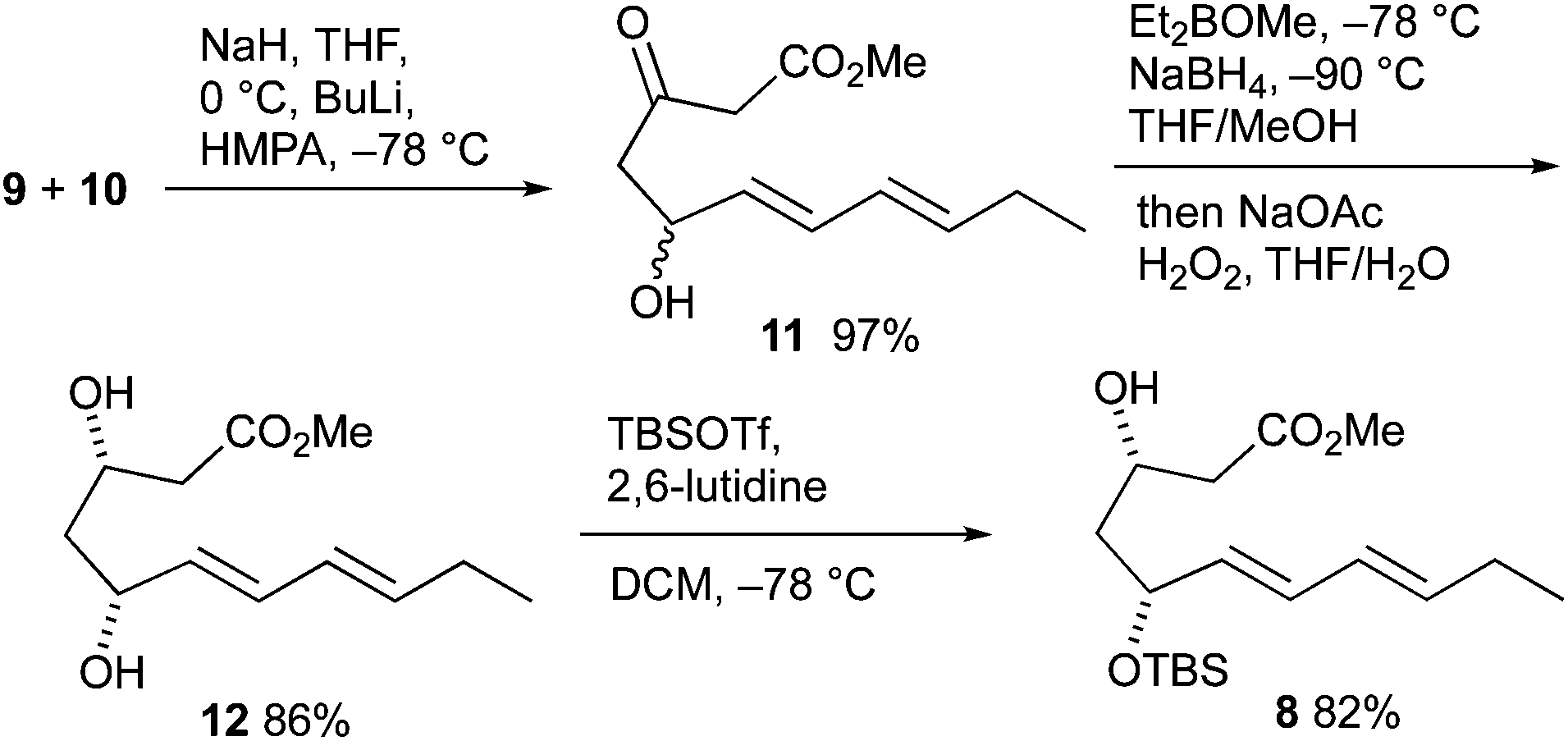 | ||
| Scheme 3 Preparation of cyclization precursor 8. | ||
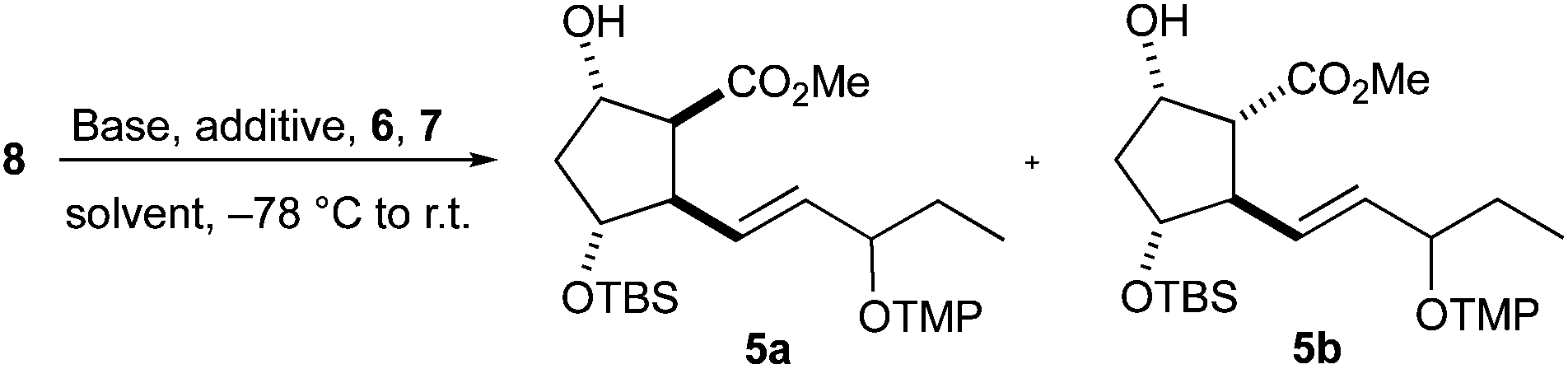
The tandem radical cyclization/oxygenation was performed by deprotonating substrate 8 with an excess of base and the resulting 1,3-enediolate was subjected to single electron transfer (SET) oxidation, cyclization and oxygenation by treatment with ferrocenium hexafluorophosphate 6 and TEMPO 7. Applying these conditions to the cyclization of 8 gave cyclopentanes 5 in 49% and 47% yield, respectively (Table 1, entries 1 and 2). The diastereomeric ratio of 5a and 5b is dependent on the applied base. The formation of PG-like structure 5b is predominant when the deprotonation was caused by the stronger chelating magnesium species (Table 1, entry 2). Decreasing the chelating ability of magnesium by the addition of HMPA prior to deprotonation led to a reversed ratio being similar to that with LDA (Table 1, entry 3 vs. 1). Pentylzinc bromide induced an increase in the 5a/5b ratio, but low conversion was observed when the deprotonation was conducted at −78 °C (Table 1, entry 4). The conversion was better at higher temperature, but the stereoselectivity dropped (Table 1, entry 5). A noticeable improvement was observed when DME was used as the solvent and the amount of HMPA was increased (Table 1, entries 8 and 9). Other attempts to affect the reaction were unsuccessful. A recently developed catalytic version of the cyclization50 proceeded with low yield and diastereoselectivity (not shown). Transmetalation of the dilithium 1,3-enediolate using Ti(OiPr)4, Al(OiPr)3 or B(OMe)3 resulted in no cyclization, but β-elimination and transesterification products were identified. No reaction occurred and 8 was recovered when 12-crown-4 was used as an additive (not shown).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Additivea (equiv.) | Solvent | 5 (%) |
a:bb |
| a LiCl is always 6 equiv. based on 8. b dr determined from the 1H NMR spectra of an inseparable mixture. c HMPA added prior to deprotonation. d Deprotonation at −78 °C. e Deprotonation at 25 °C. | |||||
| 1 | LDA | LiCl, HMPA (6) | THF | 49 | 1.6:1 |
| 2 | tBuMgCl, LDA | HMPA (6) | THF | 47 | 1:5 |
| 3 | tBuMgCl, LDA | HMPA (6)c | THF | 30 | 1.3:1 |
| 4 | PentZnBr,d LDA | HMPA (6) | THF | 16 | 3:1 |
| 5 | PentZnBr,e LDA | HMPA (6) | THF | 42 | 1.5:1 |
| 6 | LDA | LiCl, HMPA (12) | THF | 48 | 1.5:1 |
| 7 | LDA | LiCl, TMEDA (9) | THF | 51 | 1.2:1 |
| 8 | LDA | LiCl, HMPA (6) | DME | 55 | 1.6:1 |
| 9 | LDA | LiCl, HMPA (12) | DME | 64 | 2:1 |
The significant effect of the metal cation can be traced to differentially strong chelation during the course of the cyclization (Scheme 4). Both, the dilithium and the magnesium 1,3-enediolate I, are likely strongly chelated, however on SET oxidation the resulting radical anions behave significantly different. The lithium radical anion apparently equilibrates between the open form IIo and the chelated form IIc prior to cyclization, so that a mixture of cyclic radicals III and IV with low diastereoselectivity results, which stabilizes by oxygenation with 7 to cyclic esters 5a and 5b. The magnesium radical anion is likely more stable in the chelated form IIc and therefore cyclizes predominately to cyclopentanecarboxylate 5b.
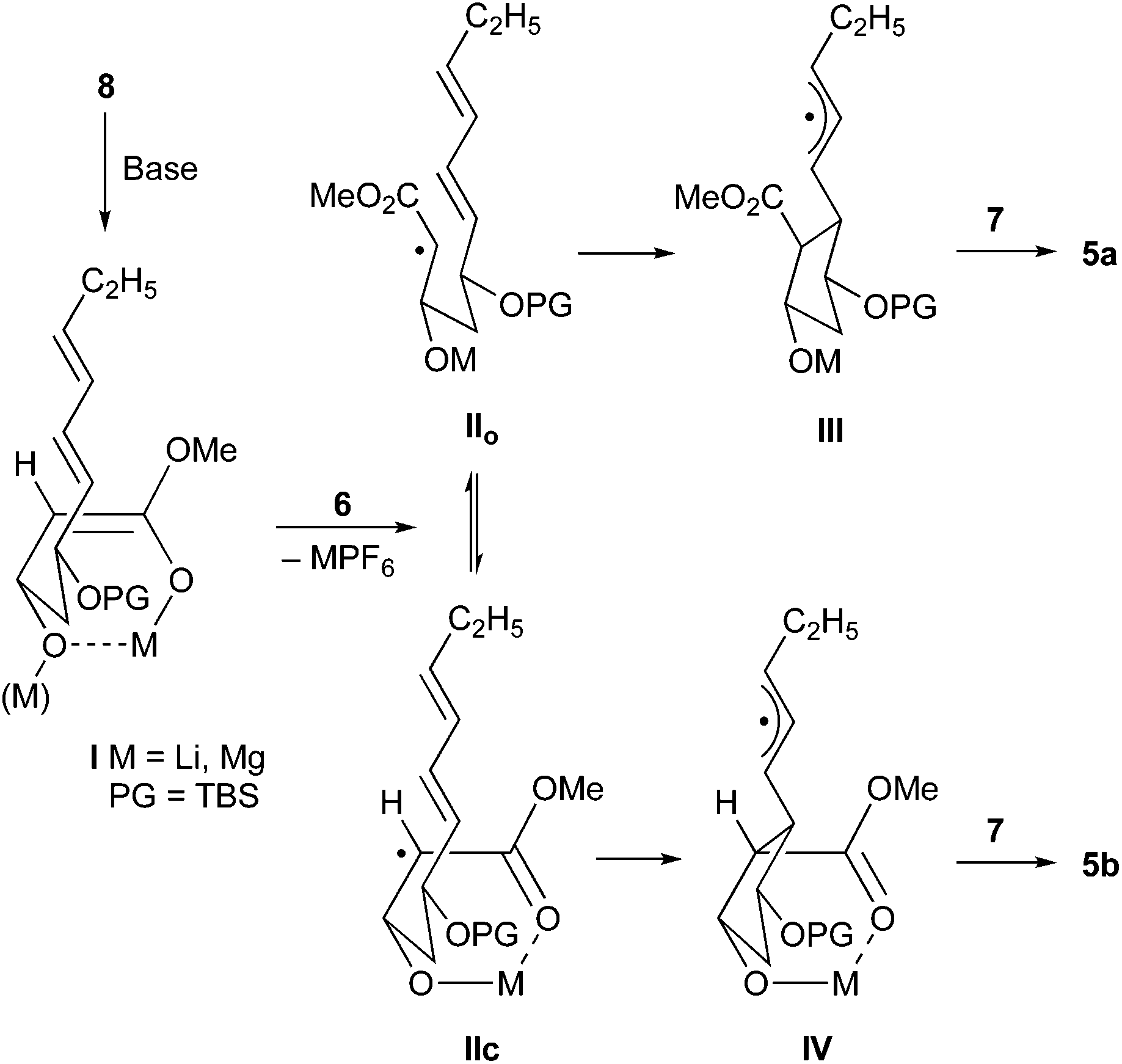 | ||
| Scheme 4 Course of the radical cyclization/oxygenation of ester 8. | ||
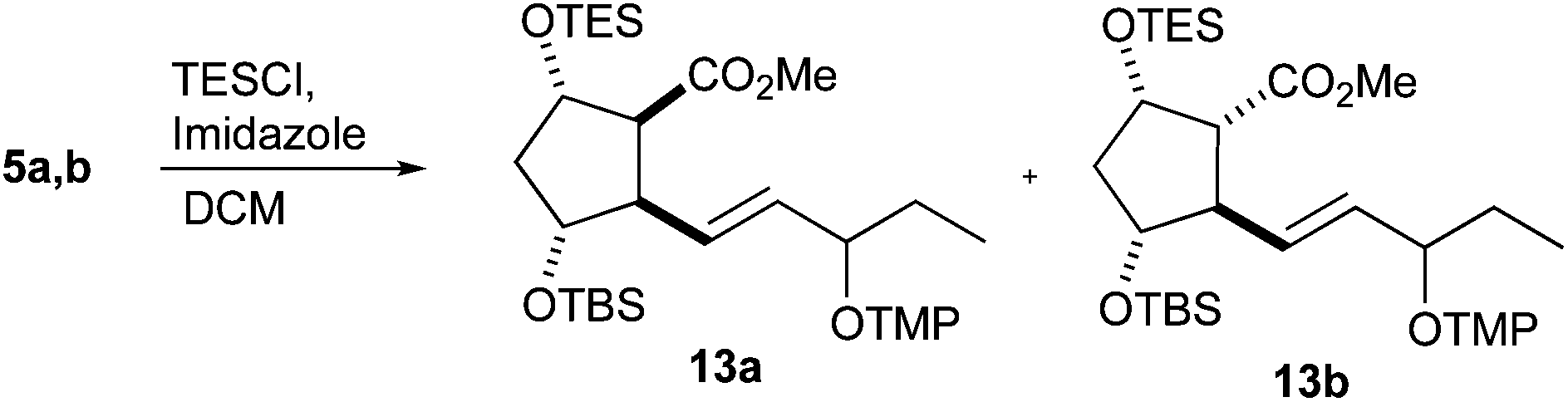
In contrast to the previous IsoP syntheses,47,48 where a free hydroxy group was mandatory, orthogonal protection was necessary to complete the unified approach to PhytoPs 1–3. Triethylsilyl (TES) protection of the free hydroxy group gave esters 13a and 13b, which were, however, only partially separable. Hypothesizing that the silylation of 5a might be significantly faster than that of 5b because of less steric interactions with the neighboring ester function, the amount of TESCl was reduced to the quantity of 5a in the mixture (Table 2). Indeed, the hydroxy group of 5a was preferably silylated and a separable mixture rich in diastereomer 13a was obtained (entry 1). A decrease in the temperature further increased the selectivity for silylation of the desired ester 5a (entries 2–4). In contrast, TES triflate reacted unselectively (not shown).
The ester group of 13a was transformed to triflate 15 in two steps (Scheme 5). Dibal-H in dichloromethane was optimal for the reduction to alcohol 14. In contrast, full deprotection of the TES group and partial deprotection of the TBS group occurred when LiAlH4 in THF was used. Alcohol 14 was converted to triflate 15 using triflic anhydride and 2,6-lutidine in good yield.
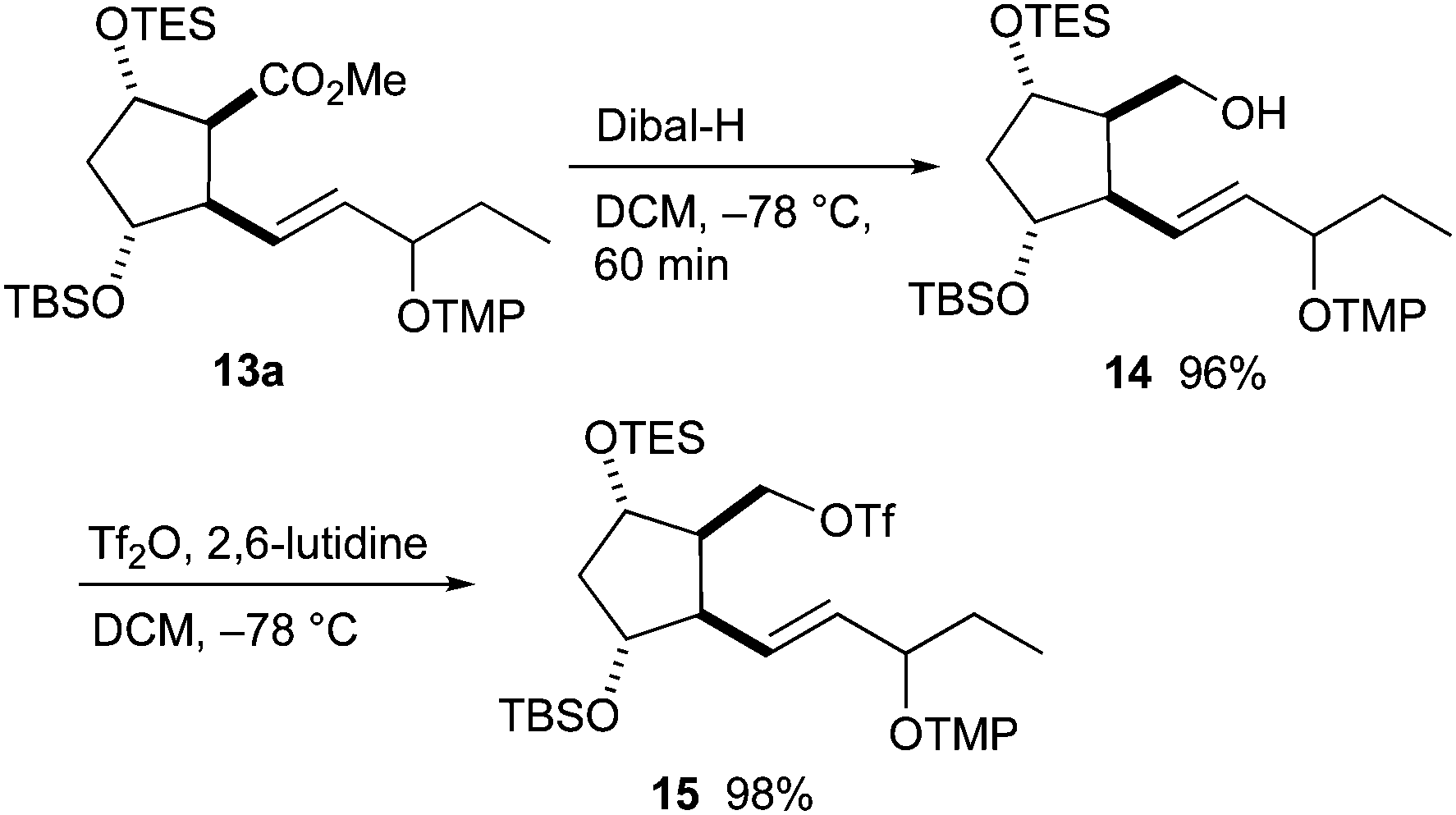 | ||
| Scheme 5 Preparation of alkyl–alkyl coupling partner 15 from ester 13a. | ||
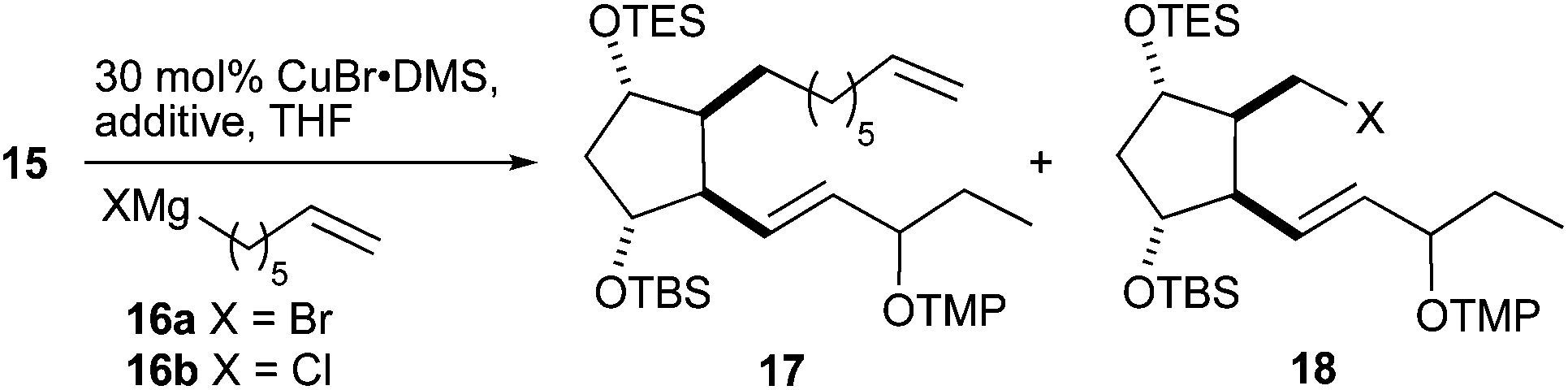
The crucial introduction of the α-chain was performed by copper(I)-catalyzed alkyl–alkyl coupling. Initial experiments were fruitless and therefore a series of model reactions was performed with cyclohexylmethyl triflate as a model substrate and several alkyl halides bearing functionality that could be later transformed to an ester group (for details, see the ESI†). Based on these results, the coupling reactions of ω-alkenyl Grignard reagents 16 with triflate 15 were further studied (Table 3). 6-Heptenylmagnesium bromide 16a gave surprisingly only a low yield of the desired PhytoP precursor 17 and bromide 18 was the main product, which was unreactive in the coupling reaction (Table 3, entries 1 and 2). A switch of the Grignard reagent to the less nucleophilic chloride 16b prevented the triflate–halogen exchange largely and compound 17 comprising the complete skeleton was obtained in a reasonable yield (Table 3, entry 3). It was subsequently found that the addition of lithium halides51 was beneficial to accelerate the coupling and to further reduce the formation of compound 18 (Table 3, entries 4 and 5). Surprisingly the bromide anion, stemming from LiBr, did not display the undesired substitution reactivity.
The central synthetic intermediate 4 was obtained from olefin 17 in four steps (Scheme 6). Hydroboration with 9-BBN using classical conditions or microwave heating provided the corresponding borane in almost quantitative yields, but the reaction time was reduced under microwave conditions. Its oxidation with basic hydrogen peroxide gave alcohol 19, which was oxidized to the corresponding acid using a two-step protocol consisting of Ley–Griffith oxidation to aldehyde 20 and subsequent Pinnick oxidation. Methyl ester 4 was prepared from the crude acid with (trimethylsilyl)diazomethane.
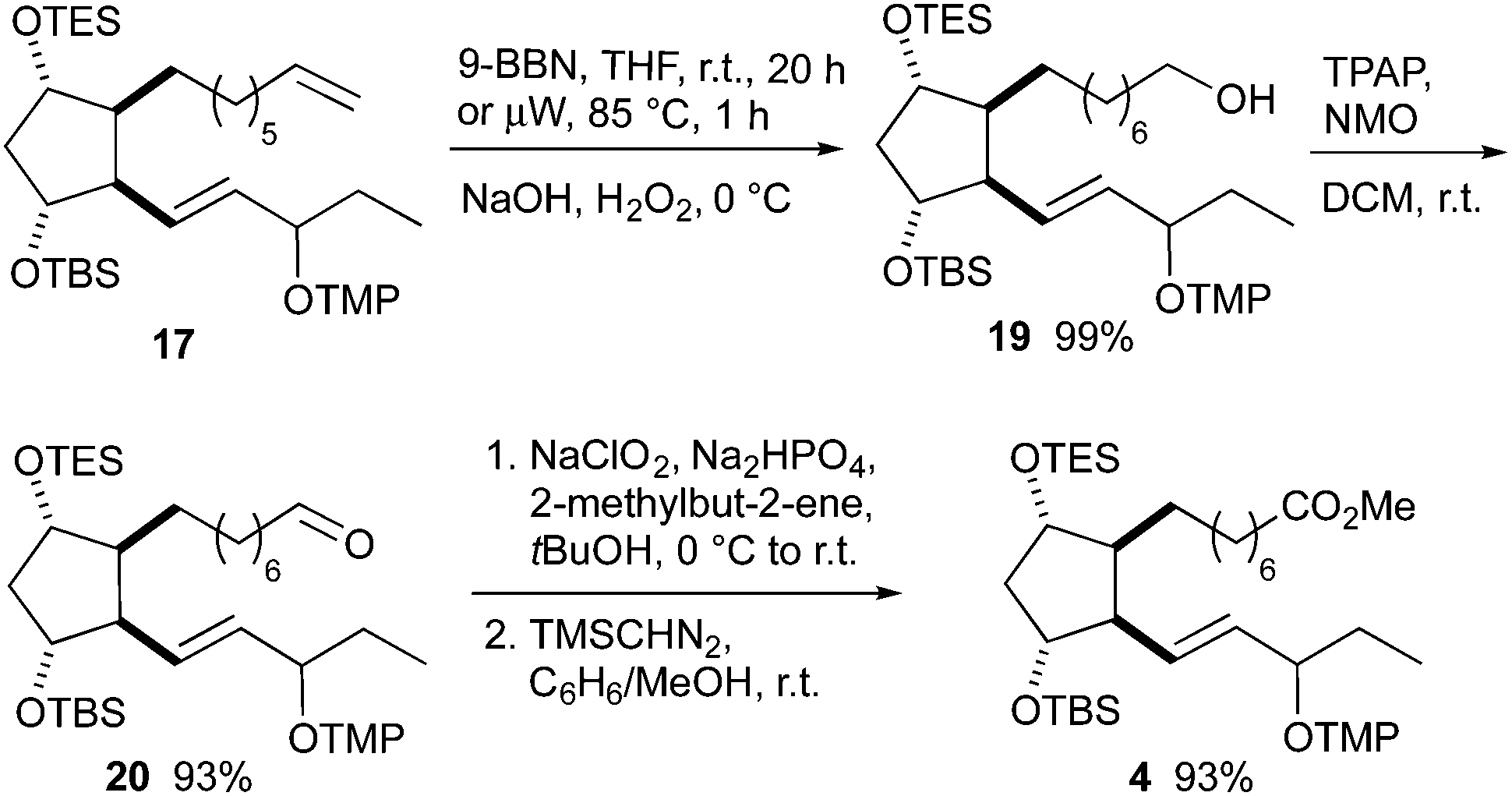 | ||
| Scheme 6 Transformation of olefin 17 to orthogonally protected ester 4. | ||
16-F1t-PhytoP methyl ester 1 was obtained from precursor 4 in three steps (Scheme 7). Oxidative removal of the tetramethylpiperidinyl group using mCPBA52 gave ketone 21 in almost quantitative yield. Although not executed here, an advantage of this strategy is the potential opportunity to reduce the carbonyl group enantioselectively.35,36 Since we focused on generating both C-16 diastereomers, the Luche reduction was applied, which gave both C-16 epimers in a 1:1.2 16-F1t-/16-epi-16-F1t-PhytoP ratio. Both silyl groups were conserved when the reduction was performed at −78 °C, however, TES deprotection occurred on warming to −10 °C. The remaining protected hydroxy groups were liberated by reaction with TBAF. The methyl esters of 16-F1t-PhytoP 1 and its 16-epimer epi-1 were separable by standard column chromatography and the correct stereochemistry at C-16 was determined by the comparison of measured and published NMR spectra.35,36 The overall yield amounted to 13% over 15 steps.
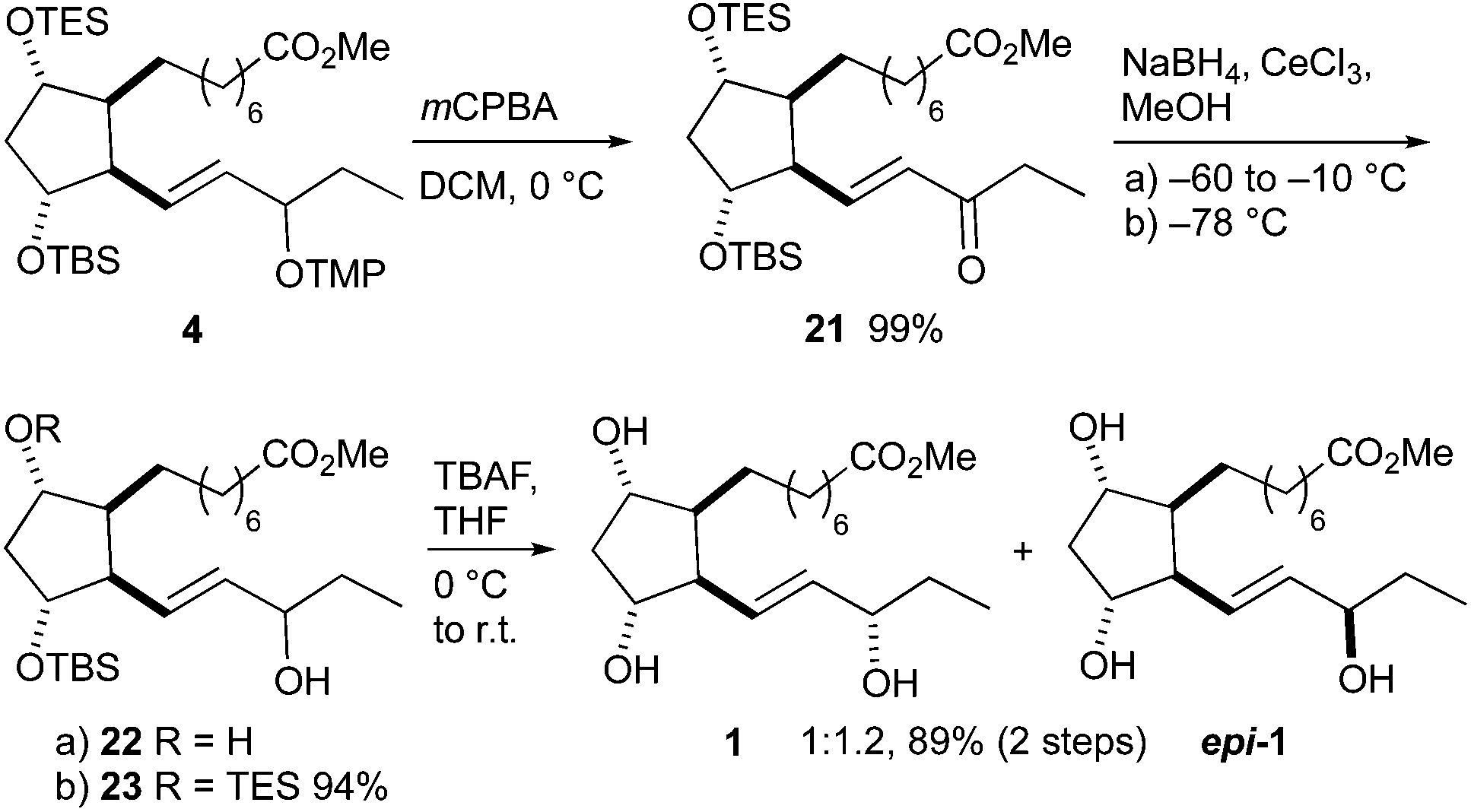 | ||
| Scheme 7 Completion of the total synthesis of 16-F1t-PhytoP 1 and 16-epi-16-F1t-PhytoP epi-1 methyl esters. | ||
The synthesis of 16-E1-PhytoP methyl esters 2 started with alcohol 23 (Scheme 8). The hydroxy group was protected as TBS ether and the TES group was subsequently selectively removed by acetic acid giving disilylated alcohol 25. Ketone 26 was prepared by Ley–Griffith oxidation; using the Dess–Martin periodinane gave a slightly lower yield of 78%. Hydrogen fluoride/pyridine proved to be the reagent of choice for the deprotection of the TBS groups. Separation of 16-E1-PhytoP 2 and its 16-epimer epi-2 by flash column chromatography was possible. The stereochemistry at C-16 was confirmed by comparison with the published NMR data.41 The epimeric ratio is in agreement with that observed in the synthesis of F1t-PhytoP methyl esters 1 and epi-1. The overall yield was 9% over 18 steps.
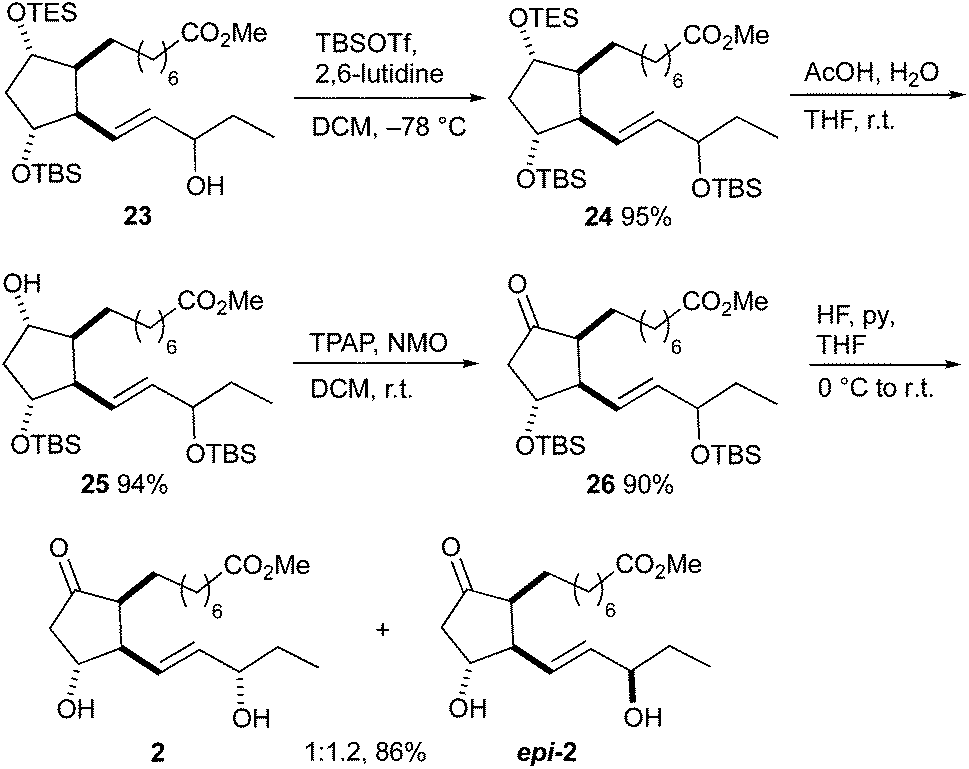 | ||
| Scheme 8 Completion of the total synthesis of 16-E1-PhytoP 2 and 16-epi-16-E1-PhytoP epi-2 methyl esters. | ||
The D-classes of IsoPs and PGs are known for their low stability caused by the sensitive β,γ-unsaturated ketone functionality,6,53–55 therefore several protecting groups removable under mild conditions were tested for the synthesis of 16-D1t-PhytoPs. The 1-ethoxyethoxy (EE) group was the only fruitful choice besides the methoxymethoxy (MOM) group. Protected 16-D1t-PhytoP derivatives 29a, b were synthesized in four steps from hydroxy ester 23 (Scheme 9). Selective removal of the TES group with acetic acid provided diol 22. Introduction of the EE group using catalytic PPTS proceeded smoothly to give 27a, whereas MOM protection giving 27b was less efficient. Removal of the remaining silyl group in 27a, b was executed by TBAF and afforded free 12-alcohols 28a, b. Their Dess–Martin oxidation to ketones 29a, b worked almost quantitatively.
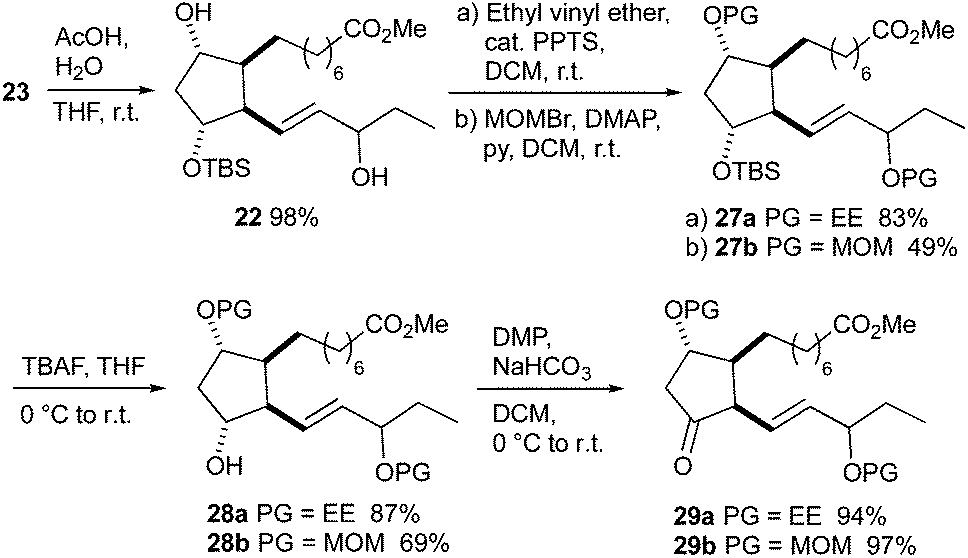 | ||
| Scheme 9 Synthesis of EE- and MOM-protected 16-D1t-PhytoP derivatives 29a and 29b. | ||
The final deprotection was performed by using catalytic amounts of PPTS (Scheme 10). Removal of the EE group of ketone 29a was completed at room temperature over three hours and gave a 1.5:1 mixture of 16-D1t-PhytoP methyl esters 3 and 16-deoxy-Δ13,15-16-D1t-PhytoP methyl ester 30. The MOM deprotection of ketone 29b proceeded very slowly at room temperature. Heating to reflux led to a hardly separable mixture, in which neither the expected D1t-PhytoP 3 nor dienyl PhytoP 30 were identified. The C-16 epimers of 3 were not separable by column chromatography and moreover, partial isomerization to more stable Δ13-16-D1t-PhytoP methyl esters 31 and epi-31 occurred during chromatographic purification. The isomerization could be promoted by treatment with silica gel and C-16 epimers 31 and epi-31 were isolated in overall 4% yield over 19 steps. The unequivocal assignment of the individual C-16 epimers was impossible. Nevertheless, we assume that the less polar epimer is Δ13-16-epi-16-D1t-PhytoP (epi-31) in analogy to the polarities of 16-epi-16-F1t-PhytoP and 16-epi-16-E1-PhytoPs. This hypothesis is supported by common signal patterns observed in the 13C NMR spectra of compounds 29b, 3 and 31 (for details see the ESI†). Moreover, epi-3 is thus the major C-16 epimer of 3 being in agreement with the epimeric ratio obtained in the Luche reduction of 21 and observed in the total syntheses of 16-F1t- and 16-E1-PhytoPs 1 and 2.
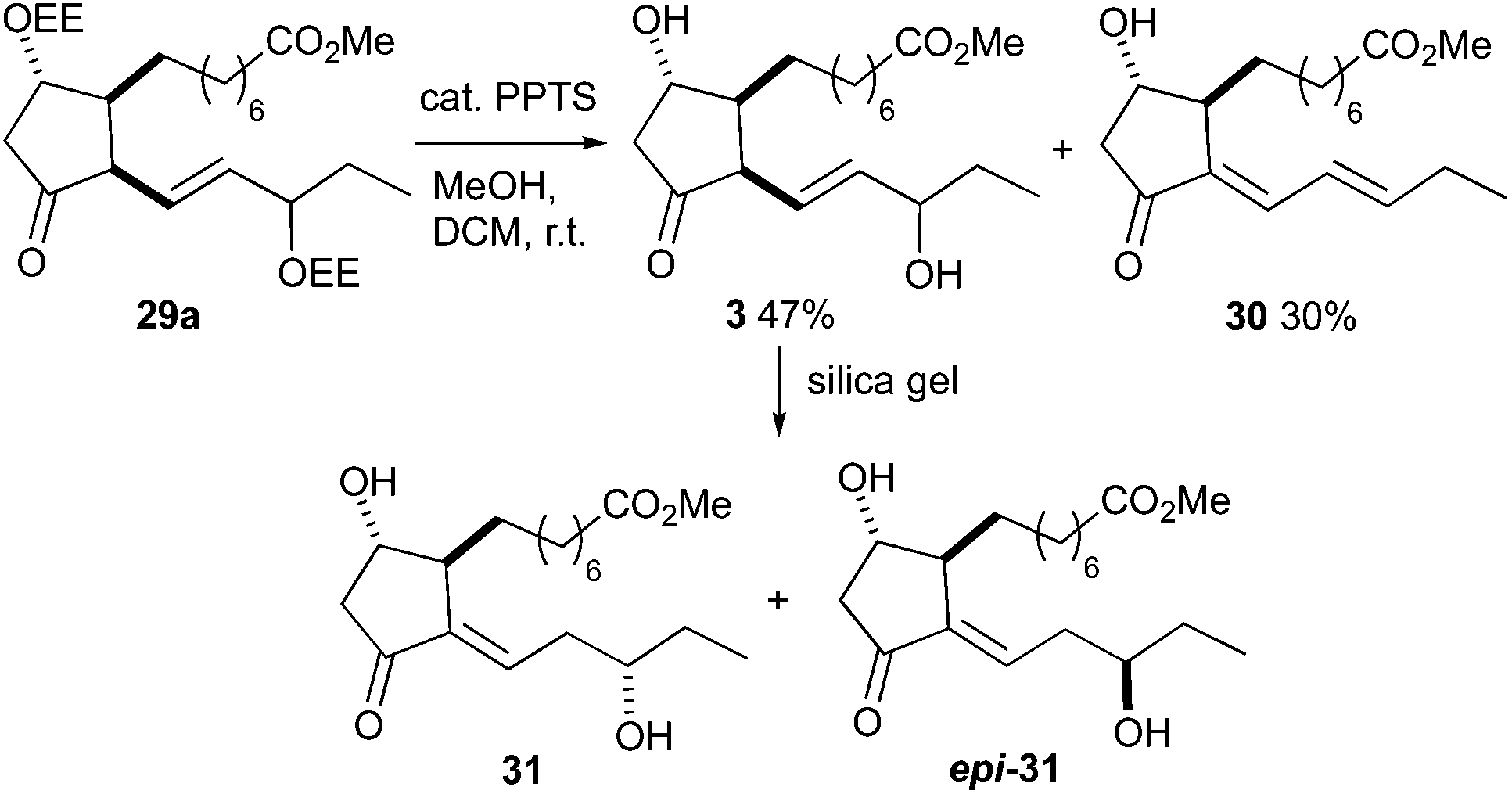 | ||
| Scheme 10 Access to 16-D1t-PhytoP methyl ester 3 by deprotection and its isomerization to Δ13-16-D1t-PhytoP methyl ester 31. | ||
Conclusions
A new strategy for the diversity-oriented synthesis of all trioxygenated 16-PhytoP classes, based on an orthogonally protected common precursor was developed. The key step for the formation of the dioxygenated cyclopentane ring was a radical oxidative cyclization mediated by ferrocenium hexafluorophosphate and TEMPO, which also introduced the oxygen functionality in the 16-position of the ω-chain. The complete carbon skeleton was assembled by a copper-catalyzed alkyl–alkyl coupling of a 6-heptenyl Grignard reagent and the properly functionalized cyclopentylmethyl triflate. 16-F1t-PhytoP methyl ester 1 and its epimer epi-1 were obtained in 13% yield over 15 steps. The synthesis of the methyl esters of 16-E1-PhytoP 2 and its C-16 epimer epi-2 took 18 steps and the overall yield amounted to 9%. The first total synthesis of 16-D1t-PhytoP methyl ester 3 resulted in an inseparable mixture of its C-16 epimers and their more stable isomerization products 31 and epi-31, which can be prepared by complete isomerization in 4% yield over 19 steps. The reported strategy will facilitate the simultaneous access to the majority of oxidatively formed cyclic metabolites derived from linolenic acid. Total syntheses of cyclopentenoic PhytoP can also be envisaged based on this approach. Moreover, the strategy will also be applicable to approach cyclic metabolites of the other major ω-3 PUFAs, eicosapentaenoic or docosahexaenoic acids. Investigations along these lines are under way in these laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support by the Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences (RVO:61388963) is gratefully acknowledged.Notes and references
- Oxidative Stress in Vertebrates and Invertebrates: Molecular Aspects on Cell Signaling, ed. T. Farooqui and A. A. Farooqui, Wiley, Hoboken, 2012 Search PubMed.
- Oxidative Stress: Diagnostics, Prevention, and Therapy, ed. S. Andreescu and M. Hepel, ACS, Washington, DC, 2011 Search PubMed.
- Prostaglandins: Biochemistry, Functions, Types, and Roles, ed. G. M. Goodwin, Nova Science Publishers, New York, 2010 Search PubMed.
- S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286–3337 CrossRef CAS PubMed.
- Prostaglandins, Leukotrienes, and Other Eicosanoids: From Biogenesis to Clinical Application, ed. F. Marks and G. Fürstenberger, Wiley-VCH, Weinheim, 1999 Search PubMed.
- U. Jahn, J.-M. Galano and T. Durand, Angew. Chem., Int. Ed., 2008, 47, 5894–5955 CrossRef CAS PubMed.
- U. Jahn, J.-M. Galano and T. Durand, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 83–86 CrossRef CAS PubMed.
- S. Parchmann and M. J. Mueller, J. Biol. Chem., 1998, 273, 32650–32655 CrossRef CAS PubMed.
- T. Durand, V. Bultel-Poncé, A. Guy, S. El Fangour, J.-C. Rossi and J.-M. Galano, Biochimie, 2011, 93, 52–60 CrossRef CAS PubMed.
- J. D. Morrow, K. E. Hill, R. F. Burk, T. M. Nammour, K. F. Badr and L. J. Roberts, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 9383–9387 CrossRef CAS.
- L. Gao, H. Yin, G. L. Milne, N. A. Porter and J. D. Morrow, J. Biol. Chem., 2006, 281, 14092–14099 CrossRef CAS PubMed.
- J. Nourooz-Zadeh, B. Halliwell and E. E. Änggård, Biochem. Biophys. Res. Commun., 1997, 236, 467–472 CrossRef CAS PubMed.
- L. J. Roberts, T. J. Montine, W. R. Markesbery, A. R. Tapper, P. Hardy, S. Chemtob, W. D. Dettbarn and J. D. Morrow, J. Biol. Chem., 1998, 273, 13605–13612 CrossRef CAS PubMed.
- J.-M. Galano, E. Mas, A. Barden, T. A. Mori, C. Signorini, C. De Felice, A. Barrett, C. Opere, E. Pinot, E. Schwedhelm, R. Benndorf, J. Roy, J.-Y. Le Guennec, C. Oger and T. Durand, Prostaglandins Other Lipid Mediators, 2013, 107, 95–102 CrossRef CAS PubMed.
- T. Durand, V. Bultel-Poncé, A. Guy, S. Berger, M. J. Mueller and J.-M. Galano, Lipids, 2009, 44, 875–888 CrossRef CAS PubMed.
- N. A. Eckardt, Plant Cell, 2008, 20, 495–497 CrossRef CAS PubMed.
- M. J. Mueller, Curr. Opin. Plant Biol., 2004, 7, 441–448 CrossRef CAS PubMed.
- J. Collado-González, T. Durand, F. Ferreres, S. Medina, A. Torrecillas and A. Gil-Izquierdo, Lipid Technol., 2015, 27, 127–130 CrossRef.
- J.-M. Galano, J. C.-Y. Lee, C. Gladine, B. Comte, J.-Y. Le Guennec, C. Oger and T. Durand, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2015, 1851, 446–455 CrossRef CAS PubMed.
- J. Collado-González, S. Medina, T. Durand, A. Guy, J.-M. Galano, A. Torrecillas, F. Ferreres and A. Gil-Izquierdo, Food Chem., 2015, 178, 212–220 CrossRef PubMed.
- M. E. Yonny, A. Rodríguez Torresi, C. Cuyamendous, G. Réversat, C. Oger, J.-M. Galano, T. Durand, C. Vigor and M. A. Nazareno, J. Agric. Food Chem., 2016, 64, 8296–8304 CrossRef CAS PubMed.
- A. M. Carrasco-Del Amor, J. Collado-González, E. Aguayo, A. Guy, J.-M. Galano, T. Durand and A. Gil-Izquierdo, RSC Adv., 2015, 5, 51233–51241 RSC.
- A. M. Carrasco-Del Amor, J. Collado-González, E. Aguayo, A. Guy, J.-M. Galano, T. Durand and A. Gil-Izquierdo, RSC Adv., 2016, 6, 25669–25669 RSC.
- A. M. Carrasco-Del Amor, E. Aguayo, J. Collado-González, A. Guy, J.-M. Galano, T. Durand and A. Gil-Izquierdo, Food Chem., 2016, 211, 869–875 CrossRef CAS PubMed.
- J. Marhuenda, S. Medina, A. Díaz-Castro, P. Martínez-Hernández, S. Arina, P. Zafrilla, J. Mulero, C. Oger, J.-M. Galano, T. Durand, F. Ferreres and A. Gil-Izquierdo, J. Agric. Food Chem., 2015, 63, 9022–9028 CrossRef CAS PubMed.
- J. Collado-González, A. Moriana, I. F. Girón, M. Corell, S. Medina, T. Durand, A. Guy, J.-M. Galano, E. Valero, T. Garrigues, F. Ferreres, F. Moreno, A. Torrecillas and A. Gil-Izquierdo, LWT–Food Sci. Technol., 2015, 64, 997–1003 CrossRef.
- A. E. Barden, K. D. Croft, T. Durand, A. Guy, M. J. Mueller and T. A. Mori, J. Nutr., 2009, 139, 1890–1895 CrossRef CAS PubMed.
- K. Karg, V. M. Dirsch, A. M. Vollmar, J.-L. Cracowski, F. Laporte and M. J. Mueller, Free Radical Res., 2007, 41, 25–37 CrossRef CAS PubMed.
- C. Traidl-Hoffmann, V. Mariani, H. Hochrein, K. Karg, H. Wagner, J. Ring, M. J. Mueller, T. Jakob and H. Behrendt, J. Exp. Med., 2005, 201, 627–636 CrossRef CAS PubMed.
- J. Gutermuth, M. Bewersdorff, C. Traidl-Hoffmann, J. Ring, M. J. Mueller, H. Behrendt and T. Jakob, J. Allergy Clin. Immunol., 2007, 120, 293–299 CrossRef CAS PubMed.
- S. Oeder, F. Alessandrini, O. F. Wirz, A. Braun, M. Wimmer, U. Frank, M. Hauser, J. Durner, F. Ferreira, D. Ernst, M. Mempel, S. Gilles, J. T. M. Buters, H. Behrendt, C. Traidl-Hoffmann, C. Schmidt-Weber, M. Akdis and J. Gutermuth, Allergy, 2015, 70, 1450–1460 CrossRef CAS PubMed.
- C. Blume, E. J. Swindle, S. Gilles, C. Traidl-Hoffmann and D. E. Davies, Tissue Barriers, 2015, 3, e1062316 CrossRef PubMed.
- E. Jahn, T. Durand, J.-M. Galano and U. Jahn, Chem. Listy, 2014, 108, 301–319 CAS.
- A. Guy, S. Flanagan, T. Durand, C. Oger and J.-M. Galano, Front. Chem., 2015, 3, 41, DOI:10.3389/fchem.2015.00041.
- S. El Fangour, A. Guy, V. Despres, J.-P. Vidal, J.-C. Rossi and T. Durand, J. Org. Chem., 2004, 69, 2498–2503 CrossRef CAS PubMed.
- S. El Fangour, A. Guy, J.-P. Vidal, J.-C. Rossi and T. Durand, Tetrahedron Lett., 2003, 44, 2105–2108 CrossRef CAS.
- A. Roland, T. Durand, D. Egron, J.-P. Vidal and J.-C. Rossi, J. Chem. Soc., Perkin Trans. 1, 2000, 245–251 RSC.
- S. El Fangour, A. Guy, J.-P. Vidal, J.-C. Rossi and T. Durand, J. Org. Chem., 2005, 70, 989–997 CrossRef CAS PubMed.
- E. Pinot, A. Guy, A.-L. Guyon, J.-C. Rossi and T. Durand, Tetrahedron: Asymmetry, 2005, 16, 1893–1895 CrossRef CAS.
- E. Pinot, A. Guy, A. Fournial, L. Balas, J.-C. Rossi and T. Durand, J. Org. Chem., 2008, 73, 3063–3069 CrossRef CAS PubMed.
- A. R. Rodríguez and B. W. Spur, Tetrahedron Lett., 2003, 44, 7411–7415 CrossRef.
- R. Beretta, M. G. Gallotti, U. Penne, A. Porta, J. F. G. Romero, G. Zanoni and G. Vidari, J. Org. Chem., 2015, 80, 1601–1609 CrossRef CAS PubMed.
- A. Porta, F. Chiesa, M. Quaroni, M. Persico, R. Moratti, G. Zanoni and G. Vidari, Eur. J. Org. Chem., 2014, 2111–2119 CrossRef CAS.
- A. Vazquez-Romero, X. Verdaguer and A. Riera, Eur. J. Org. Chem., 2013, 1716–1725 CrossRef CAS.
- E. E. Nishizawa, W. L. Miller, R. R. Gorman, G. L. Bundy, J. Svensson and M. Hamberg, Prostaglandins, 1975, 9, 109–121 CrossRef CAS.
- M. Krischke, C. Loeffler and M. J. Mueller, Phytochemistry, 2003, 62, 351–358 CrossRef CAS PubMed.
- U. Jahn and E. Dinca, J. Org. Chem., 2010, 75, 4480–4491 CrossRef CAS PubMed.
- U. Jahn and E. Dinca, Chem. – Eur. J., 2009, 15, 58–62 CrossRef CAS PubMed.
- C. Maaliki, E. Thiery and J. Thibonnet, Eur. J. Org. Chem., 2017, 209–228 CrossRef CAS.
- F. Kafka, M. Holan, D. Hidasová, R. Pohl, I. Císařová, B. Klepetářová and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 9944–9948 CrossRef CAS PubMed.
- K. Nakata, C. Feng, T. Tojo and Y. Kobayashi, Tetrahedron Lett., 2014, 55, 5774–5777 CrossRef CAS.
- T. Inokuchi and H. Kawafuchi, Tetrahedron, 2004, 60, 11969–11975 CrossRef CAS.
- Y. Brinkmann, C. Oger, A. Guy, T. Durand and J.-M. Galano, J. Org. Chem., 2010, 75, 2411–2414 CrossRef CAS PubMed.
- P. S. Foss, C. J. Sih, C. Takeguchi and H. Schnoes, Biochemistry, 1972, 11, 2271–2277 CrossRef CAS PubMed.
- G. L. Bundy, D. R. Morton, D. C. Peterson, E. E. Nishizawa and W. L. Miller, J. Med. Chem., 1983, 26, 790–799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical characterization and copies of 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c7ob02505j |
| This journal is © The Royal Society of Chemistry 2017 |