
Oligonucleotide modifications enhance probe stability for single cell transcriptome in vivo analysis (TIVA)†
S. B.
Yeldell
,
B. K.
Ruble
and
I. J.
Dmochowski
 *
*
Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA 19104-6323, USA. E-mail: ivandmo@sas.upenn.edu; Fax: +1 215-898-2037; Tel: +1 215-898-6459
First published on 13th October 2017
Abstract
Single cell transcriptomics provides a powerful discovery tool for identifying new cell types and functions as well as a means to probe molecular features of the etiology and treatment of human diseases, including cancer. However, such analyses are limited by the difficulty of isolating mRNA from single cells within biological samples. We recently introduced a photochemical method for isolating mRNA from single living cells, Transcriptome In Vivo Analysis (TIVA). The TIVA probe is a “caged” polyU![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polyA oligonucleotide hairpin designed to enter live tissue, where site-specific activation with 405 nm laser reveals the polyU-biotin strand to bind mRNA in a target cell, enabling subsequent mRNA isolation and sequencing. The TIVA method is well suited for analysis of living cells in resected tissue, but has not yet been applied to living cells in whole organisms. Adapting TIVA to this more challenging environment requires a probe with higher thermal stability, more robust caging, and greater nuclease resistance. In this paper we present modifications to the original TIVA probe with multiple aspects of enhanced stability. These newer probes utilize an extended 22mer polyU capture strand with two 9mer polyA blocking strands (“22/9/9”) for higher thermal stability pre-photolysis and improved mRNA capture affinity post-photolysis. The “22/9/9 GC” probe features a terminal GC pair to reduce pre-photolysis interactions with mRNA by more than half. The “PS-22/9/9” probe features a phosphorothioated backbone, which extends serum stability from <1 h to at least 48 h, and also mediates uptake into cultured human fibroblasts.
:polyA oligonucleotide hairpin designed to enter live tissue, where site-specific activation with 405 nm laser reveals the polyU-biotin strand to bind mRNA in a target cell, enabling subsequent mRNA isolation and sequencing. The TIVA method is well suited for analysis of living cells in resected tissue, but has not yet been applied to living cells in whole organisms. Adapting TIVA to this more challenging environment requires a probe with higher thermal stability, more robust caging, and greater nuclease resistance. In this paper we present modifications to the original TIVA probe with multiple aspects of enhanced stability. These newer probes utilize an extended 22mer polyU capture strand with two 9mer polyA blocking strands (“22/9/9”) for higher thermal stability pre-photolysis and improved mRNA capture affinity post-photolysis. The “22/9/9 GC” probe features a terminal GC pair to reduce pre-photolysis interactions with mRNA by more than half. The “PS-22/9/9” probe features a phosphorothioated backbone, which extends serum stability from <1 h to at least 48 h, and also mediates uptake into cultured human fibroblasts.
Introduction
High-throughput, next generation RNA sequencing in conjunction with common methods for mRNA isolation1,2 have enabled the bottom-up investigation of complex tissues based upon single cell transcriptomes.3 Such analyses have identified previously unknown cell types, gene regulatory mechanisms, and cellular communication networks.4 In addition to basic functional studies of healthy tissue, single cell transcriptomics also makes it possible to study the states of diseased cells both before and after drug treatment, and has been employed to profile mutations that drive drug resistance in tumors.5 We recently introduced a photochemical method for more precisely isolating mRNA from single cells within living tissue using Transcriptome In Vivo Analysis (TIVA).6The original TIVA probe was a highly functionalized polyU:polyA oligonucleotide (oligo) hairpin that was delivered into freshly resected tissue slices by means of a 5′ disulfide-linked cell-penetrating peptide (CPP, Fig. 1). One particularly versatile and protease resistant CPP is (D-Arg)9.7,8 TIVA probe loaded into tissue was designed to capture mRNA in a specific cell of interest upon 405 nm activation using confocal laser scanning microscopy (CLSM). Photoactivation cleaved two nitrobenzyl linkers in the TIVA oligo backbone, allowing both 2′-OMe 7mer polyA blocking strands to dissociate at rt and reveal the 2′-F 18mer polyU capture strand. Because the “18/7/7” hairpin termini were labeled with a Cy3–Cy5 FRET reporter pair, separation of the hairpin resulted in a readily observable (>60%) loss in Cy5 FRET emission and concomitant increase in Cy3 emission when imaged with green HeNe (543 nm) laser. The 18mer capture strand, featuring a 3′ biotin modification, subsequently bound to available mRNA polyA tails (“target strands”), thereby noncovalently attaching to mRNA one or more biotin affinity tags. “Biotinylated” mRNA was recovered by aspirating and lysing a small tissue region containing the target cell. Finally, the target mRNA was isolated by streptavidin:biotin affinity purification, linearly amplified, and sequenced.
 | ||
| Fig. 1 TIVA probe is an oligonucleotide hairpin designed to enter live tissue, where site-specific activation with 405 nm laser attaches biotin to mRNA in a target cell, enabling subsequent mRNA isolation and sequencing. | ||
Since we introduced the TIVA photochemical mRNA isolation approach in 2014,6 several new methods to probe single cell transcriptomes have been developed. Super-resolution microscopy of sequence-specific probes has yielded dazzling images of spatially-resolved transcripts in fixed tissue.9–11 Meanwhile, high-throughput microfluidic platforms can sequence up to tens of thousands of dissociated single cells by combining each cell with an mRNA capture bead. Oligos attached to these beads prime reverse transcription while adding a unique barcode so that individual transcriptomes can be reconstructed from bulk sequencing data.12–14 These techniques are powerful additions to the single-cell toolkit, and are complementary to TIVA with a focus on imaging a smaller subset of known genes in fixed tissue or achieving high-throughput analysis by using dissociated tissue. The TIVA photochemical approach remains optimal for studying the complete transcriptome of living single cells and particularly single cells within living tissue, still contextualized by their native microenvironment. Additionally, it uniquely offers the possibility of capturing mRNA from distinct subcellular regions, e.g., cytoplasm, nucleus or dendrite.
While the original “18/7/7” TIVA oligo probe was shown to be capable of isolating mRNA from cells in ex vivo tissue slices, advancing to the study of single cells in whole, living organisms presents a multitude of important challenges. In particular, the TIVA probe must remain intact and inactive (“caged”) through longer time courses in more harshly degrading, nuclease-abundant conditions. We have focused our initial redesign efforts on exploring four factors that regulate TIVA probe performance in challenging conditions: (1) increasing the hairpin's pre-photolysis thermal stability for general reliability; (2) increasing post-photolysis TIVA-mRNA capture affinity for improved pull-down; (3) improving the inertness of intact TIVA hairpin to mRNA for superior caging; and (4) increasing TIVA hairpin nuclease resistance to extend experimental time windows.
Here, we report variations of the original “18/7/7” TIVA probe that are built around lengthened hairpins, extended to as far as a 22mer polyU capture strand with two 9mer polyA blocking strands (“22/9/9”). This lengthened duplex was incorporated to improve the probe's general thermal stability pre-photolysis and to increase mRNA capture affinity post-photolysis. To improve the inertness of the hairpin in the presence of mRNA, we developed the “22/9/9 GC” probe by adding a terminal GC pair to the duplex. Finally, we explored the stronger serum stability and enhanced cellular uptake mediated by a phosphorothioate (PS) backbone in developing the “PS-22/9/9” probe. By replacing non-bridging backbone phosphodiester oxygen(s) with sulfur atom, the oligo can be made more resistant to degradation by a variety of nucleases and phosphodiesterases. In general, phosphorothioation disrupts interactions critical for coordination of these degratory enzymes to oligonucleotides by adding chiral centers to each backbone linkage, thereby increasing structural variation.15–18
Results and discussion
Optimizing duplex thermal stability
Our initial efforts in stabilizing TIVA probe focused on improving its duplex composition. We reasoned that lengthening the duplex would lead to higher thermal stability of the caged hairpin structure (Table 1) and greater target capture affinity upon photoactivation (Table 2). Furthermore, a more optimal arrangement of the blocking strands should generate a higher pre-photolysis thermal stability while still allowing for facile dissociation post-photolysis. We originally found the greatest stability enhancement by moving from an 18mer polyU capture strand with two 7mer polyA blocking strands (“18/7/7”) to a 22mer capture strand with two 9mer blocking strands (“22/9/9”).19 More recently, we also investigated the stabilizing effect of the cationic, cell-penetrating peptide, (D-Arg)9, on pre-photolysis thermal stability (Fig. 2). The role of the pendant CPP is important to understand as there are alternate methods for intracellular delivery of the TIVA probe, including ionically associated carrier peptides20 or electroporation. We found that the pendant CPP exerts a stabilizing effect that is most dramatic in low-salt (10 mM NaCl) solution, raising the Tm of the 18/7/7 and 22/9/9 probes by nearly 20 °C. In high-salt (100 mM NaCl) solution, the Tm was raised by ∼5 °C, roughly equivalent to extending the duplex by four nucleotides. While the disulfide-linked CPP is not expected to stay attached to the TIVA probe within the reducing environment of the cell, it should remain attached in the extracellular milieu where the probe is challenged by serum nucleases.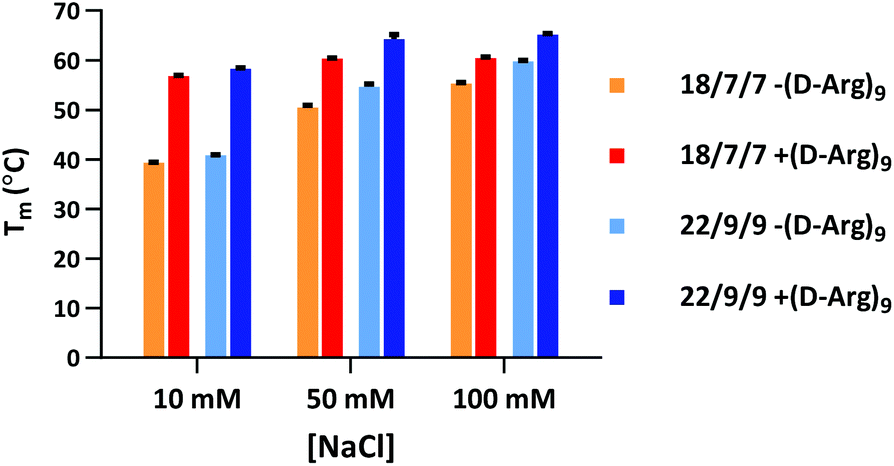 | ||
| Fig. 2 Melting temperatures of 18/7/7 and 22/9/9 TIVA probes with or without (D-Arg)9 cell-penetrating peptide in 10, 50, or 100 mM NaCl solution. Shown are averages and ranges of two forward and reverse melts for each sample. | ||
| TIVA probe configuration | Pre-photolysis Tm (°C) |
|---|---|
| 18/7/5 | 41.2 |
| 18/5/7 | 44.6 |
| 20/7/7 | 51.5 |
| 18/7/7 | 53.9 |
| 18/7/9 | 55.5 |
| 18/6/6 | 56.4 |
| 18/8/8 | 61.0 |
| 20/9/9 | 64.8 |
| 22/9/9 | 64.8 |
| TIVA probe | Target-bound Tm (°C) |
|---|---|
| 18/7/7 | 48.5 |
| 18/9/9 | 48.5 |
| 20/9/9 | 49.5 |
| 22/9/9 | 53.4 |
Improved caging
In previous TIVA experiments we have observed lower probe FRET efficiencies in the presence of mRNA or short polyA RNA model target strands, possibly corresponding to transient interactions of polyA RNA with the duplex termini where the FRET reporter pair is located or improper alignment of the non-specific hairpin sequence, leaving the FRET pair displaced by a sticky end. To better align the duplex and reduce fraying we designed a new probe with a terminal GC pair, “22/9/9 GC” (Fig. 3).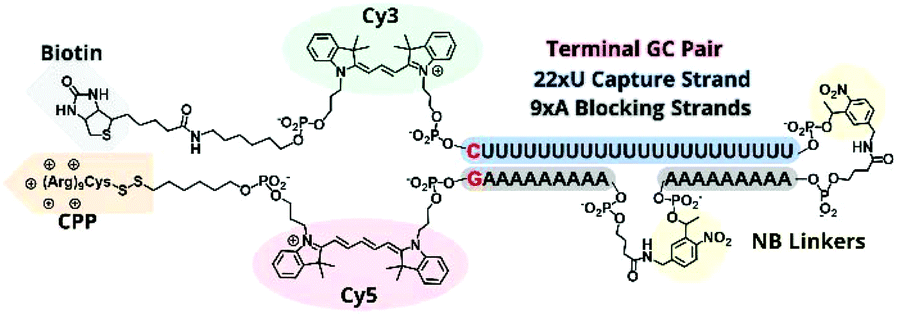 | ||
| Fig. 3 Structural scheme of 22/9/9 GC +(D-Arg)9 TIVA probe. | ||
22/9/9 GC TIVA probe was synthesized by solid-phase phosphoramidite chemistry on an ABI 394 synthesizer. Afterwards, the oligo was cleaved from its support by reaction with concentrated ammonium hydroxide for 17 h, with the terminal DMT protecting group left on. The cleaved product was then purified by RP-HPLC on a C18 column, and a representative HPLC trace is shown in Fig. S1.† To prepare for the (D-Arg)9 CPP conjugation, the oligo 5′ disulfide-linked alkyl-DMT group was removed by reduction in 500 mM TCEP for 1 h followed by separation and desalting through a Nap-10 column. The oligo – with a free 5′ thiol – was then allowed to react with 6–8 equivalents of Cys(Npys)-(D-Arg)9 in a 1:1 formamide:water solution for 24–36 h. The resulting 22/9/9 GC +(D-Arg)9 oligo was subsequently purified by AX-HPLC on a Resource Q column (Fig. S2†) and buffer-exchanged into 1× STE using Amicon 10k MWCO spin filters, concentrated to 100 μM, and stored at −20 °C.
22/9/9 GC +(D-Arg)9 characterization
The product was characterized by ESI-MS (Novatia), which confirmed the expected mass for the 17592 Da oligo-peptide conjugate (Fig. 4a). The purity of the product was estimated to be roughly 90% by RP-HPLC (Fig. 4b), with the principal impurities corresponding to product with 1–2 cyanoethyl protect groups remaining. These impurities resolve closely to the product, but are not anticipated to significantly affect the performance of the probe. Lastly, we measured the melting temperature of the probe and found the GC pair to raise the pre-photolysis Tm to 66 °C (Fig. 4c, compare with Table 1 data). The mRNA target-bound Tm was roughly 53 °C, which is in close agreement with native 22/9/9 TIVA probe (Table 2).
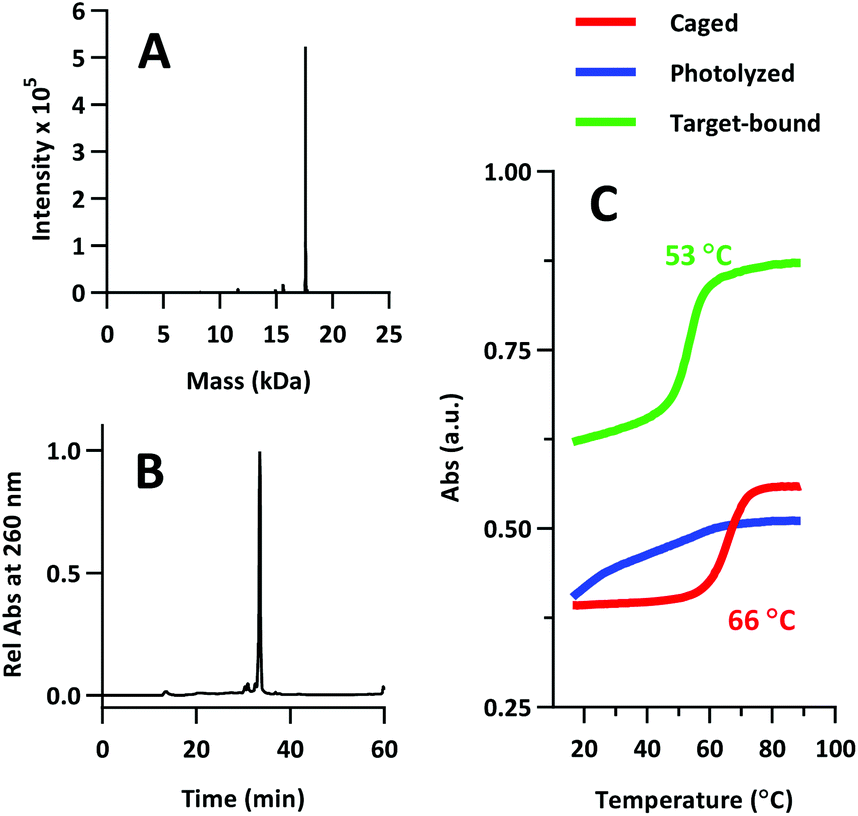 | ||
| Fig. 4 (A) ESI-MS of 22/9/9 GC TIVA probe. 17592 Da expected, 17592 Da observed. (B) Re-injection of purified 22/9/9 GC on RP-HPLC. (C) Melting curve analysis of 22/9/9 GC while caged (red), photolyzed (blue), or photolyzed and target-bound (green). | ||
We next assessed the FRET signal for both 18/7/7 +(D-Arg)9 and 22/9/9 GC +(D-Arg)9 probes at 1 μM in 1× STE, as dependent upon photolysis (“±hν”) and/or one equivalent of a 30mer 2′OMe polyA model target strand (“±polyA”) (Fig. 5). We observed slightly decreased starting FRET for the 22/9/9 GC TIVA probe (82%) relative to the original TIVA probe (87%), perhaps due to the GC pair enforcing a somewhat different probe conformation that affects relative positioning of the Cy3–Cy5 FRET pair. Because the construct has higher thermal stability than similar probes without the GC pair, we reasoned that the somewhat lower FRET efficiency may be caused by sequence-specific alterations to the stacking, isomerization, and subsequent fluorescence of the terminal dyes21 (Fig. 5, column 1). Crucially, FRET efficiency is improved for unphotolyzed 22/9/9 GC TIVA probe in the presence of polyA RNA by 13% relative to 18/7/7 TIVA probe (Fig. 5, column 2). Furthermore, the net loss of FRET after the addition of polyA RNA relative to its starting FRET has been reduced from 36.3% to 17.5%. This improvement increases confidence in the TIVA probe caging fidelity, particularly in applications that require subcellular activation or lysis of one target cell in a larger pool of unactivated cells. This more robust caging may have the side effect of marginally reducing post-photolysis dissociation of the polyA blocking strands until they are actively displaced by target mRNA (Fig. 5, column 3). However, both probes reach 6% FRET upon photoactivation in the presence of 30mer polyA RNA, so the terminal GC pair does not appear to inhibit overall activation or target binding (Fig. 5, column 4). We believe that the trends observed were driven primarily by the addition of the GC pair, as we observed no significant difference in FRET efficiency between 18/7/7 and 22/9/9 probes lacking a GC pair (Fig. S4†).
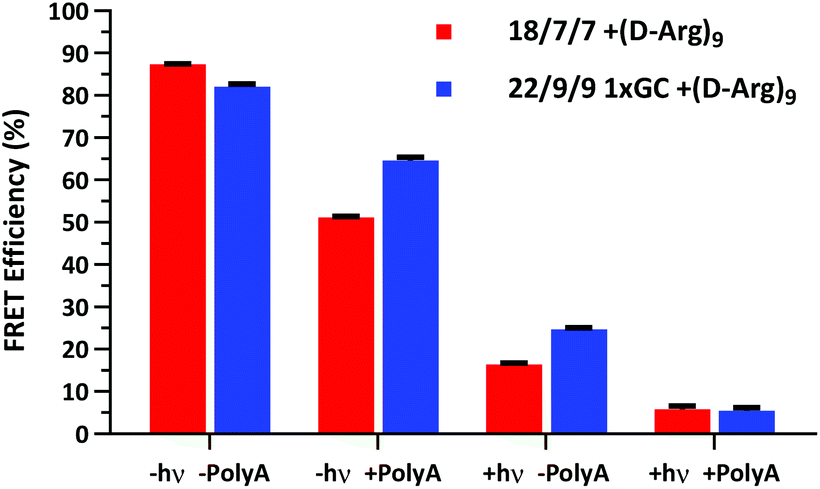 | ||
| Fig. 5 Cy3–Cy5 FRET efficiency of 1 μM 22/9/9 GC +(D-Arg)9 TIVA probe compared to 18/7/7 +(D-Arg)9 TIVA probe in 1× STE buffer with or without 1 eq. of 30mer polyA target and/or photolysis. Average and standard deviation shown for three or more samples under each condition. | ||
Enhancing nuclease resistance
To improve the nuclease resistance of the TIVA probe we phosphorothioated each backbone linkage, a well-established method for extending the serum stability of oligonucleotides. Phosphorothioated antisense oligos have been utilized in an assortment of antisense treatments,22–24 and this general topic has been reviewed recently by Frick Eckstein.25We determined that backbone sulfurs competed with the 5′ thiol modification for conjugation with (D-Arg)9 peptide, and we subsequently omitted the CPP from further PS-oligo designs. PS-22/9/9 TIVA probe (Fig. 6) was synthesized by solid-phase phosphoramidite chemistry as previously described for 22/9/9 GC TIVA probe with slight modifications: during solid-phase synthesis the oxidation step was replaced by a sulfurizing step prior to capping. After solid-phase synthesis, the oligo was cleaved and RP-HPLC purified with the DMT group off (Fig. S3†). Following HPLC purification, PS-22/9/9 TIVA probe was buffer exchanged into 1× STE and concentrated to 100 μM as previously described and stored at −20 °C.
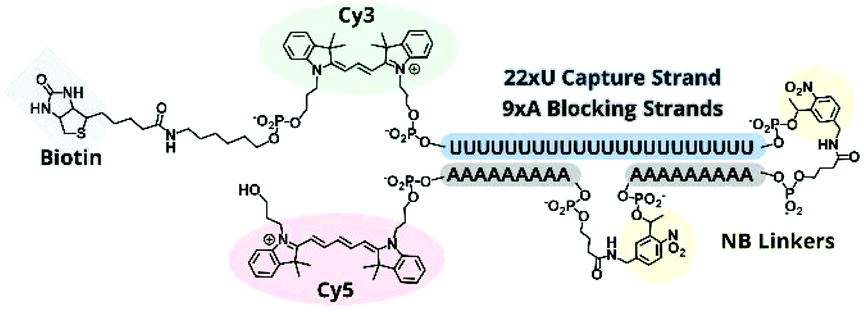 | ||
| Fig. 6 Structural scheme of PS-22/9/9 TIVA probe. All backbone linkages are phosphorothioated. | ||
PS-22/9/9 TIVA general characterization
Product was characterized by ESI-MS (Novatia), which confirmed the expected 15900 Da product with an observed 15899 Da peak (Fig. 7a). PS-22/9/9 TIVA probe purity was gauged by RP-HPLC, and found to be roughly 90% (Fig. 7b). Thermal melt analysis showed that phosphorothioation significantly decreased the thermal stability of the duplex (Tm = 48 °C), although the RNA target-bound Tm was less affected at 52 °C (Fig. 7c). This hybridization penalty has been well documented,26 and some groups have attempted to limit the penalty by reducing the number of sites that are phosphorothioated.27,28 In this case, PS-22/9/9 TIVA probe Tm values likely remain high enough for in vivo applications at 37 °C.
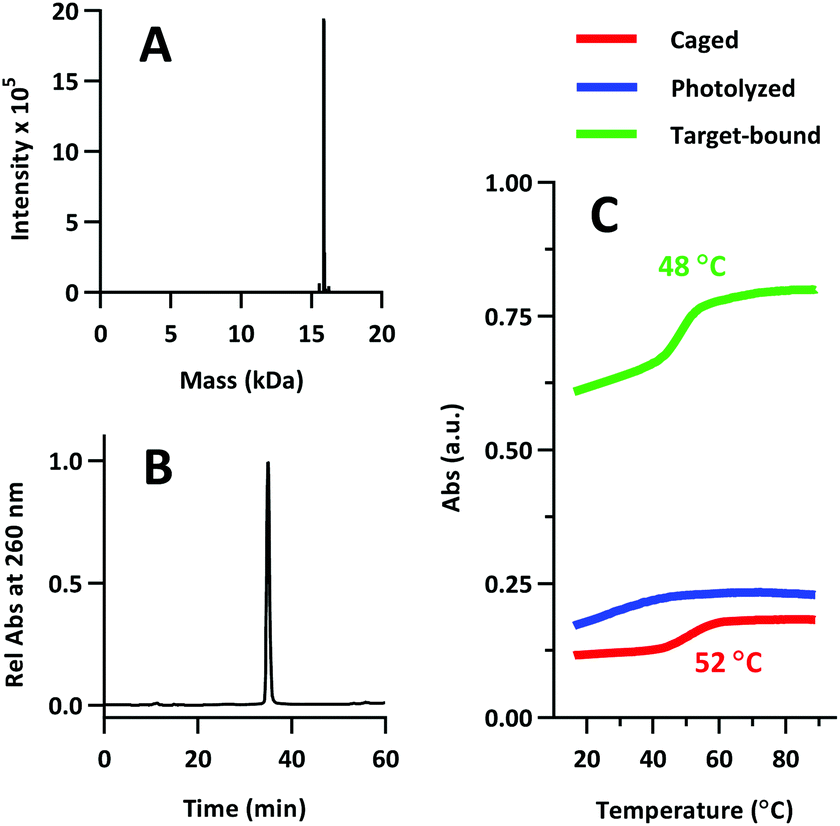 | ||
| Fig. 7 (A) ESI-MS of PS-22/9/9 TIVA probe. 15900 Da expected, 15897 Da observed. (B) Re-injection of purified PS-22/9/9 on RP-HPLC. (C) Melting curve analysis of PS-22/9/9 while caged (red), photolyzed (blue), or photolyzed and RNA target-bound (green). | ||
Fluorescence and stability assessment
The FRET efficiency of PS-22/9/9 TIVA probe was first measured in a cuvette as previously described for 22/9/9 GC (Fig. 8a). The decreased starting FRET (75%) is primarily explained by the lack of cationic CPP, but the lower thermal stability imparted by phosphorothioation may also be a contributing factor. This value drops sharply in the presence of 1 eq. 30mer polyA RNA, but can be photoactivated to achieve a final FRET efficiency of 5%, comparable to 18/7/7 TIVA probe. We noticed that the FRET efficiency of unphotolyzed PS-22/9/9 TIVA probe in the presence of 1 eq. polyA RNA was enhanced by roughly 11% when conducting the trials in PBS with 10% FBS compared to PBS alone (Fig. 8b). Phosphorothioation has been previously reported to enhance the binding of non-specific serum proteins to double-stranded oligonucleotides.29 We hypothesize that such protein interactions may further rigidify the duplex in the PS-22/9/9 TIVA probe.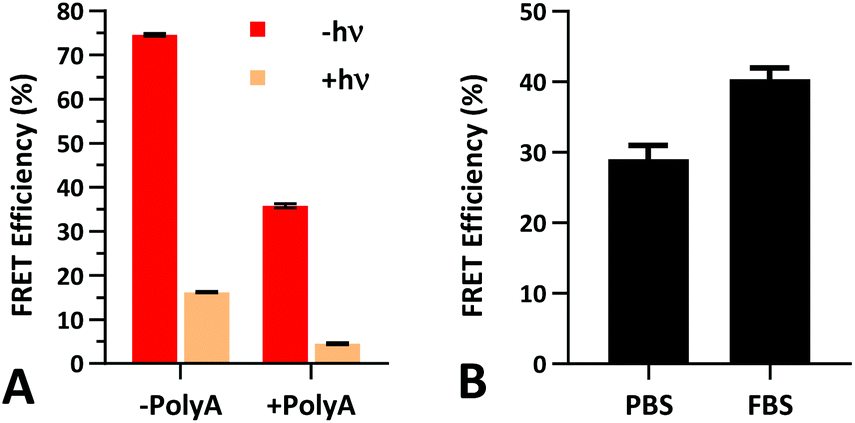 | ||
| Fig. 8 (A) Cy3–Cy5 FRET efficiency of 1 μM PS-22/9/9 TIVA probe in 1× STE buffer with or without 1 eq. of 30mer polyA target and/or photolysis. (B) Pre-photolysis FRET efficiency of PS-22/9/9 probe in the presence of 1 eq. of 30mer polyA RNA is enhanced in PBS with 10% FBS. Average and standard deviation shown for three or more samples under each condition. | ||
To assess the serum stability of PS-22/9/9 probe relative to traditional phosphodiester probe (22/9/9) we incubated both at 37 °C in PBS with 10% FBS while monitoring FRET for 24 h using a fluorimeter. Afterwards, we normalized the FRET readings for both probes relative to their starting efficiencies (Fig. 9a). We observed consistent FRET signal in PS-22/9/9 for the duration of the experiment, whereas 22/9/9 lost 40% of its starting FRET efficiency during 24 h incubation.
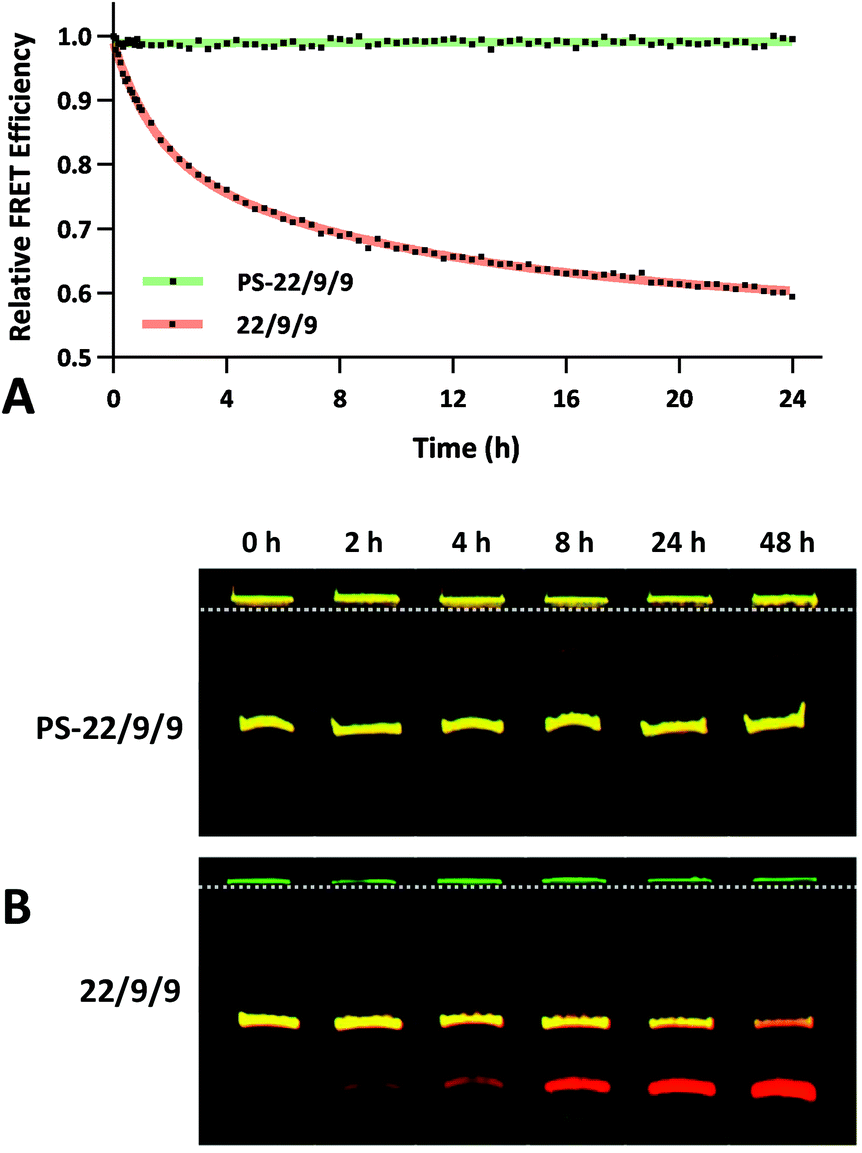 | ||
| Fig. 9 (A) 24 h stability time course of 1 μM phosphorothioate-linked PS-22/9/9 (green) or phosphodiester-linked 22/9/9 (red) TIVA probes in 10% FBS. Cy3–Cy5 FRET efficiencies were measured by fluorimeter and then normalized against starting values. (B) 48 h stability time course of 1 μM PS-22/9/9 (bottom) or 22/9/9 (top) probes in 10% FBS using gel electrophoresis. At each time point a sample was mixed with a nuclease inhibitor and flash frozen. After 48 h, all samples were loaded onto a 20% acrylamide denaturing gel and eluted using 100 V for 120 min. Cy3 fluorescence is shown in green and Cy5 fluorescence in red. | ||
To further verify that PS-22/9/9 was not being degraded by serum nucleases we performed a 48 h incubation, snap freezing aliquots at regular intervals. After 48 h, all PS-22/9/9 and 22/9/9 aliquots were purified with RNeasy spin columns and loaded onto a denaturing 7 M urea, 20% polyacrylamide gel for electrophoretic separation over 30 min at 100 V. Afterwards the gels were imaged for Cy3 and Cy5 fluorescence on a Typhoon imager (Fig. 9b). 22/9/9 exhibited partial degradation as soon as 2 h, with nearly complete degradation by 48 h. Conversely, PS-22/9/9 remained intact for the full 48 h incubation. The degradation of the 22/9/9 probe was likely mediated by a 3′ to 5′ exonuclease, the most common serum nuclease.30 However, the enzyme responsible for degradation of phosphorothioated oligonucleotides has not been identified.25
Biological assessment
In addition to improving nuclease resistance, phosphorothioation-enhanced oligo-protein adsorption has also been reported to assist in oligo delivery. The mechanism by which this modification increases cellular uptake is still under study, but is likely related to enhanced binding to proteins such as albumin, which act as a cloak and escort the oligo through various endocytotic pathways.29,31 This feature is potentially attractive as a method of delivering the probe, which lacks the 5′ CPP found in earlier TIVA constructs. We investigated this effect by incubating human fibroblast cells with 0.5 μM PS-22/9/9 or 22/9/9, without use of CPP or other delivery agent. Images of Cy3 and Cy5 FRET emission were collected using an Olympus Fluoview F1000 inverted confocal microscope at 1.5 h, 6 h, and 24 h (Fig. 10). From these images we observed uptake of the phosphorothioated probe within 1.5 h, with FRET signals that remain constant throughout the 24 h experiment. Conversely, 22/9/9 was observed to remain predominantly in solution with limited fluorescence signal within cells and significantly lower FRET signal after 24 h.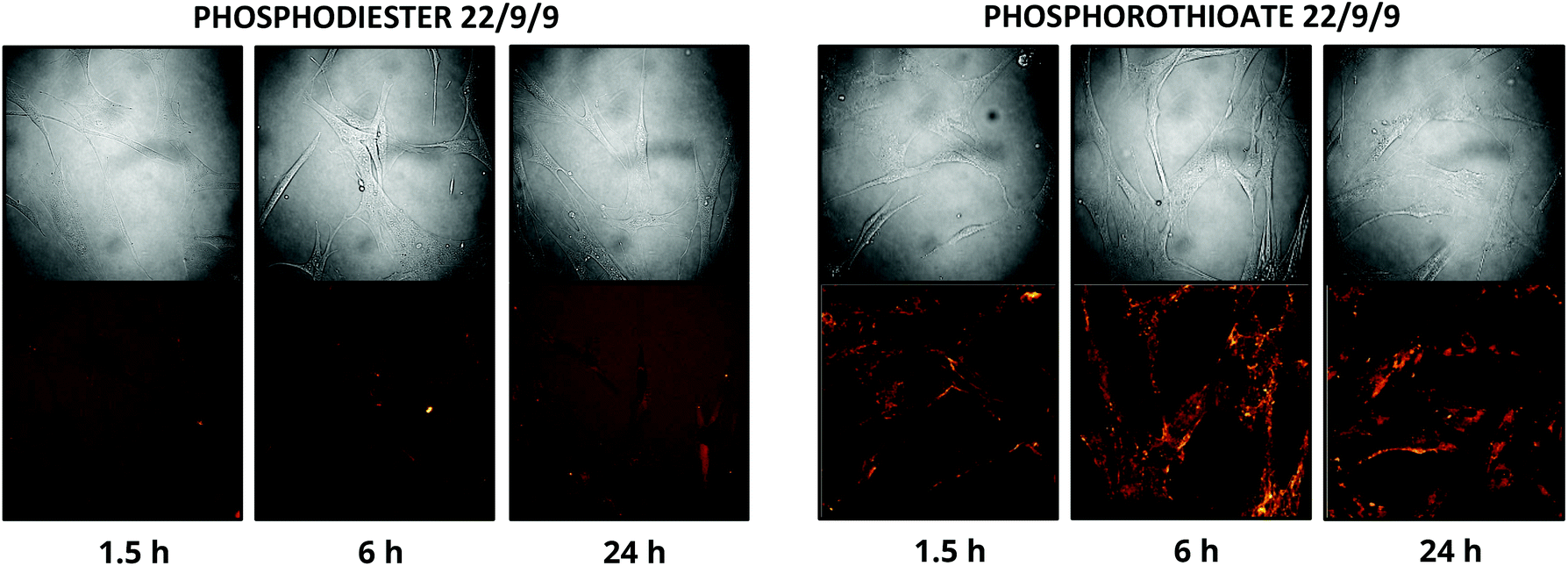 | ||
| Fig. 10 (A) 24 h uptake of 22/9/9 (left) and PS-22/9/9 (right) TIVA probes in cultured human fibroblasts. Cy3 fluorescence is shown in green and Cy5 FRET signal in red. | ||
Conclusions
In summary, we have targeted four different aspects of TIVA performance: (1) increased duplex thermal stability, (2) higher RNA capture affinity, (3) improved caging vs. target RNA binding, and (4) enhanced nuclease resistance. A longer, 22/9/9 duplex and (D-Arg)9 CPP were identified as important contributors to thermal stability, at all salt conditions tested (10–100 mM NaCl). To reduce hairpin fraying we introduced a terminal GC pair to the duplex. This addition had the effect of marginally diminishing TIVA FRET efficiency alone in buffer, but critically reduced fraying in the presence of polyA RNA by more than half.Lastly, we synthesized a phosphorothioated TIVA variant that was stable in the presence of 10% serum for 48 h and exhibited unaided cellular uptake in human fibroblast cells. These results were encouraging, and may enable PS-22/9/9 TIVA to better withstand harsh in vivo conditions. Although the stability of the duplex against nucleases may be somewhat reinforced by non-specific protein adsorption, additional measures must be taken to offset the Tm penalty imposed by phosphorothioation, such as including the terminal GC pair previously discussed. Additionally, a (D-Arg)9 peptide could be attached using alternative, sulfur-compatible conjugation chemistry such as using a 5′ amine modifier and maleimide-modified CPP. Our preliminary experiments in whole organisms suggest that phosphorothioated oligos such as PS-22/9/9 may be stable for 2–4 h but are not sufficiently robust for 24 h experiments, perhaps due to protein binding and trafficking. As needed, alternative approaches will be explored, such as employing peptide nucleic acids (PNAs) or other modified backbones to further extend TIVA probe lifetime.
Experimental
18/7/7 ±(D-Arg)9 and 22/9/9 ±(D-Arg)9 TIVA probes
Synthesis and characterization of 18/7/7, 18/7/7 +(D-Arg)9 and 22/9/9 TIVA probes were performed as described previously.6,19 22/9/9 +(D-Arg)9 probe was synthesized as described for 22/9/9 GC +(D-Arg)9 but RP-HPLC of the cleaved probe was performed on an older gradient: 10% B to 60% B over 40 min, 60% to 80% over 10 min, with the product eluting at roughly 50 min (Fig. S5†). AX-HPLC of the CPP-conjugated product was performed as described for 22/9/9 GC +(D-Arg)9 (Fig. S6†). Overall yield was estimated to be 30 nmol for a 1.0 μmol scale synthesis (3% yield) by A260 (ε = 446000 L (mol cm)−1).
Synthesis of 22/9/9 GC +(D-Arg)9 TIVA probe
TIVA probe production was conducted using phosphoramidite chemistry in a room illuminated by red lights (Tasodin B01HRMPBQQ, approx. 620–640 nm). The initial automated synthesis was performed at a 1 μmol scale on an ABI 394 synthesizer according to manufacturer's instructions except where noted. Phosphoramidites (Glen Research, Sterling, VA) were thawed in a desiccator for 10 min prior to use. To limit the time that moisture-sensitive modifier phosphoramidites spent on the instrument before coupling, the syntheses were split into segments such that modifiers were loaded onto the instrument immediately prior to use. Coupling efficiencies were further improved by dissolving the amidites in rigorously anhydrous acetonitrile (<10 ppm H2O), changing coupling reagents weekly, allowing couplings to proceed for 6 min, and performing syntheses in a chamber dehumidified to <35% atmospheric humidity. These protocol advancements have led to reproducible synthesis of TIVA probe molecules, with much higher purity and better isolated yields (Fig. S7†).After solid-phase synthesis of TIVA probe, the oligo was deprotected with the DMT group left on and cleaved from the support in 1.3 mL concentrated ammonium hydroxide at rt for 16–18 h. The ammonium hydroxide was then removed by venting for 1 h followed by drying under mild vacuum at rt for 50 min. The support was washed four times with 750 μL of 50:50 water:methanol, and the collected oligo solution was syringe filtered to 20 μm. The product was then isolated by reverse-phase (RP) HPLC (Agilent 1100S) on a C18 column at 1.0 mL min−1, 40 °C. A gradient of increasing acetonitrile (B) in 0.1 M TEAA (A) on a C18 column was employed for purification: 30% B to 40% B over 30 min, 40% B up to 90% B over 5 min, 90% B sustained for 3 min, and then back to 10% B over 8 min. The product eluted at roughly 28 min, based upon monitoring of base (254 nm), Cy3 (552 nm), and Cy5 (643 nm) absorbances (Fig. S1†), and a yield of 150 nmol (15% yield) was determined based upon A260 (ε = 463000 L (mol cm)−1 estimated via idtdna.com/calc/analyzer) and starting scale of 1.0 μmol. After RP-HPLC purification the 22/9/9 GC probe was prepared for conjugation to (D-Arg)9 through concentration to 300–350 μM by vacufuge at rt, before being split into 10 nmol aliquots and diluted to 30 μL each. Sodium phosphate buffer (10 μL of 100 mM, pH 7.1) and 4 μL of TCEP (Thermo Scientific) were added to reduce the disulfide bridge connecting the DMT group to the 5′ thiol modifier. This reduction proceeded for 1 h at rt before being eluted through Nap-10 column (GE Healthcare) to both desalt the solution and remove cleaved alkyl-DMT groups.
After concentrating the eluent TIVA probe to 100 μM by vacufuge at rt, the CPP conjugation was performed using 600 μL formamide (Fisher), 40 μL of 2.0 M TEAA (Glen Research), and 80 μL of 1 μM Cys(Npys)-(D-Arg)9 peptide (AS-61206, Anaspec). The mixture was vortexed briefly and the conjugation was allowed to proceed for 24–36 h at rt. Conjugated probe was purified by anion exchange HPLC (Agilent 1100S, 1.000 mL min−1, RT) using a Resource Q column (GE Healthcare) with a gradient of 100% buffer A (20 mM Tris-HCl (pH 6.8), 50% formamide) and 0% buffer B (20 mM Tris-HCl (pH 6.8), 50% formamide, 400 mM NaClO4) transitioning to 0% buffer A and 100% buffer B over 30 min. The conjugated product eluted at ∼23 min, prior to unconjugated material at ∼25 min (Fig. S2†). The final yield for the CPP conjugation was 58 nmol (38%), for a total yield of 5.8% relative to the 1.0 μmol starting synthesis, measured as previously described.
Isolated 22/9/9 GC +(D-Arg)9 TIVA probe was buffer exchanged into STE storage buffer (10 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl) using 10k MW cutoff Ultra-4 cellulose spin columns (Amicon) in an Eppendorf centrifuge (5810 R) cooled to 4 °C. After complete buffer exchange the probe was washed four additional times with ice-cold STE buffer and spun to a final concentration of 100 μM. Finally, the TIVA probe was separated into 3.0 nmol aliquots and stored at −20 °C.
Synthesis of PS-22/9/9 TIVA probe
Synthesis of PS-22/9/9 probe followed the protocol for 22/9/9 GC +(D-Arg)9 probe except where noted. To achieve phosphorothioation, the oxidizing reagent was replaced by a sulfurizing reagent (40-4037-xx, Glen Research) during solid-phase synthesis, with a 360 s sulfurizing step employed prior to capping per manufacturer instructions. After cleavage the DMT group was removed prior to purification. The DMT-off product was purified on a gradient of increasing acetonitrile (B) in 0.1 M TEAA (A): 30% B to 40% B over 30 min, rising to 90% B in 5 min, sustaining at 90% for 3 min, then dropping to 10% over 8 min, with the product eluting at ∼27 min (Fig. S3†). The yield was determined to be 190 nmol for the 1.0 μmol synthesis (19% yield) by A260 (ε = 446000 L (mol cm)−1). No further CPP conjugation was necessary and the probe was buffer exchanged into STE as previously described.
Mass determination
Oligonucleotide masses were determined by Novatia, LLC using a LCMS system with electrospray injection (Oligo HTCS).32 For the 22/9/9 GC +(D-Arg)9 probe the product was observed at 17591.7 Da. Minor +28 Da and +54 Da adducts were also observed, potentially cyanoethyl or Fe groups. For the PS-22/9/9 probe the product was observed at 15898.9 Da in addition to a minor +15 Da adduct likely corresponding to an unremoved cyanoethyl group. For the 22/9/9 +(D-Arg)9 probe the product was observed at 16913.30 Da, with a minor-U deletion product at −308 Da (Fig. S8†).
T m and FRET sample preparation
Four 60 μL (FRET) or 250 μL (Tm) TIVA probe samples were prepared at 1.0 μM in 1× STE buffer: two samples mixed with one equivalent of 30mer polyA 2′-OMe RNA as a model polyA tail (“+polyA”) and two samples without polyA RNA (“−polyA”). One −polyA and one +polyA sample were then photoactivated (“+hν”), while the remaining two samples were left unactivated (“−hν”). Photoactivation was performed using a TL-355R Ultraviolet Transilluminator (Spectroline, Westbury, NY), irradiating the samples at 365 nm, 9 mW cm−2 for 8 min, followed by brief mixing, and then irradiating for an additional 8 min. Finally, the samples were incubated at 37 °C for 5 min and then cooled to rt.Melting point determination
The thermal stabilities of TIVA probe samples were assessed using a Beckman Coulter DU800 UV-Vis spectophotometer and Peltier temperature controller. Samples were heated at 1.0 °C min−1 from 15 °C to 90 °C, held at 90 °C for 10 min, and then cooled at 1.0 °C min−1 from 90 °C back to 15 °C, and A260 was measured each minute. Tm values were assigned to each phase using first-derivative analysis.FRET measurement
Cy3–Cy5 FRET emission was measured on a Cary Eclipse fluorimeter (Varian) with Cary Temperature Controller (Agilent) set to 20 °C. Cy3 was excited at 552 nm and the emission spectrum was collected from 555–705 nm. FRET efficiency was approximated using the formula FRET = Ia/(Ia + (Id × γ)), where Ia is the emission intensity of the FRET acceptor at 665 nm, Id is the emission intensity of the FRET donor at 565 nm, and γ is a correction factor of 2.0 for the two fluorophores’ differing quantum yields. We chose to utilize this approximation method over more complex photobleaching or single-fluorophore probe methods as it can also be used to readily estimate the FRET signal from TIVA probe in real-time microscopy applications.Fluorimeter-based stability assay
Phosphorothioated (PS-22/9/9) or phosphodiester (22/9/9) TIVA probes were diluted to 1.0 μM in PBS with 10% FBS and incubated at 37 °C for 24 h in a Cary Eclipse fluorimeter. Cy3 emission at 565 nm and Cy5 emission at 665 nm were recorded every 5 min for the first hour and then every 20 min for hours 2–24. FRET efficiencies were approximated as previously noted, and these values were then normalized against their respective starting values to depict the relative change in FRET over time for each probe.Gel-based stability assay
PS-22/9/9 and 22/9/9 TIVA probes were prepared as 250 μL, 1.0 μM solutions in 90% PBS/10% FBS and incubated at 37 °C for 48 h. 40 μL aliquots were collected and snap-frozen in liquid nitrogen at 0, 2, 4, 8, 24, and 48 h. Each aliquot was then purified by an RNeasy Mini Kit (Qiagen) before being dried to a pellet and redissolved in 5 μL of formamide. Each sample was then mixed with 1.0 μL loading buffer before being run on a 7.0 M urea polyacrylamide denaturing gel at 300 V for 30 min. Gels were imaged for Cy3 and Cy5 fluorescence on a Typhoon FLA 7000 laser scanner (GE).Cell culture and confocal microscopy
CCD-1112Sk human neonatal foreskin-derived fibroblasts (ATCC, Manassas, VA) were cultured in IMDM with 10% FBS and penicillin/streptomycin in a 37 °C incubator with 5% CO2. For microscopy experiments split cells were seeded onto P35GC-1.5-14-C 35 mm Petri dishes with a 14 mm glass microwell (MatTek Corporation, Ashland, MA) and cultured for an additional 24 h to reach 70–90% confluency. At T0 the dishes were washed twice with DPBS and replaced with fresh serum-free media containing 0.5 μM PS-22/9/9 or 22/9/9 TIVA and then returned to the incubator. At 1.5, 6, and 24 h the dishes were removed and imaged using a Fluoview FV1000 inverted confocal microscope (Olympus) with a 1.30 NA UPLFLN 40× oil objective (Olympus), and 543 nm green HeNe laser. Images were collected for both Cy3 emission (555 nm–625 nm bandpass filter) and Cy5 FRET emission (650 nm long-pass filter).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NIH grant R01 GM083030 (to IJD) for support of this research. We acknowledge Eric Roesch (Glen Research) for his kind support in troubleshooting various matters related to solid-phase synthesis over the past several years, as well as Ryan Kubanoff for his skilled management of the UPenn Biological Chemistry Resource Center and cell culture facilities.Notes and references
- K. DeCarlo, A. Emley, O. E. Dadzie and M. Mahalingam, in Laser Capture Microdissection: Methods and Protocols, ed. G. I. Murray, Humana Press, Totowa, NJ, 2011, pp. 1–15, DOI:10.1007/978-1-61779-163-5_1.
- J. Eberwine and T. Bartfai, Pharmacol. Ther., 2011, 129, 241–259 CrossRef CAS PubMed.
- G. Donati, Immunol. Cell Biol., 2016, 94, 250–255 CrossRef CAS PubMed.
- J. P. Junker and A. van Oudenaarden, Mol. Cell, 2015, 58, 563–564 CrossRef CAS PubMed.
- M. C. Lee, F. J. Lopez-Diaz, S. Y. Khan, M. A. Tariq, Y. Dayn, C. J. Vaske, A. J. Radenbaugh, H. J. Kim, B. M. Emerson and N. Pourmand, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4726–E4735 CrossRef CAS PubMed.
- D. Lovatt, B. K. Ruble, J. Lee, H. Dueck, T. K. Kim, S. Fisher, C. Francis, J. M. Spaethling, J. A. Wolf, M. S. Grady, A. V. Ulyanova, S. B. Yeldell, J. C. Griepenburg, P. T. Buckley, J. Kim, J. Y. Sul, I. J. Dmochowski and J. Eberwine, Nat. Methods, 2014, 11, 190–196 CrossRef CAS PubMed.
- S.-S. Kim, S. Subramanya, D. Peer, M. Shimaoka and P. Shankar, in Antiviral RNAi: Concepts, Methods, and Applications, ed. R. P. van Rij, Humana Press, Totowa, NJ, 2011, pp. 339–353, DOI:10.1007/978-1-61779-037-9_21.
- J. Liu, T. Gaj, J. T. Patterson, S. J. Sirk and C. F. Barbas, 3rd, PLoS One, 2014, 9, e85755 Search PubMed.
- K. H. Chen, A. N. Boettiger, J. R. Moffitt, S. Wang and X. Zhuang, Science, 2015, 348, aaa6090 CrossRef PubMed.
- J. R. Moffitt, J. Hao, G. Wang, K. H. Chen, H. P. Babcock and X. Zhuang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11046–11051 CrossRef CAS PubMed.
- F. Chen, A. T. Wassie, A. J. Cote, A. Sinha, S. Alon, S. Asano, E. R. Daugharthy, J. B. Chang, A. Marblestone, G. M. Church, A. Raj and E. S. Boyden, Nat. Methods, 2016, 13, 679–684 CrossRef CAS PubMed.
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed.
- R. Zilionis, J. Nainys, A. Veres, V. Savova, D. Zemmour, A. M. Klein and L. Mazutis, Nat. Protocols, 2017, 12, 44–73 CAS.
- T. M. Gierahn, M. H. Wadsworth Ii, T. K. Hughes, B. D. Bryson, A. Butler, R. Satija, S. Fortune, J. C. Love and A. K. Shalek, Nat. Methods, 2017, 14, 395–398 CrossRef CAS PubMed.
- S. Verma and F. Eckstein, Annu. Rev. Biochem., 1998, 67, 99–134 CrossRef CAS PubMed.
- J. K. Frederiksen, N. S. Li, R. Das, D. Herschlag and J. A. Piccirilli, RNA, 2012, 18, 1123–1141 CrossRef CAS PubMed.
- S. M. Fica, N. Tuttle, T. Novak, N. S. Li, J. Lu, P. Koodathingal, Q. Dai, J. P. Staley and J. A. Piccirilli, Nature, 2013, 503, 229–234 CAS.
- Y. L. Zhang, F. Hollfelder, S. J. Gordon, L. Chen, Y. F. Keng, L. Wu, D. Herschlag and Z. Y. Zhang, Biochemistry, 1999, 38, 12111–12123 CrossRef CAS PubMed.
- B. K. Ruble, Doctor of Philosophy (PhD), University of Pennsylvania, 2012.
- S. E. Andaloussi, T. Lehto, I. Mager, K. Rosenthal-Aizman, I. I. Oprea, O. E. Simonson, H. Sork, K. Ezzat, D. M. Copolovici, K. Kurrikoff, J. R. Viola, E. M. Zaghloul, R. Sillard, H. J. Johansson, F. Said Hassane, P. Guterstam, J. Suhorutsenko, P. M. Moreno, N. Oskolkov, J. Halldin, U. Tedebark, A. Metspalu, B. Lebleu, J. Lehtio, C. I. Smith and U. Langel, Nucleic Acids Res., 2011, 39, 3972–3987 CrossRef PubMed.
- N. Kretschy, M. Sack and M. M. Somoza, Bioconjugate Chem., 2016, 27, 840–848 CrossRef CAS PubMed.
- S. Zell, N. Geis, R. Rutz, S. Schultz, T. Giese and M. Kirschfink, Clin. Exp. Immunol., 2007, 150, 576–584 CrossRef CAS PubMed.
- M. V. Karpuj, K. Giles, S. Gelibter-Niv, M. R. Scott, V. R. Lingappa, F. C. Szoka, D. Peretz, W. Denetclaw and S. B. Prusiner, Mol. Med., 2007, 13, 190–198 CAS.
- D. A. Kocisko, A. Vaillant, K. S. Lee, K. M. Arnold, N. Bertholet, R. E. Race, E. A. Olsen, J. M. Juteau and B. Caughey, Antimicrob. Agents Chemother., 2006, 50, 1034–1044 CrossRef CAS PubMed.
- F. Eckstein, Nucleic Acid Ther., 2014, 24, 374–387 CrossRef CAS PubMed.
- L. Kibler-Herzog, G. Zon, B. Uznanski, G. Whittier and W. D. Wilson, Nucleic Acids Res., 1991, 19, 2979–2986 CrossRef CAS PubMed.
- M. J. Damha, C. J. Wilds, A. Noronha, I. Brukner, G. Borkow, D. Arion and M. A. Parniak, J. Am. Chem. Soc., 1998, 120, 12976–12977 CrossRef CAS.
- P. P. Seth, A. Jazayeri, J. Yu, C. R. Allerson, B. Bhat and E. E. Swayze, Mol. Ther.–Nucleic Acids, 2012, 1, e47 CrossRef PubMed.
- X. H. Liang, H. Sun, W. Shen and S. T. Crooke, Nucleic Acids Res., 2015, 43, 2927–2945 CrossRef CAS PubMed.
- P. S. Eder, R. J. DeVine, J. M. Dagle and J. A. Walder, Antisense Res. Dev., 1991, 1, 141–151 CrossRef CAS PubMed.
- R. L. Juliano, X. Ming, K. Carver and B. Laing, Nucleic Acid Ther., 2014, 24, 101–113 CrossRef CAS PubMed.
- M. E. Hail, B. Elliott and K. Anderson, Am. Biotechnol. Lab., 2004, 22, 12–15 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC spectra, 6 pages; FRET comparison, 1 page; ESI-MS spectra, 1 page. See DOI: 10.1039/c7ob02353g |
| This journal is © The Royal Society of Chemistry 2017 |