
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective C–H bond electro-oxidation of benzylic acetates and alcohols to benzaldehydes†
Mateus R.
Barone
ab and
Alan M.
Jones
 *c
*c
aDepartment of Chemical Engineering, University of Sao Paulo, Brazil
bDivision of Chemistry and Environmental Science, Manchester Metropolitan University, M1 5GD, UK
cSchool of Pharmacy, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: a.m.jones.2@bham.ac.uk
First published on 14th November 2017
Abstract
A chemical oxidant-free and mediator-free, direct electro-oxidation of both benzylic alcohols and benzylic esters are reported. The scope of the reaction is explored as a function of both steric and electronic effects. Expansion of the scope to non-benzylic and heteroaryl substrates is investigated. Functionalisation of esters and alcohols selectively to the aldehyde oxidation level using a traceless electron approach is reported.
Introduction
The controlled and chemo-selective oxidation of primary alcohols to aldehydes and secondary alcohols to the corresponding ketones are fundamental reactions in organic synthesis.1 In turn, these aldehydes and ketones serve as precursors for a variety of complexity generating reactions.2 However, the controlled oxidation of primary alcohols to aldehydes can be problematic due to over-oxidation to the carboxylic acid oxidation state.3 There are a range of versatile chemical oxidants available to the academic and industrial chemist that enable this transformation to be performed on demand.4 However, the majority of these oxidation reactions are stoichiometric in nature and therefore suffer from the generation of quantities of chemical waste.5Recently, the field of electrosynthesis has undergone a renaissance6 and has found application in a variety of organic synthetic transformations, such as: C–H bond activation,7 total synthesis,8 and the Diels–Alder reaction9 amongst others.10 A fundamental advantage of the electrosynthesis approach is the replacement of the need to use stoichiometric oxidants and instead the oxidation reaction is performed on the electrode surface via quantum mechanical tunelling11 or through a mediator in solution.12
To address the challenge of identifying a cleaner oxidation, we explored the use of electrosynthesis to replace the need for both a chemical oxidant and mediator in these Redox transformations. The simultaneous removal of oxidant and mediator would minimise chemical waste associated with the reaction. We have recently investigated the Shono-type oxidation of C–H bonds adjacent to a tertiary amide13 and our initial foray into this area began with attempting to expand the scope of the amide oxidation to esters (Fig. 1). It is known that the Shono electro-oxidation of amides proceeds through an N-acyl iminium species (A).14 It was therefore postulated that a transient O-acyloxonium species (B)15 could form under similar electro-oxidative conditions in esters bearing an α-methylene or methide group.
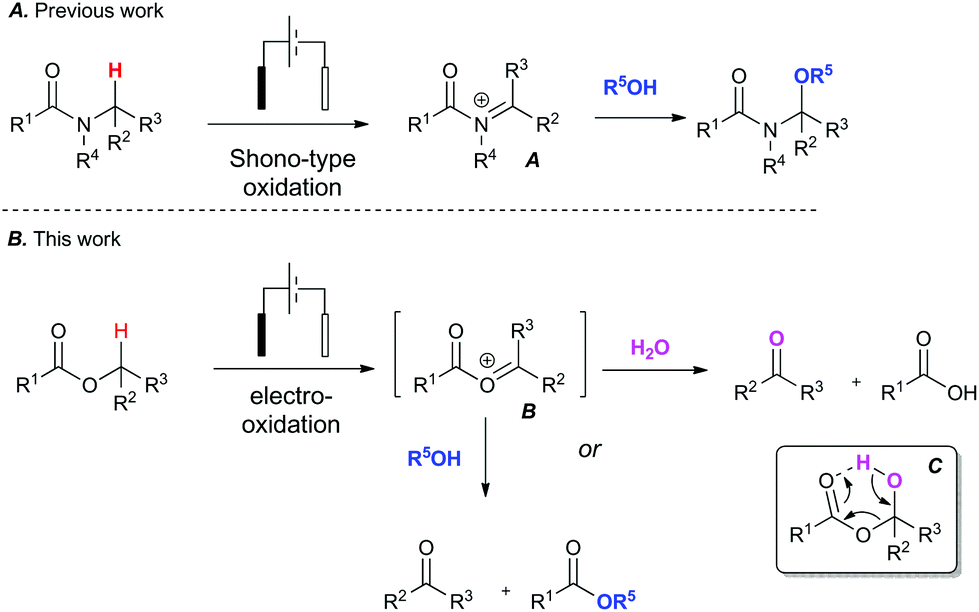 | ||
| Fig. 1 Context: Expanding the scope of amide C–H bond functionalisation from amides (A) to esters (B). | ||
This unstable O-acyloxonium species (B) would in turn react further with adventious water to form the aldehyde product and a carboxylic acid as a by-product via intermediate (C). Alternatively, an ester and aldehyde product could potentially form when conducted in an alcohol. To the best of our knowledge there are only limited reports of electro-oxidative cleavage of an ester group.16
Results and discussion
To probe whether this methodological leap was indeed feasible, a collection of benzylic acetates were prepared including the parent benzyl acetate 1a, a mild electron donating example 1b and a mild electron withdrawing example 1c (structures shown in Table 2). Cyclic voltammetry was recorded for 1a (Fig. 2).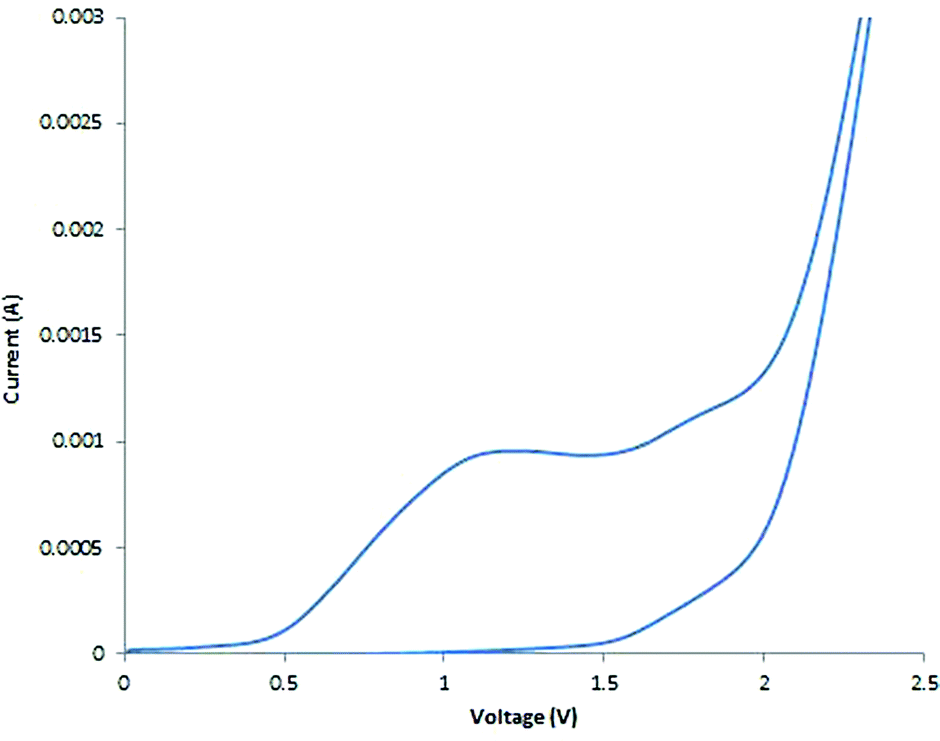 | ||
Fig. 2 Cyclic voltammetry measurement on substrate 1a (5 mM) at a scan rate of 10 mV s−1 in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile:methanol and TBAP (0.5 M) using an RVC working electrode, RVC counter electrode and a Ag/AgCl reference electrode. :1 acetonitrile:methanol and TBAP (0.5 M) using an RVC working electrode, RVC counter electrode and a Ag/AgCl reference electrode. | ||
Using a sensitive measurement of 10 mV s−1 scan rate it was observed in both electrolyte systems (LiClO4 and Bu4NClO4)17 that oxidation waves for all three substrates were observed with +1.0 V (1a), +1.2 V (1b) and +0.9 V (1c) oxidation potentials (relative to Ag/AgCl) and a slightly improved peak current (Ip) measurement in Bu4NClO4. On the basis of this positive oxidation result, screening of potential conditions to enable viable electro-oxidation of model benzylic acetate 1a was attempted (Table 1).
| Entry | Electrolyte | Solvent | Voltage (mV) | Current (mA) | Conversion (%) |
|---|---|---|---|---|---|
| 1 | Bu4NClO4 | MeCN/MeOH | +900 | — | 0 |
| 2 | Bu4NClO4 | MeCN/MeOH | +1000 | — | 0 |
| 3 | Bu4NClO4 | MeCN/MeOH | +1100 | — | <5 |
| 4 | Bu4NClO4 | MeCN/MeOH | +1200 | — | <5 |
| 5 | Bu4NClO4 | MeCN/MeOH | +1300 | — | Degradation |
| 6 | LiClO4 | MeCN/MeOH | +1100 | — | <10 |
| 7 | LiClO4 | MeCN/MeOH | +1200 | — | <10 |
| 8 | LiClO4 | MeCN/MeOH | +1300 | — | Degradation |
| 9 | Bu4NClO4 | MeCN/MeOH | — | 20 | 27 |
| 10 | Bu4NClO4 | MeOH | — | 20 | 34 |
| 11 | Bu4NClO4 | MeOH | — | 10 | 49 |
| 12 | LiClO4 | MeCN/MeOH | — | 20 | 25 |
| 13 | LiClO4 | MeOH | — | 20 | 57 |
| 14 | LiClO4 | MeOH | — | 10 | 84 |
| 15 | LiClO4 | MeOH/H2O | — | 10 | 38 |
| 16 | LiClO4 | MeOH | — | — | 0 |
In the first instance, potentiostatic conditions were screened with 1a (entries 1–8) using both Bu4NClO4 and LiClO4 as the electrolyte. The applied voltage was varied around the observed oxidation potential for 1a (cyclic voltammetry measurement, +1.0 V) with up to an extra 300 mV applied to compensate for expected iR drop across the electrode surface.18 In all cases, the reaction was performed until F mol−1 equivalent to 4 electrons per mole of substrate was passed or starting material consumption was observed. Near the oxidation potential of 1a trace conversion to the desired aldehyde 2a was observed in both electrolyte systems (entries 3, 4 and 6, 7, respectively). However, at higher applied voltages degradation products were observed (entries 5 and 8, respectively), coupled with excessive time required for sufficient charge to be passed (24 h to 5 days), a controlled voltage approach was ruled out in this system early on. Switching to a galvanostatic approach, using the same electrolyte systems and reticulated vitreous carbon (RVC) electrodes produced more promising results (entries 9–15).
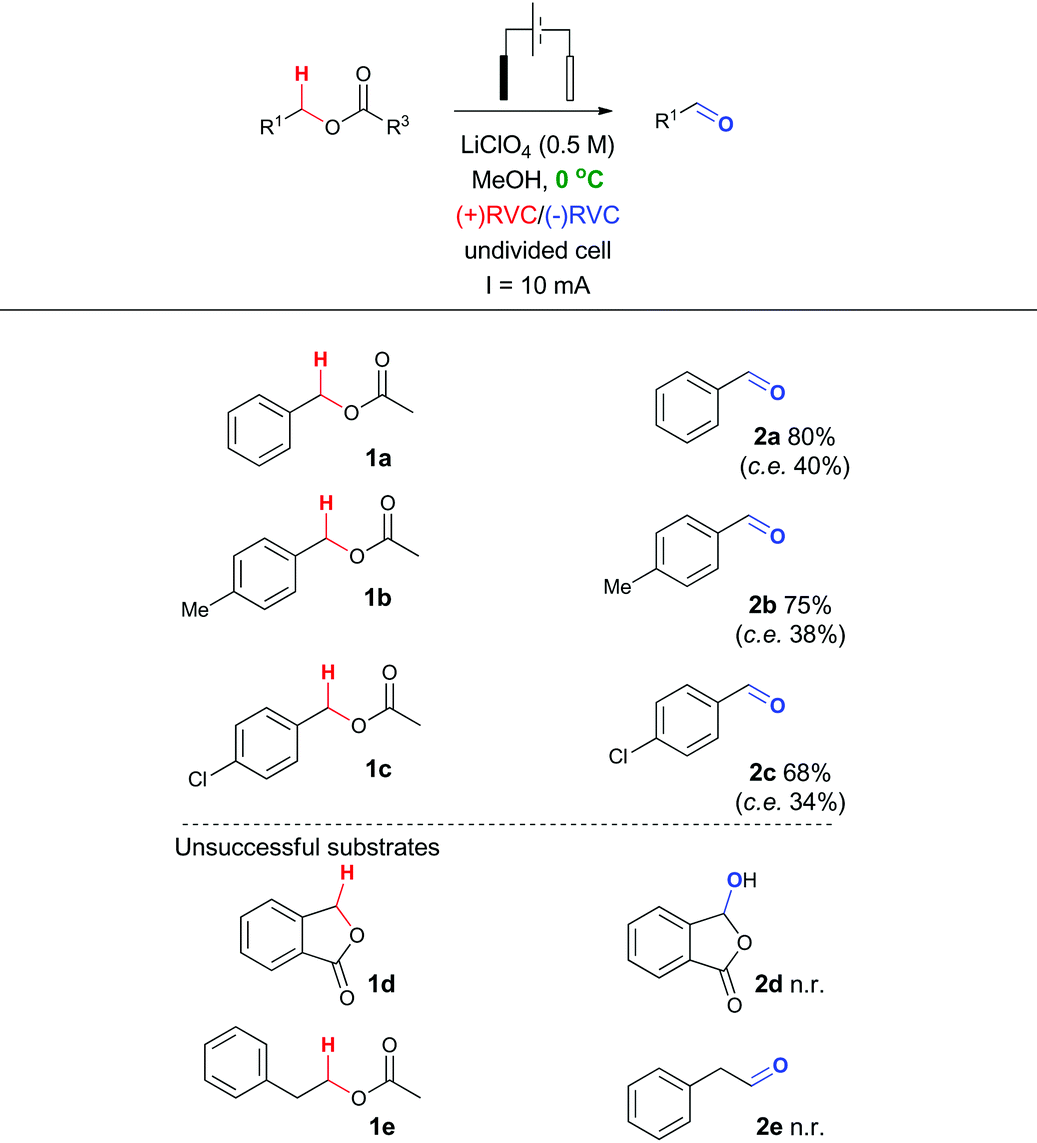
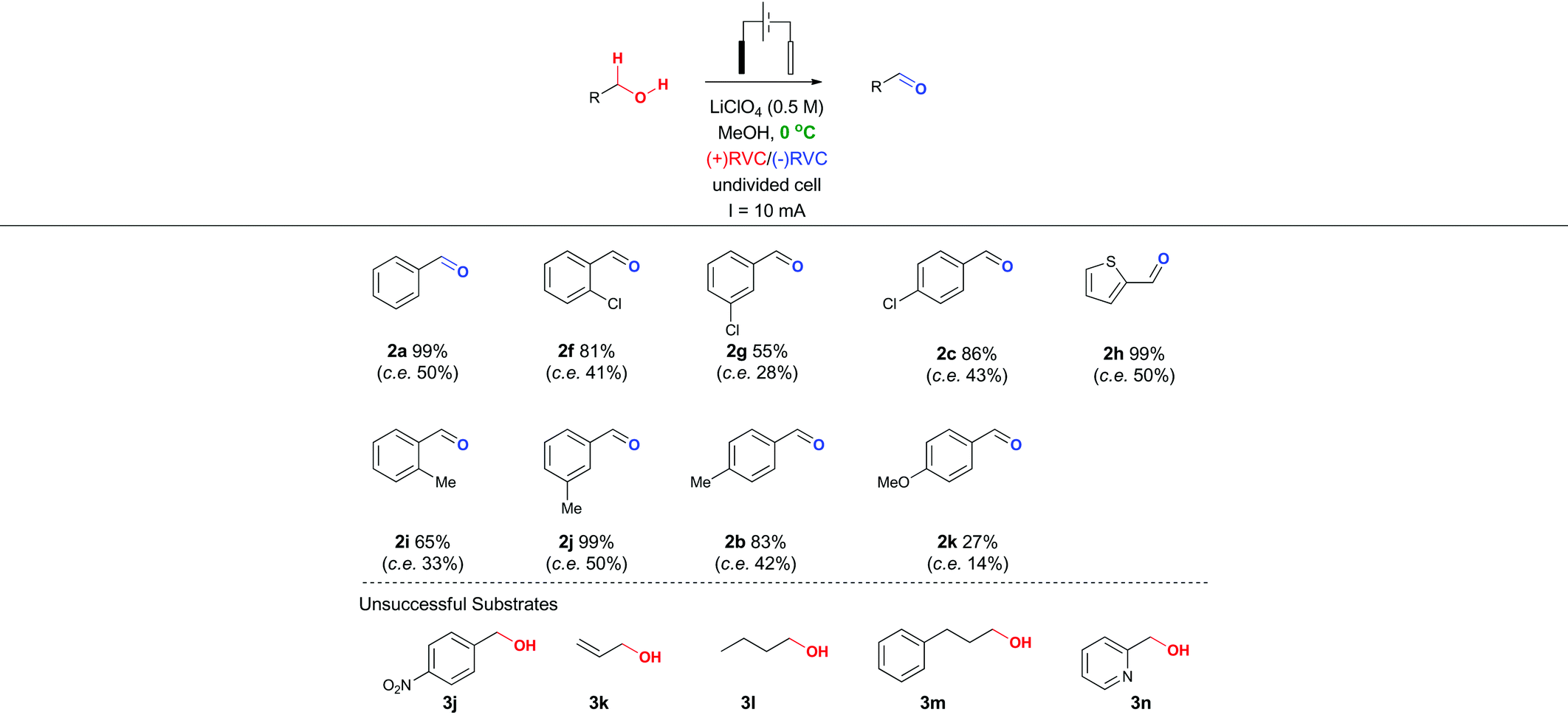
In particular, it was observed that a single solvent system gave higher yields than the previously optimized solvent system for the Shono oxidation,13 acetonitrile-methanol (entries 9 vs. 10 and 12 vs. 13, respectively). Lowering the current density across the electrode surface from an applied 20 mA to 10 mA, led to a doubling in reaction time (approx. 5–6 h) and a concomitant improvement in reaction yield in both electrolyte systems (entries 11 vs. 10 and 14 vs. 13, respectively). Lowering the current rate further led to modest improvements in conversion but unacceptable lengthening of the reaction time. It became clear that lithium perchlorate was superior to tetrabutylammonium perchlorate (entry 14 vs. 11) plus coupled with its ease of separation from the reaction products, was selected as the electrolyte. To probe whether additional water improved the reaction yield (entry 15) based on a postulated mechanism led to a reduced yield versus entry 14. Furthermore, passing no electrical current led to no reaction (entry 16). To test the hypothesis that C–H bond oxidation adjacent to an ester was possible, a collection of benzylic esters were prepared. Our initial results are detailed in Table 2. It was found using the optimised conditions, appreciable amounts of the desired aldehyde (2a–2c) were obtained from the benzylic acetates (1a–1c). To address, the mechanism issue identified in Fig. 1, a simple aqueous base wash removed the by-product. However, there were still limitations to this approach for example electro-oxidation of cyclic benzylic ester 1d or homologated ester 1e, did not afford the aldehyde 2d and 2e, respectively. These limitations coupled with the use of an acyl ancillary group still did not meet our green chemistry standards due to the additional manipulation step required to prepare the acetate. We therefore considered whether a stabilising group on the heteroatom adjacent to the C–H bond was essential for successful electro-oxidation. Promisingly, cyclic voltammetry measurements on benzyl alcohol 3a showed an oxidation wave at +1.2 V relative to Ag/AgCl.19 Using our previously optimised conditions for the ester electro-oxidation we explored the direct, mediator-free, oxidation on a range of commercially available alcohols (Table 3).
|
|
It was possible to cleanly convert benzyl alcohol (3a) to benzaldehyde (2a) under the mild conditions of lithium perchlorate in methanol in near quantitative yield and in improved yield compared to the chemical manipulation of forming the benzyl acetate (99% vs. 80%, respectively).20 The scope of this reaction was further investigated via exploration of the effects of electron withdrawing and donating group around the ring system and steric effects around the reacting centre. The use of a chlorine atom as an electron withdrawing group in the ortho- (2f, 81%), meta- (2g, 55%) and para- (2h, 86%) positions was well tolerated. The use of a methyl group as a mild electron donating group in the ortho- (2i, 66%), meta- (2j, 99%) and para- (2b, 83%) positions also afforded good to excellent yields of the aldehyde.
Intriguingly, a strong electron donating group para to the reacting centre (from para-methoxy benzyl alcohol) resulted in a greatly reduced yield of aldehyde 2k (27%). The relatively low yield of 2k compared with other benzaldehydes is likely to be due to the para-methoxy group stabilizing the carbon-centred benzylic radical leading to further unproductive reaction pathways and oxidative decomposition of 2k.
The result for a strongly electron withdrawing group in the 4-position of the substrate 3j afforded only trace conversion to the desired product as a result of the highly electron withdrawing nature of the para-nitro group. Based on the experimental results above and EPR literature21 of related systems a tentative mechanism can be proposed (Scheme 1A).
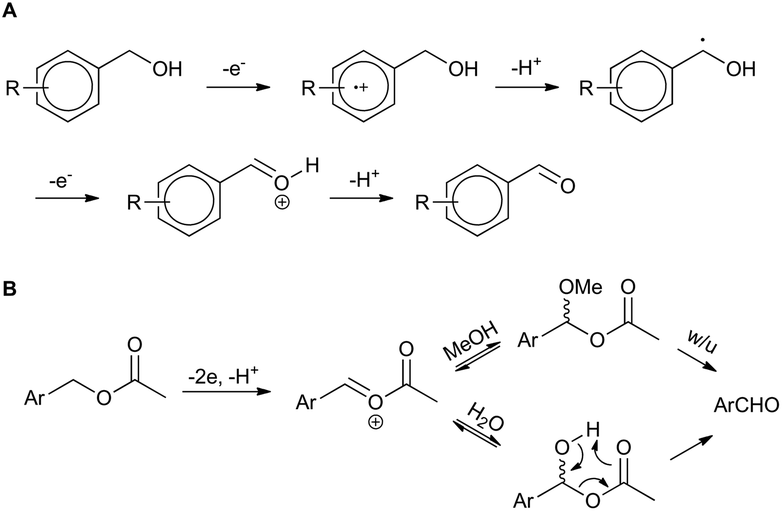 | ||
| Scheme 1 Proposed mechanisms for (a) benzyl alcohol and (b) benzyl acetate electro-oxidation (w/u = work-up). | ||
Electro-oxidation of the benzyl alcohol reveals a benzyl radical, subsequent oxidation of the hydroxyl group will deliver an aldehyde after deprotonation. Similarly, the benzylic acetate system could proceed via analogous initial steps to an O-acyloxonium species that could be trapped with water or methanol to deliver a hemiacetal or acetal species, respectively. In turn, the hemi-acetal or acetal intermediate would collapse upon work-up to deliver the aldehyde (Scheme 1B). We have shown that mild electron withdrawing and donating groups are tolerated in both systems but those that strongly donate or withdraw electrons were less compatible with direct electro-oxidation. Nitrogen containing heterocycle (3n) and non-benzylic substrates (3k–3m) gave trace conversion (<5%) to the aldehyde. However, changing the benzene ring to a thiophene was successful, affording aldehyde 2h in quantitative yield, demonstrating the utility of the approach in other classes of heterocyclic systems.
Conclusion
The use of mediator-free, galvanostatic, direct electro-oxidation of benzylic alcohols and acetates shows scope on a range of substrates and offers an alternative and complementary approach to the preparation of valuable aldehydes. Mechanistic investigations to determine the sequence of electro-oxidation steps are now underway.Experimental section
General methods
Reactions were carried out under nitrogen. Organic solutions were dried over MgSO4. Starting materials were purchased from commercial suppliers and were used without further purification. Solvents were dried over molecular sieves (3–4 Å) Flash silica chromatography was performed using Sigma-Aldrich high-purity grade, pore size 60 Å, 200–400 mesh particle size silica gel. 1H and 13C NMR spectra were recorded on a JEOL ECS 400 NMR Spectrometer at 400 MHz or Bruker AVIII 300 or 400 MHz spectrometers. Chemical shifts (δ) are reported relative to TMS (δ = 0) and/or referenced to the solvent in which they were measured. All chemical shifts (δ) are reported in parts per million, and coupling constants (J) are reported in Hertz. Low and High-resolution mass spectrometry analysis were obtained using an Agilent 6450 LC-MS/MS system. Infrared (IR) spectra were recorded on a ThermoScientific Nicolet Impact-380 ATR-FTIR spectrometer. Electrosynthesis and cyclic voltammetry were performed using a Metrohm Autolab PGSTAT100N with a 10 Amp Booster (Metrohm Autolab, UK) with an undivided electrochemical cell. Data was processed using Autolab Nova software (version 2.0). The electrode system for electrosynthesis consisted of two reticulated vitreous carbon (RVC) electrodes (supplier: ERG Aerospace; specifications: 45 pores per inch (PPI); 800 square foot per cubic foot; size 20 × 11 × 5 mm) for galvanostatic reactions plus an additional pseudo Ag/AgCl reference electrode for potentiostatic reactions. Please see ref. 13 for further details and images of the reaction set-up.General procedures and safety statements
Caution: Perchlorates – May intensify fire. In combination with flammable solvents, all sources of ignition should be avoided. During the concentration stage of the work-up, low temperature rotary evaporation with a suitable shield should be employed. The aqueous layer (after extraction) was treated with excess sodium sulphite prior to disposal.
Caution: Passing an electrical current through flammable organic solvent – The potentiostat and wiring should be regularly inspected for faults and pass all relevant electrical tests prior to use.
Spectroscopic data
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. J. thanks the Royal Society of London (United Kingdom) for the award of a research grant (RG150135). The authors thank the analytical services at Manchester Metropolitan University and the Centre for Chemical and Materials Analysis in the School of Chemistry at the University of Birmingham for analytical support. The NMR instruments used in this research were obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).Notes and references
- (a) G. Tojo and M. I. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer, New York, 2006 Search PubMed; (b) G. Franz and R. A. Sheldon, Oxidation, in Ullmann's Encyclopaedia of Industrial Chemistry, VCH-Verlag, Weinheim, 2000 Search PubMed; (c) R. C. Larock, Comprehensive Organic Transformations, Wiley, New York, 2nd edn, 1999 Search PubMed.
- (a) D. Lee, J. K. Sello and S. L. Schreiber, Org. Lett., 2000, 2, 709 CrossRef CAS PubMed; (b) A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971 CrossRef CAS.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332 Search PubMed.
- (a) D. Könning, T. Olbrisch, F. D. Sypaseuth, C. C. Tzschucke and M. Christmann, Chem. Commun., 2014, 50, 5014 RSC; (b) J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742 CrossRef CAS PubMed; (c) S. B. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639 CrossRef.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed.
- (a) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed; (b) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 78 CrossRef PubMed; (c) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed.
- (a) S. Liang, C.-C. Zeng, H.-Y. Tian, B.-G. Sun, X.-G. Luo and F. Z. Ren, J. Org. Chem., 2016, 81, 11565 CrossRef CAS PubMed; (b) R. Hayashi, A. Shimizu, Y. Song, Y. Ashikari, T. Nokami and J.-I. Yoshida, Chem. – Eur. J., 2016, 23, 61 CrossRef PubMed; (c) Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448 CrossRef CAS PubMed; (d) A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 11868 CrossRef PubMed.
- B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571 CrossRef CAS PubMed.
- J. C. Moore, E. S. Davies, D. A. Walsh, P. Sharmaab and J. E. Moses, Chem. Commun., 2014, 50, 12523 RSC.
- P. Du, J. L. Brosmer and D. G. Peters, Org. Lett., 2011, 13, 4072 CrossRef CAS PubMed.
- S. G. Christov, Ber. Bunsenges. Phys. Chem., 1975, 79, 357 CrossRef.
- (a) R. Ciriminna, M. Ghahremani, B. Karimi and M. Pagliaro, ChemistryOpen, 2017, 6, 5 CrossRef CAS PubMed; (b) A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406 CrossRef CAS PubMed; (c) C.-B. Bai, N.-X. Wang, X.-W. Lan, Y.-J. Wang, Y. Xing, J.-L. Wen, X.-W. Gao and W. Zhang, Sci. Rep., 2016, 6, 20163 Search PubMed.
- (a) A. M. Jones and C. E. Banks, Beilstein J. Org. Chem., 2014, 10, 3056 CrossRef PubMed; (b) P. Alfonso-Súarez, A. V. Kolliopoulos, J. P. Smith, C. E. Banks and A. M. Jones, Tetrahedron Lett., 2015, 56, 6863 CrossRef.
- T. Shono, Y. Matsumura and K. Tsubata, J. Am. Chem. Soc., 1981, 103, 1172 CrossRef CAS.
- A. K. Ghosh, S. S. Kulkarni, C.-X. Xu and K. Shurrush, Tetrahedron: Asymmetry, 2008, 19, 1020 CrossRef CAS PubMed.
- (a) E. Baciocchi, M. Mattioli, R. Romano and R. Ruzziconi, J. Org. Chem., 1991, 56, 7154 CrossRef CAS; (b) C.-C. Zeng and J. Y. Becker, J. Org. Chem., 2004, 69, 1053 CrossRef CAS PubMed; (c) E. A. Mayeda, L. L. Miller and J. F. Wolf, J. Am. Chem. Soc., 1972, 94, 6812 CrossRef CAS; (d) H. Lund, Acta Chem. Scand., 1957, 11, 491 CrossRef CAS.
- Scan rate sweeps for 1a–1c can be found in the ESI.†.
- W. Oelßner, F. Berthold and U. Guth, Mater. Corros., 2006, 57, 455 CrossRef.
- Cyclic voltammetry measurements of 3a are in the accompanying ESI.†.
- Exposure of 3a to lithium perchlorate in methanol without passing an electric current did not afford any 2a after 12 h.
- L. Meng, J. Su, Z. Zha, L. Zhang, Z. Zhang and Z. Wang, Chem. – Eur. J., 2013, 19, 5542 CrossRef CAS PubMed.
- J. M. Álvarez-Calero, Z. D. Jorge and G. M. Massanet, Org. Lett., 2016, 18, 6344 CrossRef PubMed.
- B. Janza and A. Studer, J. Org. Chem., 2005, 70, 6991 CrossRef CAS PubMed.
- T. Mino, Y. Hasegawa, M. Sakamoto and T. Fujita, J. Organomet. Chem., 2007, 692, 4389 CrossRef CAS.
- J. W. J. Bosco and A. K. Saikia, Chem. Commun., 2004, 1116 RSC.
- C. B. Rao, D. C. Rao, D. C. Babu and Y. Venkateswarlu, Eur. J. Org. Chem., 2010, 2855 CrossRef CAS.
- C. K. Lin and T.-J. Lu, Tetrahedron, 2010, 66, 9688–9693 CrossRef CAS.
- K. Moriyama, M. Takemura and H. Togo, J. Org. Chem., 2014, 79, 6094 CrossRef CAS PubMed.
- S. Imai and H. Togo, Tetrahedron, 2016, 72, 6948 CrossRef CAS.
- A. Kamimura, H. Komatsu, T. Moriyama and Y. Nozaki, Tetrahedron, 2013, 69, 5968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra and cyclic voltammetry measurements (PDF). See DOI: 10.1039/c7ob02300f |
| This journal is © The Royal Society of Chemistry 2017 |