
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computationally guided discovery of a reactive, hydrophilic trans-5-oxocene dienophile for bioorthogonal labeling†
William D.
Lambert
a,
Samuel L.
Scinto
a,
Olga
Dmitrenko
a,
Samantha J.
Boyd
a,
Ronald
Magboo
b,
Ryan A.
Mehl
c,
Jason W.
Chin
*d,
Joseph M.
Fox
 *a and
Stephen
Wallace
*de
*a and
Stephen
Wallace
*de
aBrown Laboratory, Department of Chemistry & Biochemistry, University of Delaware, Newark, Delaware 19716, USA. E-mail: jmfox@udel.edu
bLotus Separations LLC, Newark, DE 19711, USA
cDepartment of Biochemistry and Biophysics, Oregon State University, Corvallis, Oregon 97331, USA
dMedical Research Council Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge CB2 0QH, UK
eInstitute of Quantitative Biology, Biochemistry and Biotechnology, School of Biological Sciences, University of Edinburgh, UK
First published on 20th July 2017
Abstract
The use of organic chemistry principles and prediction techniques has enabled the development of new bioorthogonal reactions. As this “toolbox” expands to include new reaction manifolds and orthogonal reaction pairings, the continued development of existing reactions remains an important objective. This is particularly important in cellular imaging, where non-specific background fluorescence has been linked to the hydrophobicity of the bioorthogonal moiety. Here we report that trans-5-oxocene (oxoTCO) displays enhanced reactivity and hydrophilicity compared to trans-cyclooctene (TCO) in the tetrazine ligation reaction. Aided by ab initio calculations we show that the insertion of a single oxygen atom into the trans-cyclooctene (TCO) ring system is sufficient to impart aqueous solubility and also results in significant rate acceleration by increasing angle strain. We demonstrate the rapid and quantitative cycloaddition of oxoTCO using a water-soluble tetrazine derivative and a protein substrate containing a site-specific genetically encoded tetrazine moiety both in vitro and in vivo. We anticipate that oxoTCO will find use in studies where hydrophilicity and fast bioconjugation kinetics are paramount.
Introduction
Biotechnology and biomedicine have been profoundly influenced by the development of new bioorthogonal reactions – abiotic transformations that occur selectively in a biological environment.1–9 Amongst these, the cycloaddition of alkenes/alkynes and s-tetrazines has become an important member of the bioorthogonal reaction “toolbox”.10–20 Since initial reports using trans-cyclooctene (TCO)21 and norbornene derivatives,22 a complementary range of dienophiles has been developed – including cyclopropenes,23,24 cyclooctynes25,26 and simple α-olefins.27–29 However, trans-cyclooctene (TCO) still maintains the advantage of exceptional reaction kinetics in this process.3,10 For example, the cycloaddition of the equatorial diastereomer of 5-hydroxy-trans-cyclooctene and a 3,6-dipyridyl-s-tetrazine derivative occurs with a second-order rate constant of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 s−1 in H2O at 25 °C.30 Faster reactivity can be realized by using the axial diastereomer of 5-hydroxy-trans-cyclooctene (80200 M−1 s−1).11,30 However, the fastest bioorthogonal reactions described to date use the conformationally strained dienophiles s-TCO and d-TCO (Fig. 1).30,31 These bicyclic molecules adopt a half-chair conformation that is 5.6–5.9 kcal mol−1 higher in energy than the crown conformation of monocyclic TCO. Cycloaddition of these compounds with tetrazines display second-order rate constants of up to 366000 M−1 s−1 for d-TCO and 3300000 M−1 s−1 for s-TCO.
600 M−1 s−1 in H2O at 25 °C.30 Faster reactivity can be realized by using the axial diastereomer of 5-hydroxy-trans-cyclooctene (80200 M−1 s−1).11,30 However, the fastest bioorthogonal reactions described to date use the conformationally strained dienophiles s-TCO and d-TCO (Fig. 1).30,31 These bicyclic molecules adopt a half-chair conformation that is 5.6–5.9 kcal mol−1 higher in energy than the crown conformation of monocyclic TCO. Cycloaddition of these compounds with tetrazines display second-order rate constants of up to 366000 M−1 s−1 for d-TCO and 3300000 M−1 s−1 for s-TCO.
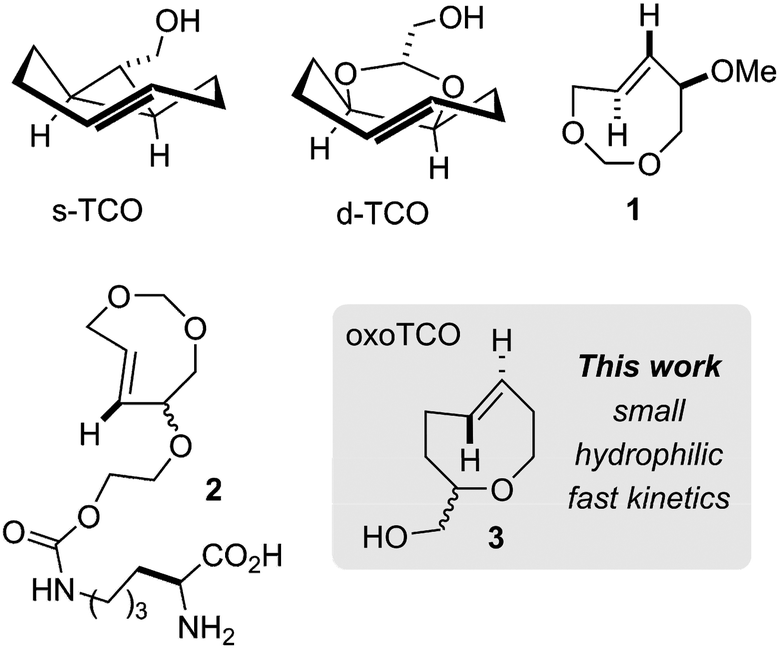 | ||
| Fig. 1 Conformationally strained and heterocyclic trans-cyclooctenes. | ||
From a kinetic standpoint, trans-cyclooctene derivatives are excellent dienophiles for studies where high reactivity is essential such as in cellular imaging and nuclear medicine.32,33 However, the hydrophobicity of TCO and s-TCO has recently been linked to high levels of non-specific background fluorescence during imaging experiments, necessitating lengthy washout protocols (>2 h) to dissociate the excess reagent from the cell.34,35 While d-TCO displays reduced lipophilicity, the compound is relatively bulky compared to the parent TCO system. The development of new, low molecular weight dienophiles for the tetrazine ligation reaction that are fast and hydrophilic is therefore an important challenge.
In seminal work, Jendrella synthesized 4,6-dioxo-TCO 1 and showed it to be 20–1000 fold faster than trans-cyclooctene in cycloadditions with cyclopentadiene, 2,3-dimethylbutadiene, mesitonitriloxide and diphenylketene.36 More recently, Dudley, Alabugin and coworkers have shown (in silico) that 3-oxocyclooctynes display fast reactivity in cycloadditions with azides, and have attributed their fast reactivity partly to the hyperconjugative effect of the allylic oxygens.37 Tomooka and Woerpel have synthesized trans-oxasilacycloalkenes, and have studied their reactivity in Diels–Alder and azide cycloadditions.38,39 Very recently, Lemke, Kele and coworkers reported the genetic incorporation of dioxo-TCO 2 and demonstrated that the lower lipophilicity of this molecule resulted in improved washout times during imaging experiments. In Diels–Alder reactions with tetrazines, the reaction rate with 2 is similar to that with the parent TCO.36,40
In the course of our synthetic studies on transannulations of cis- and trans-5-oxocenes, we queried whether such com-pounds would engage in rapid bioconjugation reactions.41,42 Here we report the computational design and synthesis of a trans-5-oxocene (“oxoTCO”, 3) – a small, hydrophilic, and highly reactive dienophile for use in the bioorthogonal tetrazine ligation reaction. The reaction of 3 (2.2:1 dr) with a water-soluble 3,6-dipyridyl-s-tetrazine-mono-succinamic acid 10 occurs with a second order rate constant of 94600 M−1 s−1 in PBS at 25 °C (Fig. 3), which is faster than either diastereomer of 5-hydroxy-trans-cyclooctene, and approaching the rate of bicyclic d-TCO. The oxoTCO heterocycle can be synthesized in seven high yielding steps from commercially available glycidol. Furthermore, oxoTCO 3 is small (MW 142) and hydrophilic with an experimental logP = 0.51. Finally, we describe the in vitro and in vivo kinetics of 3 on a recombinant protein substrate containing a site-specifically incorporated tetrazine-containing amino acid (sfGFP-150Tet-v.2.0).43 We anticipate that oxoTCO 3 will find applications in cellular imaging studies where small hydrophilic probes with fast reaction kinetics, low background fluorescence and/or rapid data acquisition are required.
Results and discussion
Computation was used to assist the design of a reactive and soluble trans-oxocene dienophile. We reasoned that the short C–O bonds in the backbone of a trans-5-oxocene would augment the olefinic strain of the trans-cycloalkene, and thereby increase the reactivity in tetrazine ligation. As shown in Fig. 2, ground state calculations were carried out for the parent trans-oxocenes 4 and 5 as well as trans-cyclooctene at the M06L/6-311+G(d,p) level. Indeed, the calculated C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C dihedral angle for 4 (134.6°) and 5 (134.4°) is significantly shorter than that for trans-cyclooctene (137.7°). M06L/6-311+G(d,p) and CAM-B3LYP/tzvp calculations were also carried out to compare the reactivity of 4 and 5 to trans-cyclooctene (Fig. 2B). These calculations were carried out with diphenyl-s-tetrazine so that they could be benchmarked against previous calculations carried our in our labs.30,31 At the M06L/6-311+G(d,p) level, the barrier for the Diels–Alder reaction of trans-cyclooctene with 3,6-diphenyl-s-tetrazine is ΔΔE‡ 13.3 kcal mol−1, ΔE‡(ZPE) 13.9 kcal mol−1, ΔH‡ 12.9 kcal mol−1. With trans-5-oxocene 4, the barrier was significantly lower, with ΔΔE‡ −1.23 kcal mol−1, ΔE‡(ZPE) −1.54 kcal mol−1 and ΔH‡ −1.44 kcal mol−1 relative to trans-cyclooctene. Interestingly, the isomeric trans-4-oxocene 5 is not predicted to be significantly more reactive than trans-cyclooctene. This computational result can be rationalized by considering the electron withdrawing nature of the allylic oxygen. Inverse electron demand Diels–Alder reactions are deactivated by electron withdrawing groups on the alkene, and the allylic oxygen of 5 is both inductively withdrawing and stereoelectronically positioned to deactivate the alkene through hyperconjugation. Thus, while the alkene of 5 is more strained than 4 (134.4° vs. 134.6° dihedral angle), the effect is attenuated by the electron withdrawing effect of the allylic oxygen.
C–C dihedral angle for 4 (134.6°) and 5 (134.4°) is significantly shorter than that for trans-cyclooctene (137.7°). M06L/6-311+G(d,p) and CAM-B3LYP/tzvp calculations were also carried out to compare the reactivity of 4 and 5 to trans-cyclooctene (Fig. 2B). These calculations were carried out with diphenyl-s-tetrazine so that they could be benchmarked against previous calculations carried our in our labs.30,31 At the M06L/6-311+G(d,p) level, the barrier for the Diels–Alder reaction of trans-cyclooctene with 3,6-diphenyl-s-tetrazine is ΔΔE‡ 13.3 kcal mol−1, ΔE‡(ZPE) 13.9 kcal mol−1, ΔH‡ 12.9 kcal mol−1. With trans-5-oxocene 4, the barrier was significantly lower, with ΔΔE‡ −1.23 kcal mol−1, ΔE‡(ZPE) −1.54 kcal mol−1 and ΔH‡ −1.44 kcal mol−1 relative to trans-cyclooctene. Interestingly, the isomeric trans-4-oxocene 5 is not predicted to be significantly more reactive than trans-cyclooctene. This computational result can be rationalized by considering the electron withdrawing nature of the allylic oxygen. Inverse electron demand Diels–Alder reactions are deactivated by electron withdrawing groups on the alkene, and the allylic oxygen of 5 is both inductively withdrawing and stereoelectronically positioned to deactivate the alkene through hyperconjugation. Thus, while the alkene of 5 is more strained than 4 (134.4° vs. 134.6° dihedral angle), the effect is attenuated by the electron withdrawing effect of the allylic oxygen.
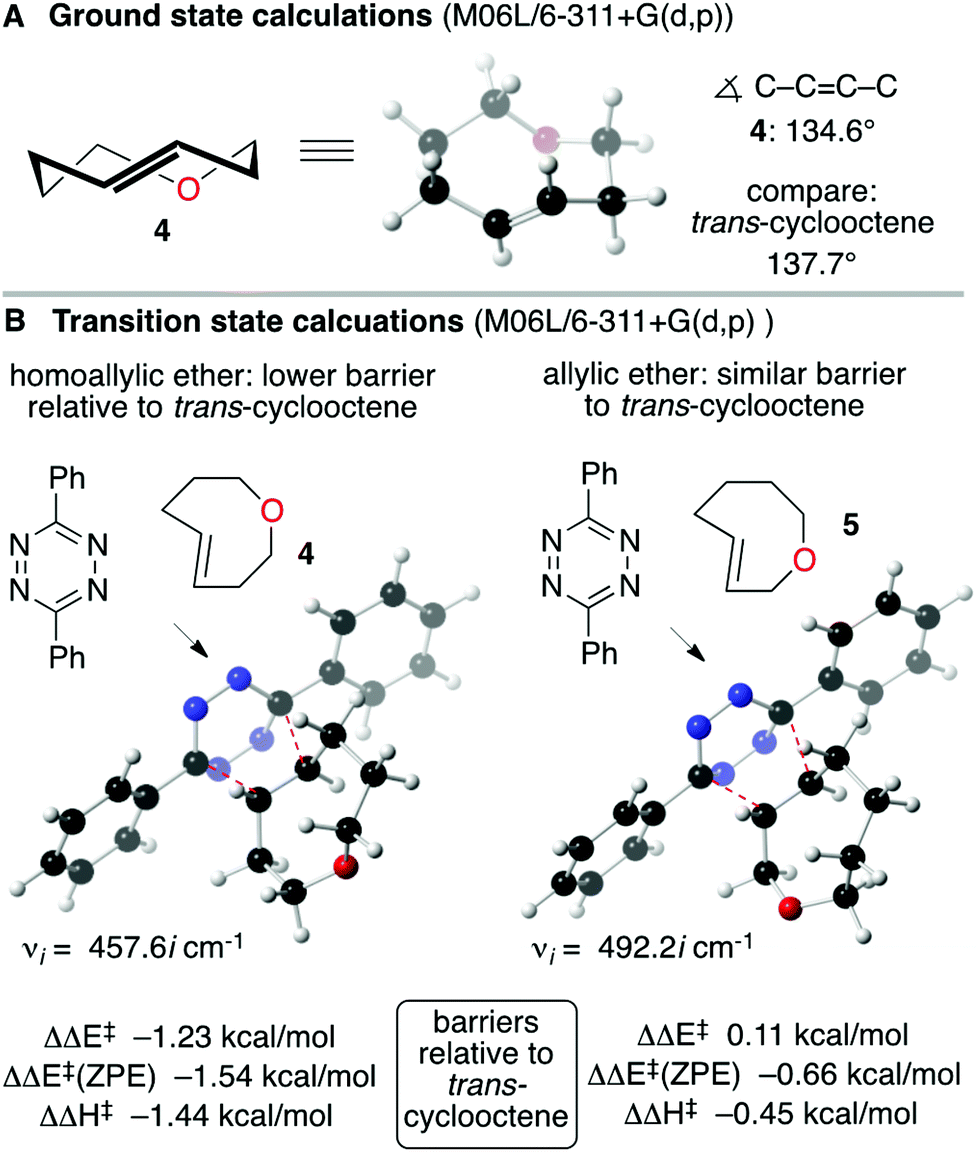 | ||
| Fig. 2 DFT transition state calculations predict that trans-5-oxocene 4, but not trans-4-oxocene 5, would be more reactive than trans-cyclooctene. While both 4 and 5 are more strained than trans-cyclooctene, that the reactivity of 5 is attenuated by the electron withdrawing allylic oxygen. | ||
Based on these computational predictions, we synthesized the alcohol-functionalized trans-5-oxocene 3 in 7 steps from commercially available glycidol 6 (Scheme 1). The synthesis began with TBS-protection and the addition of allyl magnesium chloride to provide alcohol 7. Our attempts to access 8 directly from 7via Williamson etherification or Mitsunobu chemistry were unsuccessful. Fortunately, we found that the treatment of the MOM ether of 7 with Lewis acidic stannic chloride generated a putative oxocarbenium ion that could be quenched via Sakurai allylation to afford butenyl ether 8 in 78% yield. Ring-closing metathesis of 8 using the Grubbs first-generation catalyst proceeded efficiently to afford cis-oxocene 9 in 84% yield. Finally, desilylation and photoisomerization using our closed-loop flow reactor44 afforded a 2.2:1 diastereoisomeric mixture of trans-oxocenes in 70% yield (37% overall yield over 7 steps). Separation of the diastereomers using preparative thin layer or silica gel chromatography was unsuccessful. An analytical sample of the major diastereomer of 3 was obtained by preparative supercritical fluid chromatography, however, given the difficulty of separation we continued the majority of further studies on oxoTCO using a 2.2:1 mixture of diastereomers. The logP of 3 was experimentally determined to be 0.51 whereas equatorial 5-hydroxy-trans-cyclooctene and d-TCO were both determined to be more hydrophobic with logP = 1.11 and 0.94, respectively.30
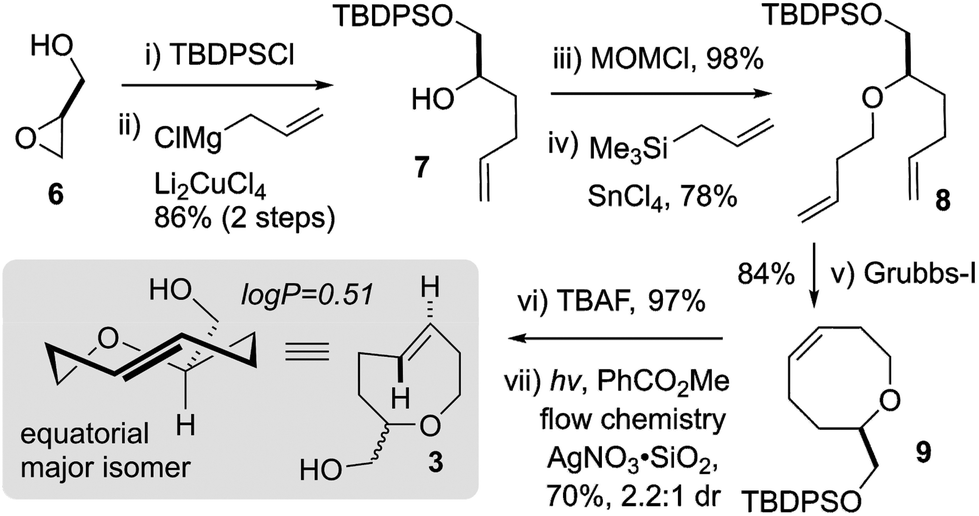 | ||
| Scheme 1 Synthesis of oxoTCO 3. | ||
The stability of oxoTCO 3 was studied under several conditions by 1H NMR spectroscopy. Over a 14 day period at room temperature, a 2.2:1 diastereoisomeric mixture of oxoTCO 3 (33 mM) showed no degradation in CD3OD. In D2O-PBS (pD = 7.4), the major, equatorial diastereomer of 3 showed less than 10% degradation after 1 week. The more reactive minor diastereomer degraded more rapidly in PBS, and decomposed with a half-life of 36 hours, with complete degradation after 9 days. oxoTCO 3 (25 mM) in the presence of mercaptoethanol (25 mM) showed only 8% isomerization in CD3OD over a 22 hours period while 92% was isomerized in phosphate buffered D2O (pD = 7.4) over the same period of time. Under similar conditions, oxoTCO stability to thiols in methanol is improved relative to d-TCO (92% isomerization after 14 h) and s-TCO (100% isomerization after 4 h).30 In D2O (pD = 7.4) containing 25 mM mercaptoethanol, the major diastereomer isomerized with a half-life of 2.2 hours, and the minor diastereomer isomerized with a half life of 1.6 hours. Overall, the stability of the oxoTCO diastereomers is similar to that of dTCO.30
We next measured the rate constant for the inverse electron-demand Diels–Alder (IEDDA) cycloaddition of oxoTCO and tetrazine 10 under pseudo-first order conditions (Fig. 3). PBS was chosen as a solvent for two reasons: aqueous solvent considerably accelerates the IEDDA reaction by the hydrophobic effect and initial kinetic studies indicated tetrazine 10, though more water soluble than 3,6-dipyridyl-s-tetrazine, was aggregating in unbuffered H2O, thus giving inconsistent first-order rates. Using a stopped-flow spectrophotometer and by following the exponential decay in tetrazine absorbance at 325 nm the second-order rate constant (k2) was determined to be 94600 ± 5700 M−1 s−1 in PBS at 25 °C for the 2.2:1 diastereomeric mixture of 3. This is faster than the reaction of a similar tetrazine with both diastereomers of 5-hydroxy-trans-cyclooctene (equatorial isomer 22600 M−1 s−1; axial isomer 80200 M−1 s−1), and is approximately ¼ as fast as a bicyclic d-TCO under comparable conditions (366000 M−1 s−1).30 The diastereomerically pure equatorial isomer of 3 was obtained by SFC, and found to react with 10 with a rate constant of 44100 ± 2600 M−1 s−1 in PBS at 25 °C. While we were unable to obtain a diasteromerically pure sample of the axial diastereomer, the rate constant can be calculated to be 310000 M−1 s−1 based on the rates observed for the diastereomer mixture and the pure equatorial isomer. The 7-fold rate acceleration for the axial isomer is consistent with prior reports for 5-hydroxy-trans-cyclooctene.11,30
 | ||
| Fig. 3 The kinetics of the cycloaddition of oxoTCO 3 with water-soluble 3,6-dipyridyl-s-tetrazine-mono-succinamic acid 10 in PBS buffer (pH 7.4). Second order rate constants (k2) were determined with a stopped-flow spectrophotometer under pseudo-first order conditions using ca. 10–30 equivalents of oxoTCO 3 (2.2:1 dr) by monitoring the decrease in tetrazine absorbance at 325 nm. | ||
We also studied the in vitro cycloaddition of oxoTCO and a green fluorescent protein encoded with an unnatrual tetrazine-containing amino acid 11 (sfGFP-150Tet-v.2.0) via the procedure of Mehl and coworkers.43 Thus, 4-(6-methyl-s-tetrazin-3-yl)phenylalanine was site-specifically introduced into a C-terminally hexahistidine-tagged GFP (sfGFP-150TAG-His6) via orthogonal translation using the evolved aminoacyl-tRNA synthetase MjRS/tRNACUA pair. Co-expression of these components in E. coli resulted in the amino acid-dependent synthesis of full-length recombinant GFP 11 which was purified by Ni-NTA chromatography and confirmed by ESI-MS. The tetrazine moiety of this protein quenches the fluorescence of the GFP chromophore, whereas the dihydropyridazine product of the TCO ligation does not. It is therefore possible to determine the kinetics of the reaction by monitoring the increase in GFP fluorescence (Fig. 4A). Accordingly, the second order rate constant of the reaction between oxoTCO and sfGFP150Tet-v.2.0 was determined to be 2030 ± 180 M−1 s−1 in phosphate buffer at room temperature (Fig. 4B). The reaction was quantitative under these conditions as determined by ESI-MS (Fig. 4C). The slower rate relative to that observed with 10 is due to the less reactive nature of the tetrazine 11 and in line with rate decreases observed with other TCOs.30
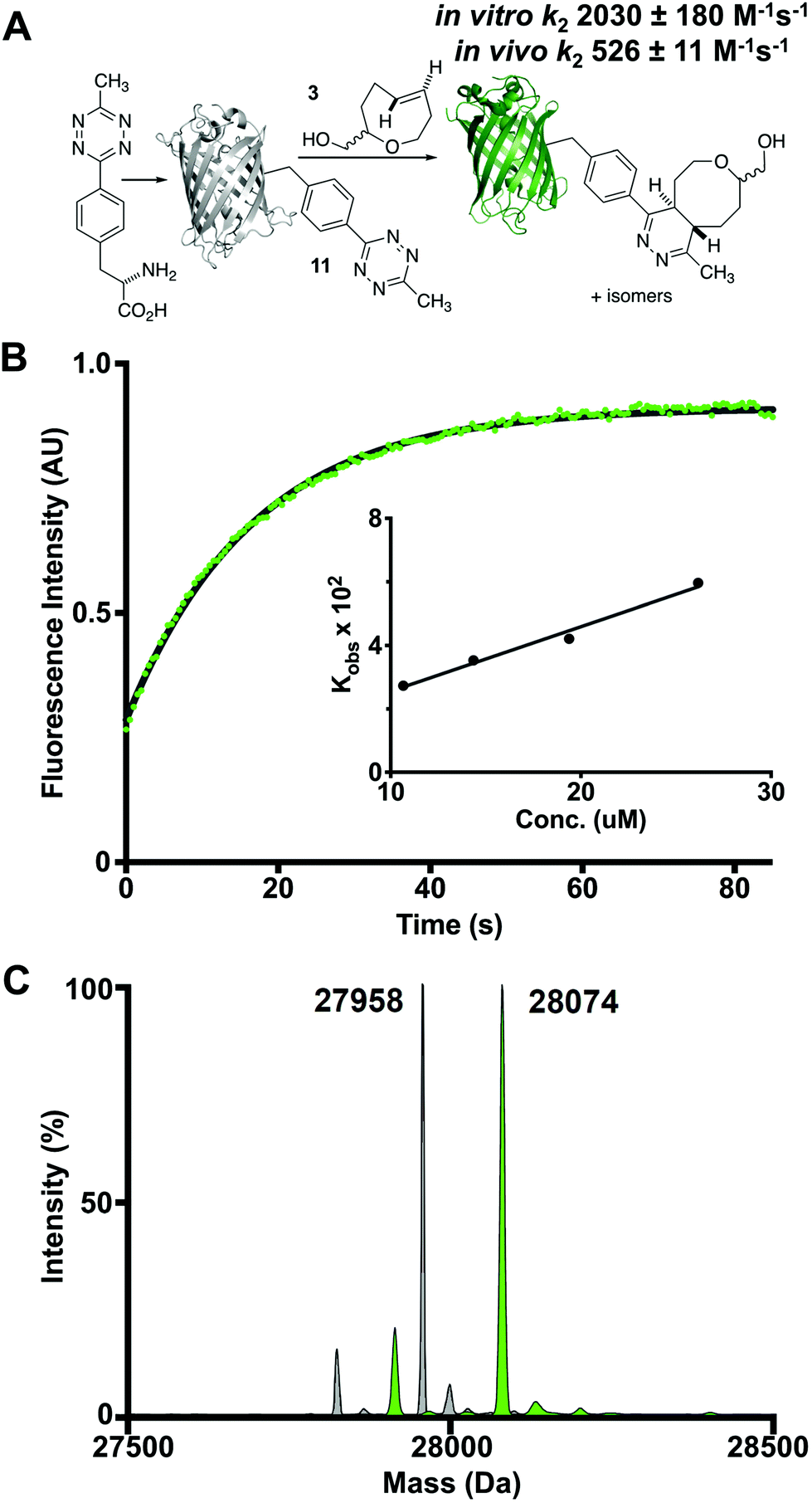 | ||
| Fig. 4 (A) The oxoTCO-tetrazine cycloaddition using a recombinant protein substrate containing a site-specifically incorporated tetrazine. Experiments were carried out both in vitro (PBS) and in vivo (E. coli) using a 2.2:1 eq/ax mixture of oxoTCO diastereomers. (B) Second order rate constants (k2) were determined under pseudo-first order conditions using ca. 100–260 equivalents of oxoTCO 3 by monitoring the increase in GFP fluorescence (in vitro study shown here). (C) Quantitative determination of the cycloadduct was confirmed by ESI-MS. | ||
Finally, the small size and good hydrophilicity of oxoTCO make it an excellent candidate for labeling in vivo. The cycloaddition was monitored in a suspension (PBS) of E. coli overexpressing sfGFP150Tet-v.2.0 by measuring the increase in whole-cell fluorescence upon addition of 3. At room temperature oxoTCO displays a second-order rate constant of 526 ± 11 M−1 s−1, which is approximately ¼ as fast as the in vitro ligation. Quantitative determination of the biorthogonal reaction was verified by ESI-MS. Cells were washed before lysis and the protein was purified via nickel affinity chromatography. The resulting protein mass was as expected for the cycloaddition product. This, alongside the whole-cell fluorescence experiment, provides evidence to suggest that oxoTCO crosses the bacterial cell membrane.
Conclusions
In summary, computation was used to design a hydrophilic 5-oxo-trans-cyclooctene derivative with high reactivity attributed to increased angle strain. A short synthesis was developed involving Sakurai allylation, olefin metathesis and flow-enabled photoisomerization as key steps. This heterocyclic trans-cyclooctene displays improved hydrophilicity, with an experimental logP value of 0.51. Kinetic analysis revealed that oxoTCO displays faster reactivity than mono-substituted TCO dienophiles, and is less bulky than bicyclic trans-cyclooctenes we have described previously. Quantitative labeling of GFP containing a genetically encoded tetrazine amino acid was studied in solution and in whole bacteria cells with complete labeling within minutes at room temperature. The high reactivity and lower hydrophobicity of oxoTCO-based probes should prove useful for in vivo applications, and in this context is the focus of active study in our labs.
Acknowledgements
This work was supported by NIH R01DC014461, R01EB014354, NSF DMR-1506613 and by Medical Research Council, UK (grants MC_U105181009 and MC_UP_A024_1008). Spectra were obtained with instrumentation supported by NIH grants P20GM104316, P30GM110758, S10RR026962, S10OD016267 and NSF grants CHE-0840401, CHE-1229234. The authors declare no competing financial interest. SLS is grateful to training at the genetic code expansion workshop at Oregon State University funded through NSF MCB-1518265.Notes and references
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- C. S. MKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- R. Rossin and M. S. Robillard, Curr. Opin. Chem. Biol., 2014, 21, 161–169 CrossRef CAS PubMed.
- J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27(12), 2791–2807 CrossRef CAS PubMed.
- M. Vrabel and T. Carell, Cycloadditions in Bioorthogonal Chemistry, Springer International Publishing, Switzerland, 2016 Search PubMed.
- I. Nikić and E. A. Lemke, Curr. Opin. Chem. Biol., 2015, 28, 164–173 CrossRef PubMed.
- R. Selvaraj and J. M. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753–760 CrossRef CAS PubMed.
- R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4964–4806 CrossRef PubMed.
- K. Wang, A. Sachdeva, D. J. Cox, N. M. Wilf, S. Wallace, R. A. Mehl and J. W. Chin, Nat. Chem., 2014, 6, 393–403 CrossRef CAS PubMed.
- T. Peng and H. C. Hang, J. Am. Chem. Soc., 2016, 138, 14426–14433 Search PubMed.
- Y. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554–561 CrossRef CAS PubMed.
- E. M. F. Billaud, E. Shahbazali, M. Ahamed, F. Cleeren, T. Noël, M. Koole, A. Verbruggen, V. Hessel and G. Bormans, Chem. Sci., 2017, 8, 1251–1258 RSC.
- S. Liu, H. Zhang, R. A. Remy, F. Deng, M. E. Mackay, J. M. Fox and X. Jia, Adv. Mater., 2015, 27, 2783–2790 CrossRef CAS PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 3 CrossRef PubMed.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- J. Yang, J. Seckute, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736–1738 RSC.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- A. Niederwieser, A. Späte, L. D. Nguyen, C. Jüngst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- Y. Lee, Y. Kurra, Y. Yang, J. Torres-Kolbus, A. Deiters and W. R. Liu, Chem. Commun., 2014, 50, 13085–13088 RSC.
- B. L. Oliveira, Z. Guo, O. Boutureira, A. Guerreiro, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2016, 128, 14903–14907 CrossRef.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS PubMed.
- H. E. Murrey, J. C. Judkins, C. W. am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS PubMed.
- R. Selvaraj, B. Giglio, S. Liu, H. Wang, M. Wang, H. Yuan, S. R. Chintala, L. Yap, P. S. Conti, J. M. Fox and Z. Li, Bioconjugate Chem., 2015, 26, 435–442 CrossRef CAS PubMed.
- C. Uttamapinant, J. D. Howe, K. Lang, V. Beránek, L. Davis, M. Mahesh, N. P. Barry and J. W. Chin, J. Am. Chem. Soc., 2015, 137, 4602–4605 CrossRef CAS PubMed.
- E. Kozma, I. Nikić, B. R. Varga, I. V. Aramburu, J. H. Kang, O. T. Fackler, E. A. Lemke and P. Kele, ChemBioChem, 2016, 17, 1518–1524 CrossRef CAS PubMed.
- H. Jendralla, Tetrahedron, 1983, 39, 1359–1363 CrossRef CAS.
- B. Gold, G. B. Dudley and I. V. Alabugin, J. Am. Chem. Soc., 2013, 135, 1558–1569 CrossRef CAS PubMed.
- K. Tomooka, S. Miyasaka, S. Motomura and K. Igawa, Chem. – Eur. J., 2014, 20, 798–7602 CrossRef PubMed.
- J. Santucci III, J. R. Sanzone and K. A. Woerpel, Eur. J. Org. Chem., 2016, 2933–2943 CrossRef.
- G. Knorr, E. Kozma, A. Herner, E. Lemke and P. Kele, Chem. – Eur. J., 2016, 22, 8972–8979 CrossRef CAS PubMed.
- M. Royzen, M. T. Taylor, A. DeAngelis and J. M. Fox, Chem. Sci., 2011, 2, 2162–2165 RSC.
- M. T. Taylor and J. M. Fox, Tetrahedron Lett., 2015, 56, 3560–3563 CrossRef CAS PubMed.
- R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044–10047 CrossRef CAS PubMed.
- M. Royzen, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760–3761 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01707c |
| This journal is © The Royal Society of Chemistry 2017 |