
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed allylic amination: a powerful tool for the enantioselective synthesis of acyclic nucleoside phosphonates†
Mariam
Azzouz
,
Sébastien
Soriano
,
Margarita
Escudero-Casao
 ,
M. Isabel
Matheu
,
Sergio
Castillón
* and
Yolanda
Díaz
*
,
M. Isabel
Matheu
,
Sergio
Castillón
* and
Yolanda
Díaz
*
Departament de Química Analítica I Química Orgànica, Facultat de Química, Universitat Rovira i Virgili, C/Marcel·lí Domingo 1, 43007 Tarragona, Spain. E-mail: yolanda.diaz@urv.cat; sergio.castillon@urv.cat
First published on 10th August 2017
Abstract
Acyclic nucleoside phosphonates have been prepared in a straightforward manner and in high yields by an enantioselective palladium-catalyzed allylic substitution involving nucleic bases as nucleophiles followed by cross-metathesis reaction with diethyl allylphosphonate.
Introduction
Acyclic nucleoside phosphonates (ANPs)1 belong to a family of modified nucleoside analogues, in which the sugar moiety has been replaced by a functionalized acyclic chain linking the nucleobase at one end and the phosphonic acid group at the other. Relevant ANPs that have been pursued as antiviral agents are cidofovir (HPMPC), adefovir (PMEA), and tenofovir2 (PMPA) (Fig. 1). Because of their limited oral bioavailability, the last two compounds have been converted to their oral prodrug forms, adefovir dipivoxil or bis(pivaloyloxymethyl)-PMEA [bis(POM)-PMEA] and tenofovir disoproxil or bis(isopropyloxycarbonyloxymethyl)-PMPA [bis(POC)-PMPA], respectively. Many analogues of these compounds have been prepared incorporating modifications in the base, alkyl chain or in the phosphonate moiety. Examples of modification in the alkyl chain are vinyl and allyl ANPs, where a CH2 group replaces the oxygen of the ether function in the alkyl chain (Fig. 1).3 | ||
| Fig. 1 Relevant reported ANPs and products 1 and 2 described in this paper. | ||
Acyclic nucleosides are commonly synthesized from nucleic bases and chiral starting materials by alkylation, Mitsunobu reaction, epoxide opening,4 and by silver-catalyzed addition of different nucleophiles to 9-allenyl-9H-purines.5 The reported asymmetric synthesis of acyclic nucleosides involves the transition metal-6 or organocatalyzed7 aza-Michael reaction of purinic bases to acrylates and asymmetric hydrogenation of purine-substituted acrylates.2
Allylic substitution has also been used in the synthesis of acyclic nucleosides to provide compounds with the nucleic base linked to the terminal carbon.8 Hartwig recently reported an enantioselective allylic amination using an iridium catalyst to afford acyclic nucleosides where the nucleic base is bonded to a stereocenter.9 Trost described the desymmetrization of meso 1,4-cyclopentenyl-dibenzoate derivatives using palladium-DACH catalysts in a straightforward synthesis of carbocyclic nucleosides.10 He found that the use of nucleobases as nucleophiles has a remarkable effect on the catalytic turnover and the enantioselectivity of desymmetrization reactions.9 Furthermore, Pd-catalyzed DYKAT of vinyl epoxide 4 is an efficient procedure for preparing 2-amino-3-buten-1-ol derivatives,11 which are attractive intermediates for synthesizing acyclic nucleosides, although for this purpose it will be necessary to explore the behavior of the different nucleic bases in the reaction. Recently, we have demonstrated that allyl carbonate 5, substituted with a hydroxymethyl group, is an appropriate substrate for the Pd-catalyzed asymmetric allylic amination reaction (Pd-AAA), affording the branched derivative with high regio- and enantioselectivities.12 In this regard, quite a striking different regioselectivity in the reaction of substrates 4 and 5 with amines was observed, with epoxide 4 furnishing equimolar mixtures of linear and branched aminated products. In this work, we report a comparative study of the Pd-catalyzed asymmetric allylic amination reaction starting from vinylepoxide 4 and carbonate 5 with different nucleic bases, and we show that the resulting derivatives 3 can be transformed into novel ANP nucleosides (1, 2), structurally related to cidofovir, tenofovir and to allyl ANP (Scheme 1). We have also extended this synthesis to the guanine precursor 10 (see Scheme 2).
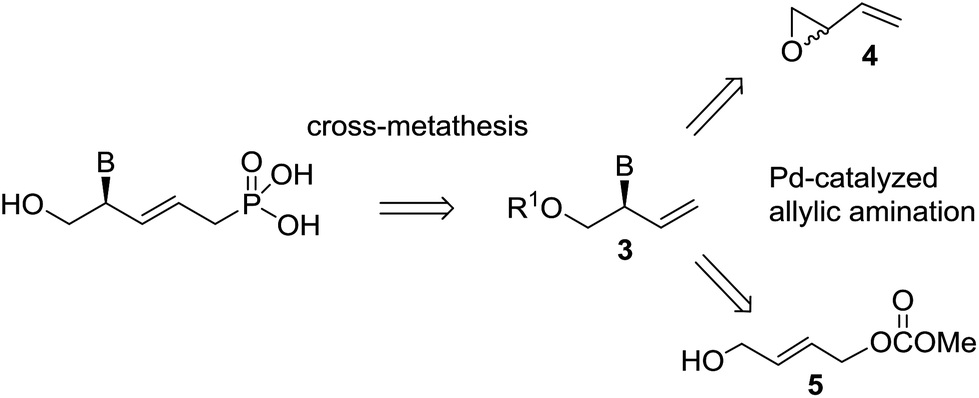 | ||
| Scheme 1 Retrosynthesis of ANPs 1 and 2. | ||
 | ||
| Scheme 2 | ||
Results and discussion
Preliminary assays starting from vinyl epoxide 4 and acetylcytosine under reaction conditions optimized in a previous study12 (6 mol% (R,R)-DACH-naphthyl Trost ligand and 2 mol% [Pd(η3-C3H5)Cl]2 as a catalytic system in CH2Cl2, at room temperature), led to the formation of a bis-allylation product containing the 3-buten-1-ol-2-yl units at position 1 and at the exocyclic NH group. This compound was obtained even by reducing the amount of acetylcytosine to 1.1 equivalents. We therefore turned our attention towards di-Boc-cytosine (6a).13 When the reaction was conducted under similar conditions using epoxide 4 and di-Boc-cytosine (6a), branched product 7a was obtained in 81% yield and 62% ee (Table 1, entry 1). Decreasing the temperature to 0 °C did not practically improve the enantioselectivity, while conducting the reaction at 35 °C eroded the enantioselectivity significantly (Table 1, entries 2 and 3). The analogous reaction of carbonate 5 with di-Boc-cytosine (6a) using the (S,S) ligand (Table 1, entry 4) furnished the enantiomer ent-7a in 92% yield and 73% ee. Interesting enough, all reactions from both 4 and 5 were highly regioselective affording exclusively the branched isomer. The use of BSA-KOAc did not improve the results. Lowering the temperature to 0 °C did not improve the enantioselectivity (Table 1, entry 5). Further optimization was focused on the precatalyst loading: a reduction from 2 mol% to 1 mol% produced a drastic decrease in the enantioselectivity (Table 1, entry 6), while an increase in the precatalyst loading to 3 mol% did not provide significant improvement (result not shown in the table). In this context, we selected room temperature and 2 mol% of the palladium precursor as standard reaction conditions.|
|
|||||
|---|---|---|---|---|---|
| Entry | Starting material | T (°C) | Conv.b (%) (yield)c (%) | eed (%) | Producte |
a Conditions: [Pd(η3-C3H5)Cl]2 (2 mol%), naphthyl ligand (6 mol%), substrate (1 equiv.), nucleophile (2 equiv.). Reaction time = 16 h. Concentration = 0.02 M in CH2Cl2.
b A branched/linear ratio >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :<2 was observed in all cases.
c Isolated yield of branched regioisomer.
d Determined by HPLC on a DAICEL CHIRALCEL OD-H column.
e The absolute configurations of the products were established by analogy with those obtained by Trost using phthalimide as analogous soft N-nucleophiles to nucleic bases (ref. 11a).
f The (R,R)-naphthyl ligand was used.
g The (S,S)-naphthyl ligand was used.
h 1 mol% [Pd(η3-C3H5)Cl]2 was used. :<2 was observed in all cases.
c Isolated yield of branched regioisomer.
d Determined by HPLC on a DAICEL CHIRALCEL OD-H column.
e The absolute configurations of the products were established by analogy with those obtained by Trost using phthalimide as analogous soft N-nucleophiles to nucleic bases (ref. 11a).
f The (R,R)-naphthyl ligand was used.
g The (S,S)-naphthyl ligand was used.
h 1 mol% [Pd(η3-C3H5)Cl]2 was used.
|
|||||
| 1f | 4 | rt | >98 (81) | 62 | 7a |
| 2f | 4 | 0 | >98 (85) | 65 | 7a |
| 3f | 4 | 35 | >98 (81) | 32 | 7a |
| 4g | 5 | rt | >98 (92) | 73 | ent- 7a |
| 5g | 5 | 0 | >98 (90) | 75 | ent- 7a |
| 6g,h | 5 | rt | >98 (90) | 42 | ent- 7a |
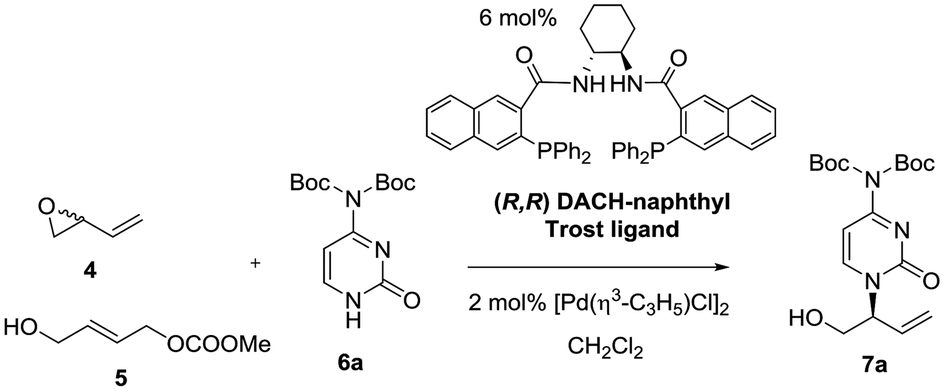
It can be concluded from Table 1 that, in analogy to reaction from vinyl epoxide 4, Pd-AAA from carbonate 5 with di-Boc-cytosine (6a) using the [Pd(η3-C3H5)Cl]2/DACH-naphthyl ligand as the catalytic system provides good yields of the branched aminated product 7a/ent-7a in complete regioselectivity and moderate enantioselectivity. We therefore decided to extend this comparative study to other purinic bases. The results are collected in Table 2.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Starting material | Nucleic base | Productb | Conv.c (yield)d | eee (%) |
|
a Conditions: [Pd(η3-C3H5)Cl]2 (2 mol%), DACH-naphthyl ligand (6 mol%), substrate (1 equiv.), nucleophile (2 equiv.), rt, 16 h. Substrate concentration = 0.02 M.
b The absolute configurations of the products were established by analogy with those obtained by Trost using phthalimide as analogous soft N-nucleophiles to nucleic bases (ref. 11a).
c A branched/linear ratio >98:<2 was observed in all cases by 1H NMR spectroscopy of the crude products.
d Isolated yield of the branched regioisomer.
e Determined by HPLC on a DAICEL CHIRALCEL OD-H column.
f (R,R)-DACH-naphthyl ligand was used.
g (S,S)-DACH-naphthyl ligand was used.
h Using [Pd(η3-C3H5)Cl]2 (2 mol%), DACH-naphthyl ligand (6 mol%), substrate (2 equiv.), nucleophile (1 equiv.), rt, 16 h. Substrate concentration = 0.04 M.
|
|||||
| 1f | 4 | 6b | 7b | >98 (92) | 92 |
| 2g | 5 | 6b | ent- 7b | >98 (92) | 88 |
| 3g | 4 | 6c | ent- 7c | >98 (89) | 91.4 |
| 4g | 5 | 6c | ent- 7c | >98 (96) | 91 |
| 5g | 4 | 6d | ent- 7d | >98 (89) | 88 |
| 6g | 5 | 6d | ent- 7d | >98 (92) | 88 |
| 7f | 4 | 6e | 7e | 31 (19) | 90 |
| 8f | 4 | 6e | 7e | >98 (64) | 90 |
| 9f | 5 | 6e | 7e | 72 (43) | 92 |
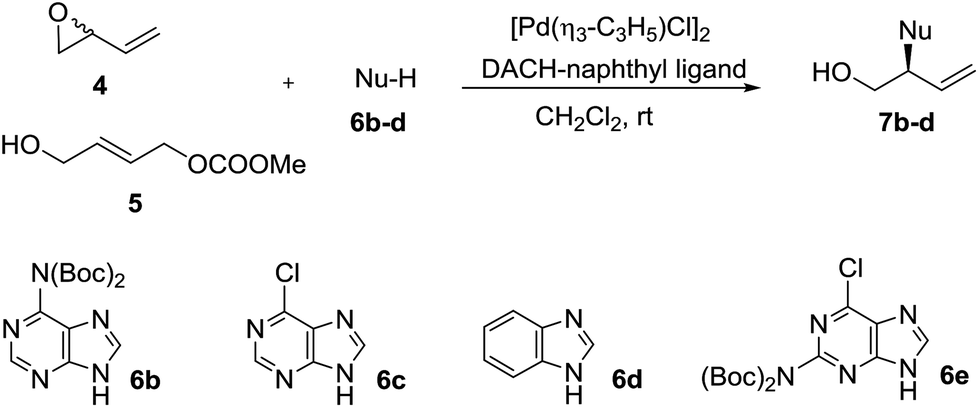
Di-Boc-adenosine (6b) reacted with epoxide 4 in the presence of the (R,R)-DACH-naphthyl ligand to provide 7b in an excellent yield of 92% and 92% ee (entry 2). When the reaction was conducted with carbonate 5 using the (S,S)-DACH-naphthyl ligand, ent-7b was obtained in an excellent yield of 92% and 88% ee (entry 2).
6-Chloropurine (6c) is a versatile starting material in nucleoside synthesis since it allows a set of useful and well-known transformations for the synthesis of nucleoside derivatives. When carbonate 5 was treated with 6c in the presence of the (S,S)-DACH-naphthyl ligand under the standard conditions, compound ent-7c was obtained in very good 91% enantioselectivity, slightly higher than that obtained from 6b (Table 2, entry 2 vs. 4). Pd-AAA of 6c from epoxide 4 furnished the allylated purine ent-7c with almost the same enantioselectivity of that obtained from 6b (Table 2, entry 1 vs. 3).
Furthermore, the reaction of epoxide 4 and carbonate 5 with benzimidazole (6d) as a nucleophile using the (S,S)-DACH-naphthyl ligand, afforded compound ent-7d in good yields and the same enantioselectivity, (88% ee) (Table 2, entries 5 and 6).
Di-Boc-2-amino-6-chloropurine 6e reacted with epoxide 4 and carbonate 5 under standard conditions to provide 7e with high enantioselectivity but with low to moderate yields (Table 2, entries 7 and 9). However, on inverting the reaction stoichiometry a substantial improvement in the yield was achieved (64%) with the same enantio-induction (Table 2, entry 8).
Contrary to their complementary behavior in terms of regioselectivity observed with hard amine nucleophiles,12 both epoxide 4 and carbonate 5 afforded valuable branched products with high degrees of enantioselectivity when reacting with nucleobases. The regioselectivity observed in the reactions of epoxide 4 with nucleobases is comparable to that already observed with other soft nitrogen nucleophiles, such as imides or imidocarboxylates, as a result of a dynamic kinetic asymmetric transformation. As for carbonate 5, the branched regioisomer is always observed as a major product, regardless of the nitrogen nucleophile used, due to an enantioselective Pd-allylic amination, where hydrogen bonding interactions play an important role both in the ionization and the nucleophilic attack steps.12
Having explored the feasibility of using nucleic bases as nucleophiles for the synthesis of chiral branched heterocycles via a Pd-AAA with the DACH-naphthyl Trost ligand, we selected compounds 7a, 7b and 7e for synthesizing model acyclic pyrimidine and purine nucleosides.
For this purpose, the primary hydroxyl group was initially protected by reaction with tert-butyldiphenylsilyl chloride in DMF in the presence of imidazole to afford compounds 8a,b,e in excellent yields (Scheme 2). According to the retrosynthetic scheme and following previous studies in the field,14 compounds 8a,b were then treated with diethyl allylphosphonate15,16 in the presence of Grubb–Nolan's catalyst to afford compounds 9a,b in excellent 92% and 90% yields, respectively, as a result of a cross-metathesis reaction. The reaction of 8e under similar conditions afforded 9e in a moderate 52% yield. Compounds with E configuration were exclusively obtained. Removal of the protecting groups15 in the nucleic base, the phosphonate moiety and the hydroxyl group, was carried out by treatment of compounds 9a,b with TMSBr15 in dichloromethane to afford the target acyclic nucleosides 1 and 2 in excellent yields. Additionally, compound 9e was treated under the deprotection conditions to afford the 2-amino-6-chloro-9H-purine derivative 10 in good yield.
The saturated nucleoside 11, with an increased conformational mobility, was prepared by the reduction of the double bond in 9a (Scheme 3). Attempts to reduce the double bond under 1 bar of hydrogen pressure and using palladium on charcoal17 as catalysts led to no conversion. At higher hydrogen pressure (10 bar) the presence of two products was observed by 1H NMR, corresponding to the expected compound 11 and to a secondary product resulting from the reduction of the double bond and partial reduction of the cytosine. Optimal conditions were found driving the reaction at room temperature and using 3 bar of hydrogen pressure. In this way, full conversion and 87% yield of compound 11 was obtained. Removal of protecting groups using TMSBr afforded acyclic nucleoside phosphonate 12 in 80% yield.
 | ||
| Scheme 3 | ||
In conclusion, acyclic nucleosides 1, 2, 10 and 12 were successfully prepared in high yields by palladium-catalyzed allylic amination from compounds 4 and 5, using pyrimidinic and purinic bases as nucleophiles, followed by ruthenium-catalyzed cross-metathesis with diethyl allylphosphonate and removal of protecting groups. Excellent enantioselectivity was obtained for the purinic derivatives 2 and 10.
Experimental section
General methods
All chemicals used were reagent grade and used as supplied unless otherwise specified. HPLC grade dichloromethane (CH2Cl2), tetrahydrofuran (THF) and dimethylformamide (DMF) were dried using a solvent purification system (Pure-Solv 400). Toluene was purified using standard procedures.181H and 13C NMR spectra were recorded on a Varian® Mercury VX 400 (400 MHz and 100.6 MHz respectively) or Varian 400-MR spectrometer in CDCl3 as a solvent, with chemical shifts (δ) referenced to the internal standard CDCl3 (7.26 ppm 1H, 77.23 ppm 13C) or Me4Si as an internal reference (0.00 ppm). 2D correlation spectra (gCOSY, NOESY, gHSQC, gHMBC) were visualized using a VNMR program (Varian®). ESI MS were run on an Agilent® 1100 Series LC/MSD instrument. Optical rotations were measured at room temperature in a PerkinElmer® 241 MC apparatus with 10 cm cells. IR spectra were recorded on a JASCO FT/IR-600 Plus Fourier transform infrared spectrometer ATR Specac Golden Gate. Optical rotations were measured at 598 nm on a Jasco DIP-370 digital polarimeter using a 100 mm cell. The enantiomeric excess was determined by an HPLC analysis using a DAICEL CHIRALCEL OD-H column. Reactions were monitored by TLC carried out on 0.25 mm E. Merck® silica gel 60F254 glass or aluminium plates. Developed TLC plates were visualized under a short-wave UV lamp (250 nm) and by heating plates that were dipped in ethanol/H2SO4 (15:1) and a basic solution of potassium permanganate. Flash column chromatography was carried out using forced flow of the indicated solvent on Fluka® or Merck® silica gel 60 (230–400 mesh). Radial chromatography was performed on 1 or 2 mm plates of Kieselgel 60 PF254 silica gel, depending on the amount of the product. Flash column chromatography (FCC) was performed using flash silica gel (32–63 μm) and employed a solvent polarity correlated with TLC mobility.
:2) provided 41 mg (90%) of compound ent-7a as a yellow syrup and 75% ee (determined by chiral HPLC (Daicel Chiralcel OD-H, n-hexane–iPrOH, 90:10, flow = 0.5 mL min−1, detection, UV 210 nm; retention times (min), 9.79, 11.42, tR(R) = 9.79 min and tR(S) = 11.42 min)). [α]25D +7.13 (c 1.06, CHCl3). FTIR-ATR (cm−1): 2918, 2849, 1775, 1741, 1665, 1460, 1369, 1313, 1253, 1154, 1131, 788. 1H NMR (CDCl3, 400 MHz,): δ (ppm) 7.67 (d, 1H, J = 7.5 Hz), 7.05 (d, 1H, J = 7.5 Hz), 6.00 (ddd, 1H, J = 17.0, 10.6, 5.9 Hz), 5.42 (dd, 1H, J = 10.6, 1.5 Hz), 5.30 (dd, 1H, J = 17.0, 1.5 Hz), 5.28–5.23 (m, 1H), 4.00–3.94 (m, 2H), 3.15 (bs, 1H), 1.55 (s, 18H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 161.9, 155.8, 149.7, 147.2, 147.0, 132.7, 120.5, 96.2, 85.1, 62.7, 61.2, 27.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H28N3O6: 382.1973, found: 382.1964.
:1) provided 93 mg (92%) of product 7b as a yellow syrup and 92% ee (determined by chiral HPLC (Daicel Chiralcel OD-H, n-hexane–iPrOH 85:15, flow = 0.5 mL min−1, detection, UV 254 nm; retention times (min), 11.77, 13.47, tR(R) = 11.77 min and tR(S) = 13.47 min)). [α]25D −15.80 (c 0.92, CHCl3). FTIR-ATR (cm−1): 3347, 2979, 2926, 1787, 1600, 1107. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.84 (s, 1H), 8.17 (s, 1H), 6.20 (ddd, 1H, J = 17.1, 10.5, 6.4 Hz), 5.40 (d, 1H, J = 10.5 Hz), 5.30–5.18 (m, 1H), 5.14 (d, 1H, J = 17.1 Hz), 4.26–4.10 (m, 2H), 3.80 (bs, 1H), 1.47 (s, 18H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 153.0, 151.7, 150.8, 150.6, 145.1, 132.7, 129.1, 119.7, 84.1, 63.8, 61.3, 27.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H28N5O5: 406.2085, found: 406.2073.
:1) provided 60 mg (89%) product ent-7c as a colourless oil and 91.4% ee (determined by chiral HPLC (Daicel Chiralcel OD-H, n-hexane–iPrOH, 90:10, flow = 1 mL min−1, detection, UV 210 nm; retention times (min), 17.12, 18.36, tR(R) = 18.36 min and tR(S) = 17.12 min)). [α]25D +10.8 (c 1.23, CHCl3). FTIR-ATR (cm−1): 3347, 2927, 1591, 1561, 1337. 1H NMR (CDCl3, 400 MHz): δ(ppm) 8.70 (s, 1H), 8.23 (s, 1H), 6.21 (ddd, 1H, J = 17.0, 10.4, 6.4 Hz), 5.43 (dd, 1H, J = 10.4, 1.4 Hz), 5.30–5.25 (m, 1H), 5.24 (dd, 1H, J = 17.0, 1.4 Hz), 4.28–4.22 (m, 1H), 4.21–4.15 (m, 1H), 3.86 (t, 1H, J = 6.3 Hz). 13C NMR (CDCl3, 100.6 MHz): 151.8, 151.5, 151.4, 145.4, 132.2, 131.8, 120.4, 63.7, 61.0. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C9H10ClN4O: 225.0538, found: 225.0532.
:1) provided 20 mg (92%) of product ent-7d as a colourless oil and 88% ee (determined by chiral HPLC (Daicel Chiralcel OD-H, n-hexane–iPrOH, 92:08, flow = 1.0 mL min−1, detection, UV 220 nm; retention times (min), 17.84, 25.57, tR(R) = 17.84 min and tR(S) = 25.57 min)). [α]25D +26.0 (c 0.80, CHCl3). FTIR-ATR (cm−1): 3089, 2922, 1492, 1457. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.84 (s, 1H), 7.53 (d, 1H, J = 7.7 Hz), 7.36 (d, 1H, J = 7.7 Hz), 7.21 (t, 1H, J = 7.7 Hz), 7.14 (t, 1H, J = 7.7 Hz), 6.11 (ddd, 1H, J = 17.0, 10.8, 5.7 Hz), 5.37 (d, 1H, J = 10.8 Hz), 5.20 (d, 1H, J = 17.0 Hz), 5.05–4.97 (m, 1H), 4.22–4.10 (m, 2H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 143.2, 142.2, 140.8, 132.9, 123.0, 122.5, 119.8, 119.4, 110.7, 63.3, 60.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H13N2O: 189.1022, found: 189.1017.
:6) provided 22 mg (43%) of compound 7e as a yellow syrup and 92% ee (determined by chiral HPLC (Daicel Chiralcel OD-H, n-hexane–iPrOH, 90:10, flow = 0.6 mL min−1, detection, UV 230 nm; retention times (min), 13.5, 15.1, tR(R) = 13.5 min and tR(S) = 15.1 min)). [α]25D −34.0 (c 0.1, CHCl3). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.25 (s, 1H), 6.17 (ddd, J = 16.9, 10.5, 6.3 Hz, 1H), 5.40 (d, J = 10.4 Hz, 1H), 5.24–5.16 (m, 2H), 4.22 (dd, J = 12, 5.9 Hz, 1H), 4.11 (dd, J = 12.0, 3.0 Hz, 1H), 3.27 (br s, 1H), 1.44 (s, 18H). 13C NMR (CDCl3, 100.6 MHz,): δ (ppm) 152.2, 151.5, 151.3, 150.4, 146.0, 132.2, 129.9, 120.0, 83.9, 63.4, 60.9, 27.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C19H26ClN5O5Na: 462.1520, found: 462.1524.
:2) to provide 137 mg (85%) of compound 8a. [α]25D −13.5 (c 1.27, CHCl3). FTIR-ATR (cm−1): 2931, 2857, 1742, 1671, 1524, 1455, 1370, 1319, 1256, 1137, 1110, 784, 701. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.75 (d, 1H, J = 7.5 Hz), 7.57 (d, 2H, J = 6.4 Hz), 7.48 (d, 2H, J = 6.4 Hz), 7.49–7.30 (m, 6H), 7.98 (d, 1H, J = 7.5 Hz), 6.01 (ddd, 1H, J = 17.6, 10.4, 6.0 Hz), 5.38–5.34 (m, 2H), 5.32 (d, 1H, J = 17.6 Hz), 3.97 (d, 2H, J = 3.6 Hz), 1.57 (s, 18H), 1.02 (s, 9H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 161.9, 155.0, 149.8, 147.1, 135.7, 135.5, 133, 132.6, 130.1, 128.0, 120.6, 95.6, 84.9, 63.9, 59.6, 27.9, 26.7, 19.3. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C34H46N3O6Si 620.3156, found 620.3171.
:2) to provide 319 mg (80%) of compound 8b as a colourless oil. [α]25D −10.93 (c 1.13, CHCl3). FTIR-ATR (cm−1): 2987, 2362, 1733, 1716, 1558, 1540, 1507, 1456, 1395, 1259, 1066, 749. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.79 (s, 1H), 8.26 (s, 1H), 7.50–7.25 (m, 10H), 6.02 (ddd, 1H, J = 17.2, 10.4, 6.4 Hz), 5.34 (dd, 1H, J = 11.6, 0.8 Hz), 5.30–5.20 (m, 1H), 5.17 (dd, 1H, J = 17.2, 0.8 Hz), 4.16 (dd, 1H, J = 11.2, 6.8 Hz), 4.05 (dd, 1H, J = 10.8, 4 Hz), 1.43 (s, 18H), 0.94 (s, 9H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 153.3, 151.8, 150.5, 150.3, 144.5, 135.5, 135.4, 132.5, 130.1, 128.9, 127.9, 119.8, 83.7, 64.8, 59.4, 27.9, 26.8, 19.1. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C35H45N5NaO5Si 666.3088, found 666.3073.
:2) to provide 244 mg (95%) of compound 8e as a colourless oil. [α]25D −11.0 (c 0.3, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.55–7.51 (m, 2H), 7.49–7.46 (m, 2H), 7.45–7.30 (m, 6H), 6.16 (ddd, J = 17.0, 10.5, 6.3 Hz, 1H), 5.34 (dd, J = 10.5, 0.8 Hz, 1H), 5.27–5.19 (m, 1H), 5.18–5.09 (m, 1H), 4.13 (dd, J = 11.1, 5.6 Hz, 1H), 4.01 (dd, J = 11.1, 3.8 Hz, 1H), 1.39 (s, 18H), 1.01–0.95 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 152.4, 151.7, 150.9, 150.4, 145.7, 135.5, 135.3, 132.3, 132.2, 132.1, 130.1, 130.0, 129.7, 127.9, 119.9, 83.4, 64.7, 59.2, 27.8, 26.7, 19.1. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C35H44ClN5O5SiNa: 700.2698, found: 700.2700.
:2) to provide 57 mg (92%) of compound 9a as a yellow liquid. [α]25D −21.4 (c 1.2, CHCl3). FTIR-ATR (cm−1): 2969, 2931, 1741, 1671, 1455, 1370, 1319, 1255, 1111, 1024, 735, 701. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.68 (d, 1H, J = 7.6 Hz), 7.56 (d, 2H, J = 6.4 Hz), 7.45 (d, 2H, J = 6.4 Hz), 7.42–7.30 (m, 6H), 6.97 (d, 1H, J = 7.6 Hz), 5.87–5.79 (m, 2H), 5.29 (brs, 1H), 4.15–4.02 (m, 4H), 3.96 (d, 2H, J = 4.0 Hz), 2.60 (dd, 2H, J = 21.0, 6.4 Hz), 1.57 (s, 18H), 1.27 (t, 6H, J = 6.8 Hz), 1.03 (s, 9H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 161.7, 154.6, 149.5, 146.9, 135.5, 135.3, 132.5, 132.4, 130, 129.9, 129.5, 129.4, 128.5, 127.8, 126.5, 126.3, 95.4, 84.7, 63.6, 62, 59.4, 30.6 (J = 140 Hz) 27.7, 26.8, 19.1, 16.4. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C39H57N3O9PSi 770.3602, found 770.3628.
:1) to provide 157 mg (90%) of product 9b as a green liquid. [α]25D −14.6 (c 1.36, CHCl3). FTIR-ATR (cm−1): 2929, 2856, 1788, 1599, 1452, 1369, 1252, 1139, 1111, 1026, 704. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.77 (s, 1H), 8.20 (s, 1H), 7.42–7.29 (m, 10H), 6.15–6.02 (m, 1H), 5.76 (ddd, 1H, J = 15.6, 10.4, 7.6 Hz), 5.25 (brs, 1H), 4.16 (dd, 2H, J = 10.4, 6.4 Hz), 4.07–3.95 (m, 4H), 2.56 (dd, 1H, J = 21.0, 7.6 Hz), 1.43 (s, 18H), 1.22 (t, 6H, J = 6.8 Hz), 0.93 (s, 9H). 13C NMR (CDCl3, 100.6 MHz): δ (ppm) 153.2, 151.9, 150.6, 150.4, 144.5, 135.2, 132.4, 130.1, 129.1, 129.0, 128.0, 126.2, 83.8, 64.8, 62.2, 62.1, 59.2, 30.6 (J = 140 Hz), 28.0, 26.9, 19.2, 16.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C40H57N5O8PSi 794.3714, found 794.3694.
:2) to provide 86 mg (52%) of compound 9e as a colourless oil. [α]25D −2.4 (c 0.4, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 7.55–7.26 (m, 10H), 6.09–5.98 (m, 1H), 5.75 (dt, J = 14.6, 7.0 Hz, 1H), 5.18 (brs, 1H), 4.10 (dd, J = 11.0, 5.6 Hz, 2H), 4.03 (ddd, J = 14.3, 9.3, 5.3 Hz, 2H), 3.96 (dd, J = 11.0, 3.8 Hz, 2H), 2.61 (d, J = 7.4 Hz, 1H), 2.55 (d, J = 7.4 Hz, 1H), 1.40 (s, 18H), 1.24 (dd, J = 12.8, 7.0 Hz, 6H), 0.97 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 152.1, 151.6, 150.9, 150.5, 145.5, 135.4, 135.3, 132.1, 130.1, 130.0, 129.8, 128.6, 128.5, 127.9, 127.8, 126.5, 126.4, 83.5, 64.7, 62.1, 61.9, 58.7, 31.2, 29.8, 27.9, 26.7, 19.0, 16.5, 16.4, 16.3. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C40H56ClN5O8PSi: 828.3324, found 828.3364.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Ministerio de Economía y Competitividad, Spain (grant DGI CTQ2014-58664-R). M. A. thanks AECI and SS thanks URV for a fellowship. MEC thanks to CONACyT-México for a post-doctoral grant.Notes and references
- (a) E. De Clercq, Collect. Czech. Chem. Commun., 2011, 76, 480 CrossRef; (b) M. Krecmerova, in Herpesviridae—A Look into This Unique Family of Viruses, ed. G. D. Magel and S. Tyring, INTECH, 2012, ch. 11, p. 245 Search PubMed; (c) E. De Clercq, Antiviral Res., 2007, 75, 1 CrossRef CAS PubMed; (d) E. De Clercq and A. Holy, Nat. Rev. Drug Discovery, 2005, 4, 928 CrossRef CAS PubMed; (e) A. Holy, Curr. Pharm. Des., 2003, 9, 2567 CrossRef CAS PubMed; (f) A. Holy, in Current Protocols in Nucleic Acid Chemistry, John Wiley and Sons, Unit 14.2, 2005 Search PubMed.
- (a) H.-L. Sun, F. Chen, M.-S. Xie, H.-M. Guo, G.-R. Qu, Y.-M. He and Q.-H. Fan, Org. Lett., 2016, 18, 2260 CrossRef CAS PubMed; (b) H.-L. Sun, F. Chen, M.-S. Xie, H.-M. Guo, G.-R. Qu, Y.-M. He and Q.-H. Fan, Org. Lett., 2014, 16, 2014 CrossRef PubMed.
- (a) K. A. Cruickshank, J. Jiricny and C. B. Reese, Tetrahedron Lett., 1984, 25, 681 CrossRef CAS; (b) H. Tanaka, H. Hayakawa, K. Obi and T. Miyasaka, Tetrahedron, 1986, 42, 4187 CrossRef CAS; (c) L. A. Agrofoglio, I. Gillaizeau and Y. Saito, Chem. Rev., 2003, 103, 1875 CrossRef CAS PubMed; (d) M. Hamada, V. Roy, T. R. McBrayer, T. Whitaker, C. Urbina-Blanco, S. P. Nolan, J. Balzarini, R. Snoeck, G. Andrei, R. F. Chinazi and L. A. Agrofolio, Eur. J. Med. Chem., 2013, 67, 398 CrossRef CAS PubMed.
- For a recent review about chemical synthesis of acyclic nucleosides see: (a) M.-S. Xie, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Tetrahedron Lett., 2014, 55, 7156 CrossRef CAS; (b) H.-M. Guo, S. Wu, H.-Y. Niu, G. Song and G.-R. Qu, in Chemical Synthesis of Nucleoside Analogues, ed. P. Merino, John Wiley and Sons, 2013, p. 103 Search PubMed.
- T. Wei, M.-S. Xie, G.-R. Qu, H.-Y. Niu and H.-M. Gou, Org. Lett., 2014, 16, 900 CrossRef CAS PubMed.
- M. Gandelman and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 2393 CrossRef CAS PubMed.
- (a) H.-M. Guo, T.-F. Yuan, H.-Y. Niu, J.-Y. Liu, R.-Z. Mao, D.-Y. Li and G.-R. Qu, Chem. – Eur. J., 2011, 17, 4095 CrossRef CAS PubMed; (b) H. Wu, Z. Q. Tian, L. L. Zhang, Y. D. Huang and Y. M. Wang, Adv. Synth. Catal., 2012, 354, 2977 CrossRef CAS.
- (a) H.-Y. Niu, C. Du, M.-S. Xie, Y. Wang, Q. Zhang, G.-R. Qu and H.-M. Guo, Chem. Commun., 2015, 51, 3328 RSC; (b) F. Amblard, S. P. Nolan, I. Gillaizeau and L. A. Agrofoglio, Tetrahedron Lett., 2003, 44, 9177 CrossRef CAS.
- L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8971 CrossRef CAS PubMed.
- (a) B. M. Trost, R. Madsen, S. G. Guile and B. Brown, J. Am. Chem. Soc., 2000, 122, 5947–5956 CrossRef CAS; (b) B. M. Trost, R. Madsen, S. G. Guile and A. E. H. Elia, Angew. Chem., Int. Ed. Engl., 1996, 35, 1569 CrossRef CAS.
- (a) B. M. Trost, D. B. Home and M. J. Weltering, Chem. – Eur. J., 2006, 12, 6607 CrossRef CAS PubMed; (b) J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillon, Org. Lett., 2009, 11, 205–208 CrossRef CAS PubMed; (c) J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillon, Eur. J. Org. Chem., 2011, 1514 CrossRef CAS; (d) S. Soriano, M. Azzouz, J. Llaveria, P. Marcé, M. I. Matheu, Y. Díaz and S. Castillon, J. Org. Chem., 2016, 81, 5217 CrossRef CAS PubMed.
- S. Soriano, M. I. Matheu, Y. Díaz and S. Castillon, Adv. Synth. Catal., 2016, 358, 4057 CrossRef CAS.
- P. Andrea, G. Giampaolo, P. Ivana, C. Mariolino and N. Giammario, Eur. J. Org. Chem., 2008, 5786 Search PubMed.
- (a) P. R. Hanson and D. S. Stoianova, Tetrahedron Lett., 1998, 39, 3939 CrossRef CAS; (b) P. R. Hanson and D. S. Stoianova, Tetrahedron Lett., 1999, 40, 3297 CrossRef CAS; (c) M. Bujard, V. Gouverneur and C. Mioskowski, J. Org. Chem., 1999, 64, 2119 CrossRef CAS PubMed; (d) M. Schuman, M. Trevitt, A. Redd and V. Gouverneur, Angew. Chem., Int. Ed., 2000, 39, 2491 CrossRef CAS.
- K. Hiroki, D. Topalis, J. Broggi, U. Pradere, V. Roy, S. Berteina-Raboin, P. S. Nolan, D. Deville-Bonne, G. Andrei, R. Snoeck, D. Garin, J. M. Crance and L. A. Agrofolio, Tetrahedron, 2008, 64, 3517 CrossRef.
- (a) U. Pradère, H. Clavier, V. Roy, S. P. Nolan and L. A. Agrofoglio, Eur. J. Org. Chem., 2011, 7324–7330 CrossRef; (b) A. Montagu, U. Pradére, V. Roy, S. P. Nolan and L. A. Agrofoglio, Tetrahedron, 2001, 67, 5319 CrossRef.
- T. Tomioka, Y. Yabe, T. Takahashi and T. K. Simmons, J. Org. Chem., 2011, 76, 4669 CrossRef CAS PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1989 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR and HPLC traces of reported products. See DOI: 10.1039/c7ob01478c |
| This journal is © The Royal Society of Chemistry 2017 |