
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2,2,2-Trifluoroethanol as a solvent to control nucleophilic peptide arylation†‡
Diana
Gimenez
a,
Anica
Dose
a,
Nicholas L.
Robson
a,
Graham
Sandford
a,
Steven L.
Cobb
 *a and
Christopher R.
Coxon
*b
*a and
Christopher R.
Coxon
*b
aDurham University, Department of Chemistry, South Road, Durham, DH1 3LE, UK. E-mail: s.l.cobb@durham.ac.uk
bSchool of Pharmacy and Biomolecular Sciences, Byrom Street Campus, Liverpool John Moores University, Liverpool, L3 3AF, UK. E-mail: c.r.coxon@ljmu.ac.uk
First published on 21st April 2017
Abstract
The SNAr arylation of peptides with perfluoroaromatics provides a route by which to install a useful chemical handle that enables both 19F-NMR analysis and further chemical modification. However, chemo-selective arylation in peptides containing multiple nucleophilic side chains currently presents a challenge to the field. Herein, we demonstrate that employing 2,2,2-trifluoroethanol (TFE) as a solvent in peptide SNAr reactions significantly improves nucleophile-selectivity when compared to N,N′-dimethylformamide (DMF).
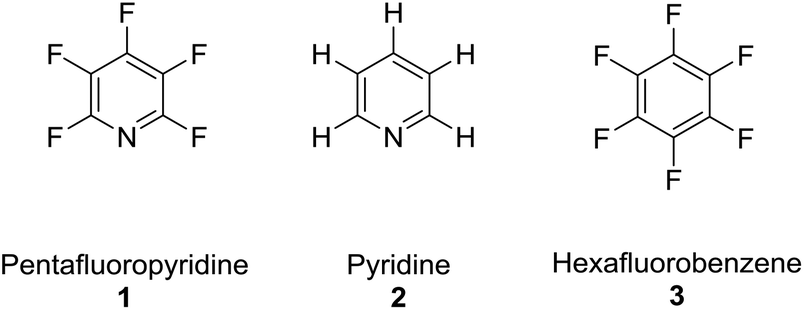
The perfluoroaromatic reagent (ArF) pentafluoropyridine (1) is a highly useful synthetic building block that can undergo multiple substitution reactions with a broad range of nucleophiles, owing to its higher reactivity when compared with other heterocycles e.g. pyridine (2) or perfluoroaromatics e.g. hexafluorobenzene (3).1–3
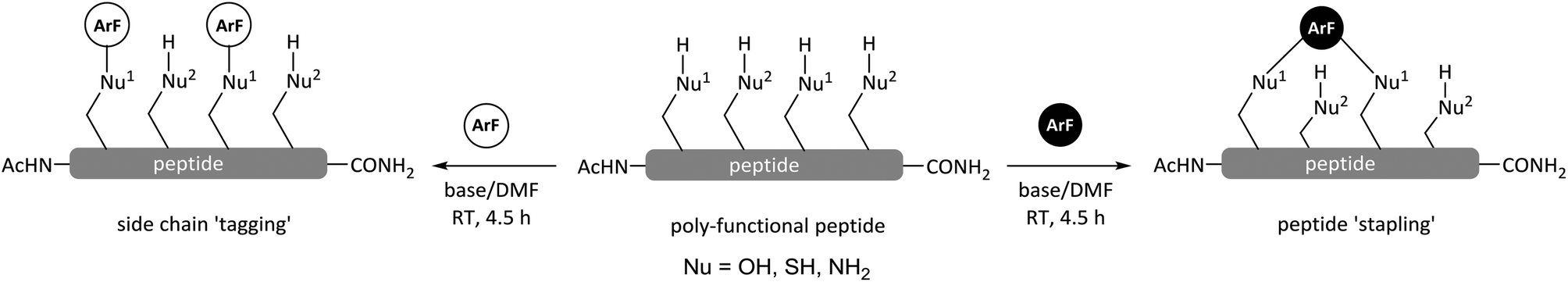 | ||
| Scheme 1 General reaction of perfluoroaromatic reagents (e.g. ArF; white = 1 or black = 3) with peptides containing more than one O, S or N centred nucleophile (Nu-H). | ||
The attachment of 1 to peptides provides a useful chemical handle that enables both 19F NMR analysis and the scope for chemical modifications such as cyclisation and ‘tagging’ for improvement of proteolytic stability.8 However, we have found that when employing N,N′-dimethylformamide (DMF) and diisopropylethylamine (DIPEA) at room temperature, reactions between 1 and peptides can lack nucleophile-selectivity, and arylation at multiple oxygen, sulfur and nitrogen-centred nucleophilic side chains can all occur. In an effort to improve side chain selectivity, the effects of reaction solvent and base on product distribution were investigated.
2,2,2-Trifluoroethanol (TFE) is undoubtedly a very useful solvent for synthetic chemistry and chemical-biology applications as it possesses an interesting set of properties compared with ethanol. It is more acidic (pKa 12.5) than ethanol (pKa 16.0)9 and it has lower nucleophilicity due to the electron-withdrawing effects of the three fluorine atoms present. Perfluorinated solvents have also been shown computationally10 and experimentally11,12 to accelerate some organic reactions. In particular, TFE has been employed to facilitate nucleophilic aromatic substitution (SNAr) reactions between halogenated heteroaromatics and nitrogen-based nucleophiles.13,14 The enhanced reactivity observed has been proposed to arise from a combination of both, the acidic properties and ability of TFE to effectively solvate the outgoing leaving group.15 TFE has been the focus of several studies where it has been shown to stabilise α-helical secondary structure in peptides and proteins by enhancing intramolecular H-bonding interactions.16–21 TFE has also been investigated as a tool for improving solid phase peptide synthesis (SPPS) protocols.22 Finally, compared with DMF, DMSO and NMP, the most frequently used solvents in peptide transformations, TFE is relatively volatile allowing its removal in vacuo even at low temperatures, and it can solubilise a broad and diverse range of polar molecules. Given all of the aforementioned properties we felt that TFE was a particularly interesting solvent to explore in peptide SNAr arylation reactions. Herein, we demonstrate that TFE can be effectively used as a solvent to tune the nucleophilic character of the side chains commonly present in peptidic systems, enhancing the chemo-selectivity of perfluoroaromatic SNAr arylation reactions.
The application of hexafluorobenzene (3) and other non-heteroatom-containing perfluoroaromatics for peptide stapling or arylation at cysteine residues has recently been reported, either employing DMF or buffered aqueous solutions.23–27 In our own studies in this area which focused on the more reactive perfluoroheteroaromatics (e.g.1) we encountered some difficulties in achieving selective arylations in peptide systems with multiple nucleophilic residues. In many cases poly-substituted products that arose due to reaction with either tyrosine (OH), lysine (NH2) and cysteine (SH) residues were obtained (Scheme 2 and Table 1).8 For this reason we were interested to investigate whether replacing DMF with TFE as our reaction solvent would afford improved chemo-selectivity between different peptide side chains and model perfluoro(hetero)aromatic reagents 1 and 3.
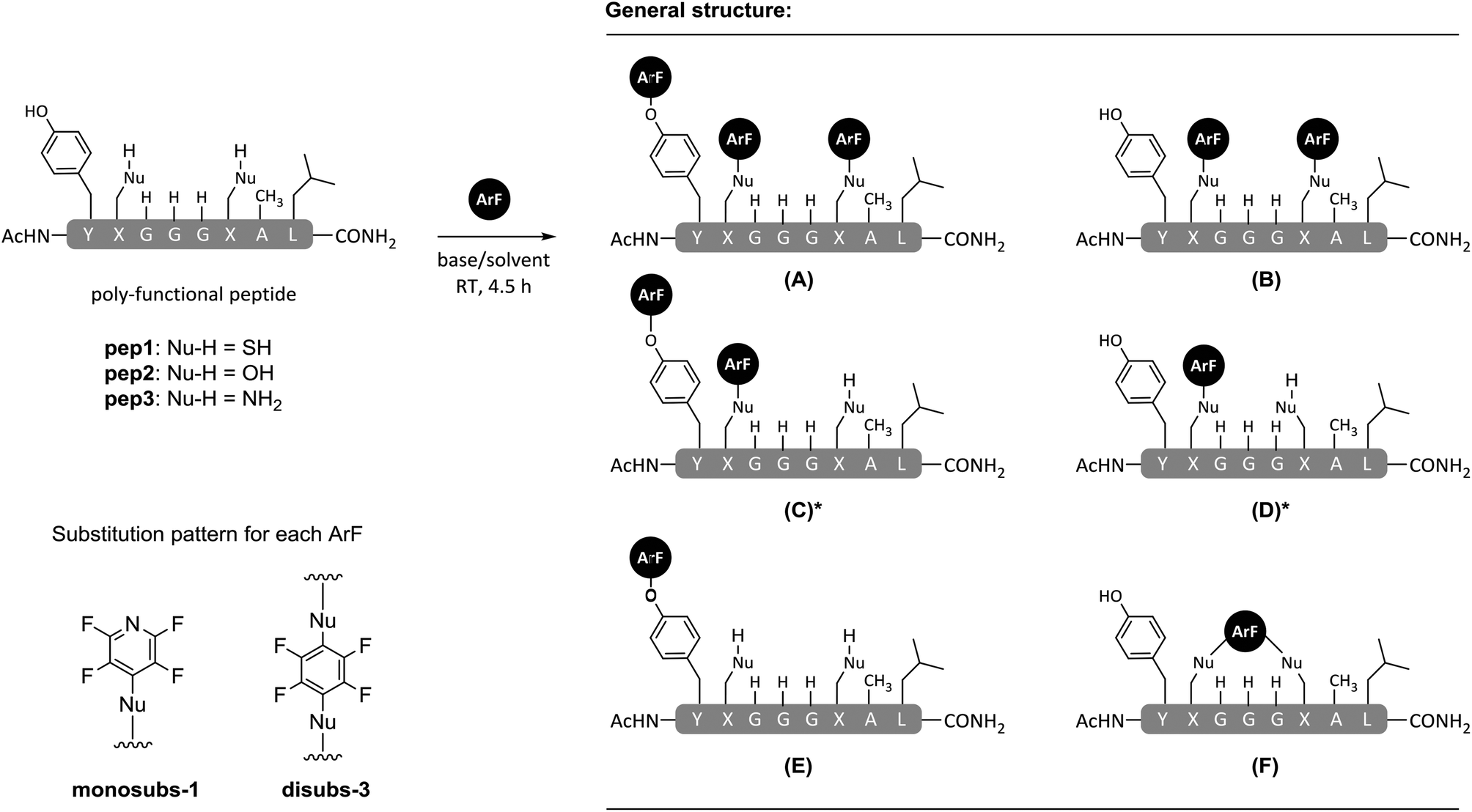 | ||
| Scheme 2 Reaction products obtained by treatment of model peptides (pep1–pep3) with ArF 1 or 3 using DIPEA/Cs2CO3 as base and DMF or TFE as solvent. *Products C and D present as non-equivalent mixture of regioisomers and were not distinguishable. | ||
| Entry | Peptide | Nu | ArF | Solvent | Base | Product structure (see Scheme 2) | Product number | Product ratiob where mixture was obtained |
|---|---|---|---|---|---|---|---|---|
| a Products present as non-equivalent mixture of regioisomers and were not distinguishable. b Ratio as calculated from LC/MS peak integration for the crude reactions at λ = 220 nm. c Reactions and compounds as characterized from previous reported work8. | ||||||||
| 1a | Pep1 | SH | 3 | DMF | DIPEA | F | 4 | — |
| 2 | Pep2 | OH | 3 | DMF | DIPEA | No reaction | — | — |
| 3 | Pep3 | NH2 | 3 | DMF | DIPEA | No reaction | — | — |
| 4a | Pep1 | SH | 1 | DMF | DIPEA | A | 5 | — |
| 5a | Pep2 | OH | 1 | DMF | DIPEA | E | 6 | — |
| 6a | Pep3 | NH2 | 1 | DMF | DIPEA | A | 7 | — |
| 7 | Pep1 | SH | 3 | TFE | DIPEA | No reaction | — | — |
| 8 | Pep2 | OH | 3 | TFE | DIPEA | No reaction | — | — |
| 9 | Pep3 | NH2 | 3 | TFE | DIPEA | No reaction | — | — |
| 10 | Pep1 | SH | 1 | TFE | DIPEA | B & Da | 8, 9 | (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) [Da: 1:2.5 ratio of regioisomers] :1) [Da: 1:2.5 ratio of regioisomers] |
| 11 | Pep2 | OH | 1 | TFE | DIPEA | No reaction | — | — |
| 12 | Pep3 | NH2 | 1 | TFE | DIPEA | No reaction | — | — |
| 13 | Pep1 | SH | 3 | TFE | Cs2CO3 | No reaction | — | — |
| 14 | Pep2 | OH | 3 | TFE | Cs2CO3 | No reaction | — | — |
| 15 | Pep3 | NH2 | 3 | TFE | Cs2CO3 | No reaction | — | — |
| 16 | Pep1 | SH | 1 | TFE | Cs2CO3 | A, Ca, Da | 5, 10, 9 | (1:43:7) [Ca: 1:1 ratio of regioisomers] |
| 17 | Pep2 | OH | 1 | TFE | Cs2CO3 | No reaction | — | — |
| 18 | Pep3 | NH2 | 1 | TFE | Cs2CO3 | No reaction | — | — |
Initially, we used the model peptide system Ac-Y![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) GGGAL-NH2; containing two nucleophilic groups, either: thiol ( = cysteine; pep1), hydroxyl ( = serine; pep2) or amine groups ( = lysine; pep3) plus one aromatic tyrosine residue that provides a competing nucleophilic site within the structure. To assess the consequences of solvent replacement, we first ran reactions using DMF (Table 1, entries 1–6), and then we used identical experimental conditions but with TFE as a solvent (Table 1, entries 7–12). The results from the aforementioned arylation reactions in both DMF and TFE are summarised in Table 1.
GGGAL-NH2; containing two nucleophilic groups, either: thiol ( = cysteine; pep1), hydroxyl ( = serine; pep2) or amine groups ( = lysine; pep3) plus one aromatic tyrosine residue that provides a competing nucleophilic site within the structure. To assess the consequences of solvent replacement, we first ran reactions using DMF (Table 1, entries 1–6), and then we used identical experimental conditions but with TFE as a solvent (Table 1, entries 7–12). The results from the aforementioned arylation reactions in both DMF and TFE are summarised in Table 1.
The reactions of pep1–pep3 with hexafluorobenzene (3) in DMF proceed to give the expected products (Table 1, entries 1–3).8,23–26 When using pentafluoropyridine (1) we observed the conversion of pep1 to a predominantly tri-substituted product, that arose through arylation at both cysteines and the tyrosine residue (Table 1, entry 4, product 5).8 The reaction of the di-lysine peptide (pep3; Nu = (CH2)3NH2) with 1 occurred predominantly at each lysine as well as the tyrosine (Table 1, entry 6, tri-substituted product 7). In the case of the serine-containing peptide (pep2; Nu = OH), reaction with 1 occurred solely at the tyrosine residue (Table 1, entry 5, product 6). The reactivity observed in these experiments was closely related to the anticipated trend with respect to the differing peptide side chain nucleophilic character i.e. SH > NH2 > OH. Contrary to our initial expectation, the use of TFE as the reaction solvent appeared to reduce rather than enhance reactivity (Table 1, entries 7–12). The effect was particularly pronounced in the case of hexafluorobenzene (3), which was observed not to undergo an SNAr arylation reaction with any of the model peptides tested, including those that had a thiol functionality present in the form of a cysteine residue (Table 1, entries 7–9). Moreover, even when employing the more electrophilic pentafluoropyridine (1), reactivity towards all but the cysteine nucleophiles was abolished. To highlight the chemo-selectivity for cysteine, it should noted that the reaction between pep1 and 1 in TFE afforded only the formation of a mixture of mono- and di-4-tetrafluoropyridinylated products in approximately a 1:2 ratio (Table 1, entry 10, products 8 and 9). No tagging of the tyrosine residue was observed in either product with both 9 (mono-ArF) and 8 (di-ArF) being isolated and characterised by 19F NMR and tandem MS/MS fragmentation to verify the proposed structures. Both, mono- (9) and di-substituted peptides (8) were confirmed to be selectively tagged through the cysteine (see ESI† for full details). The presence of two characteristic series of [bx] fragments in the MS/MS analysis of 9 arising from tetrafluoropyridine attached at either of the cysteine side chains further corroborated the compound to be a mixture of the two possible regioisomers 9 and 9′ (as shown in ESI, Fig. SI37–39†). However, the absolute identity of each could not be assigned. From 19F NMR analysis, non-equivalent integration of the relatively well-resolved multiplets at lower field allowed the determination of an estimated ratio between regioisomers of 1:2.5.
Interestingly, the SNAr reaction of pentafluoropyridine (1) in TFE not only seemed to be proceeding with enhanced chemo-selectivity with respect to the nucleophilic amino acid side chains present, but also with some degree of regioselectivity between similar nucleophiles located at different environments, at least in the case of the non-fully substituted products (e.g. formation of 9 and 9′).
We next looked to replace the organic base in the TFE reactions (DIPEA, pKa of conjugate acid (H2O) = 10.75)28 with the inorganic base Cs2CO3 (pKa of conjugate acid (H2O) = 10.33).29 Previously, we had observed that changing the base used in the reaction could have a clear effect on the amount of peptide arylation, but at the expense of chemo-selectivity.8 Using DIPEA as a base generally gave relatively clean products reactions with one major product being isolated, while the application of Cs2CO3 tended to give a much more complex product mixture. In addition Cs2CO3 (but not DIPEA) was able to activate peptides containing serine, affording for the first time novel hydroxyl tagged peptides, expanding the scope of the SNAr arylation reaction.8
Reactions of model peptides pep1–3 under the same arylation conditions previously used (Table 1, entries 7–12), but in the presence of Cs2CO3 rather than DIPEA are shown in Table 1 (entries 13–18). In accordance with our previous observations (i.e. when DIPEA was used in TFE), using Cs2CO3 and TFE as a solvent precluded any reaction between pep1–3 and the electrophile hexafluorobenzene (3). When the more reactive pentafluoropyridine (1) was used, arylation only occurred with the cysteine containing peptide pep1 (Table 1, entry 16, see ESI Fig. SI16†). Interestingly, the major product in this reaction was not the peptide in which arylation had occured on the two cysteine residues (e.g. compound 8) but rather compound 10 where arylation had occurred on one cysteine and one tyrosine residue (see ESI Fig. SI40–42† for additional details). On the basis of these results, it can be seen that as for the reactions carried out in DMF,8 changing the base used in TFE can also affect the outcome of peptide arylation.
Overall, the peptide arylation reactions summarised in Table 1 clearly highlight that TFE can be used as a solvent to modulate product formation via control of chemo-selectivity. Encouragingly, the results also suggested that the application of TFE as a reaction solvent could offer a route to achieve selective arylation of cysteine residues in the presence of competing lysine, serine and tyrosine residues. Having a simple yet effect means of controlling chemo-selectivity is highly desirable when reactive electrophiles like pentafluoropyridine (1) are utilised. To further probe whether it was possible to attain selective side chain-functionalisation via the appropriate combinations of solvent, base and perfluoro-heteroaromatic reagent, we constructed a model tetra-functional peptide sequence containing two cysteine and two lysine residues (pep4). The corresponding reactions on pep4 were carried out using either electrophile 1 or 3, with DMF or TFE as a solvent (Scheme 3). As expected, electrophile 3 was unable to react with the lysines in pep4 in either of the solvents used, and in DMF a mono-crosslinked product 11 was observed (Table 2, entry 19). The reaction of pep4 and electrophile 1 in DMF led to the expected tetra-arylated 12 as the major product (Table 2, entry 21). Some di-arylated product 13, was also observed (∼19% of the crude mixture, see ESI Fig. SI21†). By comparison, the application of TFE as a solvent in the reaction between pep4 and electrophile 1 (Table 2, entry 22), led to the predominant formation of the di-arylated cysteine product 13 and a minor amount of the mono-arylated cysteine product 14 (8:2 ratio, as estimated by LC-MS peak integration at 220 nm, see ESI Fig. SI22†). In the latter example, the fact that tetra-arylated product (12) was not seen provides very clear evidence of the chemo-selectivity that may be afforded when employing TFE as a reaction solvent (e.g. cysteine over lysine).
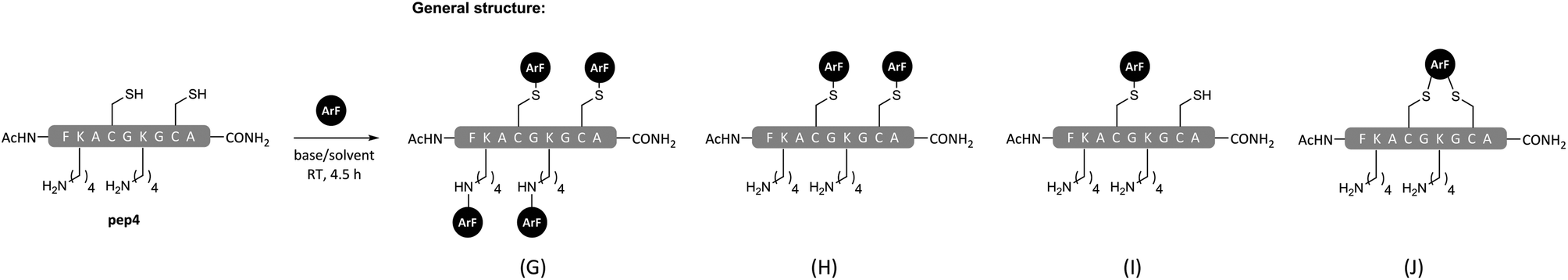 | ||
| Scheme 3 General structures of products from reactions of polyfunctional pep4 with ArF 1 or 3 in TFE or DMF with DIPEA as base (see Table 2 for details). | ||
| Entry | ArF | Solvent | Productb | Product structure (see Scheme 3) |
|---|---|---|---|---|
| a Products present as non-equivalent mixture of regioisomers and were not distinguishable. b Ratio as calculated from LC/MS peak integration for the crude reactions at λ = 220 nm. | ||||
| 19 | 3 | DMF | 11 | J |
| 20 | 3 | TFE | — | No reaction |
| 21 | 1 | DMF | 12 | G & H (3.5:1.0) |
| 13 | ||||
| 22 | 1 | TFE | 12 | G (1.1) |
| 13 | H (4.3) | |||
| 14 | Ia (1.0) | |||
Solvation of nucleophiles is known to affect the apparent nucleophilicity, and thus, reactivity of nucleophiles in substitution reactions. DMF, a dipolar aprotic solvent is less solvating than alcoholic (dipolar protic) solvents like TFE. Cysteine contains a sulfur nucleophile that is relatively large and diffuse compared with N- or O-centred nucleophiles and so it is less likely to be heavily solvated in TFE. Thus in TFE while serine and lysine reactivity is reduced due to solvation this occurs to a less extent for cysteine and hence arylation can still occur. TFE, has also been shown to slow the rate of some SNAr reactions relative to acetonitrile (aprotic solvent).30 The rate-determining step in SNAr is formation of the Meisenheimer–Jackson intermediate complex. The intermediate complex is more stable (lower energy barrier to formation) in aprotic dipolar solvents (DMF) than polar protic solvents (TFE) according to computational studies carried out in pentafluoronitrobenzene systems.31 Therefore, the observed reduction in the reactivity of the electrophiles (1 and 3) in TFE (compared with DMF) could also be due to the Meisenheimer–Jackson intermediate not being significantly stabilised during the SNAr arylation reactions.
Conclusions
Peptide arylation is emerging as a highly useful approach by which to cyclise23–26 and modify peptide6,7 systems. One of the challenges in the field is to develop methods that can be used to control chemo-selectivity. Herein, we have demonstrated that by employing 2,2,2-trifluoroethanol (TFE) as solvent in peptide arylation reactions we can impart selectivity between competing nucleophilic side chains (e.g. cysteine preference over lysine). This approach offers a mild method for controlled introduction of groups such as the tetrafluoropyridine moiety at cysteine side chains in the presence of other functionalities, such as lysines. We envisage that the new level of chemo-selectivity that this methodology offers will be a valuable addition to the field of peptide arylation chemistry, and, we are currently exploiting its application in the preparation of multi-cylic peptide systems.Acknowledgements
We acknowledgement both the European Commission, Seventh Framework Programme (FLUOR21, ITN, Gimenez) and the Engineering and Physical Sciences Research Council (EPSRC, EP/J015989/1) for funding support.Notes and references
- G. Sandford, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2005 Search PubMed.
- R. E. Banks, J. E. Burgess, W. M. Cheng and R. N. Haszeldine, J. Chem. Soc., 1965, 575–581 RSC.
- G. M. Brooke, J. Fluorine Chem., 1997, 86, 1–76 CrossRef.
- R. D. Chambers, P. R. Hoskin, A. Khalil, P. Richmond, G. Sandford, D. S. Yufit and J. A. K. Howard, J. Fluorine Chem., 2002, 116, 19–22 CrossRef CAS.
- G. Sandford, R. Slater, D. S. Yufit, J. A. K. Howard and A. Vong, J. Org. Chem., 2005, 70, 7208–7216 CrossRef CAS PubMed.
- A. S. Hudson, A. Hoose, C. R. Coxon, G. Sandford and S. L. Cobb, Tetrahedron Lett., 2013, 54, 4865–4867 CrossRef CAS.
- A. M. Webster, C. R. Coxon, G. Sandford and S. L. Cobb, Tetrahedron, 2014, 70, 4661–4667 CrossRef CAS.
- D. Gimenez, C. A. Mooney, A. Dose, G. Sandford, C. R. Coxon and S. L. Cobb, Org. Biomol. Chem., 2017 10.1039/c7ob00283a.
- P. Ballinger and F. A. Long, J. Am. Chem. Soc., 1959, 81, 1050 CrossRef CAS.
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421 CrossRef CAS PubMed.
- H. Schwertfeger, Synlett, 2010, 2971–2972 CrossRef CAS.
- J. A. Berkessel, D. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef PubMed.
- B. Carbain, H. Lebraud, C. R. Coxon, K. J. Elliott, C. J. Matheson, E. Meschini, A. R. Roberts, D. M. Turner, C. Wong, C. Cano, R. J. Griffin, I. R. Hardcastle and B. T. Golding, Chem. – Eur. J., 2013, 20, 2311–2317 CrossRef PubMed.
- H. J. Whitfield, R. J. Griffin, I. R. Hardcastle, A. Henderson, J. Meneyrol, V. Mesguiche, K. L. Sayle and B. T. Golding, Chem. Commun., 2003, 2802–2803 RSC.
- M. L. Sinnott and W. P. Jencks, J. Am. Chem. Soc., 1980, 102, 2026–2032 CrossRef CAS.
- K. Shiraki, K. Nishikawa and Y. Goto, J. Mol. Biol., 1995, 245, 180–194 CrossRef CAS PubMed.
- F. D. Sonnichsen, J. E. Van Eyk, R. S. Hodges and B. D. Sykes, Biochemistry, 1992, 31, 8790–8798 CrossRef CAS PubMed.
- P. Luo and R. L. Baldwin, Biochemistry, 1997, 36, 8413 CrossRef CAS PubMed.
- K. Gast, D. Zirwer, M. Müller-Frohne and G. Damaschun, Protein Sci., 1999, 8, 625–634 CrossRef CAS PubMed.
- P. S. Sierra, C. C. Tejuca, F. García-Blanco, C. D. Oliva, J. C. Sierra and G. E. Gorostidi, Helv. Chim. Acta, 2005, 88, 312–324 CrossRef CAS.
- R. Chitra and P. E. Smith, J. Chem. Phys., 2001, 114, 426–435 CrossRef CAS.
- D. Yamashiro, J. Blake and C. Hao Li, Tetrahedron Lett., 1976, 1469–1472 CrossRef CAS.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- Y. Zou, A. M. Spokoyny, C. Zhang, M. D. Simon, H. Yu, Y.-S. Lin and B. L. Pentelute, Org. Biomol. Chem., 2014, 12, 566–573 CAS.
- C. Zhang, P. Dai, A. M. Spokoyny and B. L. Pentelute, Org. Lett., 2014, 16, 3652–3655 CrossRef CAS PubMed.
- C. Zhang, A. M. Spokoyny, Y. Zou, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2013, 52, 14001–14005 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CAS.
- D. D. Perrin, Dissociation constants of organic bases in aqueous solution, Butterworths, London, 1965, Supplement 1972 Search PubMed.
- D. D. Perrin, Dissociation constants of inorganic acids and bases in aqueous solution, Butterworths, London, 1969 Search PubMed.
- I. H. Um, S. W. Min and J. M. Dust, J. Org. Chem., 2007, 72, 8797–8803 CrossRef CAS PubMed.
- O. Acevedo and W. L. Jorgensen, Org. Lett., 2004, 6, 2881–2884 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob00295e. |
| ‡ Underlying research data for this paper is available in accordance with the EPSRC open data policy from DOI: 10.15128/r29880vq960. |
| This journal is © The Royal Society of Chemistry 2017 |