
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The application of perfluoroheteroaromatic reagents in the preparation of modified peptide systems†‡
Diana
Gimenez
a,
Caitlin A.
Mooney
a,
Anica
Dose
a,
Graham
Sandford
a,
Christopher R.
Coxon
*b and
Steven L.
Cobb
 *a
*a
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: c.r.coxon@ljmu.ac.uk; s.l.cobb@durham.ac.uk
bSchool of Pharmacy and Biomolecular Sciences, Byrom Street Campus, Liverpool John Moores University, Liverpool, L3 3AF, UK
First published on 21st April 2017
Abstract
The perfluoroheteroaromatic reagent pentafluoropyridine has proved to be a highly reactive electrophile, undergoing SNAr arylation reactions in the presence of a range of nucleophilic peptide side chains (i.e. cysteine, tyrosine, serine and lysine) under mild conditions. Moreover, we have shown how one-step peptide-modification using perfluoroheteroaromatics can deliver enhanced proteolytic stability in pharmaceutically-relevant peptides such as oxytocin.
Introduction
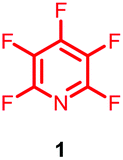
Perfluoroheteroaromatic derivatives such as pentafluoropyridine (1), tetrafluoro-pyrimidine (2) and tetrafluoropyridazine (3) are highly useful building blocks for organic synthesis because, in principle, all fluorine atoms may be displaced by nucleophiles via regioselective, sequential nucleophilic aromatic substitution reactions (SNAr). Indeed, we have extensively used 1–3 as starting materials for the synthesis of a variety of highly-substituted heterocyclic derivatives,1–3 tuned glycosyl donors4 and polyfunctional ring-fused scaffolds.5a–g In particular, 1 and 2 have been used as central multifunctional building blocks for the synthesis of a range of macrocyclic systems5h–k upon reaction with suitable bifunctional oxygen and nitrogen-centred nucleophiles (Fig. 1).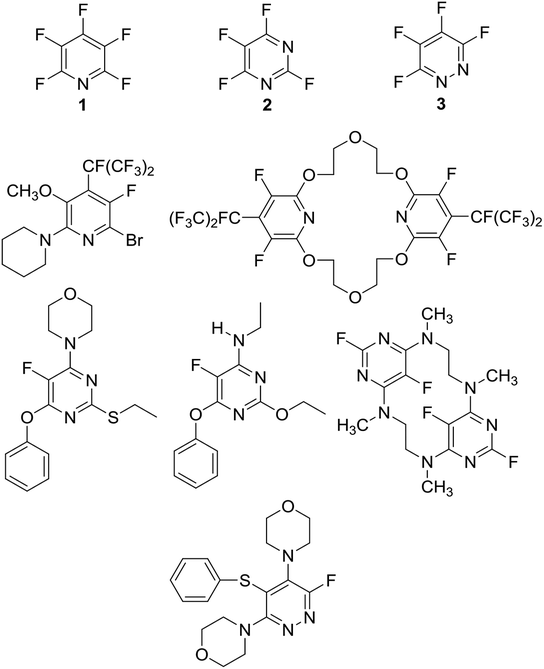 | ||
| Fig. 1 Multifunctional heterocyclic systems and macrocycles synthesised from pentafluoropyridine (1), tetrafluoropyrimidine (2) and tetrafluoropyridazine (3). | ||
Given the synthetic versatility and useful reactivity of perfluoroheteroaromatic systems with nucleophilic species, it was envisaged that they would be extremely attractive reagents to exploit within the field of peptide chemistry. We postulated that they could be reacted with nucleophilic amino acid residues (i.e. serine, cysteine and lysine) to introduce a reactive handle for peptide modification and ‘tagging’;6 and also as a route to access cyclic or “stapled” peptide scaffolds.7 To this end, as part of an ongoing wider research programme in perfluoroheterocyclic chemistry, we previously reported that orthogonally protected serine derivatives can be successfully arylated with pentafluoropyridine (1) to afford novel amino acids such as 4, which could then be utilised as a building block in peptide synthesis8a (Fig. 2).
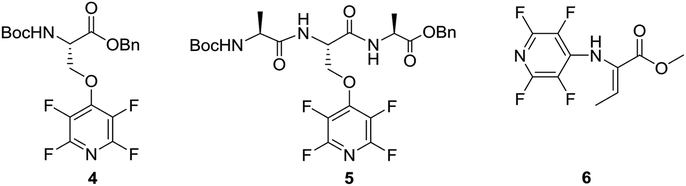 | ||
| Fig. 2 Example of pentafluoropyridine ‘tagged’ serine aminoacid 4 reported by Hudson et al.8a This modified building block was further demonstrated to be compatible with peptide elongation into a model trimeric sequence (5). N-Fluoropyrinidyl dehydrobutyrine 6, having a specific Z-alkene geometry, as synthetized by Webster et al.8b | ||
We were also able to demonstrated that (1) can be utilised as a threonine dehydrating agent to give access to novel dehydrobutyrines, such as 6, under mild reaction conditions (e.g. inorganic base, room temperature).8b
To further develop our research programme in this area, we sought to investigate the direct reaction of perfluoroheteroaromatic reagents like pentafluoropyridine (1) with various peptide substrates. Herein, we present our findings concerning the reaction of a focussed range of perfluoroheteroaromatic derivatives with model cysteine, serine and lysine containing peptides.
Within the presented work, we demonstrate the ability of pentafluoropyridine (1) to tag nucleophilic peptide side chains beyond the routinely accessed cysteine. Differences in reactivity between perfluoroheteroaromatics (e.g.1) and the known peptide arylation reagent hexafluorobenzene (7)9,10 are also highlighted and discussed. Finally, we have demonstrated the use our peptide-tagging methodology to prepare novel analogues of the well-known cyclic peptides, oxytocin and vasopressin, to probe the effects of such modifications on physicochemical properties, such as proteolytic stability.
Results and discussion
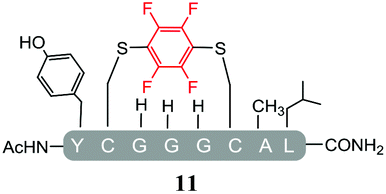
A series of simple linear model peptides based on the sequences reported by Pentelute et al.,9,10 were synthesised by Fmoc solid phase peptide synthesis. The sequences were selected to enable a comparison of the differences observed in reactivity between hexafluorobenzene (7) and pentafluoropyridine (1) based reagents. Model sequences used for these control peptides were as follows; AcNH-[YXGGGXAL]-CONH2, were X is a cysteine residue (X = Cys, pep1), a serine residue (X = Ser, pep2) or a lysine residue (X = Lys, pep3).Exploring the scope of perfluoroaromatics for the SNAr-tagging of cysteine side chains
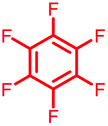
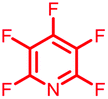
Initially, we investigated the reactivity of 1 towards sulfur-based nucleophiles (i.e. cysteine containing pep1). When peptide pep1 was reacted in DMF with 25 molar equivalents of hexafluorobenzene (7), pentafluoropyridine (1), or related perfluoropyridine-based electrophiles (8–10) in the presence of DIPEA significant differences in reaction outcomes were observed as summarised in Table 1.| Entry | Method | ArF | Comments/products formed |
|---|---|---|---|
| 1 | A |
|
|
| 2 | A |
|
|
| 3 | A |
|
No reaction |
| 4 | A |
|
No reaction |
| 5 | A |
|
|
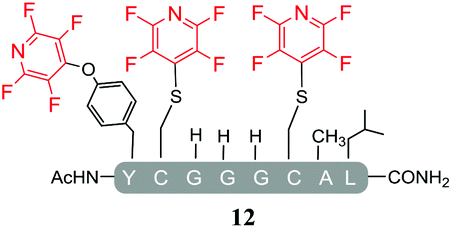
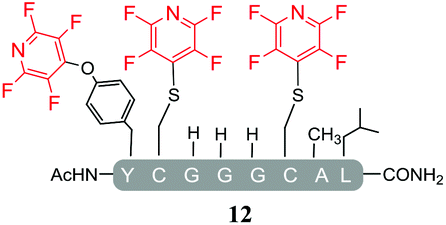
As expected, hexafluorobenzene (7) arylated thiol based pep1 to generate the corresponding “stapled” peptide 11, in full agreement with previous reports9,10 (Table 1, entry 1). In the case of pentafluoropyridine (1), the introduction of the nitrogen atom into the aromatic ring strongly increases the electron-deficient character of the electrophilic system as compared with hexafluorobenzene (7). Given this enhanced electrophilic character, (1) reacted with pep1 to give the linear tri-tagged peptide 12 as the main product. 12 is generated via1 reacting with both the cysteine residues and the tyrosine hydroxyl group (Table 1, entry 2). The formation of “stapled” peptide products via reaction with hexafluorobenzene (7) has been proposed to proceed through two sequential SNAr steps.9–13 After the initial substitution, a linear ‘mono-tagged’ intermediate is formed, where electronic charge density can be displaced from the positions ortho- and para- to the sulfur atom, further activating the intermediate towards a second SNAr reaction step. In contrast, pentafluoropyridine-tagged linear intermediates, containing the aromatic nitrogen atom para to the new substituent can only cyclise though the more sterically hindered ortho-position. Given this competition is set up between cyclisation (intramolecular) and further tagging (intermolecular). Under the reaction conditions used (method A) 1 is used in a significant excess and as such the linear peptide 12 emerges as the main product. Interestingly, reports of sulfur nucleophiles reacting with pentafluoropyridine (1) are not well document in the literature.14–16
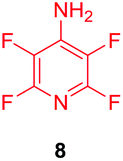
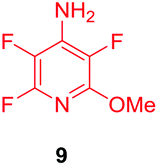
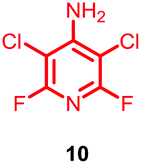
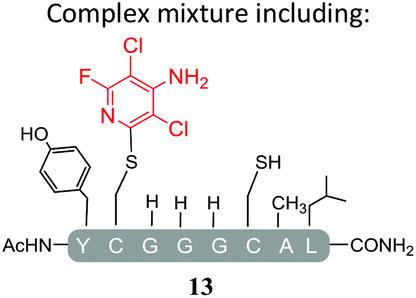
Substituents will affect the reactivity of 1 towards nucleophilic attack and to probe this, aniline variants of 1 where investigated as electrophiles in the reaction with pep1. Electrophiles 8–10 (Table 1) displayed significantly different reactivity profiles to 1, as exemplified by 4-aminotetrafluoropyridine (8), which was found to be completely unreactive (Table 1, entry 3 and ESI Fig. SI06‡); and the 3,5-dichlorinated electrophile 10, which afforded a complex mixture of products (Table 1, entry 5). From this mixture, only compound 13, a trifluoroacetate-adducted species with m/z = 1018 Da, could be isolated and successfully assigned as the mono-tagged peptide (ESI Fig. SI08‡). Site directed tandem MS/MS spectrometry corroborated that 13 exists as a mixture of regioisomers where the presence of the fluoroaromatic moiety can be located at either of the two cysteines available (ESI Fig. SI28–30‡).
Effect of peptide/electrophile ratio, concentration and temperature
Following on from the formation of the linear tri-tagged peptide 12, we were keen to investigate if reaction conditions could be optimised that would allow selective tagging with 1 at either the cysteines (di-tagged product) or tyrosine (mono-tagged product). To do this we directed our attention to probing the effects produced by changing the peptide: perfluoroaromatic ratio, concentration and reaction temperature. We hypothesised that a cyclic product would more likely be produced if the reaction was carried out in the presence of near-equimolar peptide and 1 and/or high-dilution conditions, by reducing partially the availability of the free electrophile in the reaction. Therefore, we varied the conditions initially used (method A) and ran reactions employing: only 5 equivalents of pentafluoropyridine (1) (method B), 5× dilution (method C) or elevated reaction temperature (50 °C, method D). Interestingly, none of the new reaction conditions investigated appeared to have a significant impact on the major product outcome. In all examples, where 1 was used as the electrophile the main product formed was the corresponding linear tri-tagged compound (12).Non-sulfur based nucleophiles: perfluoroaryl-tagging of serine and lysine based peptides
Having explored the application of cysteine as a nucleophile we then proceeded to analyse the reaction of hexafluorobenzene (7) and pentafluoropyridine (1) with analogous model peptides with either oxygen (X = Ser, pep2) or a nitrogen (X = Lys, pep3) centred nucleophile-containing amino acid pairs. The results obtained are outlined in Table 2.| Entry | ArF | Peptide (X =) | Products formed |
|---|---|---|---|
| 6 |
|
Ser | No reaction |
| 7 | Lys | No reaction | |
| 8 |
|
Ser |
|
| 9 | Lys |
|
|
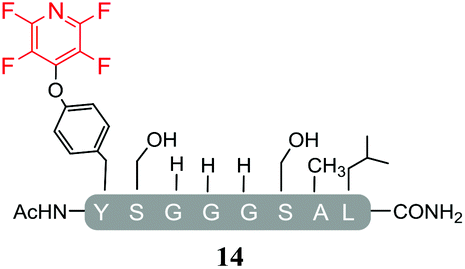
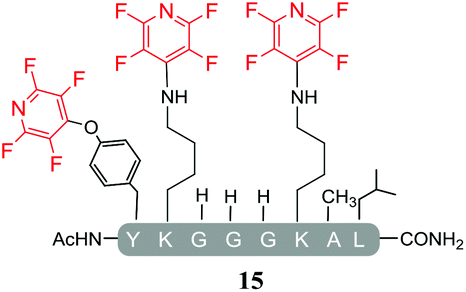
Hexafluorobenzene (7), which readily reacted with cysteine to generate a cyclic peptide 11 was found to be completely unreactive towards the nucleophilic attack by any of the non-sulphur based nucleophiles present (Table 2, entries 6, 7 and ESI Fig. SI13, 14‡). Despite this it should be noted that recently Pentelute et al. demonstrated an elegant route through which arylation at lysine could be achieved if a perfluorosulfone derivative of hexafluorobenzene is utilised.17 In comparison to 7, the more reactive pentafluoropyridine (1) was able to react with the lysine residues and the aromatic alcohol of the tyrosine residue under the same conditions (compounds 14 and 15, Table 2, entries 8 and 9 and ESI Fig. SI15, 16‡). It is noteworthy that no product derived from the reaction of pentafluoropyridine (1) with the serine aliphatic alcohol was observed. The tandem MS/MS fragmentation pattern of the isolated product 14 clearly showed that the fluoropyridine moiety was attached specifically at the tyrosine residue (ESI Fig. SI31 and SI32‡). In addition, the presence of only one single pair of 19F-NMR signals, at δ = −91.32 and −155.98 ppm, allowed the designation of the relative position of the ether group at the ring as para- to the aromatic nitrogen (ESI Fig. SI33‡). The selective formation of 14 indicated that under the reaction conditions employed (method A) tagging with 1 had specificity for aromatic hydroxyl nucleophiles (Tyr) over aliphatic hydroxyls (Ser). The formation of 14 was a somewhat unexpected result given that we had previously reported that 1 readily reacts at room temperature with individual serine and threonine amino acid derivatives7,8 (Fig. 2).
By comparing the 19F NMR signals observed for compound 14, with the corresponding spectra of compounds 12 and 15, it was possible to assign the chemical shifts arising from the fluorine atoms at the rings that have been substituted by cysteine and lysine residues. In both products, the presence of two pairs of 19F-NMR signals corresponding to the fluorine atoms located in ortho- and meta-positions relative to the aromatic nitrogen, provided evidence that also these reactions proceeded via the specific SNAr substitution of the fluorine atom located para- to the nitrogen (Table 3 and see ESI Fig. SI27,‡33 and 36 respectively).
|
|
19F-NMR observed chemical shifts δ (ppm) |
|---|---|
| X = Cys | δF1 = −93.42, δF2 = −137.49 |
| X = Lys | δF1 = −98.17, δF2 = −165.54 |
| X = Tyr | δF1 = −91.32, δF2 = −155.98 |
| X = Ser | δF1 = −97.10, δF2 = −165.92 |
Evaluating the selection of base in the SNAr reaction: organic DIPEA vs. inorganic Cs2CO3
In order to determine the effect of the base utilised on the reactivity we replaced the organic base DIPEA with an inorganic base (Cs2CO3). The isolated products from the reactions carried out using Cs2CO3 in the presence of 25 molar equivalents of hexafluorobenzene (7) or pentafluoropyridine (1) are shown in Tables 4 and 5 respectively. Swapping from DIPEA to Cs2CO3 generated several interesting results. For example, the di-serine containing peptide (pep2) was found to react with hexafluorobenzene (7) generating a mixture of products (entry 11, Table 4 and ESI Fig. SI18‡), despite no observable reaction being detected when the reaction was run in the presence of DIPEA (entry 6, Table 2). The main reaction product corresponded to the mono-tagged peptide 16 (for isolated product data see ESI‡). As expected from previous observations, it was confirmed that reaction had proceed specifically through the tyrosine and not a serine residue, as evidenced by the fact that only 3 signals were observed by 19F-NMR (ESI Fig. SI39‡).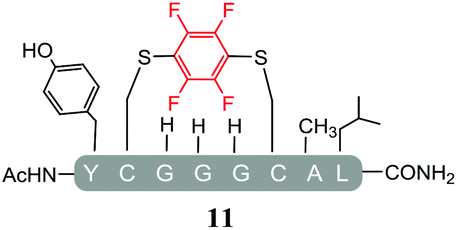
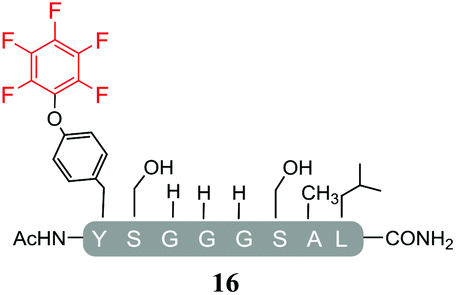
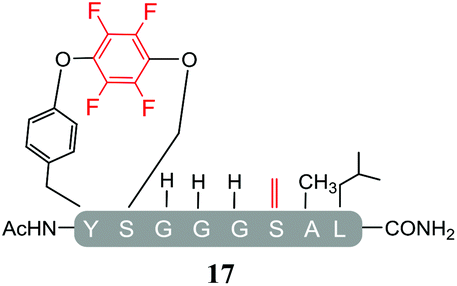
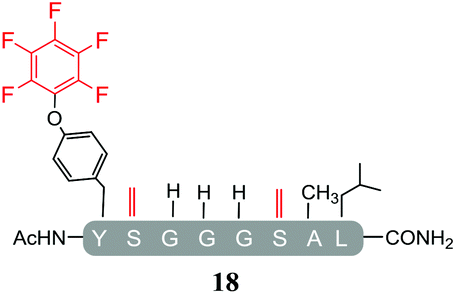
In addition, it was interesting that the dehydrated asymmetric cyclic peptide (17) and the doubly-dehydrated mono-substituted product (18) were also obtained. The presence of both suggests that cyclization prevents serine dehydration. The isolation and characterisation of both secondary products (17 see ESI Fig. SI40–42 and 18 see ESI Fig. SI043–045‡) confirmed their identity and structures (Table 4). It should also be noted that to the best of our knowledge, 17, represents the only reported example of an asymmetric perfluorobenzene stapled peptide.
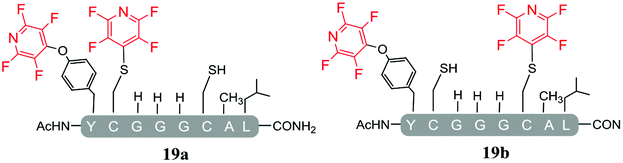
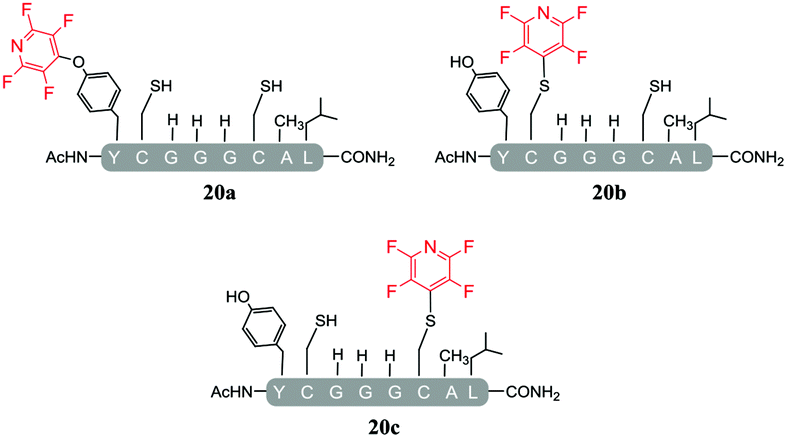
As a general trend, it was observed that in the presence of Cs2CO3, a more complex mixture of products was obtained when compared with the same reactions carried out using DIPEA. For example, in the reaction of Cs2CO3/pentafluoropyridine (1) with the di-cysteine peptide (pep1) several products were isolated (entry 13, Table 5 and ESI Fig. SI20‡) but with DIPEA (entry 2, Table 1) the linear tri-tagged product 12 was the major product formed and isolated in the reaction. Furthermore, in the presence of Cs2CO3 additional products included the di-tagged peptides 19a and 19b (ESI Fig. SI46–48‡) and the mono-tagged peptides 20a, 20b and 20c (ESI Fig. SI49–51‡), both of which were isolated as inseparable mixtures.
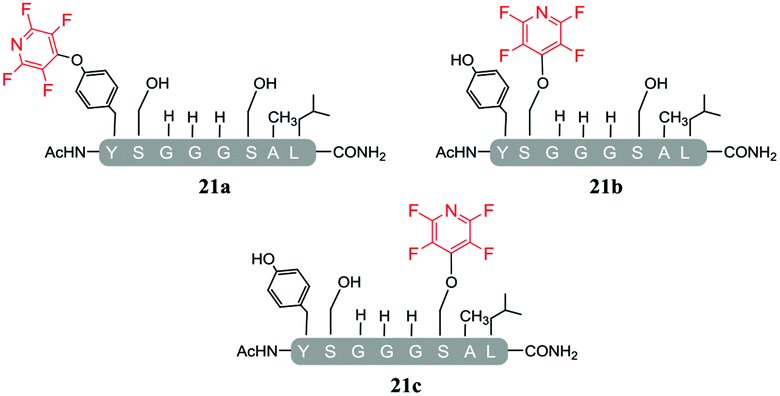
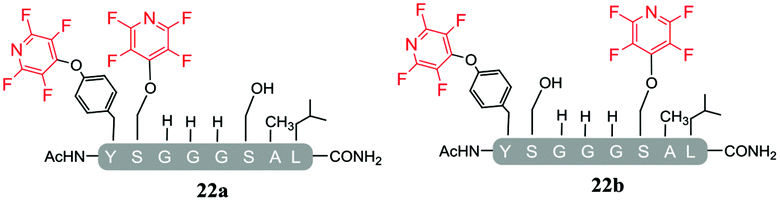
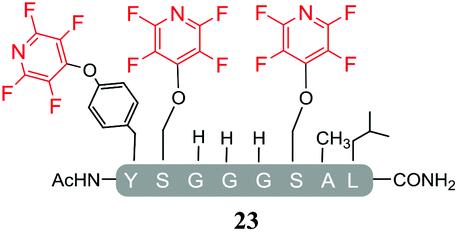
A complex mixture of products was also observed when the reaction of the di-serine peptide pep2 with electrophile 1 was analysed (entry 14, Table 5 and ESI Fig. SI21‡). In the presence of Cs2CO3, 1 was able to undergo SNAr reactions at both tyrosine and serine residues as indicated by 19F NMR analysis. For example, unlike previously observations when using DIPEA, where tagging occurred exclusively at the tyrosine residue, analysis by 19F NMR of 22a and 22b revealed two distinct sets of fluorine signals corresponding to the tetrafluoropyridine motif attached to the tyrosine (δF1 = −91.32, δF2 = −155.98 ppm) and the serine residue (δF1 = −97.10 and δF2 = −165.96 ppm). In both cases the hydroxyl nucleophile had replaced the fluorine atom located in the para-position to the ring nitrogen (ESI Fig. SI57‡). The linear tri-tagged peptide 23 (ESI Fig. SI58–60‡) was obtained in the reaction but only as a minor product.
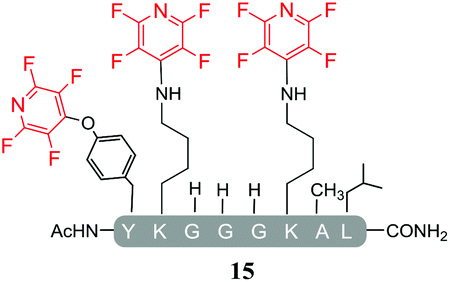
In agreement with earlier observations where DIPEA was utilised as a base, the reaction with the lysine-containing peptide, pep3 led to the formation of the linear tri-tagged peptide 15 as the major product (Table 5, entry 15 and ESI Fig. SI22‡).
Exploring the potential application of perfluoro-‘tagging’ to enhance peptide bio-stability
We hypothesised that perfluoroheteroaromatic tagging could be utilised to add steric bulk to amino acid residues and thus ‘protect’ peptides from undergoing enzymatic degradation. As such, we proceeded to synthesise novel derivatives of two bioactive peptide hormones, which typically have short half-lives in vivo.18,19Oxytocin (25) and vasopressin (26) share a common cyclic core structural motif, which has been shown to be particularly sensitive to chymotrypsin-mediated enzymatic amide bond-hydrolysis, at the aromatic Tyr and the C-terminal glycine residue, leading to the formation of inactive linear fragments.20,21
Due to their pharmacological relevance, different strategies have been employed in an effort to obtain analogues of 24 and 25 with enhanced stability, such as the incorporation of unnatural amino acids at the C-terminus,22 amide bond head to tail cyclization and replacement of the disulfide bridge.23 For example, carbetocin is an oxytocin analogue wherein the C-terminal glycine has been de-aminated, tyrosine residue alkylated (OH to OMe) and the disulfide bond chemically modified.24 While, carbetocin does display enhanced bio-stability, its synthesis compared to oxytocin, is considerably more complex. We envisaged that the “tagging” of oxytocin using perfluoroaromatics in a simple one-step process could offer a relatively mild and cost effective route by which alter its enzymatic stability (Fig. 3).
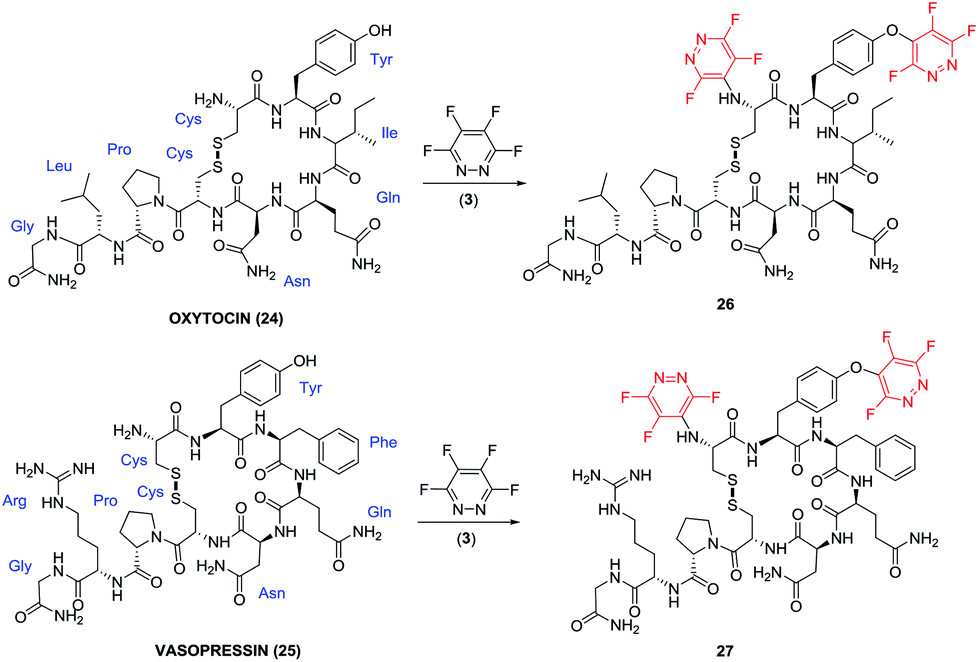 | ||
| Fig. 3 Natural cyclic peptide hormones oxytocin (24) and vasopressin (25) and their corresponding ‘perfluoroaryl-tagged’ analogues 26 and 27. | ||
The reaction of oxytocin (24) and vasopressin (25) in the presence of 5 equivalents of pentafluoropyridine (1), and DIPEA in DMF afforded a mixture of mono- and di-tagged derivatives (data given in the ESI‡). In order to afford di-tagged peptides in a more efficient manner the reaction was repeated using the more reactive tetra-fluoropyridazine (3). Reaction of peptides 24 and 25 with electrophile 3 proceeded to give the di-tagged peptides with very little trace of mono-tagging observed in the crude reaction mixture. Di-tagged peptides 26 and 27 were isolated, purified and analysed by tandem MS/MS and 19F NMR to confirm that tagging had occurred at the tyrosine residue and N-terminus (see ESI Fig. SI61–63 for compound 26, and Fig. SI64–66‡ for compound 27).
The bio-stability of 27 and 28 were analysed and compared to the non-tagged parent peptides (24 and 25 respectively) via incubation with chymotrypsin at 20 °C for 24 h (peptide/enzyme ratio 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; Tris buffer 50 mM, pH 7.4). The ratio of the remaining intact peptide/digested peptide was followed by analytical RP-HPLC (λ = 220 nm), with sample aliquots taken and analysed at different times over a 24 h period. Standard control samples containing peptides 24–27 but no enzyme were also performed (see ESI Fig. SI71A–D‡).
:1; Tris buffer 50 mM, pH 7.4). The ratio of the remaining intact peptide/digested peptide was followed by analytical RP-HPLC (λ = 220 nm), with sample aliquots taken and analysed at different times over a 24 h period. Standard control samples containing peptides 24–27 but no enzyme were also performed (see ESI Fig. SI71A–D‡).
After 24 h the digested samples were analysed by MALDI-ToF spectrometry in order to assess the identity of any peptide fragments present. Significant differences in the digestion profiles were found when we compared the non-tagged (24 and 25) vs. the tagged analogues (26 and 27) (Fig. 4 and 5).
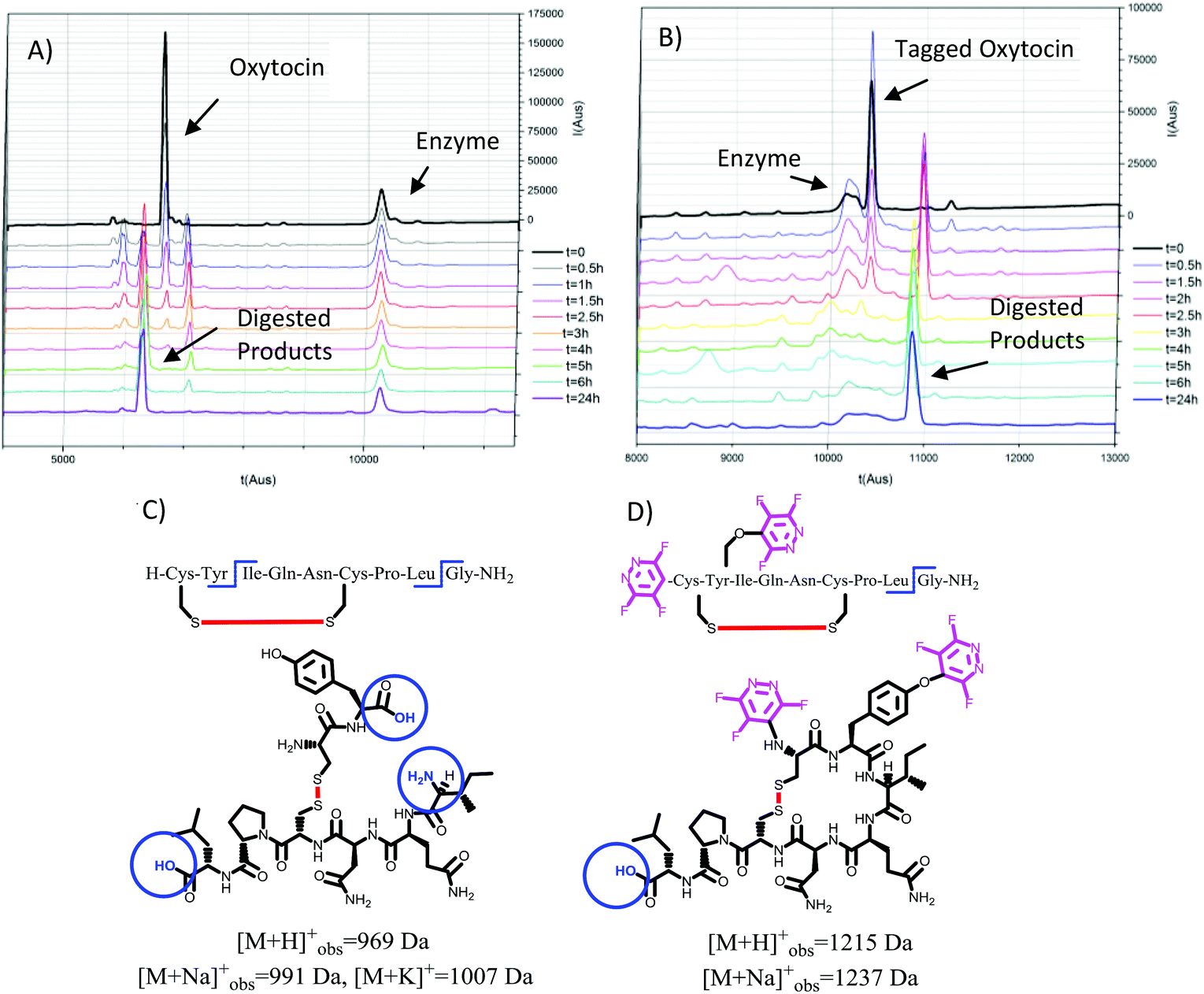 | ||
| Fig. 4 Evaluation of oxytocin (24, A) and perfluoroaryl-tagged oxytocin (27, B) digestion process in the presence of chymotrypsin, as analysed by RP-HPLC (λ = 220 nm). (C) Structures corresponding to the final digested linear product observed in parent oxytocin, showing the hydrolysis of the Tyr-Ile amide bond. In comparison, perfluoroaryl-tagged Tyr residue in compound 27 was probed to be resistant to enzymatic digestion, preserving the initial cyclic structure of the product (D). Digestion sites are circled for clarity. | ||
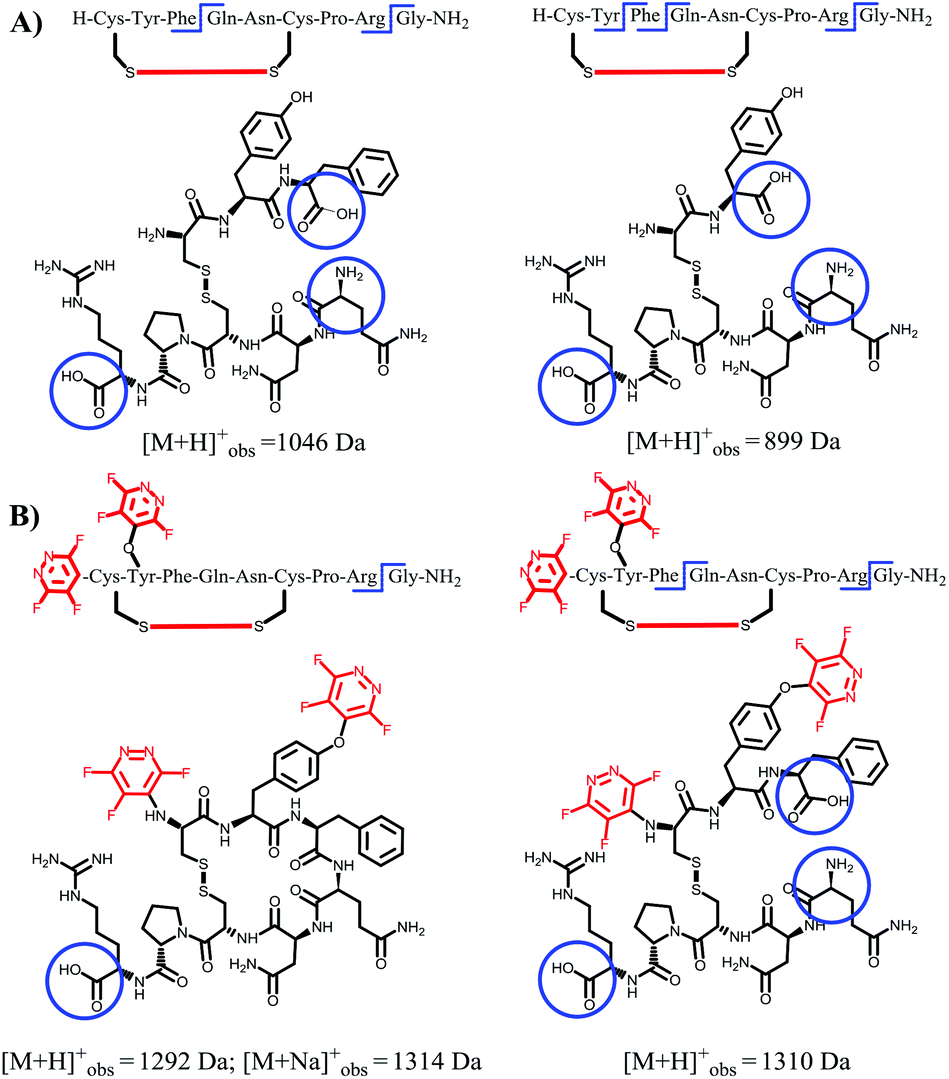 | ||
| Fig. 5 Final digested products observed in parent vasopressin (25, A) and perfluoroaryl-tagged vasopressin (27, B), as assessed by MALDI-ToF analysis after digestion (24 h). When compared to the natural sequence, the perfluoroaryl-tagged derivative could not be hydrolysed by the enzyme at the chemically modified tyrosine residue. Digestion sites are circled for clarity. | ||
As illustrated in Fig. 4A–C the natural non-tagged oxytocin 24 was found to be relatively quickly digested at the amide bond adjacent to the Tyr, resulting in the opening of the macrocyclic core motif. Further hydrolysis at the C-terminal glycine residue afforded a linear fragment (as seen in Fig. 4C). By comparison, we only observed a single digestion product when the tagged-oxytocin 26 was subjected to the same digestion process. In this system no hydrolysis of the Tyr-Ile amide bond was observed, leaving the core ring structure intact (see Fig. 4D). Cleavage of the C-terminal glycine residue was still observed but this is to be expected as it was not modified or protected. Similar results were obtained when we carried out our analysis on the cyclic peptide vasopressin 25 and its tagged analogue 27 (ESI Fig. SI69 and 70‡). The tagged peptide 27 was found to partially retain its core cyclic structure but again complete loss of the C-terminal glycine was observed. Some enzymatic degradation of the core cyclic scaffold of 27 was observed due to hydrolysis of the Phe-Gln amide bond (see structures in Fig. 5). Overall, the results obtained demonstrate that modification of the tyrosine residue offer a method to protect enzymatic cleavage at that specific site (e.g. as in case of 26), but the enhanced steric bulk does not impart full protection at adjacent residues (i.e. Phe-Gln amide bond in 27 is still partially cleaved). It should be noted that while enhanced proteolytic stability can be achieved we are yet to determine if the perfluoro-arylation of 24 and 25 results in retention of biological activity.
Conclusions
We have shown that perfluoroheteroaromatic reagents can be successfully applied to access a range of modified peptides. Pentafluoropyridine (1) has proven to be a highly reactive electrophile, allowing SNAr mediated arylations to proceed in the presence of a range of nucleophilic side chains including cysteine, tyrosine, and lysine under mild reaction conditions. As expected, hexafluorobenzene (7) which lacks a ring nitrogen, was found to be less reactive than 1, although reaction conditions were found that allowed arylation to occur at both tyrosine and serine residues (e.g. formation of product 17, Table 4). Electrophile concentration only had a minor impact on the reaction outcomes, yet the nature of the base used (organic or inorganic) did produce noticeable differences in product formation. In general when employing the inorganic base, Cs2CO3, we observed a significant increase in peptide “tagged” when compared with the use of DIPEA. The identification of reaction conditions and reagents that allow modification at non-cysteine side chains (i.e. tyrosine, serine and lysine), will add further scope to the rapidly expanding field of peptide SNAr arylation peptide chemistry.8–13,17,25,26 We were also able to demonstrated the application of perfluoroheteroaromatic reagents as means to modulate the bio-stability of two well-known cyclic hormones, oxytocin (24) and vasopressin (25). While both parent peptides (oxytocin and vasopressin) were shown to be rapidly degraded by chymotrypsin, we demonstrated that a significantly increased enzymatic stability occurred upon tagging the peptides with a perfluoroheteroaromatic. From this work, we propose that this type of site-specific perfluoroheteroaryl-tagging could offer a simple and cost effective route by which to target and block digestion sites and thus improve the in vivo properties of known peptide drugs.Experimental
Materials and general methods
All chemicals and solvents were analytical grade and used without further purification. Liquid chromatography-mass spectrometry (LC/MS; ESI+) analyses were performed on a Waters Acquity UPLC BEH C18 column (1.7 μm, 2.1 mm × 50 mm) using a Waters Acquity UPLC system equipped with a photodiode array detector, providing absorbance data from 210 nm to 400 nm. A gradient with eluent I (0.1% HCOOH in water) and eluent II (0.1% HCOOH in acetonitrile) rising linearly from 5 to 95% of II during t = 0.2–4.0 min was applied at a flow rate of 0.6 mL min−1 after 0.2 min of 95% solvent I equilibration. High-resolution QToF-LC/MS and QToF-MS/MS analyses were performed on a Acquity UPLC BEH C18 column also (1.7 μm, 2.1 mm × 50 mm) using a Waters Acquity UPLC system coupled in this case to a Micromass QToF Premier mass spectrometer, also equipped with a photodiode array detector providing absorbance data from 210 nm to 400 nm. A gradient with eluent I (0.1% HCOOH in water) and eluent II (0.1% HCOOH in acetonitrile) rising linearly from 0 to 99% of II during t = 0.0–5.0 min was applied at a flow rate of 0.6 mL min−1. 19F-NMR spectra studies were recorded at 376 MHz in a Bruker Advance spectrometer at 298 K, using 8 scans with a relaxation delay of 1 s. All data has been processed using Mestrenova® software.Solid phase synthesis of model peptides pep1–pep3
Each of the described peptide sequences was prepared using automated Fmoc-SPPS methods on a Liberty 1 peptide synthesiser (CEM) with microwave-assisted couplings (single coupling per amino acid; 10 min, 75 °C or 50 °C for Fmoc-Cys(Trt)-OH coupling). Solid phase synthesis was conducted using Rink amide resin (0.7 mol g−1 loading) on a 0.1 mmol scale, employing HBTU and DIPEA as the activator and base, respectively. Standard acid-sensitive amino acid side chain protecting groups e.g. Boc, tBu, trityl were employed. Following the on-resin growing of the appropriate sequence, N-terminal acetylation was achieved by using a solution of Ac2O in DMF (20% v/v, 5 mL) while shaking at room temperature for 15 min. Once completed, the beads were washed with DMF three times and the acetylation step repeated. The resin was then carefully washed with DMF, MeOH and DCM (3 × 10 mL). Finally, peptides were cleaved from the resin as the corresponding C-terminal amides by treatment with a mixture of TFA/TIPS/H2O 95:5:5% v/v (2 mL) at room temperature for 4 h. After removal of volatiles under vacuum, the product was triturated and carefully washed with Et2O (3 × 10 mL). The final precipitated solid residue was then re-dissolved in H2O/TFA 99:1 (15 mL) and lyophilised affording the corresponding peptides as a white/yellow powder. The crude peptides were used without further purification.
General procedure for solution phase peptide tagging and stapling with perfluoroaromatics
Solid crude peptides pep1–3 (2 mg, approx. 2.5 μmol) were dissolved in DMF (0.5 mL) in a 1.5 ml plastic Eppendorf tube, to which a Cs2CO3 or DIPEA stock solution (50 mM in appropriate solvent, 0.5 mL) was then added. 25 equivalents of the corresponding perfluoroaromatic were slowly added and the tube was shaken vigorously at room temperature for 4.5 h (method A, unless otherwise stated). After removal of volatiles under vacuum, all products were dissolved in an 8:1:1 mixture of DMF/H2O/MeCN (1 mL) and characterised by LC/MS (ESI+). When formation of novel compounds was observed, 10-fold scaled reactions were employed in all cases for product isolation in order to afford a complete characterisation.
Product isolation and characterization
Products from the large-scale reactions were purified and isolated by semi-preparative reverse phase HPLC performed on a Discovery Bio wide pore C18-5 column (Supelco; 5 μm, 25 cm × 10 mm), using a Pelkin-Elmer 200 LC pump coupled to a Waters 486 tunable absorbance detector settled up at λ = 220 nm. A gradient with eluent A (95:5:0.1% H2O:MeCN:TFA) and eluent B (5:95:0.1% H2O:MeCN:TFA) was applied, where eluent B increased linearly from 0 to 100% over 40 min. Purified pooled fractions were then freeze-dried and analysed by LC/MS. The desired pure peptide products were then further characterised by 19F NMR (2 mg mL−1 in H2O/CD3CN 1:1, unless otherwise stated), high resolution QToF-LC/MS and ion directed tandem mass spectrometry (MS/MS).
General procedure for oxytocin and vasopressin-tagging
Commercially available C-teminal amide oxytocin (24) was obtained from Cayman Chemical Co., and vasopressin (25) was purchased form Apollo Scientific Ltd. Both peptides were used as starting materials without any further purification. In both cases 10 mg of the peptide was dissolved in 5 mL of DMF to which another 5 mL of a freshly made stock solution of DIPEA 50 mM in DMF was then added. Solutions where placed in argon flushed chemically-resistant syringes, and 25 equivalents of either pentafluoropyridine (1) or tetrafluoropyridazine (3) were slowly added. The systems were reacted with vigorously shaking at room temperature for 4.5 h. After removal of DMF and volatiles under vacuum, the remaining oily residue was dissolved in H2O/MeCN (10 mL) and freeze-dried. Crude materials were characterised by LC/MS (ESI+) to confirm the presence of the desired products and further purified by semi-preparative HPLC (as described previously). Isolated compounds were further characterised by 19F-NMR (2 mg mL−1 in CD3CN), high resolution QToF-LC/MS and ion directed tandem mass spectrometry (MS/MS).General procedure for enzymatic digestion using chymotrypsin
For the bio-stability assays α-chymotrypsin, from bovine pancreas, was purchased from Sigma-Aldrich. Peptides were dissolved in a stock solution of Tris buffer (25 mM, pH 7.6) up to a concentration of 1 mg mL−1 concentration. Simultaneously a freshly made stock solution of enzyme in the same chilled buffer was prepared. The samples were immediately prepared by mixing in a polypropylene microcentrifuge tube an aliquot of 100 μL of peptide solution with the corresponding amount of enzyme stock solution to afford a final peptide to enzyme ratio of 25:1. The sample mixtures were then incubated in a thermally controlled water bath at 20 °C for a minimum of 24 h. During the incubation time, 15 μL aliquots of sample mixture were withdrawn at different times, quenched at once with 15 μL of 30% MeCN/TFA solution and analyzed by analytical reverse phase HPLC, employing a X-Bridge C18 column (5.3 μm, 4.6 × 100 mm, 40 °C) in a Pelkin-Elmer 200 series LC system equipped with auto-sampler, UV/Vis-detector and a Peltier column oven. In all samples a linear gradient rising from eluent A (95:5:0.05% H2O:MeCN:TFA) to 100% of eluent B (5:95:0.03% H2O:MeCN:TFA) over t = 30 min plus 5 min of isocratic B elution was applied at a constant flow rate of 1 mL min−1. Detection was performed at λ = 220 nm. The signals corresponding to both peptide and enzyme in the raw chromatograms were baseline corrected and then fitted into separate Gaussian equations using Origin8 software. The area enclosed by each function was taken as the peak neat integrated area. The ratio of non-digested peptide is then estimated as: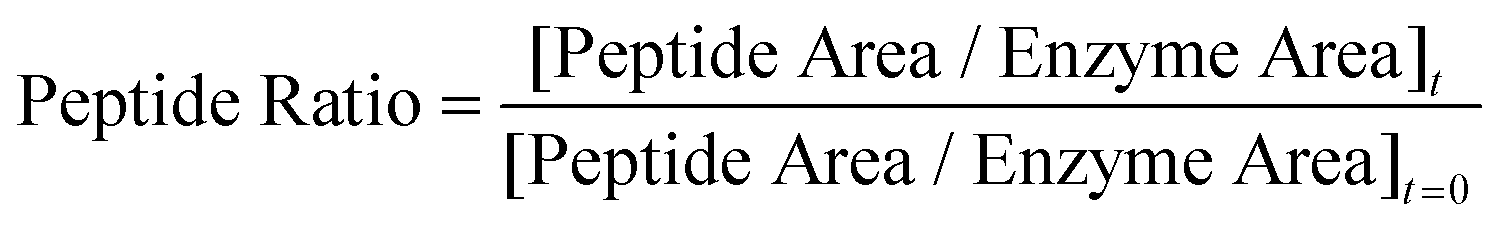
For each peptide a minimum of two independent assays were performed, and the final Peptide Ratio vs. Incubation time analysed in terms of an exponential decay behaviour where:
| Peptide Ratio = y0 + Ae−kt. |
Control samples were run by subjecting each peptide to the same procedure and conditions using the corresponding volume of Tris buffer in the absence of enzyme.
Acknowledgements
We acknowledgement both the European Commission, Seventh Framework Programme (FLUOR21, ITN, Gimenez) and the Engineering and Physical Sciences Research Council (EPSRC, Durham University DTG, Mooney) for funding support.Notes and references
- R. D. Chambers, P. R. Hoskin, G. Sandford, D. S. Yufit and J. A. K. Howard, J. Chem. Soc., Perkin Trans. 1, 2001, 2788 RSC.
- E. L. Parks, G. Sandford, D. S. Yufit, J. A. K. Howard, J. A. Christopher and D. D. Miller, Tetrahedron, 2010, 66, 6195–6204 CrossRef CAS.
- G. Sandford, Top. Heterocycl. Chem., 2012, 27, 1–32 CAS.
- C. A. Hargreaves, G. Sandford and B. G. Davis, Fluorinated Synthons, ed. V. A. Soloshonok, ACS Symposium Series 949, ACS, Washington D.C., 2007, pp. 323–336 Search PubMed.
- (a) G. Sandford, R. Slater, D. S. Yufit, J. A. K. Howard and A. Vong, J. Org. Chem., 2005, 70, 7208–7216 CrossRef CAS PubMed; (b) G. Sandford, C. A. Hargreaves, R. Slater, D. S. Yufit, J. A. K. Howard and A. Vong, Tetrahedron, 2007, 63, 5204–5211 CrossRef; (c) G. Pattison, G. Sandford, D. S. Yufit, J. A. K. Howard, J. A. Christopher and D. D. Miller, J. Org. Chem., 2009, 74, 5533–5540 CrossRef CAS PubMed; (d) G. Pattison, G. Sandford, D. S. Yufit, J. A. K. Howard, J. A. Christopher and D. D. Miller, Tetrahedron, 2009, 65, 8844–8850 CrossRef CAS; (e) M. R. Cargill, K. E. Linton, G. Sandford, D. S. Yufit and J. A. K. Howard, Tetrahedron, 2010, 66, 2356–2362 CrossRef CAS; (f) M. W. Cartwright, E. L. Parks, G. Pattison, R. Slater, G. Sandford, I. Wilson, D. S. Yufit, J. A. K. Howard, J. A. Christopher and D. D. Miller, Tetrahedron, 2010, 66, 3222–3227 CrossRef CAS; (g) G. Pattison, G. Sandford, I. Wilson, D. S. Yufit, J. A. K. Howard, J. A. Christopher and D. D. Miller, Tetrahedron, 2017, 73(5), 437–454 CrossRef CAS; (h) G. Sandford, Chem. – Eur. J., 2003, 9, 1464 CrossRef CAS PubMed; (i) R. D. Chambers, P. R. Hoskin, A. R. Kenwright, P. Richmond, G. Sandford, M. S. Yufit and J. A. K. Howard, Org. Biomol. Chem., 2003, 1, 2137 RSC; (j) R. D. Chambers, A. Khalil, P. Richmond, G. Sandford, D. S. Yufit and J. A. K. Howard, J. Fluorine Chem., 2004, 125, 715–720 CrossRef CAS; (k) R. Ranjbar-Karimi, G. Sandford, D. S. Yufit and J. A. K. Howard, J. Fluorine Chem., 2008, 129, 307–313 CrossRef CAS.
- For recent reviews of methods for chemically tagging peptides and proteins see (a) N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CAS; (b) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- For a recent review of stapled peptides see Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- (a) A. S. Hudson, A. Hoose, C. R. Coxon, G. Sandford and S. L. Cobb, Tetrahedron Lett., 2013, 54, 4865–4867 CrossRef CAS; (b) A. M. Webster, C. R. Coxon, A. M. Kenwright, G. Sandford and S. L. Cobb, Tetrahedron, 2014, 70, 4661–4667 CrossRef CAS.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- Y. Zou, A. M. Spokoyny, C. Zhang, M. D. Simon, H. Yu, Y. Lin and B. L. Pentelute, Org. Biomol. Chem., 2014, 12, 566–573 CAS.
- C. Zhang, P. Dai, A. M. Spokoyny and B. L. Pentelute, Org. Lett., 2014, 16, 3652–3655 CrossRef CAS PubMed.
- C. Zhang, A. M. Spokoyny, Y. Zou, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2013, 52, 14001–14005 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CAS.
- S. L. Cobb, C. R. Coxon and G. Sandford, PCT/GB2015/051543.
- S. L. Cobb, C. R. Coxon and G. Sandford, PCT/GB2015/051542.
- S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. M. Nuss and R. Derda, Chem. Sci., 2016, 7, 3785–3790 RSC.
- G. Lautrette, F. Touti, H. G. Lee, P. Dai and B. L. Pentelute, J. Am. Chem. Soc., 2016, 138, 8340–8343 CrossRef CAS PubMed.
- N. Cort, S. Einarsson and G. Aström, Am. J. Vet. Res., 1982, 43, 1283–1285 CAS.
- R. Walter, T. Yamanaka and S. Sakakibara, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1901–1905 CrossRef CAS.
- V. Pliška and T. Barth, Collect. Czech. Chem. Commun., 1970, 35, 1576–1586 CrossRef.
- T. Barth, V. Pliska and I. Rychlik, Collect. Czech. Chem. Commun., 1967, 32, 1058–1063 CrossRef CAS.
- J. Hlaváček and I. Frič, Collect. Czech. Chem. Commun., 1989, 54, 2261–2270 CrossRef.
- J. J. Ferrie, J. J. Gruskos, A. L. Goldwaser, M. E. Decker and D. A. Guarracino, Bioorg. Med. Chem. Lett., 2013, 23, 989–995 CrossRef CAS PubMed.
- H. Vilhardt, A. Atke, J. Barthova, K. Ubik and T. Barth, Pharmacol. Toxicol., 1997, 81, 147–150 CAS.
- T. Lühmann, S. K. Mong, M. D. Simon, L. Meinel and B. L. Pentelute, Org. Biomol. Chem., 2016, 14, 3345–3349 Search PubMed.
- D. T. Cohen, C. Zhang, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 9784–9787 CrossRef CAS PubMed.
Footnotes |
| † Underlying research data for this paper is available in accordance with the EPSRC open data policy from DOI:10.15128/r2w95050466. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob00283a |
| This journal is © The Royal Society of Chemistry 2017 |