
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapidly accessible “click” rotaxanes utilizing a single amide hydrogen bond templating motif†
Beth E.
Fletcher
,
Michael J. G.
Peach
 and
Nicholas H.
Evans
*
and
Nicholas H.
Evans
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@lancaster.ac.uk
First published on 6th March 2017
Abstract
The synthesis of hydrogen bond templated rotaxanes using the CuAAC click reaction has been achieved in yields of up to 47%, employing near stoichiometric equivalents of macrocycle and readily prepared azide and alkyne half-axle components. Interlocked structure formation has been confirmed by NMR spectroscopy and mass spectrometry. Density functional theory calculations support 1H NMR spectroscopic analysis that the macrocycle resides over the amide of the axle component, rather than the newly formed triazole, as a result of more favourable hydrogen bond interactions.
Introduction
Rotaxanes,1 interlocked molecules consisting of macrocyclic ring(s) trapped on stoppered axle(s), have been applied to a number of nanotechnological applications,2 some of which rely on the relative motion of their ring and axle components.3 Alongside metal cations4 and π–π donor–acceptor interactions,5 hydrogen bonding,6 sometimes augmented with additional cationic7 or anionic8 assistance, has proven a popular choice of template to generate rotaxanes. While a large number of protocols for rotaxane synthesis have now been disclosed, new synthetic routes that allow rapid access to rotaxanes in good yield and using only stoichiometric amounts of precursor components – that minimises waste – are still of considerable interest.Little used to date has been an elegantly simple hydrogen bond templation system reported by Philp and co-workers in 2008.9 This relies upon the hydrogen bond templated threading of an amide containing component through a 2,6-bis-amide pyridyl macrocycle, followed by covalent capture of the resulting pseudo-rotaxane by either maleimide/nitrone 1,3-dipolar cycloaddition9–11 or aza-Wittig reaction.12
Inspired by this work, we chose to investigate and are able to report the isolation of rotaxanes in good yields by use of the straightforward and versatile CuAAC click reaction13 to stopper an azide half-axle, that contains the critical amide functional group required for the pre-requisite pseudo-rotaxane formation with a polyether-isophthalamide macrocycle (Fig. 1). Interlocked structure formation was confirmed by 1H NMR spectroscopy and mass spectrometry. Density functional theory calculations support 1H NMR spectroscopic analysis which suggests that in the prepared rotaxanes the macrocyclic ring remains over the amide of the axle component rather than translating to the newly formed triazole.
 | ||
| Fig. 1 Schematic representation of the rapid synthesis of rotaxanes utilizing a single amide hydrogen bond templating motif and the CuAAC click reaction. | ||
Results and discussion
Synthesis of rotaxane precursors
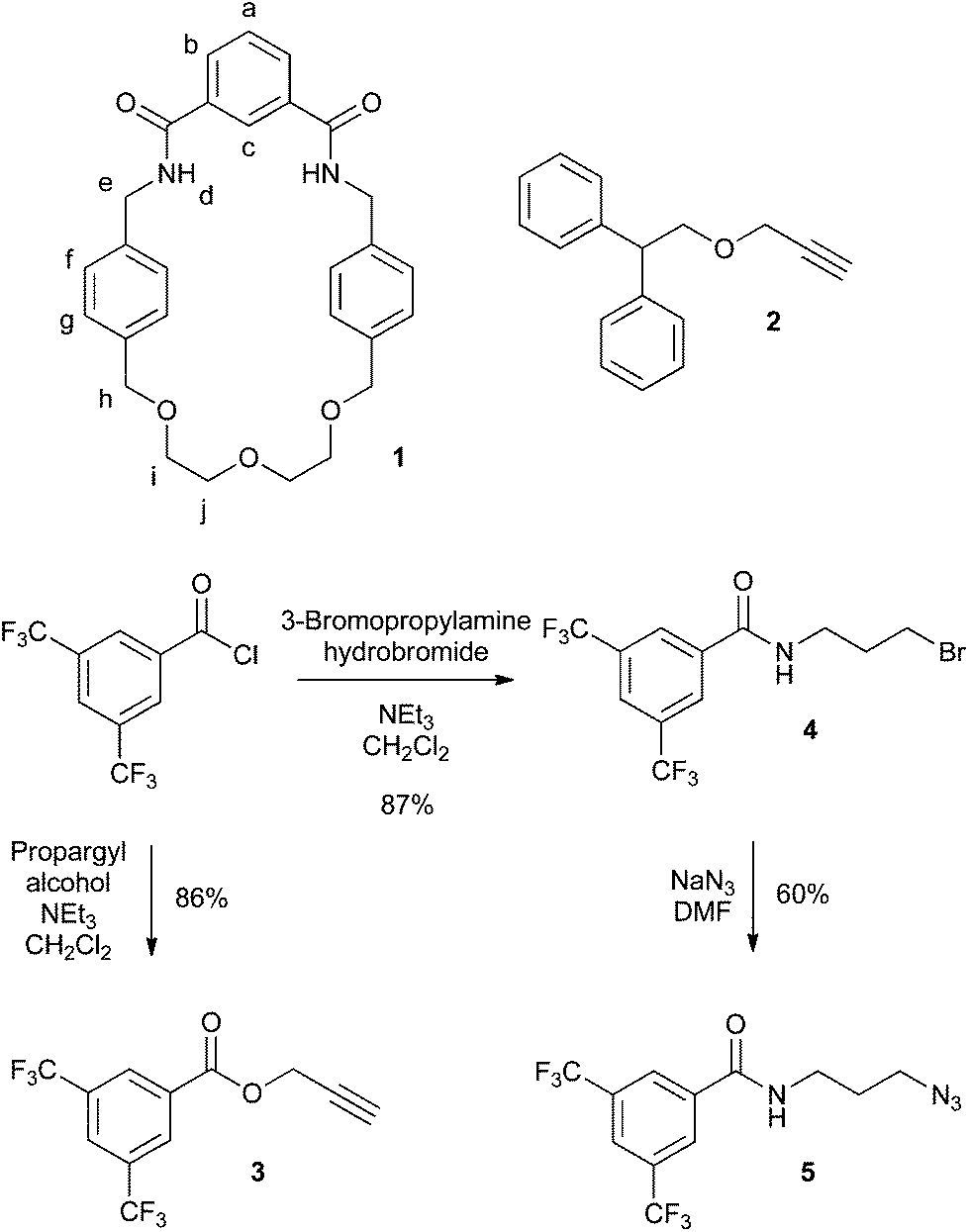
The structures of all rotaxane precursors used in this study, along with the syntheses of novel compounds, are presented in Scheme 1. Previously reported macrocycle 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 has now been prepared in higher yield by use of a template method (see ESI†), while diphenyl alkyne 215 was prepared according to the previously reported procedure. Novel alkyne stopper 3 was prepared by simply reacting equimolar amounts of 3,5-bis(trifluoromethyl)benzoyl chloride and propargyl alcohol, with excess NEt3 in dry CH2Cl2. After aqueous workup, 3 was isolated in 86% yield. The azide click precursor was prepared in two steps. First bromo-amide 4 was prepared by reacting equimolar amounts of 3,5-bis(trifluoromethyl)benzoyl chloride and 3-bromopropylamine hydrobromide, with excess NEt3 in dry CH2Cl2. After aqueous workup, 4 was isolated in 87% yield. Conversion to azide 5 was accomplished by heating a solution of 4 in dry DMF with excess NaN3 at 80 °C. After dilution, and extraction, 5 was isolated in 60% yield. The successful preparation of novel compounds 3–5 was confirmed by NMR and IR spectroscopies and mass spectrometry (see ESI†).
14 has now been prepared in higher yield by use of a template method (see ESI†), while diphenyl alkyne 215 was prepared according to the previously reported procedure. Novel alkyne stopper 3 was prepared by simply reacting equimolar amounts of 3,5-bis(trifluoromethyl)benzoyl chloride and propargyl alcohol, with excess NEt3 in dry CH2Cl2. After aqueous workup, 3 was isolated in 86% yield. The azide click precursor was prepared in two steps. First bromo-amide 4 was prepared by reacting equimolar amounts of 3,5-bis(trifluoromethyl)benzoyl chloride and 3-bromopropylamine hydrobromide, with excess NEt3 in dry CH2Cl2. After aqueous workup, 4 was isolated in 87% yield. Conversion to azide 5 was accomplished by heating a solution of 4 in dry DMF with excess NaN3 at 80 °C. After dilution, and extraction, 5 was isolated in 60% yield. The successful preparation of novel compounds 3–5 was confirmed by NMR and IR spectroscopies and mass spectrometry (see ESI†).
 | ||
| Scheme 1 Structures of rotaxane precursors and syntheses of novel intermediates and precursors used in this study. | ||
“Click” synthesis and spectral characterisation of rotaxanes
Synthesis of rotaxanes 6 and 7 were attempted as according to Scheme 2. In each case, to a solution of macrocycle 1 in dichloromethane, 1.1 equivalents of azide 5 and 1.1 equivalents of alkyne (2 or 3) were added. These were followed by catalytic Cu(CH3CN)4BF4 and TBTA, and 1.2 equivalents of DIPEA. The reactions were stirred overnight at room temperature under an inert atmosphere, and then submitted to aqueous workup, before purification on silica. By analogous reactions (in the absence of macrocycle 1), non-interlocked axles 8 and 9 were also prepared (in 88% and 66% yield respectively) to aid 1H NMR spectroscopic interpretation. | ||
| Scheme 2 Syntheses of rotaxanes 6 and 7 and non-interlocked axles 8 and 9. Reaction conditions: (i) cat. Cu(CH3CN)4BF4, cat. TBTA, DIPEA, CH2Cl2. | ||
It was not possible to separate rotaxane 6 and unthreaded macrocycle 1 by silica gel column chromatography. Purification using prep TLC, allowed for isolation of pure rotaxane 6 in a reasonable 32% yield. Pleasingly, rotaxane 7 could be purified by careful column chromatography, being isolated in a yield of 47%.
The 1H NMR spectrum of rotaxane 6, along with that of non-interlocked macrocycle 1 and axle 8 for comparison, is shown in Fig. 2.16 The upfield shift and splitting of protons f and g in rotaxane 6 compared to macrocycle 1 is consistent with intercalation of the axle component between the aromatic rings of the macrocycle. The downfield shift of protons c and d in the rotaxane are indicative of interactions with a hydrogen bond acceptor on the axle. The functional group of the axle residing within the macrocyclic ring in rotaxane 6 is believed to be the templating amide, and not the newly formed triazole. Evidence for this conclusion is provided by the large upfield shifts in axle protons m and n and in particular the lack of a significant shift in proton r. Another notable feature of the 1H NMR spectrum of rotaxane 6 is the splitting of protons e, h, i and j, which arise from the two faces of the macrocycle no longer being equivalent in the rotaxane due to the directionality of the threaded axle. Comparison of the 1H NMR spectra of macrocycle 1, rotaxane 7 and axle 9 reveals similar trends, indicative of the macrocycle also residing over the axle amide in rotaxane 7, rather than the triazole or ester (see ESI†).16
 | ||
| Fig. 2 1H NMR spectra of (a) macrocycle 1, (b) rotaxane 6 and (c) axle 8 (CDCl3, 400 MHz, 298 K). For atom labels see Schemes 1 and 2. | ||
The interlocked natures of rotaxanes 6 and 7 are further supported by the appearance of through-space correlations being observed in the 1H–1H ROESY NMR spectra between signals arising from protons in the macrocyclic and axle components (see ESI†). In addition, molecular ion peaks for the rotaxanes are observed in the positive-ion electrospray mass spectra (see ESI†).
Computational modelling of rotaxanes
While reasonably confident in our deduction that the macrocycle is residing over the axle amide, and not the triazole functionality, in both rotaxanes, we sought further evidence to confirm the co-conformational behaviour of the rotaxanes. Unable to grow single crystals for X-ray structure determination, we therefore turned our attention to computational modelling. Density-functional theory (DFT) calculations were undertaken on both 6 and 7.17 The calculations were performed using the Gaussian 09 program,18 using the B3LYP exchange–correlation functional19 together with the 6-31G* basis set for all atoms. Solvent effects (CHCl3) were modelled using a polarisable continuum model, using the default solvent cavity parameters as defined in Gaussian 09.For each of the rotaxanes, geometry optimisations were run with the macrocycle starting over (i) the amide and (ii) the triazole functional groups of the axle component.20 Distinct optimised structures were obtained in each case (see Fig. 3 and ESI†), but for both rotaxanes there was an energetic preference for the macrocycle being over the amide “station” (41 kJ mol−1 for 6 and 76 kJ mol−1 for 7).21
 | ||
| Fig. 3 Minimum energy structures of rotaxane 7 with macrocycle occupying (a) amide and (b) triazole “stations”. Hydrogen bonds are represented by green dashed lines. Hydrogen atoms not participating in hydrogen bonding are omitted for clarity. | ||
Inspection of the optimised structures suggests that this preference is due to more favourable hydrogen bonding interactions being present when the amide “station” is occupied by the macrocycle.21 In this case the isophthalamide of the macrocycle adopts a syn–syn conformation and presents three hydrogen bond donors to the O atom of the amide “station”. If the macrocycle resides at the triazole “station”, the isophthalamide adopts a syn–anti conformation, resulting in only one N–H hydrogen bond donating to the N atom of the triazole.22 Also, in the preferred amide “station” co-conformation, the triazole C–H hydrogen bonds to a polyether oxygen on the macrocyclic ring, further stabilising this conformation. A structural consequence of this additional hydrogen bonding interaction is to bend the axle, such that protons o and p are partially within the macrocycle cavity, which is consistent with the significant upfield shifts of these protons in the rotaxane 1H NMR spectra compared to the spectra of the free axles (see Fig. 2 and ESI†). Finally, there is a substantially greater difference in energies for the two co-conformations of rotaxane 7 (compared to 6). We attribute this to the disruption in the planarity of the ester and bulky aromatic stopper group in the co-conformation of 7 when the macrocycle occupies the neighbouring triazole “station” (Fig. 3b).
Conclusions
This study has demonstrated it is possible to generate rotaxanes by short, straightforward and highly yielding reaction sequences, employing CuAAC click chemistry to stopper Philp-style self-assembled pseudo-rotaxanes of an amide half-axle threaded through an aryl bis-amide macrocycle. Density functional theory calculations provide strong supporting evidence for the co-conformational behaviour of the prepared rotaxanes, with the macrocycle remaining over the templating amide of the axle, rather than over the newly formed triazole functionality.We are confident that the synthetic ease in generating these rotaxanes will not only be appealing to those already preparing rotaxanes, but will encourage other chemists to begin using rotaxanes in their own research. We ourselves are undertaking further study of this class of rotaxane with respect to synthetic methodology development and molecular motion and host–guest applications.
Experimental
General information
Commercially available solvents and chemicals were used without further purification unless stated. Dry solvents, NEt3 and DIPEA were purchased dry and stored under an inert atmosphere. Cu(CH3CN)4BF4 was stored in a desiccator over P4O10. Deionised water was used in all cases. Previously reported macrocycle 114 has now been prepared in higher yield by use of a template method, details of which may be found in the ESI.† Alkyne 2 was prepared as according to the previously reported procedure.15
Silica gel with a 60 Å particle size was used as the stationary phase for column chromatography. Analytical TLC was used to monitor the progress of column chromatography and analytical TLC plates were typically examined under short wavelength (λ = 254 nm) UV light. If required, ceric ammonium molybdate or potassium permanganate stains were used to develop the analytical TLC plates. Preparatory TLC was carried out on silica gel possessing a fluorescent indicator to allow for examination with short wavelength UV light.
IR spectra were recorded on an Agilent Technologies Cary 630 FTIR spectrometer. NMR spectra were recorded on a Bruker Ultrashield 400 Plus spectrometer at 298 K, with the NMR data reported below assigned according to the atom labels to be found in the Schemes 1 and 2. Mass spectra were recorded on a Waters Xevo G2-S or Thermofisher LTQ Orbitrap XL at the EPSRC UK National Mass Spectrometry Facility at Swansea University, UK, or a Shimadzu LCMS IT ToF instrument at Lancaster University, UK. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
Experimental procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1360, 1280, 1240, 1180, 1130. δH(400 MHz; CDCl3) 8.53 (2H, s, ArH), 8.10 (1H, s, ArH), 5.01 (2H, d, 4J = 2.5 Hz, CH2), 2.59 (1H, t, 4J = 2.5 Hz, CH2). δC(100 MHz; CDCl3) 163.2 (CO), 132.3 (quar, 2J = 34 Hz, ArCCF3), 131.6 (ArC), 129.9 (ArC), 126.7 (ArC), 122.8 (quar, 1J = 288 Hz, CF3), 75.9 (CH), 53.5 (OCH2). δF(377 MHz; CDCl3) −63.0. m/z (ASAP) 296.0277 ([M]+ C12F6H6O2 requires 296.0272).
O), 1620, 1550, 1450, 1390, 1320, 1280, 1120. δH(400 MHz; CDCl3) 8.23 (2H, s, ArH), 8.01 (1H, s, ArH), 6.70 (1H, br s, NH), 3.66–3.71 (2H, app quar, NHCH2), 3.52 (2H, t, 3J = 6.4 Hz, CH2Br), 2.22–2.28 (2H, app quin, NHCH2CH2). δC(100 MHz; CDCl3) 164.9 (CO), 136.3 (ArC), 132.2 (quar, 2J = 32 Hz, ArCCF3), 127.3 (ArC), 125.1 (ArC), 122.8 (quar, 1J = 271 Hz, CF3), 39.2 (NHCH2), 31.9 (CH2Br), 30.7 (CH2). δF(377 MHz; CDCl3) −63.0. m/z (ES) 399.9741 ([M + Na]+ C12F6H10NNaO requires 399.9742).
NN), 1650 (CO), 1550, 1470, 1380, 1270, 1170, 1130. δH(400 MHz; CDCl3) 8.23 (2H, s, ArH), 8.02 (1H, s, ArH), 6.61 (1H, br s, NH), 3.59–3.64 (2H, app quar, NHCH2), 3.50 (2H, t, 3J = 6.4 Hz, CH2N3), 1.92–1.99 (2H, app quin, NHCH2CH2). δC(100 MHz; CDCl3) 164.7 (CO), 136.4 (ArC), 132.2 (quar, 2J = 34 Hz, ArCCF3), 127.2 (ArC), 125.1 (ArC), 122.8 (quar, 1J = 271 Hz, CF3), 49.7 (CH2N3), 38.4 (NHCH2), 28.5 (CH2). δF(377 MHz; CDCl3) −62.9. m/z (APC) 339.0680 ([M − H]− C12F6H9N4O requires 339.0686).
:1 to 98:2 CH2Cl2/CH3OH), which allowed for isolation of the product contaminated with macrocycle 1. Pure rotaxane 6 was obtained by use of prep TLC (repeated running in 98:2 CH2Cl2/CH3OH) to give the title compound as a transparent colourless film (14 mg, 32%). Rf = 0.20, 98:2 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3320 (N–H), 3060 (C–H), 2920 (C–H), 2870 (C–H), 1650 (CO), 1530, 1450, 1360, 1280, 1180, 1130, 1080. δH(400 MHz; CDCl3) 8.47 (1H, s, Hc), 8.27 (2H, dd, 3J = 7.8 Hz, 4J = 1.6 Hz, Hb) 8.09 (2H, s, Hl), 8.04 (1H, s, Hk), 7.62 (1H, t, 3J = 7.8 Hz, Ha), 7.39 (2H, br s, Hd), 7.18–7.30 (11H, m, Hq & phenyl H), 6.86 (4H, d, 3J = 8.0 Hz, Hf), 6.78 (1H, br s, Hm), 6.65 (4H, d, 3J = 8.0 Hz, Hg), 4.62 (2H, s, Hr), 4.52–4.57 (2H, m, He), 4.40–4.45 (2H, m, He), 4.31 (1H, t, 3J = 7.2 Hz, Ht), 4.24 (2H, d, 3J = 10.0 Hz, Hh), 4.10 (2H, d, 3J = 7.2 Hz, Hs), 4.03 (2H, d, 3J = 10.0 Hz, Hh), 3.95 (2H, t, 3J = 6.8 Hz, Hp), 3.50–3.71 (8H, m, Hi & Hj), 2.55–2.59 (2H, app quar, Hn), 1.61–1.68 (2H, app quin, Ho). δC(100 MHz; CDCl3) 166.6, 164.2 (2 × CO), 141.8, 137.8, 136.0, 135.5, 134.1, 131.7, 131.3, 129.3, 128.7, 128.5, 128.4 128.2, 126.6, 124.6, 124.4, 123.6, 121.7, 74.2(Cs), 73.6 (Ch), 70.7, 69.6, 64.0 (Cr), 51.0 (Ct), 48.0 (Cp), 44.1 (Ce), 37.4 (Cn), 28.1 (Co). δF(377 MHz; CDCl3) −62.5. m/z (ES) 1073.4014 ([M + Na]+ C57F6H56N6NaO7 requires 1073.4007).
:2 CH2Cl2/CH3OH) to give the title compound as a transparent colourless film (22 mg, 47%). Rf = 0.17, 98:2 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3330 (N–H), 3060 (C–H), 2920 (C–H), 2870 (C–H), 1730 (ester CO), 1650 (amide CO), 1520, 1460, 1380, 1360, 1280, 1240, 1170, 1130, 1080. δH(400 MHz; CDCl3) 8.48 (2H, s, Hs), 8.45 (1H, s, Hc), 8.28 (2H, dd, 3J = 7.8 Hz, 4J = 1.6 Hz, Hb), 8.08 (2H, s, Hl), 8.05 (2H, app br s, Hk & Ht), 7.17 (1H, s, Hq), 7.64 (1H, t, 3J = 7.8 Hz, Ha), 7.30 (2H, t, 3J = 4.6 Hz, Hd), 6.87 (4H, d, 3J = 8.1 Hz, Hf), 6.80 (1H, t, 3J = 5.3 Hz, Hm), 6.65 (4H, d, 3J = 8.1 Hz, Hg), 5.51 (2H, s, Hr), 4.61–4.66 (2H, m, He), 4.32–4.37 (2H, m, He), 4.28 (2H, d, 3J = 9.6 Hz, Hh), 4.00–4.04 (4H, m, Hh & Hp), 3.54–3.77 (8H, m, Hi & Hj), 2.57–2.62 (2H, app quar, Hn), 1.65–1.73 (2H, app quin, Ho). δC(100 MHz; CDCl3) 166.3, 164.1, 163.8 (3 × CO), 141.3, 137.6, 136.1, 135.4, 134.0, 132.3 (quar, 2J = 34 Hz, ArCCF3), 131.8, 131.4 (quar, 2J = 34 Hz, ArCCF3), 129.9, 129.4, 128.8, 128.3, 126.6, 125.2, 124.6, 123.3, 123.1 (quar, 1J = 271 Hz, CF3), 122.7 (quar, 1J = 271 Hz, CF3), 73.7 (Ch), 70.8, 69.6, 58.7 (Cr), 48.0 (Cp), 44.3 (Ce), 37.4 (Cn), 28.7 (Co). δF(377 MHz; CDCl3) −62.5, −63.0. m/z (ES) 1133.3077 ([M + Na]+ C52F12H46N6NaO8 requires 1133.3078).
:1 CH2Cl2/CH3OH) to give the title compound as a transparent colourless film (75 mg, 88%). Rf = 0.33, 98:2 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3300 (N–H), 3060 (C–H), 3030 (C–H), 2930 (C–H), 2860 (C–H), 1650 (CO), 1540, 1450, 1380, 1280, 1170, 1130, 1050. δH(400 MHz; CDCl3) 8.29 (2H, s, Hl), 8.01 (1H, s, Hk), 7.18–7.29 (12H, m, Hm, Hq & phenyl H), 4.62 (2H, s, Hr), 4.39 (2H, t, 3J = 6.5 Hz, Hp), 4.27 (1H, t, 3J = 7.2 Hz, Ht), 4.04 (3J = 7.2 Hz, Hs), 3.47–3.51 (2H, app quar, Hn), 2.17–2.23 (2H, app quin, Ho). δC(100 MHz; CDCl3) 164.8 (CO), 145.4, 141.9, 136.1, 132.1 (quar, 2J = 34 Hz, ArCCF3), 128.4, 128.2, 127.5, 126.6, 125.0, 123.1, 122.9 (quar, 1J = 272 Hz, CF3), 73.9 (Cs), 64.3 (Cr), 51.0 (Ct), 48.1 (Cp), 37.4 (Cn), 29.4 (Co). δF(377 MHz; CDCl3) −62.8. m/z (ES) 599.1840 ([M + Na]+ C29F6H26N4NaO2 requires 599.1852).
:2 to 1:1 petrol 40–60/EtOAc) to give the title compound as a white solid (122 mg, 66%). Rf = 0.34, 98:2 CH2Cl2/CH3OH. Mp 130–131 °C. νmax/cm−1 (neat) 3340 (N–H), 3130 (C–H), 3090 (C–H), 1720 (ester CO), 1650 (amide CO), 1620, 1560, 1470, 1430, 1370, 1340, 1270, 1250, 1180, 1120. δH(400 MHz; CDCl3) 8.47 (2H, s, Hs), 8.24 (2H, s, Hl), 8.07 (1H, s, Ht), 8.01 (1H, s, Hk), 7.87 (1H, s, Hq), 6.98 (1H, br s, Hm), 5.52 (2H, s, Hr), 4.54 (2H, t, 3J = 6.5 Hz, Hp), 3.55–3.60 (2H, app quar, Hn), 2.29–2.35 (2H, app quin, Ho). δC(100 MHz; CDCl3) 164.9, 163.8 (2 × CO), 142.4, 136.0, 132.3 (quar, 2J = 34 Hz, 2 × ArCCF3), 131.7, 129.8, 127.3, 126.6, 125.2, 124.9, 122.8 (quar, 1J = 272 Hz, CF3), 122.8 (quar, 1J = 272 Hz, CF3, sic), 58.8 (Cr), 48.1 (Cp), 37.5 (Cn), 29.7 (Co). δF(377 MHz; CDCl3) −62.9, −63.0. m/z (ES) 659.0926 ([M + Na]+ C24F12H16N4NaO requires 659.0923).
O), 1360, 1280, 1240, 1180, 1130. δH(400 MHz; CDCl3) 8.53 (2H, s, ArH), 8.10 (1H, s, ArH), 5.01 (2H, d, 4J = 2.5 Hz, CH2), 2.59 (1H, t, 4J = 2.5 Hz, CH2). δC(100 MHz; CDCl3) 163.2 (CO), 132.3 (quar, 2J = 34 Hz, ArCCF3), 131.6 (ArC), 129.9 (ArC), 126.7 (ArC), 122.8 (quar, 1J = 288 Hz, CF3), 75.9 (CH), 53.5 (OCH2). δF(377 MHz; CDCl3) −63.0. m/z (ASAP) 296.0277 ([M]+ C12F6H6O2 requires 296.0272).
O), 1620, 1550, 1450, 1390, 1320, 1280, 1120. δH(400 MHz; CDCl3) 8.23 (2H, s, ArH), 8.01 (1H, s, ArH), 6.70 (1H, br s, NH), 3.66–3.71 (2H, app quar, NHCH2), 3.52 (2H, t, 3J = 6.4 Hz, CH2Br), 2.22–2.28 (2H, app quin, NHCH2CH2). δC(100 MHz; CDCl3) 164.9 (CO), 136.3 (ArC), 132.2 (quar, 2J = 32 Hz, ArCCF3), 127.3 (ArC), 125.1 (ArC), 122.8 (quar, 1J = 271 Hz, CF3), 39.2 (NHCH2), 31.9 (CH2Br), 30.7 (CH2). δF(377 MHz; CDCl3) −63.0. m/z (ES) 399.9741 ([M + Na]+ C12F6H10NNaO requires 399.9742).
NN), 1650 (CO), 1550, 1470, 1380, 1270, 1170, 1130. δH(400 MHz; CDCl3) 8.23 (2H, s, ArH), 8.02 (1H, s, ArH), 6.61 (1H, br s, NH), 3.59–3.64 (2H, app quar, NHCH2), 3.50 (2H, t, 3J = 6.4 Hz, CH2N3), 1.92–1.99 (2H, app quin, NHCH2CH2). δC(100 MHz; CDCl3) 164.7 (CO), 136.4 (ArC), 132.2 (quar, 2J = 34 Hz, ArCCF3), 127.2 (ArC), 125.1 (ArC), 122.8 (quar, 1J = 271 Hz, CF3), 49.7 (CH2N3), 38.4 (NHCH2), 28.5 (CH2). δF(377 MHz; CDCl3) −62.9. m/z (APC) 339.0680 ([M − H]− C12F6H9N4O requires 339.0686).
:1 to 98:2 CH2Cl2/CH3OH), which allowed for isolation of the product contaminated with macrocycle 1. Pure rotaxane 6 was obtained by use of prep TLC (repeated running in 98:2 CH2Cl2/CH3OH) to give the title compound as a transparent colourless film (14 mg, 32%). Rf = 0.20, 98:2 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3320 (N–H), 3060 (C–H), 2920 (C–H), 2870 (C–H), 1650 (CO), 1530, 1450, 1360, 1280, 1180, 1130, 1080. δH(400 MHz; CDCl3) 8.47 (1H, s, Hc), 8.27 (2H, dd, 3J = 7.8 Hz, 4J = 1.6 Hz, Hb) 8.09 (2H, s, Hl), 8.04 (1H, s, Hk), 7.62 (1H, t, 3J = 7.8 Hz, Ha), 7.39 (2H, br s, Hd), 7.18–7.30 (11H, m, Hq & phenyl H), 6.86 (4H, d, 3J = 8.0 Hz, Hf), 6.78 (1H, br s, Hm), 6.65 (4H, d, 3J = 8.0 Hz, Hg), 4.62 (2H, s, Hr), 4.52–4.57 (2H, m, He), 4.40–4.45 (2H, m, He), 4.31 (1H, t, 3J = 7.2 Hz, Ht), 4.24 (2H, d, 3J = 10.0 Hz, Hh), 4.10 (2H, d, 3J = 7.2 Hz, Hs), 4.03 (2H, d, 3J = 10.0 Hz, Hh), 3.95 (2H, t, 3J = 6.8 Hz, Hp), 3.50–3.71 (8H, m, Hi & Hj), 2.55–2.59 (2H, app quar, Hn), 1.61–1.68 (2H, app quin, Ho). δC(100 MHz; CDCl3) 166.6, 164.2 (2 × CO), 141.8, 137.8, 136.0, 135.5, 134.1, 131.7, 131.3, 129.3, 128.7, 128.5, 128.4 128.2, 126.6, 124.6, 124.4, 123.6, 121.7, 74.2(Cs), 73.6 (Ch), 70.7, 69.6, 64.0 (Cr), 51.0 (Ct), 48.0 (Cp), 44.1 (Ce), 37.4 (Cn), 28.1 (Co). δF(377 MHz; CDCl3) −62.5. m/z (ES) 1073.4014 ([M + Na]+ C57F6H56N6NaO7 requires 1073.4007).
:2 CH2Cl2/CH3OH) to give the title compound as a transparent colourless film (22 mg, 47%). Rf = 0.17, 98:2 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3330 (N–H), 3060 (C–H), 2920 (C–H), 2870 (C–H), 1730 (ester CO), 1650 (amide CO), 1520, 1460, 1380, 1360, 1280, 1240, 1170, 1130, 1080. δH(400 MHz; CDCl3) 8.48 (2H, s, Hs), 8.45 (1H, s, Hc), 8.28 (2H, dd, 3J = 7.8 Hz, 4J = 1.6 Hz, Hb), 8.08 (2H, s, Hl), 8.05 (2H, app br s, Hk & Ht), 7.17 (1H, s, Hq), 7.64 (1H, t, 3J = 7.8 Hz, Ha), 7.30 (2H, t, 3J = 4.6 Hz, Hd), 6.87 (4H, d, 3J = 8.1 Hz, Hf), 6.80 (1H, t, 3J = 5.3 Hz, Hm), 6.65 (4H, d, 3J = 8.1 Hz, Hg), 5.51 (2H, s, Hr), 4.61–4.66 (2H, m, He), 4.32–4.37 (2H, m, He), 4.28 (2H, d, 3J = 9.6 Hz, Hh), 4.00–4.04 (4H, m, Hh & Hp), 3.54–3.77 (8H, m, Hi & Hj), 2.57–2.62 (2H, app quar, Hn), 1.65–1.73 (2H, app quin, Ho). δC(100 MHz; CDCl3) 166.3, 164.1, 163.8 (3 × CO), 141.3, 137.6, 136.1, 135.4, 134.0, 132.3 (quar, 2J = 34 Hz, ArCCF3), 131.8, 131.4 (quar, 2J = 34 Hz, ArCCF3), 129.9, 129.4, 128.8, 128.3, 126.6, 125.2, 124.6, 123.3, 123.1 (quar, 1J = 271 Hz, CF3), 122.7 (quar, 1J = 271 Hz, CF3), 73.7 (Ch), 70.8, 69.6, 58.7 (Cr), 48.0 (Cp), 44.3 (Ce), 37.4 (Cn), 28.7 (Co). δF(377 MHz; CDCl3) −62.5, −63.0. m/z (ES) 1133.3077 ([M + Na]+ C52F12H46N6NaO8 requires 1133.3078).
:1 CH2Cl2/CH3OH) to give the title compound as a transparent colourless film (75 mg, 88%). Rf = 0.33, 98:2 CH2Cl2/CH3OH. νmax/cm−1 (neat) 3300 (N–H), 3060 (C–H), 3030 (C–H), 2930 (C–H), 2860 (C–H), 1650 (CO), 1540, 1450, 1380, 1280, 1170, 1130, 1050. δH(400 MHz; CDCl3) 8.29 (2H, s, Hl), 8.01 (1H, s, Hk), 7.18–7.29 (12H, m, Hm, Hq & phenyl H), 4.62 (2H, s, Hr), 4.39 (2H, t, 3J = 6.5 Hz, Hp), 4.27 (1H, t, 3J = 7.2 Hz, Ht), 4.04 (3J = 7.2 Hz, Hs), 3.47–3.51 (2H, app quar, Hn), 2.17–2.23 (2H, app quin, Ho). δC(100 MHz; CDCl3) 164.8 (CO), 145.4, 141.9, 136.1, 132.1 (quar, 2J = 34 Hz, ArCCF3), 128.4, 128.2, 127.5, 126.6, 125.0, 123.1, 122.9 (quar, 1J = 272 Hz, CF3), 73.9 (Cs), 64.3 (Cr), 51.0 (Ct), 48.1 (Cp), 37.4 (Cn), 29.4 (Co). δF(377 MHz; CDCl3) −62.8. m/z (ES) 599.1840 ([M + Na]+ C29F6H26N4NaO2 requires 599.1852).
:2 to 1:1 petrol 40–60/EtOAc) to give the title compound as a white solid (122 mg, 66%). Rf = 0.34, 98:2 CH2Cl2/CH3OH. Mp 130–131 °C. νmax/cm−1 (neat) 3340 (N–H), 3130 (C–H), 3090 (C–H), 1720 (ester CO), 1650 (amide CO), 1620, 1560, 1470, 1430, 1370, 1340, 1270, 1250, 1180, 1120. δH(400 MHz; CDCl3) 8.47 (2H, s, Hs), 8.24 (2H, s, Hl), 8.07 (1H, s, Ht), 8.01 (1H, s, Hk), 7.87 (1H, s, Hq), 6.98 (1H, br s, Hm), 5.52 (2H, s, Hr), 4.54 (2H, t, 3J = 6.5 Hz, Hp), 3.55–3.60 (2H, app quar, Hn), 2.29–2.35 (2H, app quin, Ho). δC(100 MHz; CDCl3) 164.9, 163.8 (2 × CO), 142.4, 136.0, 132.3 (quar, 2J = 34 Hz, 2 × ArCCF3), 131.7, 129.8, 127.3, 126.6, 125.2, 124.9, 122.8 (quar, 1J = 272 Hz, CF3), 122.8 (quar, 1J = 272 Hz, CF3, sic), 58.8 (Cr), 48.1 (Cp), 37.5 (Cn), 29.7 (Co). δF(377 MHz; CDCl3) −62.9, −63.0. m/z (ES) 659.0926 ([M + Na]+ C24F12H16N4NaO requires 659.0923).
Computational methodology
The Gaussian 09 program was used to conduct all density functional theory calculations.18 In each case, geometry optimisations using the B3LYP/6-31G* model chemistry were undertaken,19 with CHCl3 solvent effects modelled using the default polarisable continuum model (and default solvent cavity parameters) as defined in Gaussian 09. To obtain reasonable starting structures, the structures were pre-optimised using a combination of molecular mechanics and semi-empirical PM6 methods. Several ‘starting points’ were considered for the optimisations, to minimise bias in the final structures.Acknowledgements
We thank Lancaster University for financial support of this project. For a sample of alkyne 2 we thank Charles Gell (Lancaster University). For the recording of mass spectrometry data we thank the EPSRC UK National Mass Spectrometry Facility (NMSF) at Swansea University, UK (samples of 3 and 4) and Dr David Rochester at Lancaster University, UK (samples of 5, 6, 7, 8, and 9). For assistance in the recording of 1H–1H ROESY NMR spectra we thank Dr Geoff Akien (Lancaster University).All underlying data for this paper are provided in the experimental section and the ESI.† Electronic copies of NMR spectra (including fid files) are available from DOI: 10.17635/lancaster/researchdata/132.
Notes and references
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- (a) C. Wu, P. R. Lecavalier, Y. X. Shen and H. W. Gibson, Chem. Mater., 1991, 3, 569 CrossRef CAS; (b) J.-C. Chambron, V. Heitz and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1992, 1131–1133 RSC; (c) L. Hogg, D. A. Leigh, P. J. Lusby, A. Morelli, S. Parsons and J. K. Y. Wong, Angew. Chem., Int. Ed., 2004, 43, 1218–1221 CAS; (d) A. M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914–3918 CrossRef CAS PubMed; (e) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed; (f) H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS PubMed; (g) A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC; (h) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- (a) P. R. Ashton, M. Grognuz, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1991, 32, 6235–6238 CrossRef CAS; (b) R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS; (c) M. J. Gunter, N. Bampos, K. D. Johnstone and J. K. M. Sanders, New J. Chem., 2001, 25, 166–173 RSC; (d) G. Barin, A. Coskun, M. M. G. Fouda and J. F. Stoddart, ChemPlusChem, 2012, 77, 159–185 CrossRef CAS.
- (a) F. Vögtle, M. Händel, S. Meier, S. Ottens-Hildebrandt, F. Ott and T. Schmidt, Liebigs Ann., 1995, 739–743 CrossRef; (b) D. A. Leigh, A. Murphy, J. P. Smart and A. M. Z. Slawin, Angew. Chem., Int. Ed., 1997, 36, 728–732 CrossRef CAS; (c) F. G. Gatti, D. A. Leigh, S. A. Nepogodiev, A. M. Z. Slawin, S. J. Teat and J. K. Wong, J. Am. Chem. Soc., 2001, 123, 5983–5989 CrossRef CAS PubMed; (d) Y.-L. Huang, W.-C. Hung, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2007, 46, 6629–6633 CrossRef CAS PubMed; (e) D. M. D′Souza, D. A. Leigh, L. Mottier, K. M. Mullen, F. Paolucci, S. J. Teat and S. Zhang, J. Am. Chem. Soc., 2010, 132, 9465–9470 CrossRef PubMed; (f) A. Altieri, V. Aucagne, R. Carrillo, G. J. Clarkson, D. M. D′Souza, J. A. Dunnett, D. A. Leigh and K. M. Mullen, Chem. Sci., 2011, 2, 1922–1928 RSC; (g) R. Ahmed, A. Altieri, D. M. D′Souza, D. A. Leigh, K. M. Mullen, M. Papmeyer, A. M. Z. Slawin, J. K. Y. Wong and J. D. Wollins, J. Am. Chem. Soc., 2011, 133, 12304–12310 CrossRef CAS PubMed; (h) A. Tron, P. J. Thornton, M. Rocher, H.-P. J. de Rouville, J.-P. Desvergne, B. Kauffmann, T. Buffeteau, D. Cavagnat, J. H. R. Tucker and N. D. McClenaghan, Org. Lett., 2014, 16, 1358–1361 CrossRef CAS PubMed.
- (a) P. R. Ashton, P. T. Glink, J. F. Stoddart, P. A. Tasker, A. J. P. White and D. J. Williams, Chem. – Eur. J., 1996, 2, 729–736 CrossRef CAS; (b) H. Zheng, W. Zhou, J. Lv, X. Yin, Y. Li, H. Liu and Y. Li, Chem. – Eur. J., 2009, 15, 13253–13262 CrossRef CAS PubMed; (c) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed.
- (a) G. M. Hübner, J. Gläser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383–386 CrossRef; (b) C. A. Schalley, G. Silva, C. F. Nising and P. Linnartz, Helv. Chim. Acta, 2002, 85, 1578–1596 CrossRef CAS; (c) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS PubMed; (d) E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS PubMed; (e) L. M. Hancock, L. C. Gilday, S. Carvalho, P. J. Costa, V. Félix, C. J. Serpell, N. L. Kilah and P. D. Beer, Chem. – Eur. J., 2010, 16, 13082–13094 CrossRef CAS PubMed.
- A. Vidonne and D. Philp, Tetrahedron, 2008, 64, 8464–8475 CrossRef CAS.
- T. Kosikova, N. I. Hassan, D. B. Cordes, A. M. Z. Slawin and D. Philp, J. Am. Chem. Soc., 2015, 137, 16074–16083 CrossRef CAS PubMed.
- A. Vidonne, T. Kosikova and D. Philp, Chem. Sci., 2016, 7, 2592–2603 RSC.
- N. I. Hassan, V. del Amo, E. Calder and D. Philp, Org. Lett., 2011, 13, 458–461 CrossRef CAS PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- C. N. Marrs and N. H. Evans, Org. Biomol. Chem., 2015, 13, 11021–11025 CAS.
- N. H. Evans, C. E. Gell and M. J. G. Peach, Org. Biomol. Chem., 2016, 14, 7972–7981 CAS.
- See ESI† for further NMR, IR and high resolution mass spectra of rotaxanes 6 & 7 and axles 8 & 9.
- See ESI† for coordinate data files from simulations.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford Ct, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (d) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1998, 37, 785–789 CrossRef.
- In the case of rotaxane 7 similar geometry optimisations were also run with the macrocycle starting over the ester functional group, but repeatedly the macrocycle would translate along the axle over the course of the optimisation, to one of the other “stations”. This result is as would be expected as the ester group is unable to act as a hydrogen bond donor.
- See ESI† for a full tabulated summary of energies and hydrogen bond distances in the simulated rotaxane structures.
- As detailed in the ESI† for both rotaxanes, when the macrocycle resides at the amide “station”, the axle N–H⋯O polyether hydrogen bond is significantly longer than the equivalent triazole C–H⋯O hydrogen bond when the macrocycle occupies the triazole “station”. However, we hypothesise that this difference (which would favour macrocycle occupancy of the triazole “station”) is outweighed by the interactions that favour occupancy of the amide “station” detailed in the main text.
Footnote |
| † Electronic supplementary information (ESI) available: Additional notes on experimental procedures; copies of characterisation spectra of novel compounds and further computational modelling data. See DOI: 10.1039/c7ob00284j |
| This journal is © The Royal Society of Chemistry 2017 |