
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA yellowish-green-light-controllable nitric oxide donor based on N-nitrosoaminophenol applicable for photocontrolled vasodilation†
Hana
Okuno
,
Naoya
Ieda
,
Yuji
Hotta
,
Mitsuyasu
Kawaguchi
,
Kazunori
Kimura
and
Hidehiko
Nakagawa
 *
*
Graduate School of Pharmaceutical Science, Nagoya City University, 3-1, Tanabe-dori, Mizuho-ku, Nagoya, Aichi 467-8603, Japan. E-mail: deco@phar.nagoya-cu.ac.jp
First published on 8th March 2017
Abstract
Nitric oxide (NO) has been known as a gaseous chemical mediator, which modulates several physiological functions. Spatial and temporal control of NO release facilitates further study and medical application of NO. Herein, we report design and synthesis of a novel NO donor, NO-Rosa. NO-Rosa has a rosamine moiety, which absorbs yellowish green light. Upon irradiation with yellowish green light (530–590 nm), NO is released from NO-Rosa, presumably via photoinduced electron transfer from the N-nitrosoaminophenol moiety to the rosamine moiety. NO release from NO-Rosa was detected by ESR spin trapping and a NO fluorescent probe. Cellular NO release control was achieved in HEK293 cells using a NO fluorescent probe, DAF-FM DA. Furthermore, temporally controlled NO-induced vasodilation was demonstrated by treatment of a rat aortic strip with NO-Rosaex vivo and irradiation by yellowish green light. NO-Rosa is expected to be utilized for further study of NO-related physiological functions, utilizing its ability of spatiotemporal release of NO as a photocontrollable compound with harmless yellowish-green light.
Introduction
Nitric oxide (NO) is biosynthesized from L-arginine by nitric oxide synthase (NOS) in humans1 and is an essential mediator in multiple physiological processes such as vasodilation,2 neurotransmission,3 and biodefence.4 NO exists as a gas under ambient conditions, and is an unstable free radical with a half-life of only a few seconds under physiological conditions.5 Therefore, NO donor molecules are required to investigate the physiological effects of NO and as candidate chemotherapeutic agents. Spontaneous NO donors such as NONOates6 and SNAP7 are frequently used in biological research, but do not allow spatiotemporally controlled NO release; in contrast, the physiological actions of NO are tightly spatiotemporally controlled. Some photo-controllable NO donors have been reported,8–10 but application of many of them is limited by factors such as cell damage due to activating UV light,8 metal toxicity,9 or the universality of two-photon excitation.10 Therefore, there is a need for more practical photo-controllable NO donors.We previously developed a blue light-controllable NO donor, NOBL-1 (1, Fig. 1), and showed that it was suitable for temporal control of vasodilation.11NOBL-1 consists of an NO-releasing N-nitrosoaminophenol moiety and a blue light-absorbing cyano-BODIPY moiety, which serves as an antenna moiety. Upon photoirradiation, NO release is triggered by photoinduced electron transfer (PeT)12,13 from the electron-rich N-nitrosoaminophenol moiety to the electron-deficient antenna moiety, generating an unstable phenoxyl radical moiety that releases NO to form a relatively stable quinoneimine (Fig. S1†).11,14 In this work, we designed, synthesized, and evaluated a novel NO donor, NO-Rosa (2, Fig. 1), in which rosamine dye is used as the antenna moiety in place of the cyano-BODIPY moiety of NOBL-1. The choice of rosamine as the dye was motivated by the fact that rosamine is excited by yellowish green light (λmax ≈ 550 nm); we expected that the new dye would be practically superior to NOBL-1, which is excited by blue light (λmax ≈ 500 nm), because light in the former wavelength range would be less harmful15 to biological samples and more penetrating.16
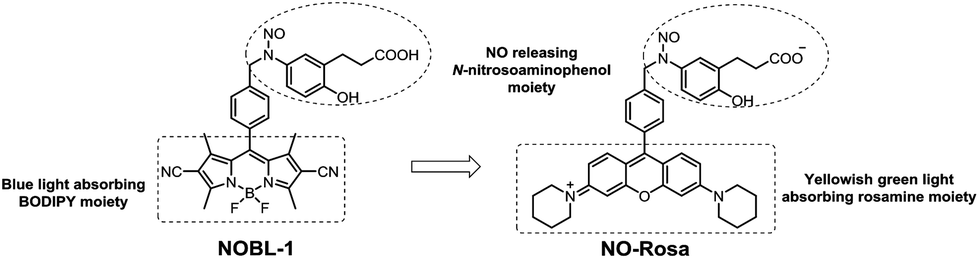 | ||
| Fig. 1 Structures of NOBL-1 (1) and NO-Rosa (2). | ||
Results
NO-Rosa was synthesized as shown in Scheme 1. After protection of 2-hydroxy-5-nitrobenzaldehyde (3), the Wittig reaction gave the t-butyl cinnamate derivative 5. Reduction with Pd–C/H2 afforded protected aminophenol 6. Rosamine 9 was obtained by lithium halogen exchange from an acetal-protected aldehyde (7)17 and 3,6-bispiperidinoxanthone (8), which was synthesized as reported.18 Reductive amination of 9 with protected aminophenol 6 gave 10, which was deprotected and N-nitrosylated to afford NO-Rosa (2). The structure and purity of NO-Rosa were confirmed by means of 1H NMR, 13C NMR, mass spectrometry, and HPLC. Water solubility of NO-Rosa was practically efficient, and it did not disturb any experiments in this report.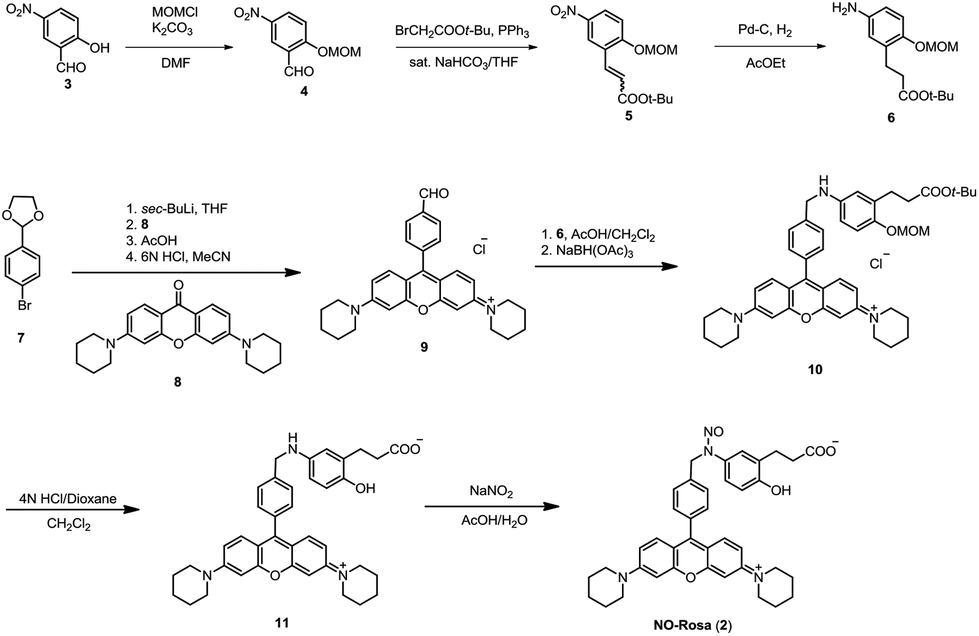 | ||
| Scheme 1 Synthesis of NO-Rosa (2). | ||
With NO-Rosa in hand, we first examined photoinduced NO release by means of ESR spin-trapping with an iron ion and an N-methylglucamine dithiocarbamate complex (Fe–MGD), which forms an NO–Fe–MGD complex that exhibits a distinctive three-line spectrum at around 330 mT in 1 GHz ESR spectrometry.19 Since the absorption spectra of NO-Rosa showed a maximum at 564 nm (Fig. 2), irradiation was performed with a MAX-302 apparatus (Asahi Spectra) equipped with a 530–590 nm band pass filter. After irradiation (100 mW cm−2) of an aqueous solution of Fe–MGD and NO-Rosa (100 μM) for 15 min, the ESR spectrum showed the distinctive triplet signal of the NO–Fe–MGD complex (Fig. 3). In the absence of irradiation, this signal was not observed (Fig. S2†).
 | ||
Fig. 2 Absorption spectrum of NO-Rosa (10 μM) in MilliQ water containing 0.1% DMSO, λmax = 564 nm, ε = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 318 M−1 cm−1. 318 M−1 cm−1. | ||
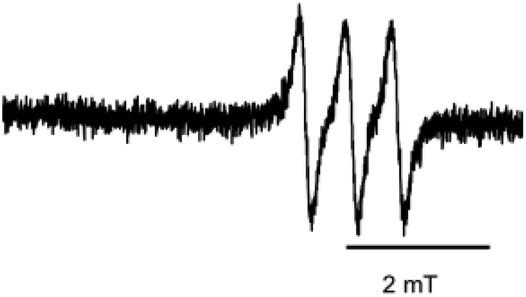 | ||
| Fig. 3 ESR spectrum of a solution of Fe–MGD and NO-Rosa after photoirradiation. NO-Rosa (100 μM), N-methyl-D-glucamine dithiocarbamate (6 mM), and FeSO4 (1.5 mM) were dissolved in MilliQ water containing 15% DMSO. The ESR spectrum of the solution was measured after irradiation with yellowish green light (530–590 nm, 100 mW cm−2, 15 min). ESR conditions: microwave power, 10 mW; frequency, 9.4 GHz; field, 330 mT; sweep width, 7.5 mT; sweep time, 4 min; modulation width, 0.125 mT; time constant, 0.10 s; g = 2.040. | ||
To evaluate the amount of released NO, quantitative NO analysis was conducted by using 2,3-diaminonaphthalene (DAN). DAN is converted to naphtho[2,3-d]triazole (NAT) upon reaction with the nitrite ion, an oxidation product of NO, under acidic conditions.20 Although NO can be oxidized to both nitrite and nitrate, nitrate was converted to nitrite by nitrate reductase before the reaction with DAN. By measuring the fluorescence due to NAT, we determined that NO-Rosa released NO efficiently after photoirradiation; 9.8 μM of NO was released from 10 μM of NO-Rosa (Fig. S3†). These results indicated that NO release from NO-Rosa was efficiently controllable with yellowish green light.
Photodecomposition of NO-Rosa was confirmed by HPLC analysis (Fig. S4†). After photoirradiation (530–590 nm, 60 mW cm−2, 15 min), 84% of NO-Rosa was decomposed. LC-ESI-MS (Fig. S5†) revealed the formation of three major photodecomposition products (m/z 452, 453, and 616), which were assigned as the intermediates 9, S1, and 11, respectively (Fig. S6†). These results are consistent with a PeT-based NO release mechanism through a radical intermediate, in accordance with our previous decomposition analysis of NOBL-1.11
Next, to test the suitability of this compound for cellular applications, light-induced NO release from NO-Rosa in HEK293 cells was examined with DAF-FM DA21 (Fig. 4). HEK293 cells were treated with DAF-FM DA (10 μM) and either NO-Rosa (10 μM) or DMSO (vehicle), and then irradiated at 530–590 nm (60 mW cm−2, 15 min). The fluorescence intensity was clearly increased after photoirradiation in the presence of NO-Rosa (Fig. 4a and b), while little fluorescence was observed in the absence of NO-Rosa (Fig. 4d). These results suggested that photo-controlled NO release from NO-Rosa also occurs intracellularly. We have also recorded red fluorescence images in the experiment with HEK293 cells in addition to the green fluorescence range (Fig. S7†). Interestingly, the red fluorescence was increased after photoirradiation. This result implies that the emission from decomposed products of NO-Rosa would be potentially usable as an NO release tracer. Additionally, we compared the light-toxicity in the same light intensity (40 mW cm−2) between blue light (470–500 nm) which was utilized for NOBL-1, and yellowish-green light (530–590 nm) by means of the Cell Counting Assay Kit (Dojindo, Kumamoto). As shown in Fig. S8,† a larger number of cell-death was induced by blue light irradiation (470–500 nm) than yellowish-green light (530–590 nm). In terms of this light toxicity, NO-Rosa is more suitable for biological application than NOBL-1.
 | ||
| Fig. 4 Photocontrolled NO release in HEK293 cells. Fluorescence imaging of NO release from NO-Rosa in HEK293 cells was performed by using DAF-FM DA. Cultured HEK293 cells were treated with DAF-FM DA (10 μM) and either NO-Rosa (10 μM) or vehicle (DMSO). The dishes were then photoirradiated with yellowish green light (530–590 nm, 60 mW cm−2 for 15 min). The cells were observed with a differential interference contrast microscope and a confocal microscope. (a) Before photoirradiation with NO-Rosa, (b) after photoirradiation with NO-Rosa, (c) before photoirradiation without NO-Rosa, (d) after photoirradiation without NO-Rosa. Left: DIC images; Right: Fluorescence images. The scale bar represents 40 μm. | ||
NO is known to induce vasodilation via the sGC-cGMP pathway.2 Therefore, we next examined whether vasodilation can be temporally controlled by the combination of NO-Rosa and photoirradiation in an ex vivo system. A strip of rat aorta was placed in a Magnus tube filled with Krebs buffer. The aortic strip was pretreated with L-NAME22 to block endogenous NO formation by NOS, and then tensioned by exposure to noradrenaline. After equilibration, NO-Rosa was added to the incubation buffer and the strip was irradiated at 530–590 nm. We found that vasodilation was induced during the photoirradiation, and the tension quickly recovered when the light was turned off (Fig. 5). It has been reported that the half-life of the sGC-NO complex is a few seconds even in vivo, so the quick tension recovery is consistent with previous findings.23 This vasodilation effect was dependent on the light intensity, and significant vasodilation was induced even at light intensity as low as 4 mW cm−2. Addition of a sGC inhibitor, ODQ,24 completely blocked the vasodilation. When the aortic strip was treated with the photodecomposition product instead of NO-Rosa, no distinct vasodilatory response to photoirradiation was observed (Fig. S9†). These results suggested that NO release from NO-Rosa was finely controlled by yellowish green light under the ex vivo conditions, and induced vasodilation via the NO-sGC-cGMP pathway.
 | ||
| Fig. 5 Photo-induced vasodilation with NO-Rosa. A rat aortic strip was placed in a Magnus tube filled with Krebs buffer at 37 °C. The strip was pretreated with L-NAME (10 μM) and noradrenaline (10 μM). After equilibration, NO-Rosa (10 μM) was added to the tube. The strip was irradiated with a light source (MAX-302, Asahi Spectra) equipped with a 530–590 nm band-pass filter for 1 min each time. After several cycles of photoirradiation, ODQ (10 μM) was added and the photoirradiation was performed again. Light intensity (mW cm−2): (a) 97, (b) 32, (c) 12, (d), 4, (e) 32, (f) 32, and (g) 32. | ||
Conclusions
In conclusion, we designed and synthesized NO-Rosa as an NO releaser controllable by photo-irradiation in the yellowish green wavelength range, and we confirmed that it works well in cells and ex vivo. NO release was confirmed by ESR spin trapping and with a fluorescent probe in vitro. Photoinduced NO release from NO-Rosa in cells was also confirmed with another fluorescent NO probe, DAF-FM DA. Furthermore, this system enabled fine temporal control of NO-dependent vasodilation in rat aortic strips ex vivo. Thus, our PeT-based strategy was applicable to rosamine, which has a relatively long absorption wavelength (λmax = 564 nm). Light in this wavelength range (530–590 nm) is less harmful to biological samples than blue light which was utilized for our previously reported NO donor, NOBL-1, and NO-Rosa should be more practically useful as a tool for detailed studies of NO-related physiological functions, as well as a candidate for the treatment of conditions such as ischemic heart disease after further optimization in future.Experimental
General methods
Proton nuclear magnetic resonance spectra (1H NMR) and carbon nuclear magnetic resonance spectra (13C NMR) were recorded on a JEOL JNM-LA500, JNM-A500, Varian VNMRS 500 spectrometer or a BRUKER AVANCE 600 spectrometer in the indicated solvent. Chemical shifts (δ) were reported in parts per million relative to the internal standard tetramethylsilane (TMS). High-resolution mass spectra (HRMS, ESI+) were recorded on a JEOL JMS-T100LP AccuTOF LC-plus 4G. Elemental analysis was performed with a Yanaco CHN CORDER NT-5 analyzer. Purity test using analytical HPLC was performed with a Shimadzu instrument equipped with an ODS-3 (4.6 × 150 nm, GL Science). Ultraviolet-visible-light absorption spectra were recorded on an Agilent 8453 spectrometer or a Shimadzu UV-1800 spectrometer. Fluorescence intensity was recorded on a Shimadzu RF-5300PC spectrophotometer or ARVO-X5 (PerkinElmer). Photoirradiation was performed by using the light source of Asahi Spectra MAX-302 or MAX-303 irradiation apparatus. The NO2/NO3 assay was conducted using the NO2/NO3 Assay Kit-FX (Fluorometric) 2,3-diaminonaphthalene kit (DOJINDO LABORATORIES, Kumamoto, Japan). The cell viability assay was conducted using the Cell Counting Kit-8 (DOJINDO LABOLATORIES, Kumamoto, Japan). ESR spectra were recorded on a JES-RE2X spectrometer (JEOL Co. Ltd, Tokyo, Japan). MGD (N-(dithiocarbamoyl)-N-methyl-D-glucamine, sodium salt) was obtained from DOJINDO LABORATORIES. All other reagents and solvents were purchased from Aldrich, Tokyo Kasei Kogyo, Wako Pure Chemical Industries, Nacalai Tesque, Kanto Chemical, Junsei Chemical, and Apollo Chemical, and used without purification. Flash column chromatography was performed using silica gel 60 (particle size 0.046–0.063 mm) supplied by Taiko-Shoji.Synthesis of 4
K2CO3 (183 mg, 1.32 mmol, 2.2 equiv.) was added to a solution of 5-nitrosalicylaldehyde (101 mg, 0.603 mmol) in DMF (4 mL), and the mixture was stirred at room temperature. Chloromethyl methyl ether (100 μL, 1.32 mmol, 2.2 equiv.) was added at room temperature. The reaction was quenched with H2O (12 mL) after stirring for 18 h. After ether extraction (50 mL) and washing with H2O (2 × 50 mL), 1 N NaOH (2 × 50 mL) and brine (2 × 50 mL), the organic layer was dried over NaSO4, filtered and evaporated in vacuo to afford 4 as a yellow solid (86 mg, 0.41 mmol, 68%): 1H NMR (CDCl3, 500 MHz, δ; ppm) 10.49 (1H, s), 8.72 (1H, d, J = 3.0 Hz), 8.41 (1H, dd, J = 9.3 Hz, 3.0 Hz), 7.38 (1H, d, J = 9.3 Hz), 5.42 (2H, s), 3.56 (3H, s).Synthesis of 5
To a slurry of 4 (825 mg, 3.91 mmol) and tert-butyl bromoacetate (1.03 mL, 7.04 mmol, 1.8 equiv.) in sat. NaHCO3 (22 mL) and THF (8 mL) was added PPh3 (1.54 g, 5.87 mmol, 1.5 equiv.). The reaction mixture was stirred at room temperature for 80 min, then diluted with water and extracted with CHCl3. The organic layer was washed with brine and dried over Na2SO4. Filtration, evaporation in vacuo and purification by silica gel flash chromatography (AcOEt/n-hexane = 1/8) gave 5 (1.10 g, 3.57 mmol, 91%) as a yellow oil: 1H NMR (CDCl3, 500 MHz, δ; ppm, trans/cis = 2/1) trans; 8.43 (1H, d, J = 2.9 Hz), 8.18 (1H, td, J = 9.6 Hz, J = 2.9 Hz), 7.91 (1H, d, J = 16.2 Hz), 7.26 (1H, d, J = 9.6), 6.53 (1H, d, J = 16.2 Hz), 5.34 (2H, s), 3.51 (3H, s), 1.58 (9H, s)/cis; 8.36 (1H, d, J = 2.9 Hz), 8.18 (1H, td, J = 9.6 Hz, J = 2.9 Hz), 7.20 (1H, d, J = 9.6 Hz), 6.99 (1H, d, J = 12.4 Hz), 6.03 (1H, d, J = 12.4 Hz), 5.29 (2H, s), 3.49 (3H, s), 1.40 (9H, s).Synthesis of 6
A slurry of 5 (906 mg, 2.93 mmol) and 10% Pd/C (312 mg) in AcOEt (10 mL) was stirred at room temperature under H2 for 2 h, and then filtered on Celite. The filtrate was evaporated in vacuo. The residue was purified by silica gel flash chromatography (AcOEt/n-hexane = 1/2) to obtain 722 mg (2.57 mmol, 88%) of 6 as a yellow oil: 1H NMR (CDCl3, 500 MHz, δ; ppm) 6.88 (1H, d, J = 8.6 Hz), 6.53 (1H, d, J = 2.9 Hz), 6.49 (1H, dd, J = 8.6 Hz, J = 2.9 Hz), 5.09 (2H, s), 3.48 (3H, s), 3.41 (2H, s), 2.84 (2H, t, J = 7.9 Hz), 2.50 (2H, t, J = 7.9 Hz), 1.43 (9H, s).Synthesis of 9
7 (8.52 g, 37.3 mmol, 5.0 equiv.) was dissolved in dry THF (276 mL) and the solution was cooled to −78 °C. To the solution s-BuLi was then added dropwise (140 mL, 1.02 M in hexane, 41.0 mmol, 5.5 equiv.) under an Ar atmosphere. The mixture was stirred for 30 min, and then a solution of 8 (2.70 g, 7.44 mmol) in dry THF (84 mL) was added. The solution was immediately warmed to rt, and further stirred for 1 h. Acetic acid was slowly added to the reaction mixture on an ice bath until the color changed, and then the mixture was evaporated in vacuo. To the residue were added acetonitrile (120 mL) and 6 N HCl (180 mL). The mixture was stirred at room temperature for 15 h, and then evaporated to remove acetonitrile. The residue was neutralized with sat. NaHCO3 and aqueous 2 N NaOH and extracted with CH2Cl2/iPrOH. The organic layer was washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by silica gel flash column chromatography (CH2Cl2/MeOH = 20/1 → 10/1 → 7/1) to afford crude 9 (1.79 g) as a purple solid: 1H NMR (CD3OD, 500 MHz, δ; ppm) 10.17 (1H, s), 8.20 (2H, d, J = 8.2 Hz), 7.70 (2H, d, J = 8.2 Hz), 7.31 (2H, d, J = 9.7 Hz), 7.23 (2H, dd, J = 9.7 Hz, J = 2.4 Hz), 7.16 (2H, d, J = 2.5 Hz), 3.80–3.78 (8H, m), 1.82–1.75 (12H, m).Synthesis of 10
A solution of crude 9 (1.79 g), 6 (1.13 g, 4.04 mmol) and AcOH (14 mL) in CH2Cl2 (70 mL) was stirred at room temperature for 23 h. NaBH(OAc)3 (2.33 g, 11.0 mmol) was added, and the mixture was stirred for 15 min, then poured into sat. NaHCO3 and extracted with CH2Cl2. The organic layer was washed with brine and dried over Na2SO4. Filtration, evaporation and purification by silica gel flash chromatography (CH2Cl2/MeOH = 20/1 → 15/1 → 10/1 → 7/1) gave crude 10 (1.81 g) as a purple solid: 1H NMR (CD3OD, 500 MHz, δ; ppm) 7.67 (2H, d, J = 8.1 Hz), 7.43–7.39 (4H, m), 7.21 (2H, dd, J = 9.7 Hz, J = 2.5 Hz), 7.12 (2H, d, J = 2.5 Hz), 6.89 (1H, d, J = 8.7 Hz), 6.54 (1H, d, J = 2.8 Hz), 6.49 (1H, dd, J = 8.8 Hz, J = 2.9 Hz), 5.08 (2H, s), 4.45 (2H, s), 3.78–3.76 (8H, m), 3.46 (3H, s), 2.80 (2H, t, J = 7.5 Hz), 2.47 (2H, t, J = 8.0 Hz), 1.80–1.75 (12H, m), 1.40 (9H, s).Synthesis of 11
To a solution of crude 10 (1.81 g) in CH2Cl2 (18 mL) was added 4 N HCl/dioxane (42 mL) under an Ar atmosphere. The reaction mixture was stirred at room temperature for 7 h, and the reaction was quenched with 2 N NaOH and sat. NaHCO3. The whole was extracted with CH2Cl2/iPrOH. The organic layer was washed with brine, dried over Na2SO4, filtered and evaporated in vacuo. Purification by silica gel flash chromatography (CH2Cl2/MeOH = 15/1 → 14/1 → 12/1 → 10/1 → 7/1 → 5/1) gave crude 11 (103 mg) as a purple solid.Synthesis of NO-Rosa (2)
To a solution of crude 11 (95 mg) in AcOH (22 mL) was added a solution of NaNO2 (11 mg, 0.16 mmol) in water (22 mL) on an ice bath under an Ar atmosphere. The mixture was stirred on the ice bath for 20 min, then poured into sat. NaHCO3 and extracted with CH2Cl2. The organic layer was washed with brine and dried over Na2SO4. Filtration, evaporation and purification by silica gel flash chromatography (CH2Cl2/MeOH = 20/1 → 14/1 → 12/1) gave the crude product. Further purification by HPLC (0.1 M TEAA buffer/CH3CN = 50/50) gave NO-Rosa (5 mg) as a purple solid: 1H NMR (CD3OD, 500 MHz, δ; ppm) 7.41 (4H, s), 7.32–7.29 (4H, m), 7.22 (2H, dd, J = 9.6 Hz, J = 2.4 Hz), 7.10 (2H, d, J = 2.3 Hz), 6.87 (1H, d, J = 8.7 Hz), 5.43 (2H, s), 3.77 (8H, m), 2.90 (2H, t, J = 7.1 Hz), 2.49 (2H, t, J = 7.1 Hz), 1.81–1.74 (12H, m); 13C NMR (CD3OD, 150 MHz, δ; ppm) 159.91, 158.10, 156.82, 138.81, 134.88, 133.07, 132.61, 131.72, 131.12, 129.36, 124.47, 121.54, 117.63, 116.04, 114.79, 98.12, 49.90, 49.69, 39.05, 30.68, 28.08, 27.13, 25.32; HRMS (ESI+) calcd: 645.3077, found: 645.3079; HPLC tR = 16.7 min [A is MeCN containing 0.1% FA, B is MilliQ water containing 0.1% FA; gradient conditions: A conc. 30–50% (0–15 min), 50% (15–22 min); purity was 87.4% based on the absorbance at 254 nm].ESR analysis
N-Methyl-D-glucamine dithiocarbamate (6 mM), FeSO4 (1.5 mM), and NO-Rosa (100 μM) were dissolved in MilliQ water containing DMSO as a cosolvent. The ESR spectrum of the solution was measured after irradiation with MAX-303 (Asahi Spectra) equipped with a 530–590 nm band pass filter under an argon atmosphere. ESR conditions: microwave power, 10 mW; frequency, 9.4 GHz; field, 330 mT; sweep width, 7.5 mT; sweep time, 4 min; modulation width, 0.125 mT; time constant, 0.10 s.Photocontrolled NO release from NO-Rosa in HEK293 cells
Fluorescence imaging of NO release from NO-Rosa in HEK293 cells was performed by using DAF-FM DA. Cultured HEK293 cells were treated with DAF-FM DA (10 μM) and either NO-Rosa (10 μM) or vehicle (DMSO). The dishes were then photoirradiated with yellowish green light (530–590 nm, 60 mW cm−2 for 15 min). The cells were observed with a differential interference contrast microscope and a confocal microscope (Olympus, IX71).Photoinduced vasodilation with NO-Rosa
A rat aortic strip was placed in a Magnus tube filled with Krebs buffer at 37 °C. The strip was pretreated with L-NAME (10 μM) and noradrenaline (10 μM). After equilibration, NO-Rosa (10 μM) was added to the tube. The strip was irradiated with a light source (MAX-302, Asahi Spectra) equipped with a 530–590 nm band-pass filter for 1 min each time. After several cycles of photoirradiation, ODQ (10 μM) was added and the photoirradiation was performed again.Acknowledgements
This work was supported by a JSPS KAKENHI Grant No. JP26111012 (H. N.), as well as by a JSPS KAKENHI Grant No. JP16H05103 (H. N.) and JSPS KAKENHI Grant No. JP16 K15693 (Y. H., N. I.).Notes and references
- W. K. Alderton, C. E. Cooper and R. G. Knowles, Biochem. J., 2001, 357, 593 CrossRef CAS PubMed.
- L. J. Ignarro, G. M. Buga, K. S. Wood, R. E. Byrns and G. Chaudhuri, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9265 CrossRef CAS.
- R. A. Hopper and J. Garthwaite, J. Neurosci., 2006, 26, 11513 CrossRef CAS PubMed.
- C. Bogdan, Nat. Immunol., 2001, 2, 907 CrossRef CAS PubMed.
- S. Moncada, R. M. Palmer and E. A. Higgs, Pharmacol. Rev., 1991, 43, 109 CAS.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, J. Org. Chem., 1993, 58, 1472 CrossRef CAS.
- L. Field, V. R. Dilts, R. Ravichandran, G. P. Lenhert and E. G. Carnahan, J. Chem. Soc., Chem. Commun., 1978, 249 RSC.
- L. R. Makings and R. Y. Tsien, J. Biol. Chem., 1994, 269, 6282 CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289 CrossRef CAS PubMed.
- K. Hishikawa, H. Nakagawa, T. Furuta, K. Fukuhara, H. Tsumoto, T. Suzuki and N. Miyata, J. Am. Chem. Soc., 2009, 131, 7488 CrossRef CAS PubMed.
- N. Ieda, Y. Hotta, N. Miyata, K. Kimura and H. Nakagawa, J. Am. Chem. Soc., 2014, 136, 7085 CrossRef CAS PubMed.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597 CrossRef CAS PubMed.
- T. Miura, Y. Urano, K. Tanaka, T. Nagano, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 8666 CrossRef CAS PubMed.
- S. Namiki, T. Arai and K. Fujimori, J. Am. Chem. Soc., 1997, 119, 3840 CrossRef CAS.
- S. Wäldchen, J. Lehmann, T. Klein, S. van de Linde and M. Sauer, Sci. Rep., 2015, 5, 15348, DOI:10.1038/srep15348.
- R. R. Anderson and J. A. Parrish, J. Invest. Dermatol., 1981, 77, 13 CrossRef CAS PubMed.
- B. Wang, P. Li, F. Yu, P. Song, X. Sun, S. Yang, Z. Lou and K. Han, Chem. Commun., 2013, 49, 1014 RSC.
- P. Štacko, P. Šebej, A. T. Veetil and P. Klán, Org. Lett., 2012, 14, 4918 CrossRef PubMed.
- T. Yoshimura and Y. Kotake, Antioxid. Redox Signaling, 2004, 6, 639 CrossRef CAS PubMed.
- P. Damiani and G. Burini, Talanta, 1986, 33, 649 CrossRef CAS PubMed.
- H. Kojima, Y. Urano, K. Kikuchi, T. Higuchi, Y. Hirata and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 3209 CrossRef CAS PubMed.
- G. A. Gray, C. Schott, G. Julou-Schaeffer, I. Fleming, J. R. Paratt and J. Stoclet, Br. J. Pharmacol., 1991, 103, 1218 CrossRef CAS PubMed.
- M. Russwurm, E. Mergia, F. Mullershausen and D. Koesling, J. Biol. Chem., 2002, 277, 24883 CrossRef CAS PubMed.
- J. Garthwaite, E. Southam, C. L. Boulton, E. B. Nielsen, K. Schmidt and B. Mayer, Mol. Pharmacol., 1995, 48, 184 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob00245a |
| This journal is © The Royal Society of Chemistry 2017 |