
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition-metal-free synthesis of 3-(1-pyrrolidinyl)quinolines and 3-(1-pyrrolidinyl)quinoline 1-oxides via a one-pot reaction of 3-(1-pyrrolidinyl)crotonates with nitrobenzenes†
Robert
Bujok
 *,
Piotr
Cmoch
,
Zbigniew
Wróbel
and
Krzysztof
Wojciechowski
*,
Piotr
Cmoch
,
Zbigniew
Wróbel
and
Krzysztof
Wojciechowski
Institute of Organic Chemistry Polish Academy of Sciences, ul. Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: robert.bujok@icho.edu.pl; krzysztof.wojciechowski@icho.edu.pl
First published on 21st February 2017
Abstract
A carbanion of tert-butyl 3-(1-pyrrolidinyl)crotonate adds to nitrobenzenes to form σH-adducts, which in the presence of pivaloyl chloride and triethylamine are converted into 3-(1-pyrrolidinyl)quinolines or 3-(1-pyrrolidinyl)quinoline 1-oxides depending on the nitrobenzene structure. This is the first methodology in which a quinoline ring is constructed from a substrate bearing a pyrrolidinyl ring. Starting from optically pure enamines, the method allows synthesis of the corresponding chiral products without racemisation.
Introduction
Quinoline is a key building block in many naturally occurring and synthetically prepared compounds that have important practical applications.1 Thus, the synthesis of substituted quinolines is still of substantial interest.2 In recent years quinoline derivatives bearing a 1-pyrrolidinyl substituent at the 3-position have attracted attention due to their biological activity and potential applications as antimicrobials (antimalarial activity),3 hepatitis C virus polymerase inhibitors,4 antineoplastic (antitumor) agents,5 as well as compounds for osteoporosis treatment.6 Representative structures are shown in Fig. 1. | ||
| Fig. 1 Selected 3-(1-pyrrolidinyl)quinolines exhibiting biological activity. | ||
General methods for the synthesis of 3-(1-pyrrolidinyl)-quinolines consist of transition-metal-catalyzed replacement of halogen (mostly bromine) (Fig. 2, path a),4,7 reaction of 3-aminoquinoline with 4-chlorobutyryl chloride followed by reduction with LiAlH4 (path b),3 ring-closing metathesis in 3-diallylaminoquinolines followed by hydrogenation (path c),3 and alkylation of 3-aminoquinoline with 1,4-dibromobutane (path d).3 One of the drawbacks of the recently developed methods for the synthesis of heterocycles, including quinolines, is the use of transition metal catalysts, which often requires arduous removal of the catalyst, particularly if the product has potential pharmaceutical applications.8 Thus new transition-metal-free methodologies are still in demand.
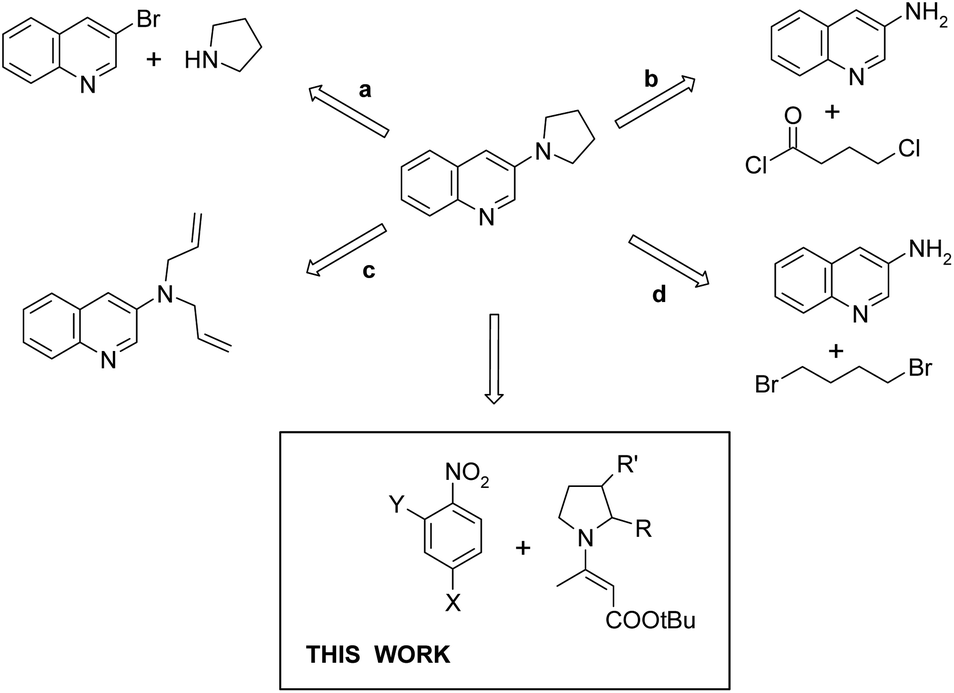 | ||
| Fig. 2 Synthetic strategies towards 3-(1-pyrrolidinyl)-quinolines. | ||
Quinoline 1-oxides are usually obtained via direct oxidation of quinolines with peracids. Such oxidation will be ineffective if the quinoline derivative contains an amino group. Less widespread are methods in which the quinoline 1-oxides are formed as a result of interaction of the nitro group with adjacent substituents in ortho-substituted nitroarenes.9 To the best of our knowledge, the synthesis of 3-(1-pyrrolidinyl)quinoline-1-oxides is still unexplored.
During our studies on reactions of carbanions with nitroarenes we have developed numerous methods for the synthesis of heterocycles, especially indoles and quinolines.10,11 Particularly effective for the synthesis of quinolines were methods in which the σH-adducts formed by addition of allyl carbanions to nitroarenes were subsequently treated with a silylating agent12 such as trialkylchlorosilane or bis-trimethylsilylacetamide (BSA) or with an acyl chloride, preferably pivaloyl chloride.12c
Results and discussion
In this paper we present an efficient one-pot method for the synthesis of 3-(1-pyrrolidinyl)quinoline-2-carboxylic acid esters and the corresponding quinoline 1-oxides directly from nitrobenzenes. Unlike all the literature methods for the synthesis of 3-(1-pyrrolidinyl)quinoline, in this procedure a quinoline ring is constructed. Nitrobenzenes are cheap, commercially available starting materials, while quinoline derivatives are less abundant, so this synthesis is an interesting alternative to literature methodologies.As a carbanion precursor we have chosen tert-butyl 3-(1-pyrrolidinyl)crotonate 1 prepared from tert-butyl acetoacetate and pyrrolidine in an excellent yield by simple mixing of equimolar amounts of the reagents without solvent (Scheme 1). 1H NMR and nuclear Overhauser effect (1D-NOE) experiments proved the exclusive formation of the E-isomer.
 | ||
| Scheme 1 Synthesis of enamine 1. | ||
As a model nitroarene for testing the reaction conditions we have chosen 2,4-dichloronitrobenzene, because earlier studies on vicarious nucleophilic substitution of hydrogen in nitroarenes revealed its relatively high electrophilicity.13
In the initial experiment (Table 1) we used potassium tert-butoxide as a base for the generation of the carbanion and pivaloyl chloride to acylate the formed σH-adduct. The reaction was carried out in THF at −70 °C. Unfortunately, this attempt was unsuccessful – no reaction with 2,4-dichloronitrobenzene occurred. Probably, potassium tert-butoxide is too weak a base to deprotonate the enamine 1. Indeed, when we used a stronger base, namely lithium diisopropylamide, the expected tert-butyl 6,8-dichloro-3-(1-pyrrolidinyl)quinoline-2-carboxylate 2a was obtained, but the yield was rather low (24%) and a large amount of by-products was formed. In the case of n-butyllithium only traces of the product 2a (TLC) were observed in the absence of any compounds, decreasing the degree of aggregation of the base.
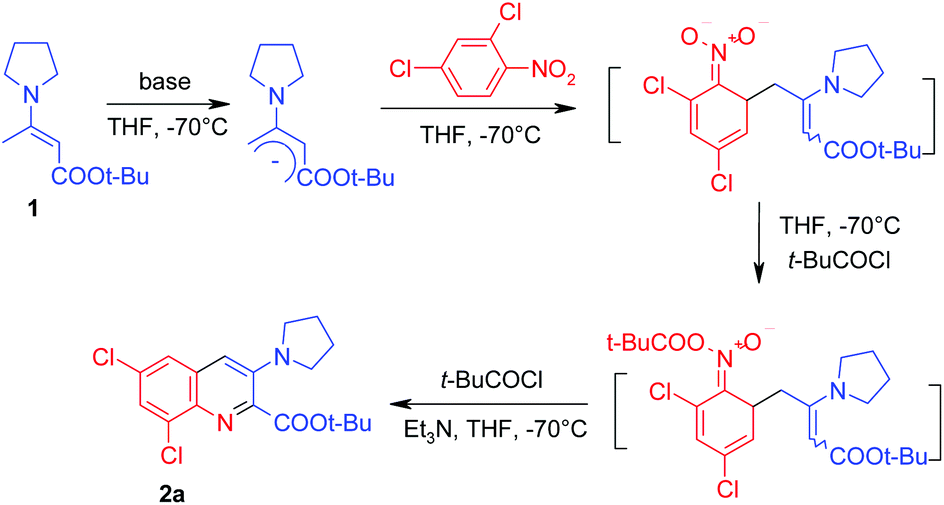
n-Butyllithium exists as a tetramer14 in hexane/THF solution and HMPA is known to be the most effective reagent14 that breaks organo-lithium oligomers and increases the activity of the organolithium compound. Taking this into consideration, we carried out the reaction with n-butyllithium in the presence of 1 equivalent of HMPA. In this case, treatment of the formed σH-adduct with triethylamine and pivaloyl chloride resulted in the formation of the ester 2a in 57% yield. Attempts to replace HMPA by other reagents (N-methylpyrrolidone (NMP) and TMEDA) were not successful – yields have not exceeded 20%. Because of the multistep nature of the investigated reaction, we have found the result obtained in the presence of HMPA to be satisfactory and applied these conditions15 to screen the scope of the reaction (Table 2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15
15

The reactions of 2,4-disubstituted nitrobenzenes with the enamine 1 led to the expected quinoline derivatives 2a–2e in 54–67% yields. As mentioned above, these are the overall yields of several steps.
Surprisingly, 4-substituted nitrobenzenes with no substituents at the 2-position react under analogous conditions to the enamine 1 to give tert-butyl 6-substituted 3-pyrrolidinyl-2-carboxylate-quinoline 1-oxides 3a–3h instead of the expected quinolines. The reaction is of general character – 4-substituted nitrobenzenes with electron-withdrawing (CF3, Cl) and electron-donating groups (CH3, t-Bu) give quinoline 1-oxides, usually in ca. 50% yield.
In the case of the 2-chloronitrobezene complex a mixture of compounds was formed and no attempt was made to separate them. The mixture was formed probably due to the attack of the carbanion on the 4-position of 2-chloronitrobezene.
To show the utility of our approach we have obtained carbanion precursors containing chiral pyrrolidines. Commercially available N-Boc-L-prolinol and N-Boc-D-prolinol were chosen for this purpose. Optically pure enamines (S)-1a and (R)-1a were prepared in very good overall yields (Scheme 5; for details see the ESI†). Some of the biologically active quinolines contain substituents at the 3-position of the pyrrolidine ring, so the corresponding enamines (S)-1b and (R)-1b were also obtained (Scheme 2).
 | ||
| Scheme 2 Synthesis of optically pure enamines (S)-1a, (R)-1a, (S)-1b and (R)-1b (details are given in the ESI). | ||
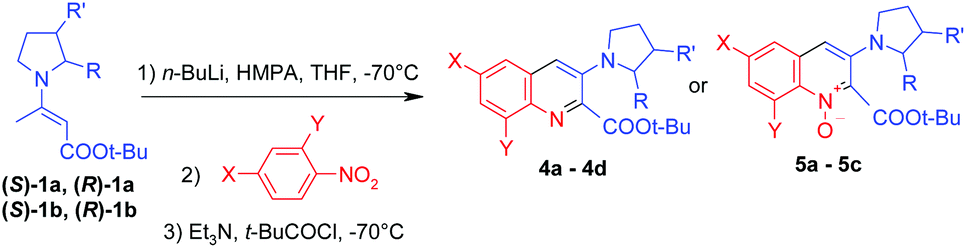
Chiral enamines (S)-1a, (R)-1a, (S)-1b and (R)-1b react with nitrobenzenes in a similar manner to achiral enamine 1. Reaction with 2,4-disubstituted nitrobenzenes led to the corresponding non-racemic quinolines 4a–4d and reaction with 4-substituted nitrobenzenes led to non-racemic quinoline 1-oxides 5a–5c. The yields are somewhat lower than for achiral enamine 1, but the results are still acceptable considering the complex nature of the obtained structures (Table 3).
15
To verify the optical purity of the non-racemic products, the R-enantiomer of quinoline 4b and the R-enantiomer of quinoline-1-oxide 5b were synthesized and both enantiomers of the examined compounds were converted to diastereomeric salts with R-Mosher's acid. Diagnostic signals of each salt formed from the corresponding enantiomer could be clearly recognized in 1H NMR spectra. Only one diastereoisomer was detected in each case, which proved that all the examined compounds are optically pure (Fig. 3).
 | ||
| Fig. 3 Diagnostic signals of diastereoisomeric salts formed from 5b, 5c and R-Mosher's acid. | ||
The surprising selectivity of the reactions (in all cases only quinoline or quinoline 1-oxide was obtained depending on the structure of the nitrobenzene; a mixture of these products was never observed) suggests that two different mechanisms operate. To gain an insight into the mechanisms, a few control experiments were carried out.
First of all, reactions of 2,4-dichloronitrobenzene and 4-chloronitrobenzene with enamine 1 under standard conditions but in the absence of pivaloyl chloride were carried out. In both cases no reaction was observed. These results indicate that σH-adducts formed from these nitrobenzenes do not undergo further reactions in the absence of t-BuCOCl and substrates are recovered after the hydrolysis of σH-adducts during aqueous work-up. So in the presence of pivaloyl chloride both σH-adducts are acylated and these intermediates are subsequently transformed into quinolines or quinoline 1-oxides depending on the character of the Y substituent (Scheme 3).
 | ||
| Scheme 3 Behaviour of σH-adducts in the absence of pivaloyl chloride and in the presence of the reagent. | ||
Acylated σH-adduct (I) probably undergoes elimination of the pivalate anion. The formed nitroso group enters an intramolecular Ehrlich-Sachs12a,e reaction resulting in the formation of the corresponding 1-pivaloyloxy-dihydroquinoline derivative (II) (Scheme 4). When Y ≠ H due to steric interaction of the Y substituent and the bulky pivaloyloxy group, elimination of the pivalate anion takes place and quinoline 2a is formed.
 | ||
| Scheme 4 Plausible mechanism for the synthesis of quinoline 2a. | ||
The formation of quinolone 1-oxides seems more difficult to explain. It could arise from nitro compound (III) (Scheme 5) via its intramolecular cyclization;12c,16 however, experiments conducted in the absence of t-BuCOCl proved that the addition of the carbanion to nitroarene was reversible and the spontaneous oxidation of the σH-adduct did not occur (Scheme 3).
 | ||
| Scheme 5 Attempted conversion of nitrocompound (III) into quinoline 1-oxide 3a under conditions employed for of synthesis of quinolines. | ||
On the other hand the intermediate (III) might be formed from other than σH-adduct intermediates and then it could cyclize to quinoline 1-oxide 3a. To check this hypothesis an appropriate nitrocompound (III) was synthesized and exposed to reaction conditions (Scheme 5). Product 3a was not formed under these conditions – only the substrate and a moderate amount of polar by-products were observed. Thus quinoline 1-oxides are not formed via nitro-compound (III).
Based on these data, we suppose that 1-pivaloyloxy-dihydroquinoline derivative (II) is oxidized to 1-pivaloyloxy-quinoline (IV) by a nitrocompound (always used in excess) and then the pivaloyl group is attacked by a nucleophile (e.g. t-BuCOO−) leading to product 3a (Scheme 6).
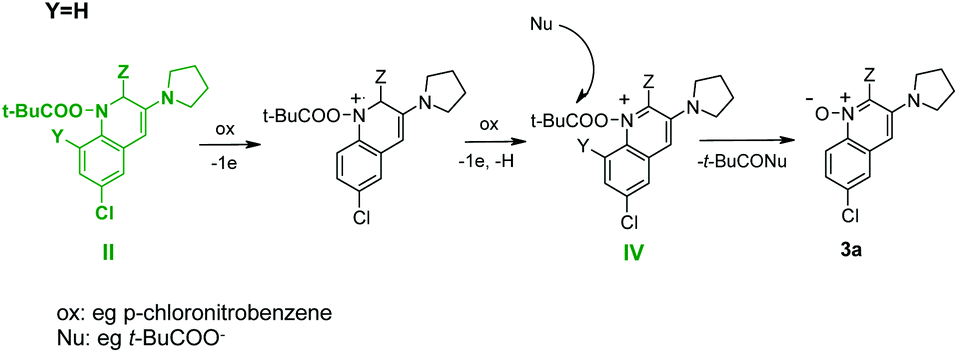 | ||
| Scheme 6 Plausible mechanism for synthesis of quinoline 1-oxide 3a. | ||
The tert-butoxycarbonyl group can be easily removed from compounds 2–3 by refluxing in 20% aqueous sulfuric acid. Hydrolysis and subsequent decarboxylation occurred, giving the expected products in good to excellent yields (81–97%) (Table 4).
17
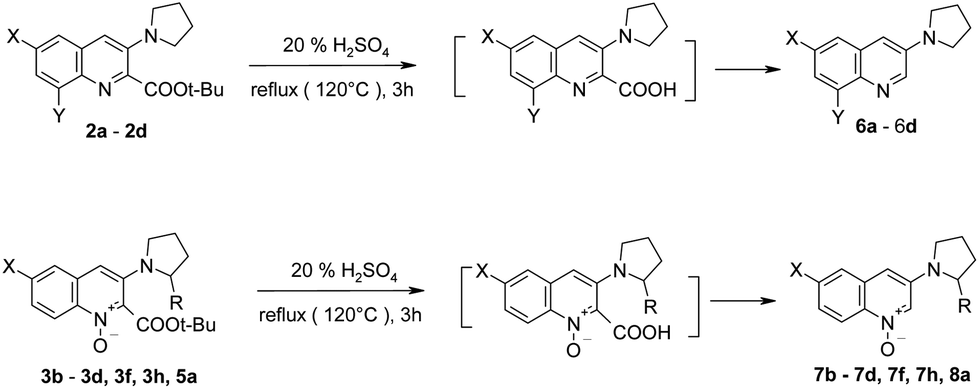
The hydrolysis/decarboxylation domino reaction was also successfully performed for non-racemic quinoline 1-oxide 5a – the expected product 8a was obtained in 79% yield.
It is worth noting that treating a solution of tert-butyl esters 2a, 2e in dichloromethane with trifluoroacetic acid at room temperature resulted in the formation of the corresponding acids 9a, 9e in good yields of ca. 80% (Scheme 7). Elemental analysis indicated that compound 9a was obtained as a free base (no fluorine present). In the case of compound 9e, elemental analysis and 19F NMR spectroscopy suggest that the product contains “0.5 molecules” of trifluoroacetic acid. Probably, 2 molecules of compound 9e are coordinated by 1 molecule of trifluoroacetic acid.
 | ||
| Scheme 7 Transformation of COOt-Bu group into COOH group (the isolated yields are given).18 | ||
Conclusions
We have developed a one-pot, transition-metal-free method for the synthesis of 3-(1-pyrrolidinyl)quinolines or 3-(1-pyrrolidinyl)-quinoline 1-oxides depending on the structure of the starting materials (2,4-disubstituted or 4-substituted nitrobenzenes). This is the first methodology in which a quinoline ring is constructed from a substrate bearing a pyrrolidinyl ring. The method allows direct synthesis of complicated structures from easily available enamines and commercial nitrobenzenes. Starting from optically pure enamines the method allows synthesis of the corresponding chiral products without racemisation.Acknowledgements
This work was supported by National Scientific Center Grant No. 2012/07/B/ST5/00813. We would like to thank Dr Mikołaj Chromiński (also known as the Chemistry Wizard) for helpful discussions.Notes and references
- (a) L. A. Mitscher, Chem. Rev., 2005, 105, 559 CrossRef CAS PubMed; (b) M. Robert, J. Josef, K. Katarina and D. R. Richardson, Bioorg. Med. Chem., 2007, 15, 1280 CrossRef PubMed; (c) V. V. Kouznetsov, L. Y. V. Mendez and C. M. M. Gomez, Curr. Org. Chem., 2005, 9, 141 CrossRef CAS; (d) K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245 CrossRef CAS PubMed; (e) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488 CrossRef CAS PubMed; (f) O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871 CrossRef CAS PubMed; (g) A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1 CrossRef PubMed.
- (a) M. Jose, P. Elena, S. Abdelouahid, C. Mariado and S. Elena, Chem. Rev., 2009, 109, 2652 CrossRef PubMed; (b) G. Liu, M. Yi, L. Liu, J. Wang and J. Wang, Chem. Commun., 2015, 51, 2911 RSC; (c) Y. Matsubara, S. Hirakawa, Y. Yamaguchi and Z. Yoshida, Angew. Chem., Int. Ed., 2011, 50, 7670 CrossRef CAS PubMed; (d) G.-L. Gao, Y.-N. Niu, Z.-Y. Yan, H.-L. Wang, G.-W. Wang, A. Shaukat and Y.-M. Liang, J. Org. Chem., 2010, 75, 1305 CrossRef CAS PubMed; (e) S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463 RSC; (f) Y. Zhou, C. Wu, X. Dong and J. Qu, J. Org. Chem., 2016, 81, 5202–5208 CrossRef CAS PubMed; (g) R. Bujok, Z. Wróbel and K. Wojciechowski, Tetrahedron Lett., 2016, 57, 1014–1018 CrossRef CAS.
- S. Vandekerckhove, S. V. Herreweghe, J. Willems, B. Danneels, T. Desmet, C. de Kock, P. J. Smith, K. Chibaleand and M. D'hooghe, Eur. J. Med. Chem., 2015, 92, 91 CrossRef CAS PubMed.
- J. de Vicente Fidalgo, J. Li, R. C. Schoenfeld, F. X. Talamas and J. P. G. Taygerly, US2010/311760A1, 2010 Search PubMed.
- A. G. Novartis, H. Deng, X. Fu, H. Guo, F. He, Y. Mi, X. Yan, H. Yu and J. Y. Zhang, WO2012/107500A1, 2012 Search PubMed.
- J.-K. Kim, S.-W. Kim, S.-B. Shim, S.-Y. Ko, S. Chang, J.-S. Lee, D. P. Kang, H. C. Sung, J.-A. Park and Y. S. Yang, WO2007/114672A1, Osotec Inc., 2007 Search PubMed.
- (a) E. F. DiMauro, J. L. Buchanan, A. Cheng, R. Emkey, S. A. Hitchcock, L. Huang, M. Y. Huang, B. Janosky, J. H. Lee, X. Li, M. W. Martin, S. A. Tomlinson, R. D. White, X. M. Zheng, V. F. Patel and R. T. Fremeau Jr., Bioorg. Med. Chem. Lett., 2008, 18, 4267 CrossRef CAS PubMed; (b) F. X. Talamas, S. C. Abbot, S. Anand, K. A. Brameld, D. S. Carter, J. Chen, D. Davis, J. de Vicente, A. D. Fung, L. Gong, S. F. Harris, P. Inbar, S. S. Labadie, E. K. Lee, R. Lemoine, S. Le Pogam, V. Leveque, J. Li, J. McIntosh, I. Najera, J. Park, A. Railkar, S. Rajyaguru, M. Sangi, R. C. Schoenfeld, L. R. Staben, Y. Tan, J. P. Taygerly, A. G. Villasenor and P. E. Weller, J. Med. Chem., 2014, 57, 1914 CrossRef CAS PubMed; (c) J. Cheng and M. L. Trudell, Org. Lett., 2001, 3, 1371 CrossRef CAS PubMed.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS.
- P. N. Preston and G. Tennant, Chem. Rev., 1972, 72, 627 CrossRef CAS.
- (a) M. Mąkosza and K. Wojciechowski, Heterocycles, 2001, 54, 445 CrossRef; (b) M. Mąkosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631 CrossRef PubMed; (c) M. Mąkosza and K. Wojciechowski, Heterocycles, 2014, 88, 75 CrossRef.
- (a) M. Mąkosza and K. Wojciechowski, Top. Heterocycl. Chem., 2014, 37, 51 Search PubMed; (b) M. Mąkosza and K. Wojciechowski, Chem. Heterocycl. Compd., 2015, 51, 210 CrossRef.
- (a) K. Anczkiewicz, M. Królikiewicz, Z. Wróbel and K. Wojciechowski, Tetrahedron, 2015, 71, 3924 CrossRef CAS; (b) M. Nowacki and K. Wojciechowski, RSC Adv., 2015, 5, 94296 RSC; (c) M. Bobin, A. Kwast and Z. Wróbel, Tetrahedron, 2007, 63, 11048 CrossRef CAS; (d) Z. Wrobel, Synlett, 2004, 1929 CrossRef CAS; (e) Z. Wróbel, Eur. J. Org. Chem., 2000, 521 CrossRef; (f) R. Bujok, A. Kwast, P. Cmoch and Z. Wróbel, Tetrahedron, 2010, 66, 698 CrossRef CAS.
- (a) S. Błażej and M. Mąkosza, Chem. – Eur. J., 2008, 14, 11113 CrossRef PubMed; (b) F. Seelinger, S. Błażej, S. Bernhardt, M. Mąkosza and H. Mayr, Chem. – Eur. J., 2008, 14, 6108 CrossRef PubMed.
- J. Clayden, Organolithium: Selectivity for Synthesis, Elsevier Science, 2002, pp. 2–7 Search PubMed.
- To a solution of the enamine 1 (0.317 g, 1.5 mmol) and HMPA (0.35 mL, 0.357 g, 2.0 mmol) in dry THF (10 mL) at −70 °C under argon, 2.5 M BuLi in hexane (0.67 mL, 1.67 mmol) was added in portions in 4 min and the solution was stirred at −70 °C for 30 min. A solution of the nitroarene (3.0 mmol) in THF (1.7 mL) was added and the resultant mixture was stirred for 10 min at −70 °C. Et3N (1.0 mL, 7.2 mmol) was added and then pivaloyl chloride (0.85 mL, 6.9 mmol) was added in portions (7 min). After the addition was complete, the solution was stirred at −70 °C for 2 h. The cooling bath was removed, water (10 mL) was added and the mixture was stirred for 5 min, then extracted with AcOEt (30 mL), dried and evaporated. The products were purified by column chromatography (SiO2, hexane/AcOEt).
- (a) Z. Wróbel, Tetrahedron Lett., 1997, 38, 4913 CrossRef; (b) Z. Wróbel, A. Kwast and M. Mąkosza, Synthesis, 1993, 31 CrossRef; (c) Z. Wróbel and M. Mąkosza, Tetrahedron, 1993, 49, 5315 CrossRef.
- A suspension of compounds 2–5 (0.2–0.4 mmol) in 20% H2SO4 (2–4 mL) was heated to reflux for 3 h. The mixture was cooled to RT and diluted with water (15 mL) and solid K2CO3 was added in portions to achieve pH = 10. The mixture was extracted with AcOEt (2 × 25 mL), and the combined extracts were dried and evaporated. The crude products were washed with pentane or Et2O. The obtained products were pure according to 1H NMR.
- To a solution of compound 2a (0.358 mmol) in CH2Cl2 (3.8 mL), trifluoroacetic acid (1.0 mL) was added and the solution was stirred at RT for 1 h. The solvent was evaporated and the crude product was washed with Et2O. The obtained product was pure according to 1H NMR.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02658c |
| This journal is © The Royal Society of Chemistry 2017 |