
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature atomic layer deposition delivers more active and stable Pt-based catalysts†
Hao
Van Bui‡
 a,
Fabio
Grillo‡
*a,
Sri Sharath
Kulkarni
a,
Ronald
Bevaart
a,
Nguyen
Van Thang
b,
Bart
van der Linden
a,
Jacob A.
Moulijn
a,
Michiel
Makkee
a,
Michiel T.
Kreutzer
a and
J. Ruud
van Ommen
a
a,
Fabio
Grillo‡
*a,
Sri Sharath
Kulkarni
a,
Ronald
Bevaart
a,
Nguyen
Van Thang
b,
Bart
van der Linden
a,
Jacob A.
Moulijn
a,
Michiel
Makkee
a,
Michiel T.
Kreutzer
a and
J. Ruud
van Ommen
a
aDepartment of Chemical Engineering, Delft University of Technology, 2628 BL, Delft, The Netherlands. E-mail: f.grillo@tudelft.nl
bDepartment of Radiation Science and Technology, Delft University of Technology, 2628 BL, Delft, The Netherlands
First published on 12th July 2017
Abstract
We tailored the size distribution of Pt nanoparticles (NPs) on graphene nanoplatelets at a given metal loading by using low-temperature atomic layer deposition carried out in a fluidized bed reactor operated at atmospheric pressure. The Pt NPs deposited at low temperature (100 °C) after 10 cycles were more active and stable towards the propene oxidation reaction than their high-temperature counterparts. Crucially, the gap in the catalytic performance was retained even after prolonged periods of time (>24 hours) at reaction temperatures as high as 450 °C. After exposure to such harsh conditions the Pt NPs deposited at 100 °C still retained a size distribution that is narrower than the one of the as-synthesized NPs obtained at 250 °C. The difference in performance correlated with the difference in the number of facet sites as estimated after the catalytic test. Our approach provides not only a viable route for the scalable synthesis of stable supported Pt NPs with tailored size distributions but also a tool for studying the structure–function relationship.
1 Introduction
Noble metals have always played a pivotal role in catalysis as they boast high activity and selectivity across a wide range of industrially relevant reactions.1–7 Noble metals have been dispersed on high-surface-area supports as nanoparticles (NPs) to maximize the number of active surface sites.5 Recently, the advent of nanotechnology has brought new methods and techniques that promise better control over the NP size distribution, such that the relation between NP size and function can be experimentally explored and used in fields beyond conventional catalysis such as microelectronics, and energy storage and conversion.4,8–12 Yet, depositing controlled amounts of metal in the form of size-selected NPs, and thus achieving the desired functionality while retaining low metal utilization, remains a challenge, especially on an industrial scale.4,9,13 For example, NPs synthesized via liquid-phase routes based on ligand-stabilization typically undergo loss of size-selection upon exposure to reactive environments due to ligand removal and residual solvent decomposition.9 Furthermore, gas-phase routes for NP size-selection are typically line-of-sight techniques (e.g., pulsed laser deposition, electron beam lithography, and cluster beam deposition) and thus not suitable to high-surface-area substrates.9,14,15Atomic layer deposition (ALD) is a gas-phase and solvent-free technique that is a promising route for the controlled deposition of NPs, subnanometer clusters, and even single atoms.13,16–21 Briefly, the advantage of ALD is that the deposition proceeds stepwise through cyclic self-terminating surface reactions, such that (1) each step can run to completion, even in hard-to-reach places in high-surface-area substrates,14,16,19,20,22 (2) the amount deposited in each cycle of alternating reactions is tightly controlled, and (3) programming different precursors in sequences of cycles allow for more complex NPs such as bimetallic particles and core/shell and overcoated NPs.17 In particular, when carried out in fluidized bed reactors (FBRs), ALD lends itself to the deposition of noble metals on bulk quantities of high-surface-area powders with hardly any loss of metal precursors.18,23,24 As a result, ALD has the potential to fabricate supported-noble-metal catalysts reproducibly and on an industrially relevant scale.
Even if the amount deposited is precisely controlled by the self-limiting reactions, control over particle size and stability is more elusive. For example, increasing the number of cycles in thermal ALD of Pt and Pd has been reported to not only vary the average NP size but also broaden the size distribution.21,25–27 We have recently described the fate of adatoms on supports during ALD, showing that they indeed migrate, form clusters and sinter.27 These processes are highly size-dependent (large clusters are less mobile), temperature-dependent (as holds for most sintering processes1,2), and crucially depend also on the extent to which both surface reactions have run to completion. In fact, one can fabricate single-atom catalysts by not running the oxidative removal of precursor ligands to completion. Such a strategy may work for applications where the adsorbed metal atom, surrounded by ligands, is stable. For instance, room-temperature electrocatalytic applications of such strategy have been shown to exhibit limited deactivation.21 However, exposing such materials to reaction conditions at higher temperatures burns off the ligands and immediately renders these single-atoms mobile, which can then sinter into larger NPs.27 The crucial outstanding question is whether ALD techniques can produce supported NPs of desirable narrow size distributions that remain stable in catalytic reaction conditions.
Here, we show that Pt nanoparticles deposited by ALD on graphene nanoplatelets (GNP) at low temperatures (100 °C) using a fluidized bed operated at atmospheric pressure not only have a narrower size distribution but are also more stable than their high-temperature counterparts. In particular, high-loading Pt/GNP composites synthesized at low temperature are more active than their high-temperature counterparts. We demonstrate the activity and the stability of the Pt/GNP composites during the propene oxidation reaction at high reaction temperatures (T > 200 °C) for prolonged periods of time (>24 hours). We chose the propene oxidation reaction since it has been widely used as a model reaction for the Pt-catalyzed combustion of volatile organic compounds and, in particular, because it has been shown to be strongly affected by the Pt NP size while being relatively insensitive to the nature of the support.28–32 The graphene nanoplatelets were chosen because they are a promising support for catalytic applications.33–36 Moreover, Pt adatoms and nanoparticles tend to migrate and sinter more readily on graphene than on other supports,37–40 such that our system can be considered especially challenging in this respect. Finally, we show that our method, by producing bulk quantities of supported-NPs with narrow size distributions, is also an excellent tool for studying the size-dependent catalytic properties of supported noble-metal NPs.
2 Experimental
2.1 Materials
Graphene nanoplatelets (6–8 nm thick and 15 μm wide, surface area of about 150 m2 g−1) and trimethyl(methylcyclo-pentadienyl)platinum(IV) (MeCpPtMe3) were obtained from Strem Chemicals and used as received.2.2 ALD experiments
The ALD experiments were carried out in a home-built fluidized bed reactor operating at atmospheric pressure as described elsewhere.16,18 In brief, the system consists of a glass column (26 mm in internal diameter and 500 mm in height) placed on top of a single motor Paja PTL 40/40–24 vertical vibration table to assist the fluidization. MeCpPtMe3 was used as the Pt and synthetic air (20 wt% oxygen) was used as the oxygen source. The Pt precursor, contained in a stainless steel bubbler, was heated and maintained at 70 °C. The stainless steel tubing connecting the bubbler and the reactor were maintained at 80 °C to avoid precursor condensation. The reactor was heated by an infrared lamp placed parallel to the column with feedback control to maintain a constant temperature during ALD. In each experiment, 0.75 g of graphene nanoplatelets (GNP) were loaded in the reactor. A gas flow of 0.5 L min−1 was introduced through a distributor plate at the bottom of the reactor column in order to fluidize the powders. Prior to ALD, the powders were dried in air at 120 °C for 1 h and then pretreated in ozone-enriched air at 200 °C for 30 min in order to remove adventitious carbon and provide reproducible surface conditions, by flowing synthetic air (0.5 L min−1) through an OAS Topzone ozone generator. The ozone-enriched air had an ozone content of about 1.5 wt%. The ALD process consisted of sequential exposures of the powders to the Pt precursor (4–5 min) and synthetic air (5–10 min), separated by a purging step (5 min) using nitrogen as an inert gas (N2, 99.999 vol%).2.3 Material characterization
Elemental analysis was carried out by means of instrumental neutron activation analysis (INAA). 30 mg of each as-synthesized sample were loaded into high purity polyethylene capsules. The samples as well as a standard sample (reference) were then packaged and irradiated by a constant neutron flux. The INAA used in this work enables a detection limit in the range of 10–100 nanogram.A Mettler Toledo TGA/SDTA 851e thermogravimetric analyzer was used for studying the thermal behavior of the synthesized composites. 3 mg of Pt/GNP were used for each TGA measurement. The TGA curves were recorded while ramping up the powders from 25 to 800 °C with a ramping rate of 5 °C min−1 in a synthetic air flow of 100 ml min−1.
The morphology of the deposited Pt nanoparticles was investigated by means of transmission electron microscopy (TEM) and high resolution TEM (HRTEM). HRTEM micrographs were obtained using a FEI Titan G2 60–300 transmission electron microscope operated at 300 kV. TEM micrographs were obtained by using a JEOL JEM1400 transmission electron microscope operating at a voltage of 120 kV. As-synthesized composites were suspended in ethanol and transferred to regular transmission electron microscopy grids (3.05 mm in diameter). Number-based particle size distributions were obtained by image analysis of 10–30 TEM micrographs taken at different locations and at different magnifications (e.g., 50k and 100k). The image analysis was carried out by using the ImageJ software. For each sample of the Pt/GNP composites, the equivalent projected diameter (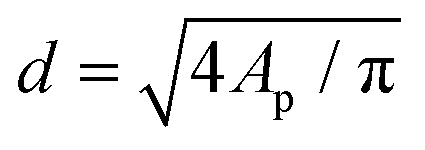 ) of a number of NPs in the range of 2500–7000 was estimated by means of a semiautomatic process involving several steps, including background subtraction, Fast-Fourier-Transform (FFT) band pass filter, treshholding and outline detection. The number-based PSD (F1(d)) was defined in terms of probability density as follows:
) of a number of NPs in the range of 2500–7000 was estimated by means of a semiautomatic process involving several steps, including background subtraction, Fast-Fourier-Transform (FFT) band pass filter, treshholding and outline detection. The number-based PSD (F1(d)) was defined in terms of probability density as follows:
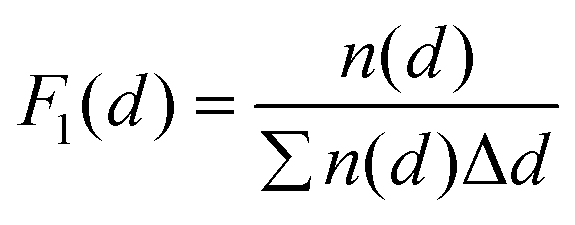 | (1) |
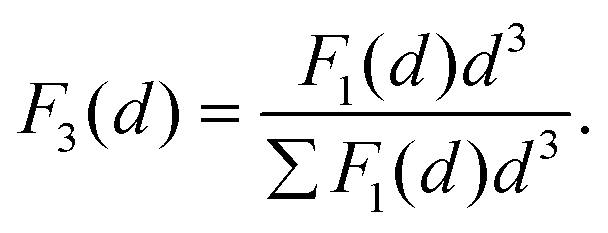 | (2) |
In particular, we define the span or width of the mass-based distribution as the range of sizes where 80% of the Pt mass lies:
| PSD span = d3(90%) − d3(10%) | (3) |
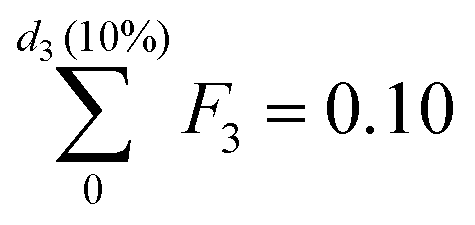 and
and 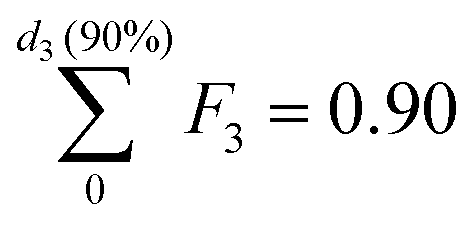 , respectively.
, respectively.
2.4 Catalytic tests
The catalytic tests of the Pt/GNP composites were carried out by means of a fixed bed reactor coupled with an analysis unit already described elsewhere.41 In brief, the experimental apparatus consisted of three sections: feed mixing, reactor, and analysis. In each experiment the feed to the reactor consisted of He containing 1000 ppm of propene (C3H6) and 1 vol% O2. The flow rate was maintained at 30 ml min−1. The reactor consists of a quartz column 4 mm in diameter and 25 cm in length. The reactor was loaded with 15.4 mg of Pt/GNP composite. To avoid catalyst entrainment, the reactor was first loaded with 50 mg of silicon carbide. The reactor was placed in a furnace capable of withstanding temperatures up to 1000 °C and equipped with a temperature controller. The analysis section connected to the outlet of the reactor was used to monitor the concentration of propene, CO2, and H2O. It consisted of a gas chromatograph Chrompack CP 9001, a thermal conductivity detector (TCD), and a flame ionization detector (FID). The apparatus was controlled via a dedicated program run on the Labview software. Prior to the catalytic test, the catalytic bed was exposed to H2 for 5 min. Each catalytic test consisted of at least two heating and cooling cycles from room temperature to 450 °C with a ramp of 2 °C min−1. Pt/GNP composites obtained after 10 ALD cycles at 100 °C and 200 °C were further tested at constant reaction temperatures on the order of 200 °C for about 20 hours. The apparent reaction rate kapp (molC3H6 molPt−1 s−1) was calculated from measurements of the C3H6 conversion XC3H6 during the cooling phase of the second heating and cooling cycle of each catalytic test as follows: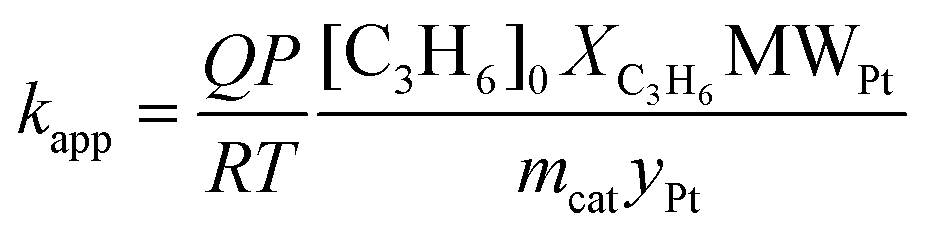 | (4) |
3 Results and discussion
We synthesized several model catalysts consisting of Pt NPs supported on graphene nanoplatelets (Pt/GNP) by using the atmospheric-pressure variant of the MeCpPtMe3/O2 ALD process described in our previous work.27 The reader is referred to the latter for a comprehensive characterization of the composites obtainable in the cycle and temperature range used here. In brief, such ALD process, by relying on high oxygen partial pressures (i.e., 0.2 bar) and oxygen exposures on the order of minutes, enables the deposition of metallic Pt NPs at temperatures (e.g., 100 °C) at which conventional ALD would otherwise lead to negligible deposition without resorting to powerful oxidizers such as ozone and oxygen plasma.18,25,42,43 The Pt/GNP composites were obtained after 1, 3, and 10 ALD cycles carried out at both 100 °C and 200 °C. In addition, we also carried out 10 ALD cycles at 250 °C to establish a worse case reference for the initial particle size distribution (PSD) span.The same number of cycles resulted in Pt/GNP composites characterized by the same Pt loading and yet different PSDs. The Pt loadings estimated via INAA were 0.5 wt% (∼4 ng cm−2), 1.5 wt% (∼12 ng cm−2), and 6 wt% (∼48 ng cm−2) after 1, 3, and 10 cycles, respectively. As the evolution of the PSD with the number of cycles was already discussed in detail in our previous work,27 here we now elaborate on the evolution with the deposition temperature of the mass-based PSD of the Pt/GNP obtained after 10 cycles (see Fig. 1), that is, the composites with the highest loading considered here. As we shall point out later, the mass-based PSDs are crucial to the interpretation of the results of the catalytic test. Instead, the PSDs of the Pt/GNP composites obtained after 1 and 3 cycles will be discussed in a later section dedicated to the sintering of the Pt NPs during the catalytic test.
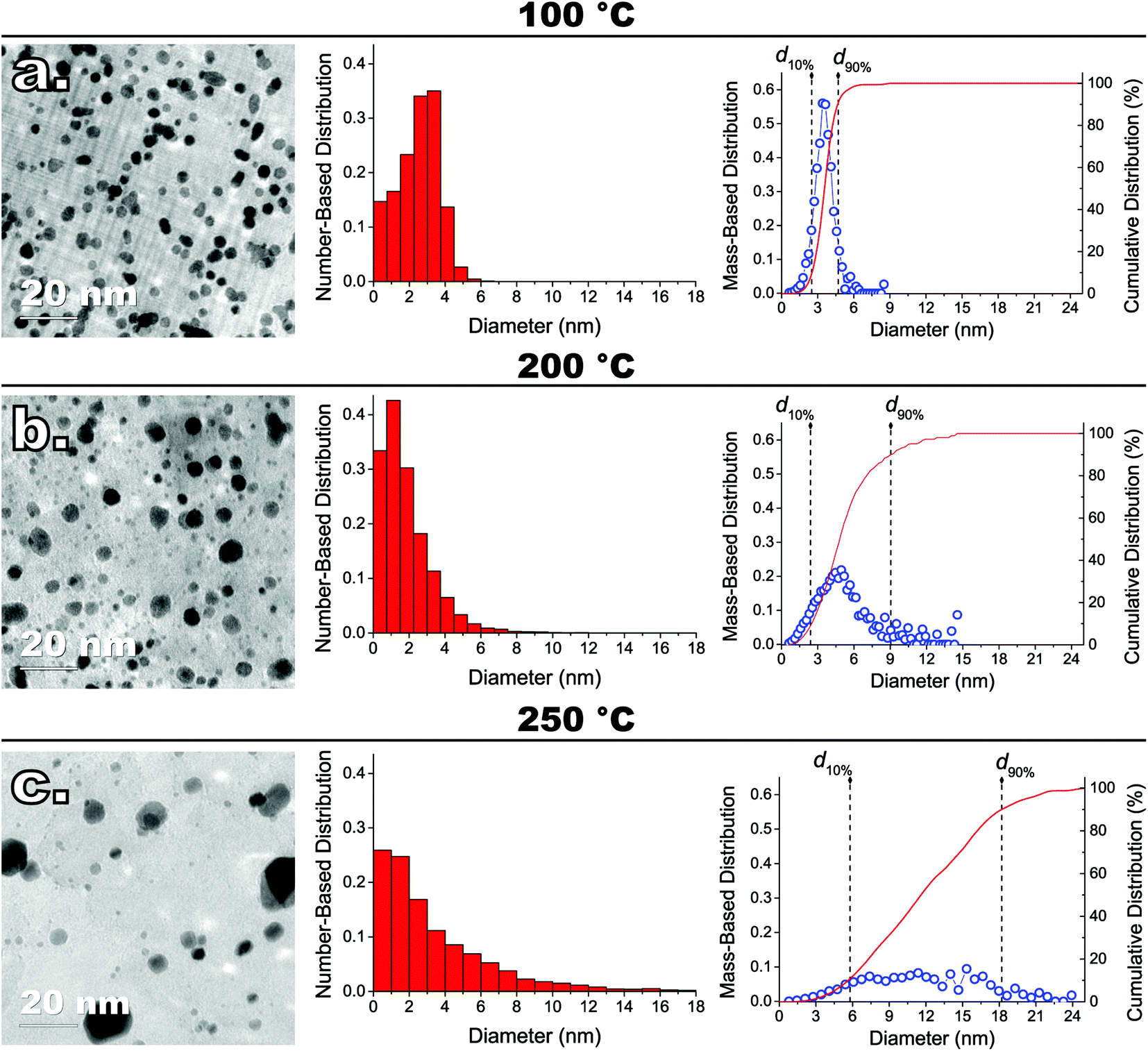 | ||
| Fig. 1 Representative TEM images of ALD-grown Pt nanoparticles (NPs) on graphene nanoplatelets (GNP) after 10 cycles at (a) 100 °C, (b) 200 °C, (c) 250 °C, and respective (middle) number-based and (right-hand-side) mass-based size distributions, that is, F1(d) and F3(d), respectively. F1(d) was plotted in the form of histogram by fixing the bin size to about 1 nm in all cases, whereas F3(d) was plotted in terms of probability density function by calculating the number of bins/points with the Freedman–Diaconis’ rule. The platinum loading of the Pt/GNP composites of (a), (b), and (c) was about 6 wt% (∼48 ng cm−2) in all cases. | ||
10 cycles of conventional thermal Pt ALD, that is, ALD carried out at 250 °C,18,21,27,42 gave rise to a very broad right-skewed number-based PSD characterized by a numerous population of small NPs of about 1 nm coexisting next to NPs as large as 24 nm. The analysis of the mass-based PSD shows that, at such conditions, 80% of the platinum mass lies in NPs whose size range spans over more than 12 nm (see Fig. 1c). Carrying out the same number of cycles below the 250 °C threshold resulted in a dramatic reduction in the metal aggregation. Decreasing the ALD temperature by 50 °C, in fact, brought about a two-fold reduction in the span of the mass-based PSD, which went from about 12 nm to 6 nm (see Fig. 1b). Further reducing the ALD temperature to 100 °C led to (I) a six-fold decrease in the span of the mass-based PSD, which shrank to about 2 nm, (II) virtually no NPs larger than 6 nm, and (III) a nearly symmetric number- and mass-based PSD (see Fig. 1a).
We put to test the performance and the stability of the Pt/GNP composites by using propene oxidation as a benchmark reaction. The influence of the ALD temperature on the catalytic performance was investigated by testing the Pt/GNP composites obtained at 100 °C and at 200 °C. The catalytic test consisted of three consecutive steps: (I) two heating/cooling cycles where the temperature of the catalytic bed was increased up to 450 °C and then decreased to room temperature; (II) heating up of the catalytic bed to the temperature at which 100% propene conversion is achieved, which is then maintained for approximately 10 hours, after which the bed is cooled down to room temperature, (III) heating up of the catalytic bed to a temperature at which an intermediate conversion is obtained, which is then maintained for approximately another 10 hours, after which the bed cooled down to room temperature for the last time. The conversion data from the cooling down phase of the second cycle of each step (I) was used to obtain the apparent reaction rate kapp, that is, the moles of propene converted per moles of Pt per unit time.
The deposition temperature had little to no effect on the catalytic performance of the composites obtained after 1 and 3 cycles (see Fig. S3†). Furthermore, higher number of cycles, and thus loading, translated into higher conversions at a given temperature, which is in agreement with other reports on the relationship between platinum loading and propene conversion.28–32 On the other hand, despite the same Pt loading, the Pt/GNP composite obtained at low temperature (Pt/GNP/10L) after 10 cycles outperformed its high-temperature counterpart (Pt/GNP/10H). Fig. 2a shows the catalytic behavior of Pt/GNP/10L and Pt/GNP/10H against temperature cycling in terms of conversion of propene. During the whole duration of the two temperature cycling, which, given the 2 °C min−1 ramp, amounted to about 14 hours in total, the Pt/GNP/10L composite delivered higher conversions at lower temperatures than the Pt/GNP/10H (ΔT50% ≃ 20 °C), even after exposure to the reacting environment at temperatures as high as 450 °C. To further test the resilience of the gap in performance, in addition to the two temperature cycling, we exposed the composites to the reacting environment for two consecutive periods of 11 hours each at temperatures in the range of 190–220 °C (see Fig. S4†). As shown in Fig. 2b, the Pt/GNP/10L composite retains its high activity compared to Pt/GNP/10H, with virtually no deactivation, even when exposed to 200 °C for more than 11 hours.
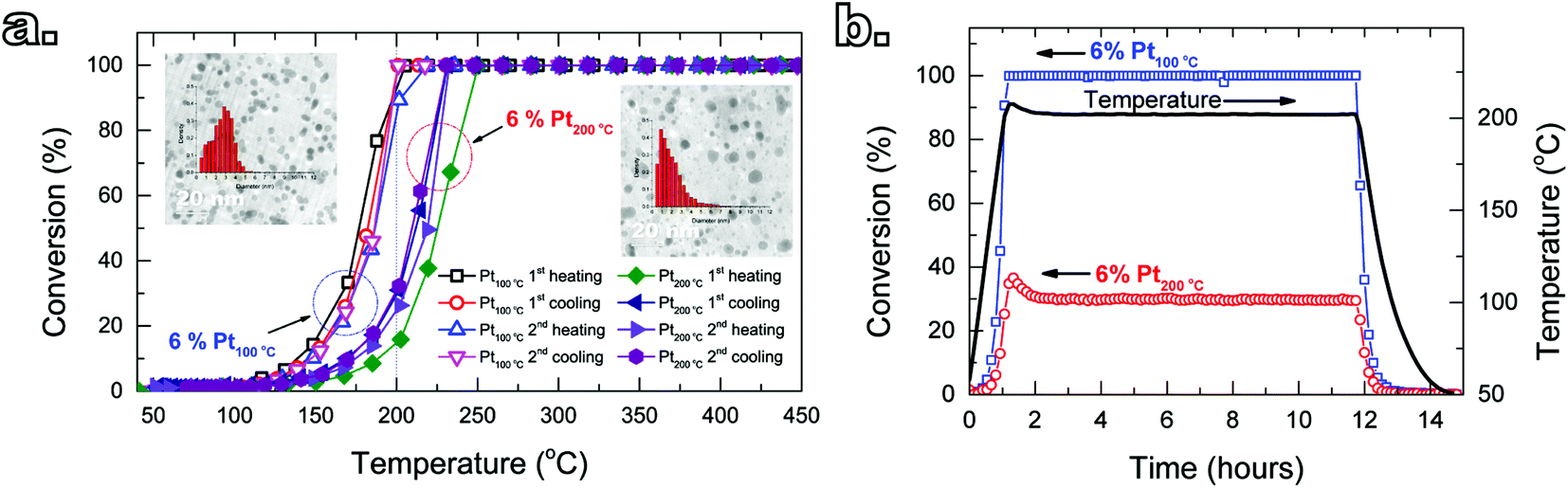 | ||
| Fig. 2 Catalytic performance of Pt/GNP obtained after 10 cycles at 100 °C and 200 °C towards propene oxidation. (a) Propene conversion as a function of temperature during two consecutive heating and cooling cycles from room temperature to 450 °C (ramp: 2 °C min−1, total time 14 hours). (b) Conversion vs. time at a reaction temperature of 200 °C, which was maintained constant for about 11 hours. | ||
To obtain further insights into the reasons behind the gap in activity between Pt/GNP/10L and Pt/GNP/10H, we constructed the Arrhenius plot shown in Fig. 3 by using the experimental values of the conversion in the range of 2.5–35% obtained from the cooling part of the second temperature cycle of the catalytic test of Fig. 2a. By using linear regression we obtained an apparent activation energy of about 68–70 kJ mol−1 (16–17 kcal mol−1) for all the composites. This value is within the range of activation energies reported in the literature for propene oxidation over Pt-based catalysts.31 While the activation energy is approximately the same for all composites, Pt/GNP/10L is still 2.9–2.6 times more active than its high-temperature counterpart in the temperature range of 125–220 °C, that is, in the conversion range of 0–100%. Interestingly, the reaction rate per unit mass of platinum at a given temperature was approximately the same for the composites obtained after 1 and 3 cycles and Pt/GNP/10L. Furthermore, the fact that the activation energy is virtually the same for all composites suggests that the gap in performance between Pt/GNP/10L and Pt/GNP/10H arises from a different number of active sites rather than from active sites of different nature.
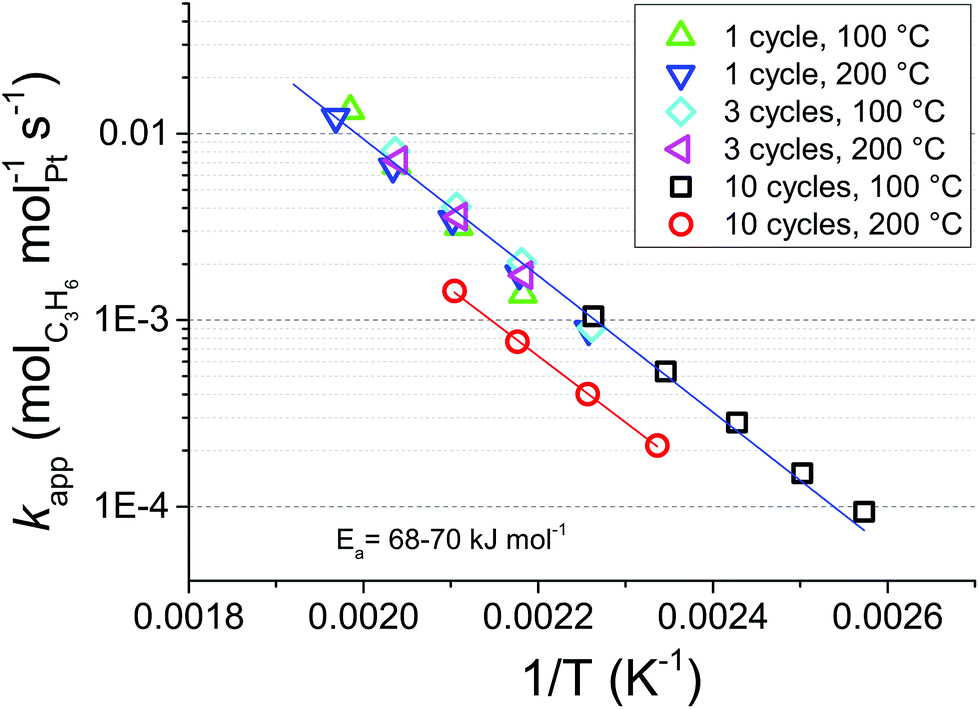 | ||
| Fig. 3 Arrhenius plot for kapp of propene oxidation over the Pt/GNP composites obtained after 1, 3, and 10 cycles at both 100 °C and 200 °C. | ||
To understand whether the results of the catalytic test can be interpreted in terms of differences in the PSDs, we also analyzed the composites after the catalytic test since the PSDs obtained after synthesis and prior to the catalytic test are not representative of the state of the catalyst during reaction, as this can induce NP sintering.29 The Pt NPs in both composites, in fact, did sinter during the catalytic test. Fig. 4 shows a box-and-whisker plot and data overlap representation of the number-based PSDs of the Pt NPs obtained after 3 and 10 cycles at both 100 °C and 200 °C before and after the catalytic test (see also Fig. 5a–b and d–e for representative TEM micrographs of the composites after the catalytic test). While both the low-temperature and the high-temperature composites did sinter, the low-temperature composites retained a narrow PSD compared to their high-temperature counterparts. In particular, the catalytic test resulted in virtually no change in the average NP size and the span of the number-based PSD of the low-temperature composite obtained after 3 cycles, whereas its high-temperature counterpart saw a twofold increase in the PSD span and in the average size as well as the appearance of a long tail on the large-size side. Analogously, while the Pt/GNP/10L composite saw a slight increase in the PSD span and in the average size, the Pt/GNP/10H composite saw a three-fold increase in the PSD span and in the average size, and the appearance of a long tail on the large-size side.
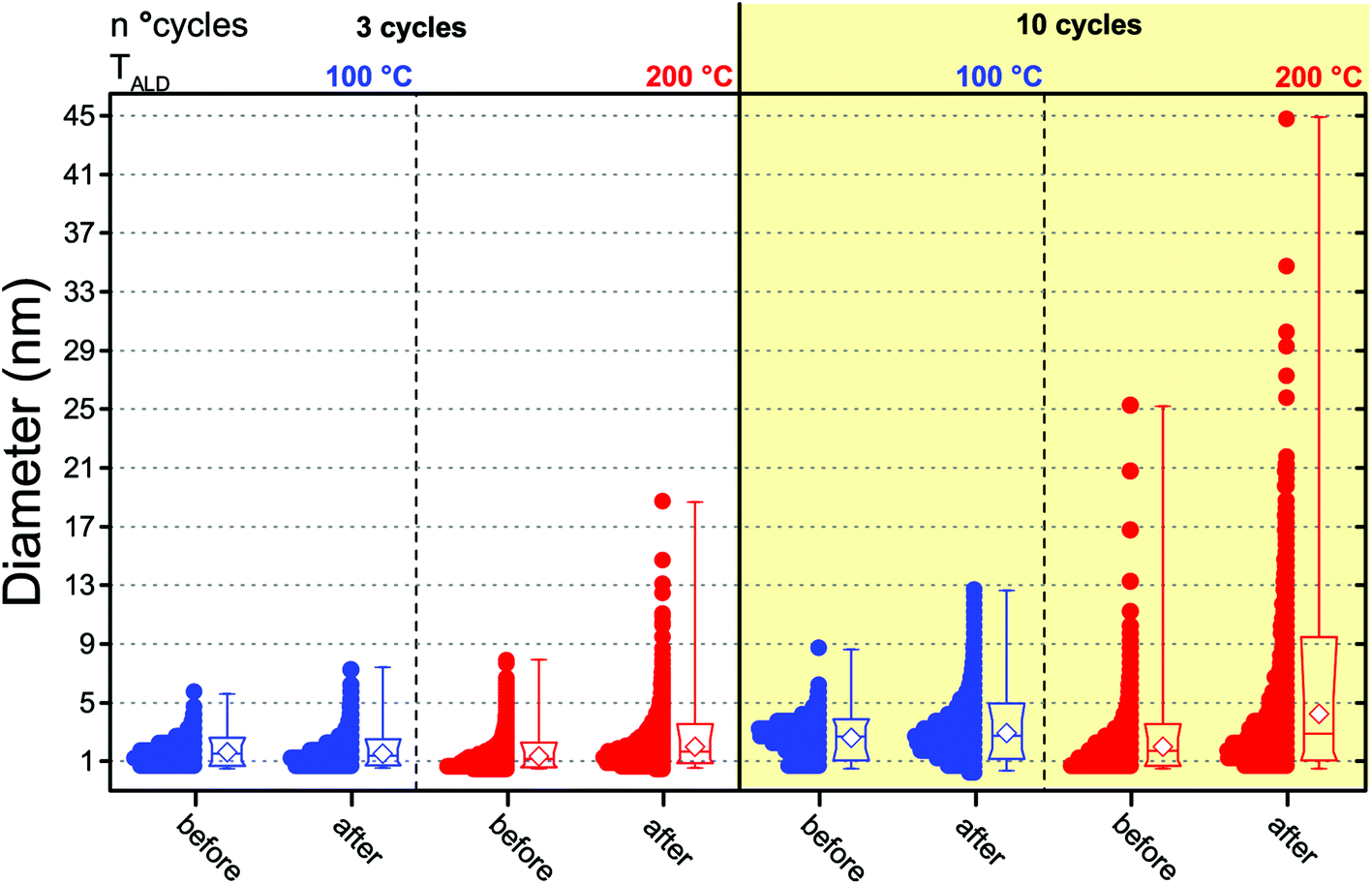 | ||
| Fig. 4 Box-and-whisker plots and data overlap of the number-based size distributions of the Pt nanoparticles obtained after 3 and 10 cycles at both 100 °C and 200 °C before and after the catalytic test. The boxes indicate the 10th, 25th, 75th, and 90th percentiles of the population, the whiskers indicate the minimum and the maximum size, and the white diamonds indicate the average size. | ||
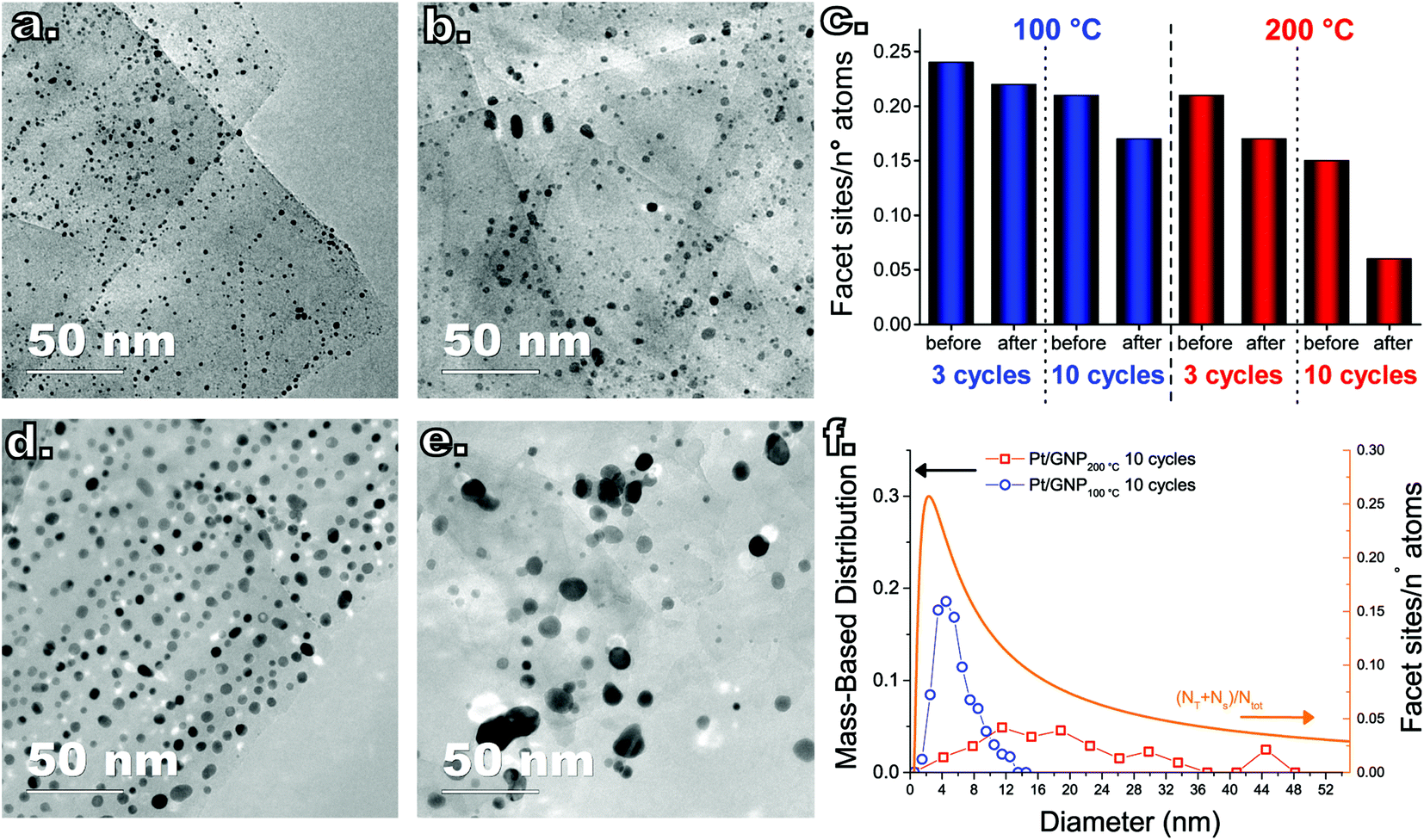 | ||
| Fig. 5 TEM micrographs taken after the catalytic test of the Pt/GNP composites obtained after 3 cycles at (a) 100 °C and (b) 200 °C, and after 10 cycles at (d) 100 °C and (e) 200 °C. (c) Number of facet sites, that is, surface sites minus edge and corner sites, divided by the total number of platinum atoms estimated by using the size distributions and assuming the NPs to assume a cubo-octahedral geometry. (f) Mass-based size distributions of the Pt NPs after the catalytic test of the Pt/GNP composites obtained after 10 cycles plotted together with the fraction of facet sites a cubo-octahedral platinum nanoparticle. | ||
Despite the sintering, the Pt/GNP/10L (Fig. 5d) still retained a homogeneous spatial and size distribution. On the other hand, the NPs in the Pt/GNP/10H composite (Fig. 5e) sintered dramatically, giving rise to a considerable reduction in NP density (∼94%), loss of spatial homogeneity, the formation of aggregates of NPs as well as of individual NPs as large as 45 nm. While the span of the mass-based PSD of the NPs in the Pt/GNP/10L composite increased to about 6 nm from an initial value of 2 nm, the Pt/GNP/200 composite saw a more than five-fold increase in the same span, which went from 6 nm to more than 32 nm (see Fig. 5f). It is worth noting that the span of the mass-based PSD of the Pt/GNP/10L composite after the catalytic test, and thus exposure to temperatures as high as 450 °C for prolonged periods of time, is still two times smaller than the span of the PSD of the as-synthesized NPs deposited at 250 °C (see Fig. 5c and Fig. 1c). Finally, the fact that such a drastic difference between the PSDs before and after the catalytic test was not reflected in an appreciable loss of activity during the long phases of the catalytic test (see Fig. S4† and Fig. 2) suggests that NP sintering took place only in the very beginning of the test.
To gain further insights into the different sintering and catalytic behaviours of the Pt/GNP composites we devised a series of computer simulations of the sintering process and a geometrical model for the catalytic activity of the NPs. To model the evolution of the PSDs due to sintering, we used the rate-equation approach already described in our previous work.27 This model was used here to model the evolution of the PSDs of the as-synthesized Pt/GNP composites due to both gas-phase-mediated Ostwald ripening and NP diffusion and coalescence. The purpose of the simulations was to give a qualitative description of the effect of different initial PSDs at a given metal loading on the sintering process and not to reproduce the exact PSDs after the catalytic test.
The resistance to sintering of the low-temperature composites and the different effect of the temperature during the synthesis and the catalytic test can arise from a number of reasons. The tendency of an ensemble of NPs to sinter depends, in fact, not only on the temperature but also on the shape of the initial PSD, the reaction environment, and the presence of carbonaceous species adsorbed on the catalyst surface.1,2,27,44–46 The initially narrow PSD of the Pt NPs synthesized at low temperature can effectively suppress sintering due to Ostwald ripening, given that the driving force of such mechanism lies in the PSD span.44 Furthermore, the PSD of the Pt/GNP/10L NPs was not only narrow but also symmetric, whereas the Pt/GNP/10H composite had a large number of small NPs coexisting next to large ones. Sintering simulations using the experimental PSDs of both the Pt/GNP/10L and the Pt/GNP/10H composites as the initial condition clearly show that the latter sinters much faster than the former (see Fig. S10a†) both in conditions that promote Ostwald ripening and in conditions in which the ensemble sinters via simultaneous Ostwald ripening and NP diffusion and coalescence. However, while we could achieve a qualitative description of the different stability of the low- and high-temperature composites, given a set of simulation parameters, we could not reproduce the exact discrepancy between the composites. The presence of large particles (d > 5 nm) in the high-temperature composites before the catalytic test might explain this discrepancy if such NPs become more mobile than smaller ones upon exposure to the reacting mixture. Yet, at this moment, we do not have enough evidence to substantiate such mechanism. Finally, we argue that the effect of temperature during synthesis is more severe than it is during the catalytic test since the former relies on the cyclic combustion of the organic ligands remaining after the precursor chemisorption, and this process promotes sintering via NP diffusion and coalescence.27
The geometrical model for describing the catalytic activity of an ensemble of Pt NPs builds on both the already available body of literature on the structure sensitivity of Pt-catalyzed propene oxidation and the evidence presented here. It has already been shown that the reaction rate per unit of mass of platinum is approximately the same across catalysts characterized by relatively low loadings (∼1 wt%) and different NP sizes,31 which is in agreement with our results for Pt/GNP/10L and the Pt/GNP composites obtained after 1 and 3 cycles. Since larger NPs have lower dispersions, that is, surface sites vs. total number of atoms, the reaction rates per unit area of platinum, often referred to as turnover frequencies (TOF), are higher for larger NPs. As a result, there is a general agreement on the size dependence of the catalytic activity of Pt NPs towards the combustion of hydrocarbons, and in particular of propene: the surface of large NPs is more active than the one of small NPs.28–32,47 Such size dependence has been attributed to the fact that large NPs exhibit weaker Pt–O bonding, given that loosely bound oxygen is more active towards the oxidation of hydrocarbons.28,30,31,47 However, while this simple picture is in agreement with most of our results, it still cannot explain the gap in performance between Pt/GNP/10L and Pt/GNP/10H. Hence, we propose a simple geometrical model that can account for all the observed results. We assume that oxygen is strongly bound to the atoms sitting at the corners and the edges of the exposed facets of an NP, which are thus not active towards propene oxidation.9,31 Instead, the number of moles of propene converted per unit time is assumed to be directly proportional to the number of facet sites and thus:
| kapp ∝ NF/Ntot = DF | (5) |
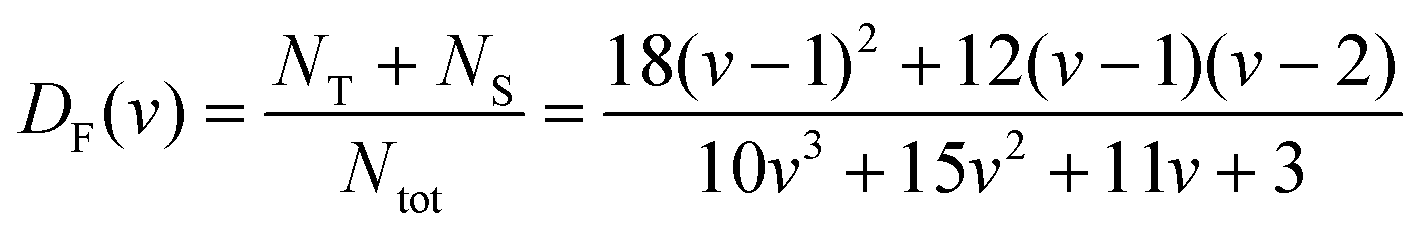 | (6) |
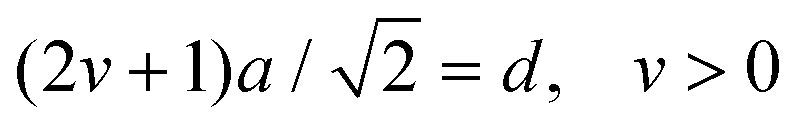 | (7) |
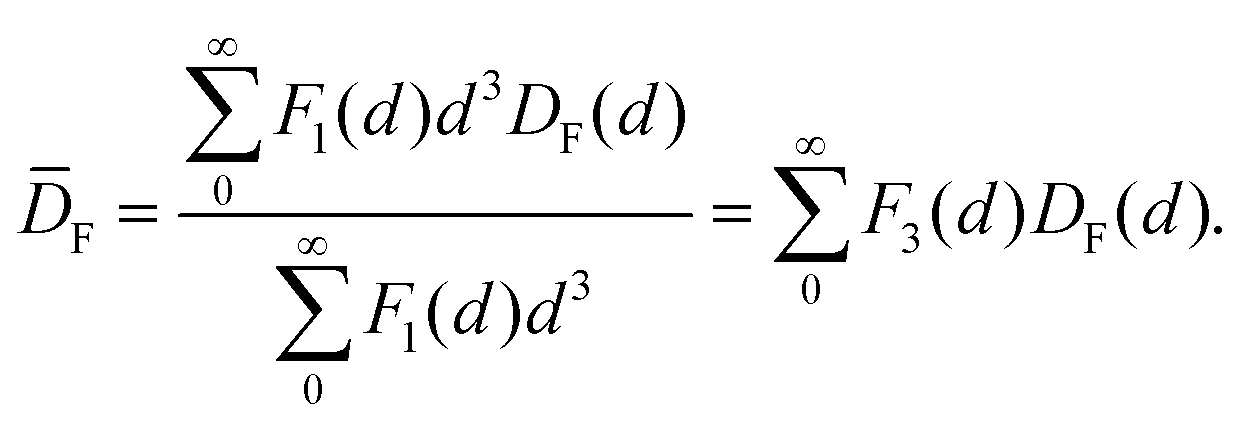 | (8) |
By using eqn (8) and the experimental mass-based PSDs before and after the catalytic test we could estimate the ratio between the number of facet sites and the total number of platinum atoms in the Pt/GNP composites (see Fig. 5c). Moreover, eqn (8) was also used to calculate the fraction of facet sites of the simulated PSDs (see Fig. S9 and S10†). Indeed, the Pt/GNP composites obtained after 3 cycles and the Pt/GNP/10L composite have about the same fraction of facet sites, which is in agreement with their having virtually the same reaction rate per unit of mass of platinum. Crucially, the Pt/GNP/10H composite has about 63% less facet sites than the other composites, which is in agreement with the Pt/GNP/10L being about 2.9–2.6 times more active than Pt/GNP/10H (see Fig. 3, 5c and f). Finally, sintering simulations of the Pt NPs obtained for a low number of cycles and thus low loadings, show that the fraction of facet sites remains fairly constant even after the NPs ensemble has lost more than 90% of the initial number of NPs and the dispersion has dropped by more than 2–3 times and the surface-averaged diameter, that is the equivalent diameter obtained from CO and H2 chemisorption, has increased by the same figure (see Fig. S9†). This is again in agreement with the fact that catalysts with low loadings and different NP size exhibit approximately the same reaction rate per unit mass of platinum. The reason why the fraction of facet sites of an ensemble of NPs remains fairly constant at low coverages, regardless of the average size, is that the fraction of facet sites is a nonlinear function of the Pt NP size (see Fig. 5f) that approaches zero for small NPs and has a maximum at about 2.5 nm after which it decreases and then steadily levels off at large sizes. Clearly, there exists an infinite number of mass-based PSDs that, once multiplied by DF(d) and integrated (see eqn (8)), lead to the same fraction of facet sites and thus the same reaction rate per unit mass of platinum. However, at relatively high loadings (large average sizes), broad right-skewed mass-based PSDs, such as the one of the Pt/GNP/10H composite after the catalytic test, fall under the descending side of DF(d) and thus lead to small facet sites fractions.
4 Conclusions
In conclusion, we have demonstrated that, compared to conventional Pt ALD, low-temperature ALD enables the synthesis of supported Pt nanoparticles that have (I) narrow size distributions, (II) higher activity at high metal loadings, and (III) higher stability at a given Pt loading. Crucially, the gap in performance between the low-temperature composite obtained after 10 cycles and its high-temperature counterpart towards propene oxidation was retained even after prolonged period of times (>24 hours) at temperatures as high as 450 °C. Furthermore, the Pt NPs synthesized at 100 °C temperature, once exposed to such harsh conditions, retained a size distribution that is narrower than the one of the Pt NPs synthesized at 250 °C. Hence, the temperature history is more important than the absolute temperature to which the Pt NPs are exposed. The difference in catalytic activity was attributed to different fractions of surface active sites, as corroborated by a simple geometrical model based on the analysis of the size distributions after the catalytic test. Our analysis emphasizes that when the reaction takes place mostly on the facets of the NPs, dispersion and average size are poor descriptors of the catalytic performance. Instead, one should take into account the whole size distribution to make meaningful comparisons. The ability to tailor the size distribution of NPs at a given metal loading coupled with the analysis of the performance presented here exemplifies how ALD can be used as a tool for elucidating the relationship between size and functionality in NP-based materials. Such advantages, demonstrated here by using graphene nanoplatelets, can find application in the study and the optimization of the activity, selectivity, and stability of carbon-supported Pt NPs for fuel cells electrodes. Finally, we anticipate that the use of high oxygen partial pressure can push the lower temperature limit of thermal ALD of noble metals on other carbon supports way below 100 °C, that is, the lowest ALD temperature explored here. However, whether the advantages of our approach can be extended to oxide supports merits further study.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant, Agreement No. 279632.References
- P. Wynblatt and N. Gjostein, Prog. Solid State Chem., 1975, 9, 21–58 CrossRef CAS.
- P. J. F. Harris, Int. Mater. Rev., 1995, 40, 97–115 CrossRef CAS.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed.
- S. Guo and E. Wang, Nano Today, 2011, 6, 240–264 CrossRef CAS.
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS.
- H. Gandhi, G. Graham and R. McCabe, J. Catal., 2003, 216, 433–442 CrossRef CAS.
- L. Li, A. H. Larsen, N. A. Romero, V. A. Morozov, C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 222–226 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS PubMed.
- B. R. Cuenya and F. Behafarid, Surf. Sci. Rep., 2015, 70, 135–187 CrossRef.
- N. Satoh, T. Nakashima, K. Kamikura and K. Yamamoto, Nat. Nanotechnol., 2008, 3, 106–111 CrossRef CAS PubMed.
- H. Ye and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 4930–4934 CrossRef CAS PubMed.
- F. N. Gür, F. W. Schwarz, J. Ye, S. Diez and T. L. Schmidt, ACS Nano, 2016, 10, 5374–5382 CrossRef PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- C. Detavernier, J. Dendooven, S. Pulinthanathu Sree, K. F. Ludwig and J. A. Martens, Chem. Soc. Rev., 2011, 40, 5242–5253 RSC.
- D. Longrie, D. Deduytsche and C. Detavernier, J. Vac. Sci. Technol., A, 2014, 32, 010802 Search PubMed.
- H. Van Bui, F. Grillo and J. R. van Ommen, Chem. Commun., 2017, 53, 45–71 RSC.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef.
- A. Goulas and J. R. van Ommen, J. Mater. Chem. A, 2013, 1, 4647 CAS.
- S. M. George, Chem. Soc. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- V. Miikkulainen, M. Leskelä, M. Ritala and R. L. Puurunen, J. Appl. Phys., 2013, 113, 021301 CrossRef.
- F. Grillo, M. T. Kreutzer and J. R. van Ommen, Chem. Eng. J., 2015, 268, 384–398 CrossRef CAS.
- D. M. King, J. A. Spencer II, X. Liang, L. F. Hakim and A. W. Weimer, Surf. Coat. Technol., 2007, 201, 9163–9171 CrossRef CAS.
- A. J. M. Mackus, M. J. Weber, N. F. W. Thissen, D. Garcia-Alonso, R. H. J. Vervuurt, S. Assali, A. A. Bol, M. A. Verheijen and W. M. M. Kessels, Nanotechnology, 2015, 27, 034001 CrossRef PubMed.
- H. V. Bui, F. Grillo, R. Helmer, A. Goulas and J. R. van Ommen, J. Phys. Chem. C, 2016, 120, 8832–8840 Search PubMed.
- F. Grillo, H. Van Bui, J. A. Moulijn, M. T. Kreutzer and J. R. van Ommen, J. Phys. Chem. Lett., 2017, 8, 975–983 CrossRef CAS PubMed.
- S. Benard, M. Ousmane, L. Retailleau, A. Boreave, P. Vernoux and A.-F. Giroir-Fendler, Can. J. Civ. Eng., 2009, 36, 1935–1945 CrossRef CAS.
- P. Denton, A. Giroir-Fendler, H. Praliaud and M. Primet, J. Catal., 2000, 189, 410–420 CrossRef CAS.
- L. Liotta, Appl. Catal., B, 2010, 100, 403–412 CrossRef CAS.
- L. M. Carballo and E. E. Wolf, J. Catal., 1978, 53, 366–373 CrossRef CAS.
- P. Marécot, A. Fakche, B. Kellali, G. Mabilon, P. Prigent and J. Barbier, Appl. Catal., B, 1994, 3, 283–294 CrossRef.
- N. Zhang, Y. Zhang and Y.-J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54–75 CAS.
- H. D. Ozaydin, H. Sahin, R. T. Senger and F. M. Peeters, Ann. Phys., 2014, 526, 423–429 CrossRef CAS.
- C. Feng, J. Wang, Y. Cheng, P. He and K. M. Liew, RSC Adv., 2014, 4, 60711–60719 RSC.
- E. J. Lamas and P. B. Balbuena, J. Phys. Chem. B, 2003, 107, 11682–11689 CrossRef CAS.
- J. Chen and K.-Y. Chan, Mol. Simul., 2005, 31, 527–533 CrossRef CAS.
- S. Sartipi, H. Jansma, D. Bosma, B. Boshuizen, M. Makkee, J. Gascon and F. Kapteijn, Rev. Sci. Instrum., 2013, 84, 124101 CrossRef PubMed.
- A. J. M. Mackus, M. A. Verheijen, N. Leick, A. A. Bol and W. M. M. Kessels, Chem. Mater., 2013, 25, 1905–1911 CrossRef CAS.
- T. Aaltonen, M. Ritala, T. Sajavaara, J. Keinonen and M. Leskelä, Chem. Mater., 2003, 15, 1924–1928 CrossRef CAS.
- K. Wettergren, F. F. Schweinberger, D. Deiana, C. J. Ridge, A. S. Crampton, M. D. Rtözer, T. W. Hansen, V. P. Zhdanov, U. Heiz and C. Langhammer, Nano Lett., 2014, 14, 5803–5809 CrossRef CAS PubMed.
- Y. Chu and E. Ruckenstein, Surf. Sci., 1977, 67, 517–540 CrossRef CAS.
- K.-J. Hu, S. R. Plant, P. R. Ellis, C. M. Brown, P. T. Bishop and R. E. Palmer, J. Am. Chem. Soc., 2015, 137, 15161–15168 CrossRef CAS PubMed.
- N. Radic, B. Grbic and A. Terlecki-Baricevic, Appl. Catal., B, 2004, 50, 153–159 CrossRef CAS.
- A. Le Valant, C. Comminges, F. Can, K. Thomas, M. Houalla and F. Epron, J. Phys. Chem. C, 2016, 120, 26374–26385 CAS.
- J. Montejano-Carrizales, F. Aguilera-Granja and J. Morán-López, Nanostruct. Mater., 1997, 8, 269–287 CrossRef CAS.
- C. P. Poole, F. J. Jones and F. J. Owens, Introduction to Nanotechnology, John Wiley & Sons, Inc., New York, NY, USA, 2003 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Platinum loading vs. temperature; TGA curves; extended catalytic test results; supplementary TEM micrographs; HRTEM micrographs and lattice constant estimation; surface-based size distributions after catalytic test; sintering simulation results; evaluation of mass transfer limitations. See DOI: 10.1039/C7NR02984E |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |