
Pyrrolizidine alkaloids: occurrence, biology, and chemical synthesis
Jeremy
Robertson
 * and
Kiri
Stevens
* and
Kiri
Stevens
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: jeremy.robertson@chem.ox.ac.uk; Tel: +44 (0)1865275660
First published on 26th October 2016
Abstract
Covering: 2013 up to the end of 2015
This review covers the isolation and structure of new pyrrolizidines; pyrrolizidine biosynthesis; biological activity, including the occurrence of pyrrolizidines as toxic components or contaminants in foods and beverages; and formal and total syntheses of naturally-occurring pyrrolizidine alkaloids and closely related non-natural analogues.
 Jeremy Robertson | Jeremy Robertson worked in Oxford with Professor Sir Jack Baldwin, FRS on free radical ring-expansion reactions for his D.Phil. then moved to Columbia University, New York, to work with Professor Gilbert Stork on the development of new synthetic routes to taxol. He returned to Oxford in 1992 to take up his current academic position, where he is now Professsor of Chemistry. His research interests span mechanistic organic chemistry, synthetic methodology, natural product synthesis, and biology-driven collaborative projects. |
 Kiri Stevens | Kiri Stevens received M.Chem. and D.Phil. degrees from the University of Oxford, conducting research into the synthesis of complex natural products under the supervision of Dr Jeremy Robertson. Following postdoctoral positions at the Institute of Cancer Research and University College London, she now works as a research scientist at Vertex Pharmaceuticals, UK. |
1 Introduction
This review concerns chemical, biological, and environmental aspects of the class of pyrrolizidine alkaloids (PAs); natural products and their close structural analogues (mainly stereoisomers) that contain the pyrrolizidine motif, as shown below with the conventional atom numbering indicated.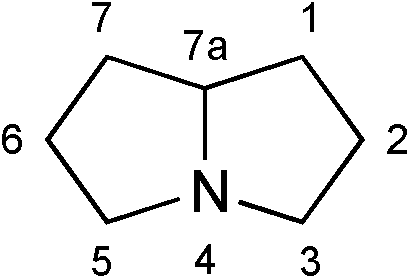
The coverage picks up from where our previous review1 left off and encompasses the literature published up to the end of December 2015.
During this three-year period our knowledge of the broader chemistry and biology of PAs and their place in the environment has advanced substantially on several fronts. For example, although the field of PA research originated when the symptoms of poisoning were associated with the ingestion by humans and domestic animals of certain plants, the details of how toxicity arises remains an area of active investigation. Recently, the molecular mechanisms involved in PA-induced toxicity have become more refined through both computation, to understand the origin of toxic dehydropyrrolizidines, and by experiment, to determine the fate in vivo of reactive iminium ions derived from them. A second major recent advance is the elucidation of the biosynthetic pathways leading to bacterial pyrrolizidines of the vinylogous urethane type. This work has led to the identification of new members of this class and a prediction based on genetic relationships that many more PAs will be discovered by further examining the metabolite profiles of diverse bacterial species. These aspects are outlined in the first part of the review which also includes a summary of the large research effort to confirm the presence of and quantify toxic PAs in foods, beverages, and medicinal formulations, which has obvious implications for human health.
The second part of the review highlights the fascination that these alkaloids continue to hold for synthetic chemists who are attracted by the biological activity and, being relatively simple in structure, the opportunities for developing, testing, and showcasing new synthetic methods. The majority of these targets contain a hydroxymethyl substituent, most commonly at C(1) or C(3), but coverage extends to simple polyhydroxypyrrolizidines, aminopyrrolizidines, and more exotic structures that contain the pyrrolizidine fragment. Compounds in which the pyrrolizidine is the minor structural feature or which have been deemed by the authors to be of lesser interest are not covered.
This survey of the synthetic chemistry shows that some genuinely new strategies have emerged for application to PAs. There has been a reduced focus on enantiospecific syntheses from chiral pool materials, an increase in convergent approaches, and new methods for stereoselective C–C and C–N bond construction including the exploitation of two relatively recent additions to the synthetic chemist's toolbox: asymmetric organocatalysis and C–H activation. Coverage includes essentially all the reported total and formal syntheses but where particular methodologies or strategies were described in the previous review, discussion is kept to a minimum. As in the previous review, the syntheses are mainly described chronologically within each sub-section except where an alternative grouping provides a more natural flow of the discussion (e.g. routes based on nitrone chemistry).
2 Non-synthetic aspects
2.1 Metabolism and toxicity
Hepatotoxicity (especially veno-occlusive disease) is a potentially serious result of ingestion of PAs. Following ingestion, those PAs that bear a C(1)–C(2) double bond may become metabolised to pyrrolizine intermediates 2 (Scheme 1) which then rapidly eject a carboxylate leaving group from either the C(1)-acyloxymethyl or C(7)-acyloxy substituents giving extended iminium ions 3 or 4, respectively. Such iminium intermediates are reactive alkylating agents that are readily trapped by cellular components leading ultimately to acute liver damage and, in some animal models, genotoxicity and carcinogenesis.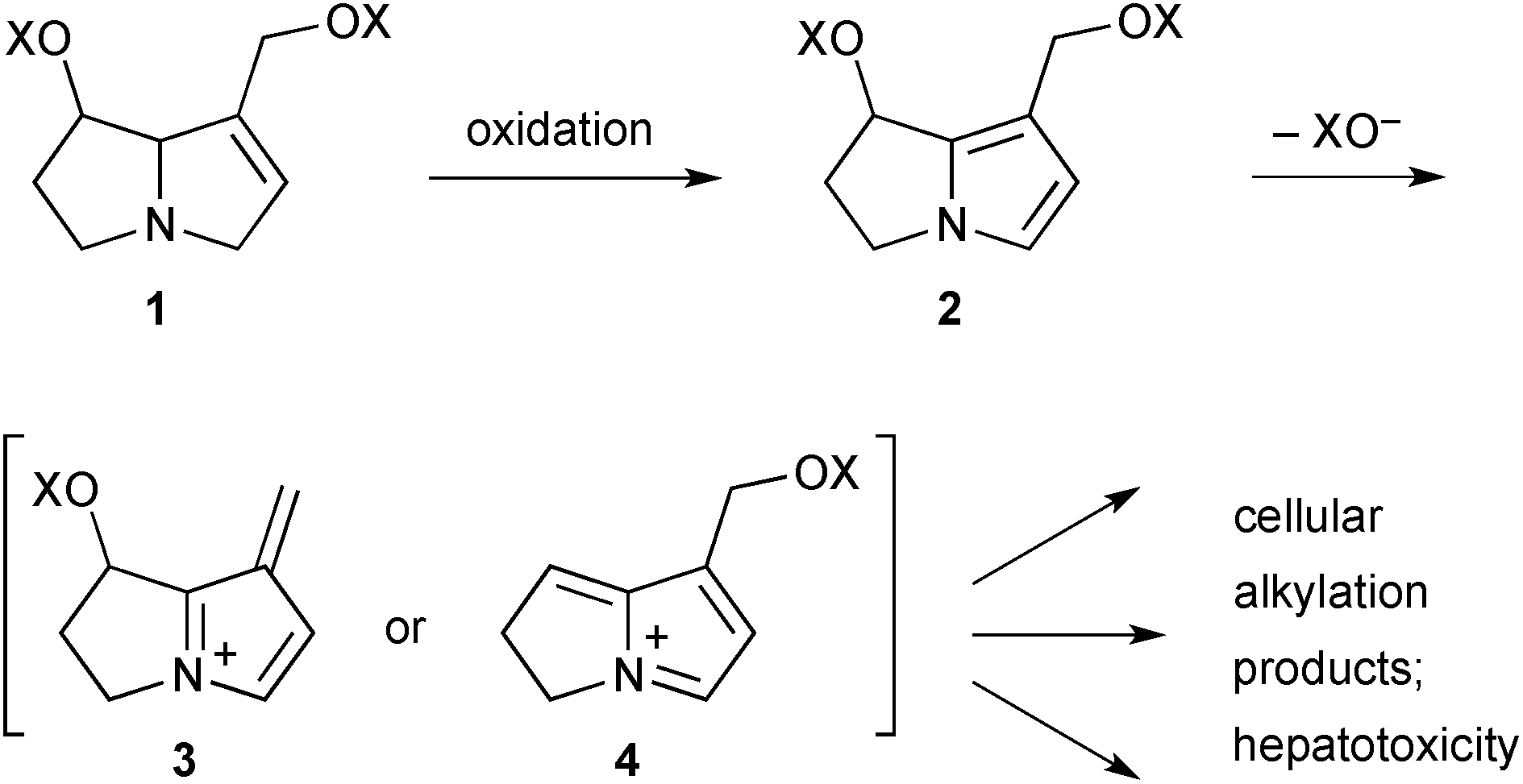 | ||
| Scheme 1 Metabolic activation of C(1)–C(2) unsaturated PAs results in toxicity. | ||
Work has continued in order to gain insight into each step of this general scheme. With reference to the first step (‘oxidation’, Scheme 1), application of the electrophilic Fukui function was combined with computed bond dissociation energies and molecular docking simulations to predict the initial site of human cytochrome P450 (CYP3A4) mediated activation of toxic pyrrolizidines of the heliotrine, retronecine, and otonecine classes.2 As might be expected, the C(3)-, C(5)- and C(7a)- [numbered C(8) in the paper] positions in the first two classes, and the C(3)-, C(5)- and N-methyl sites in the otonecines were shown to be the most susceptible by the first two measures; molecular docking results showed more variation between the three classes. On this basis, the authors presented three hydroxylation mechanisms (Scheme 2): hydrogen abstraction and rebound hydroxylation at C(3)- (→6) or C(7a)- (→7) followed by dehydration (→8); or, for the otonecines 9, demethylation by hydroxylation (→10) and loss of formaldehyde that leads, following transannular cyclisation, to the same type of C(7a)-hydroxypyrrolizidine intermediate 7 obtained from the heliotrine/retronecine classes. Representatives of the three classes – lasiocarpine, retrorsine, and senkirkine – were then incubated with either human CYP3A4 or human liver microsomes, in both cases trapping the dehydropyrrolizidines with glutathione (GSH). The rate of formation of the mono-GSH adduct in both in vitro studies was highest for lasiocarpine (krel = 19), then retrorsine (krel = 7.6) and senkirkine (krel = 1).
 | ||
| Scheme 2 Formation of dehydropyrrolizidines by cytochrome P450-mediated hydroxylation and dehydration, with oxidative demethylation in the otonecine series. | ||
A further study examined the metabolic profile of lasiocarpine when exposed to liver microsomes from human, pig, rat, mouse, rabbit, and sheep.3 The study found that the distribution of twelve metabolites was broadly similar in the non-human cases. With human liver microsomes, while the same major metabolite, M9, was produced, the product of O-demethylation, a second metabolite, M7, was formed to almost the same extent. Comparisons were carried out with recombinant human CYP3A4 to support the involvement of the CYP3A enzyme family in lasiocarpine metabolism. The results are interpreted in relation to the relative toxicity of PAs in different mammalian species.
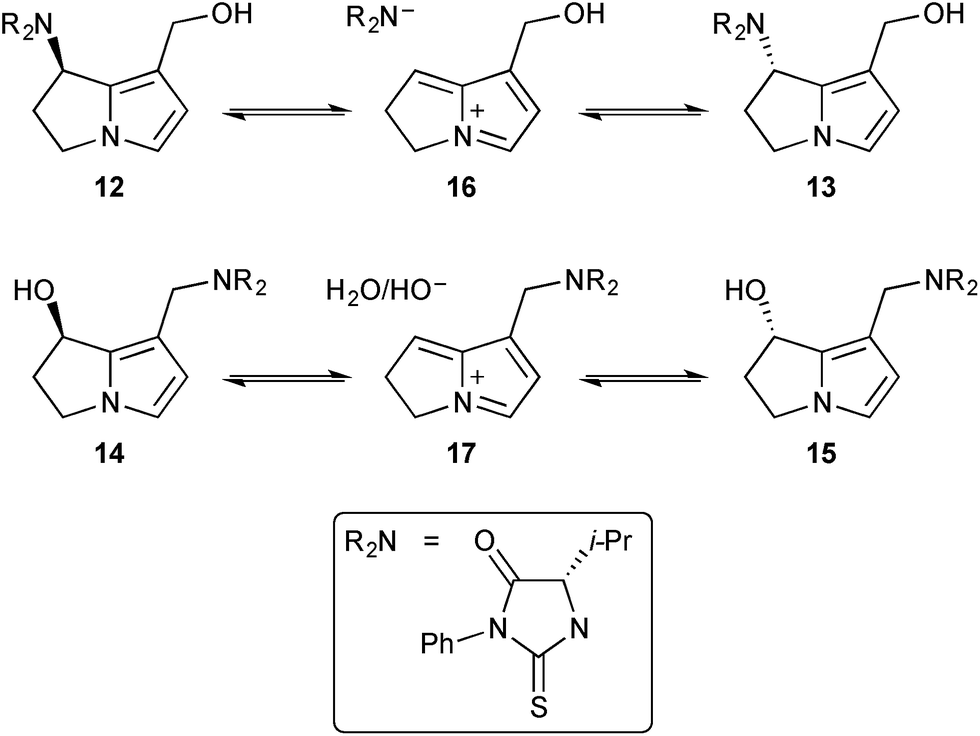
With reference to the second step (‘–XO−’) in Scheme 1, and building on the synthesis of standards DHP-dG-3, DHP-dG-4, DHP-dA-3, and DHP-dA-4,4 Fu's group showed that all four adducts were produced in the livers of rats dosed with hepatotoxic PAs (riddelliine and its N-oxide, retrorsine, monocrotaline, lasiocarpine, heliotrine, clivorine, and senkirkine).5 For non-hepatoxic alkaloids, and within the limits of detection, these adducts were either present at very low concentrations (lycopsamine) or absent (retronecine, platyphilline). The authors concluded that the four adducts act as a biomarker for PA-induced tumour formation. In related work, the group add to knowledge of how toxic PAs interact with cellular constituents following activation to the dehydro-forms.6 In this study, dehydromonocrotaline 11 was treated with valine to yield four adducts, characterised as their phenylisothiocyanate (PITC) adducts 12/13 and 14/15.
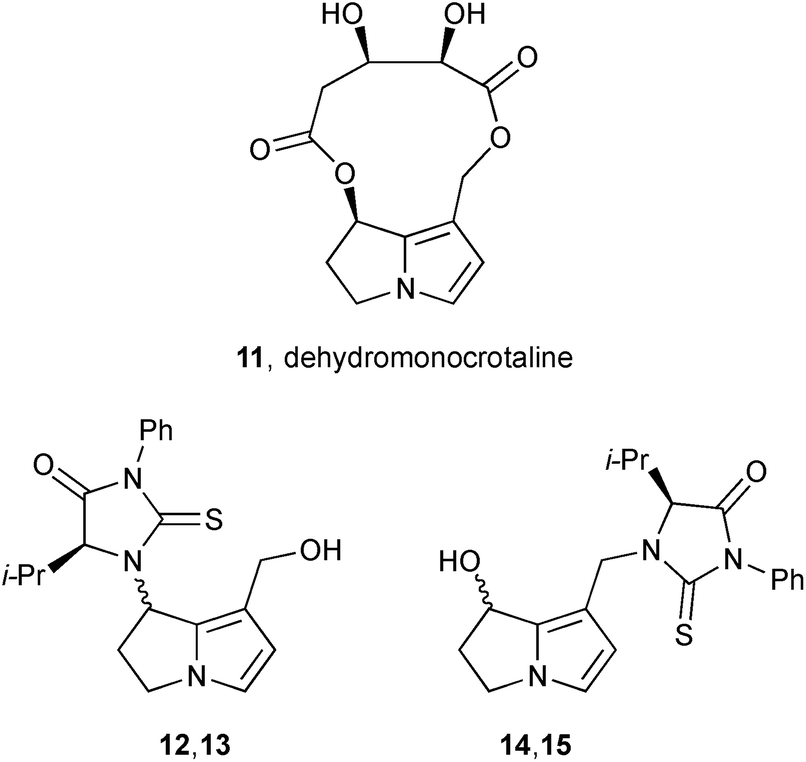
The group then prepared these adducts discretely and studied the mechanism of interconversion of epimeric pairs 12/13 and 14/15 in 18O-labelled water.7 The equilibration of 12 and 13 (Scheme 3) was accompanied by incorporation of 18OH into the C(1)-hydroxymethyl substituent whereas the equilibration of 14 and 15 (and thence the C(7)–18OH derivatives) did not, apparently, lead to hydrolysis of the C(1)–CH2NR2 group.
 | ||
| Scheme 3 SN1-type equilibration of DHP-valine-PITC adducts. | ||
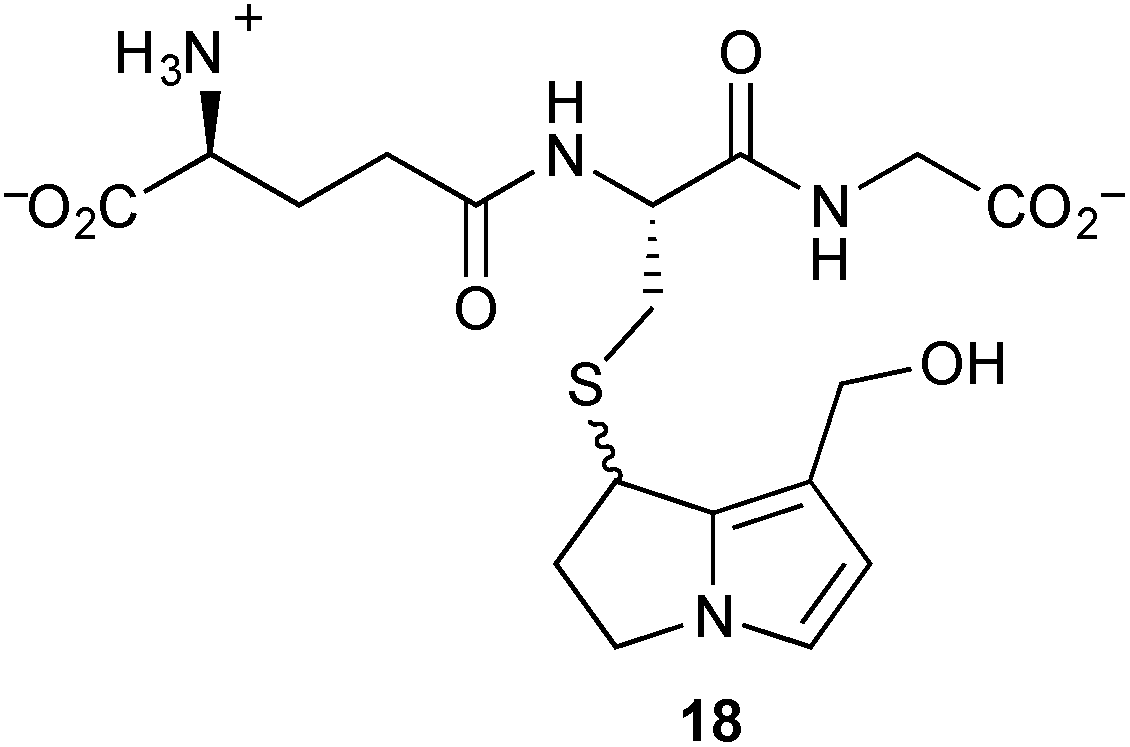
To shed further light on potential metabolic pathways of toxic pyrrolizidines, incubation of dehydromonocrotaline 11 with a sub-stoichiometric quantity of glutathione (GSH) in the presence of spleen phosphodiesterase gave the C(7)–GS adduct 18; use of a large excess of GSH gave the C(7,9)–bis-GS adduct (not shown).8 Shaking 18 with either 2′-deoxyguanosine (dG) or 2′-deoxyadenosine (dA) produced all four mono-adducts in both series, DHP-dG-1–4 or DHP-dA-1–4, over a period of hours to days. Similar reactions with the C(7,9)–bis-GS adduct resulted in, at most, traces of these mono-adducts. It is concluded that the conjugation of dehydropyrrolizidines (DHPs) with glutathione is not, as was previously considered, a detoxification pathway; indeed, the C(7)–GS adducts are proposed as a relatively stable ‘reservoir’ of the DHPs themselves.
In the same context, oxidative degradation of hepatotoxic PAs, such as retrorsine, by human liver microsomes (or in an electrochemical cell) gave a variety of dehydropyrrolizidines, among which the novel metabolite 22 (Scheme 4) was observed.9 The structure of this compound was confirmed by comparison with an authentic synthetic sample, prepared from pyrrole as shown. In the presence of glutathione (GSH), this metabolite and a minor alkene regioisomer formed GSH adducts (cf.18) most likely via protonation at C(6) and subsequent trapping of the so-formed extended iminium ion.
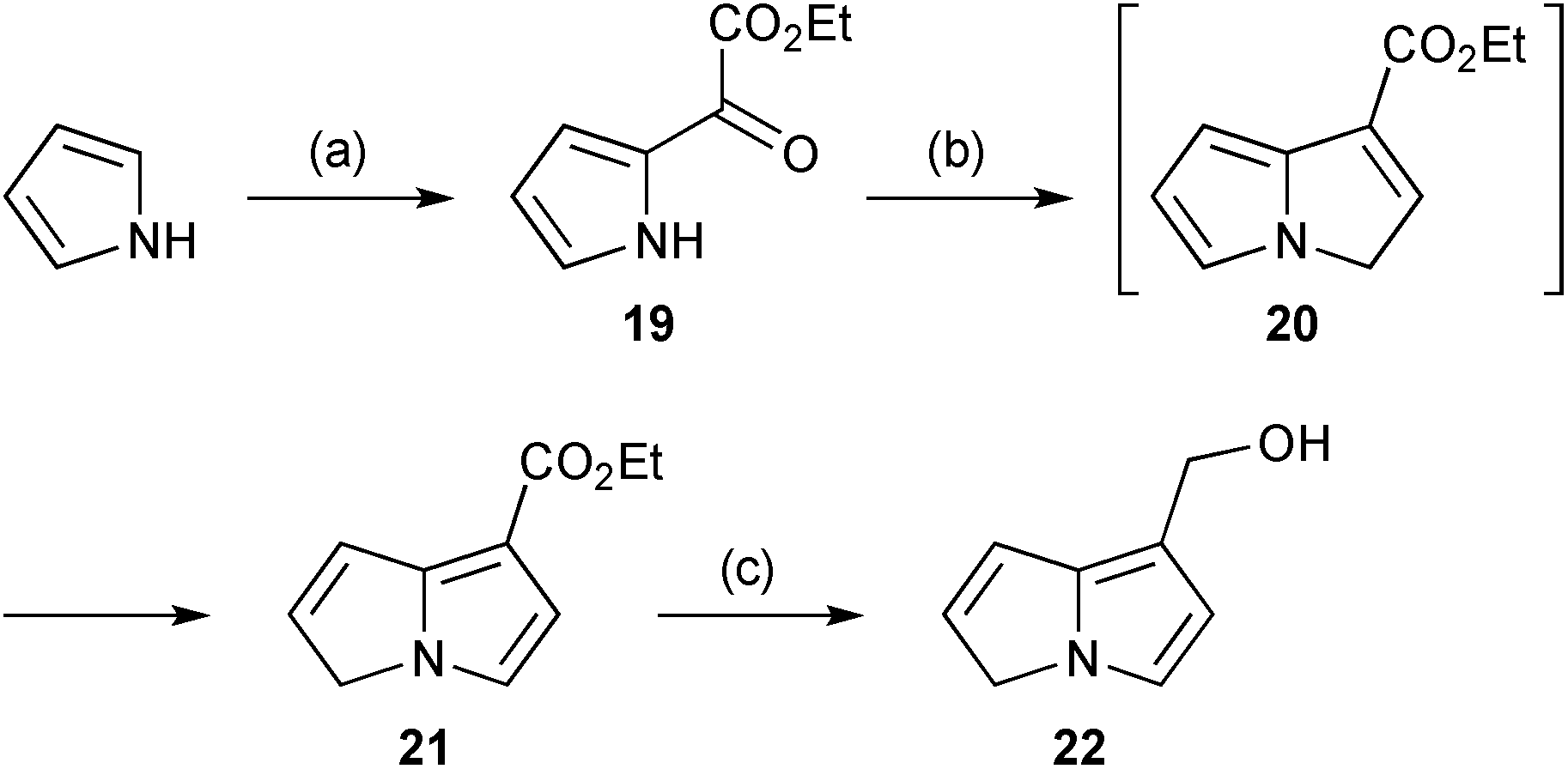 | ||
| Scheme 4 Reagents and conditions: (a) ethoxalyl chloride, pyridine, CH2Cl2, −80 °C; (b) NaH, vinyl triphenylphosphonium bromide, Et2O, reflux; (c) LiAlH4, Et2O, reflux. | ||
Stegelmeier developed an in vitro cell model to take up and activate dehydropyrrolizidine alkaloids and their N-oxides in order to arrive at a toxicity ranking of small quantities of these molecules present in a variety of samples.10 It was found, in a pilot study, that chicken hepatocellular carcinoma (CRL-2118) cells are highly susceptible to exposure to riddelliine and these, in combination with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, were used to rank the toxicity of eleven pyrrolizidines. Three groups were identified: (1) highly toxic – lasiocarpine, seniciphylline, senecionine, and heliotrine; (2) moderately toxic – riddelliine, monocrotaline, and riddelliine N-oxide; (3) least toxic – intermedine, lycopsamine, lasiocarpine N-oxide, and senecionine N-oxide.
A combined metabolomic and genomic study of senecionine toxicity in rats demonstrated that the observed toxicity is associated with compromised bile acid metabolism through a series of interconnected pathways.11 The paper's introduction contains a concise overview of the global impact of toxic pyrrolizidines on human health (see below).
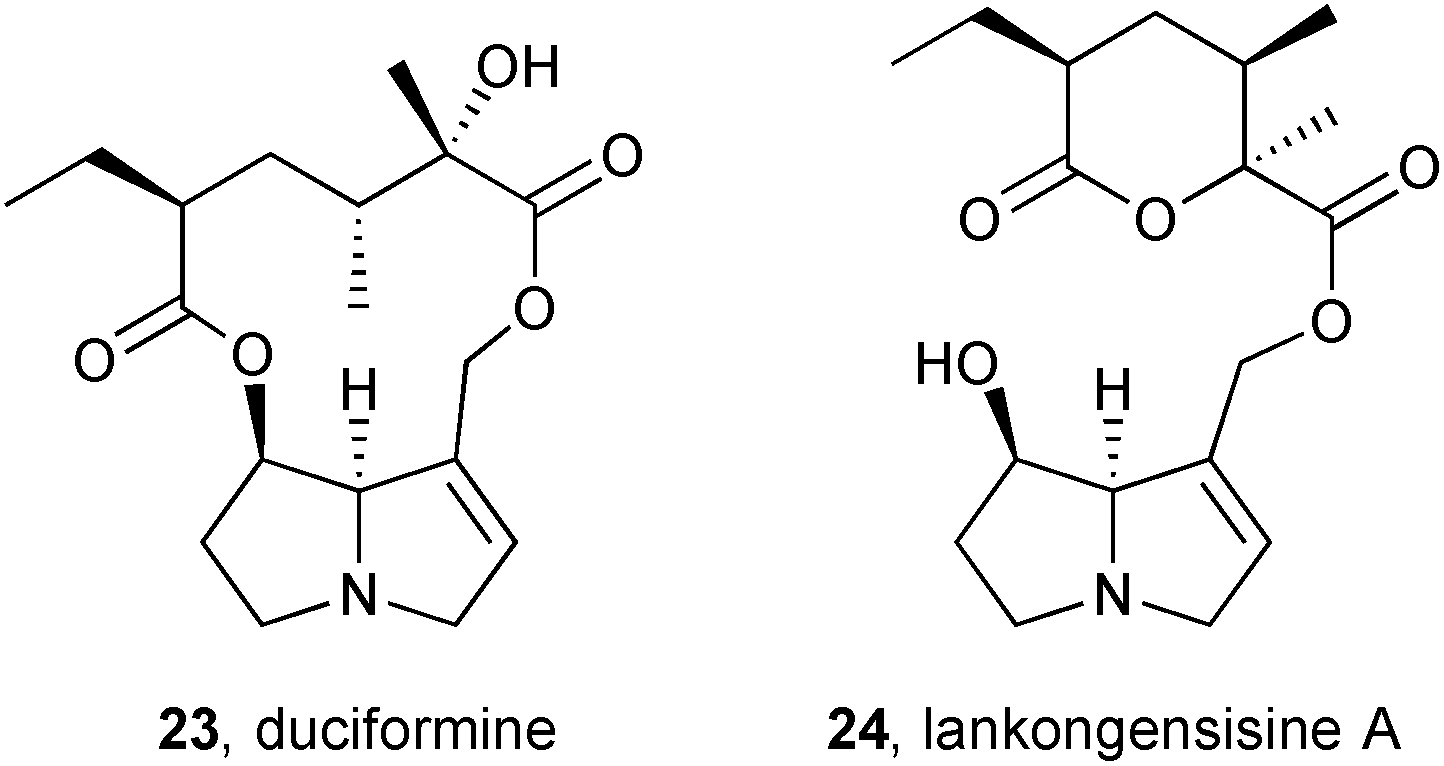
A continuation of an investigation into the rat liver microsomal metabolism of PAs from Ligularia duciformis (Asteraceae) showed that 12-O-acetylduciformine gave both the deacetyl parent 23 and the product 24 of intramolecular transacylation (lankongensisine A; δ-lactone stereochemistry assumed from duciformine).12
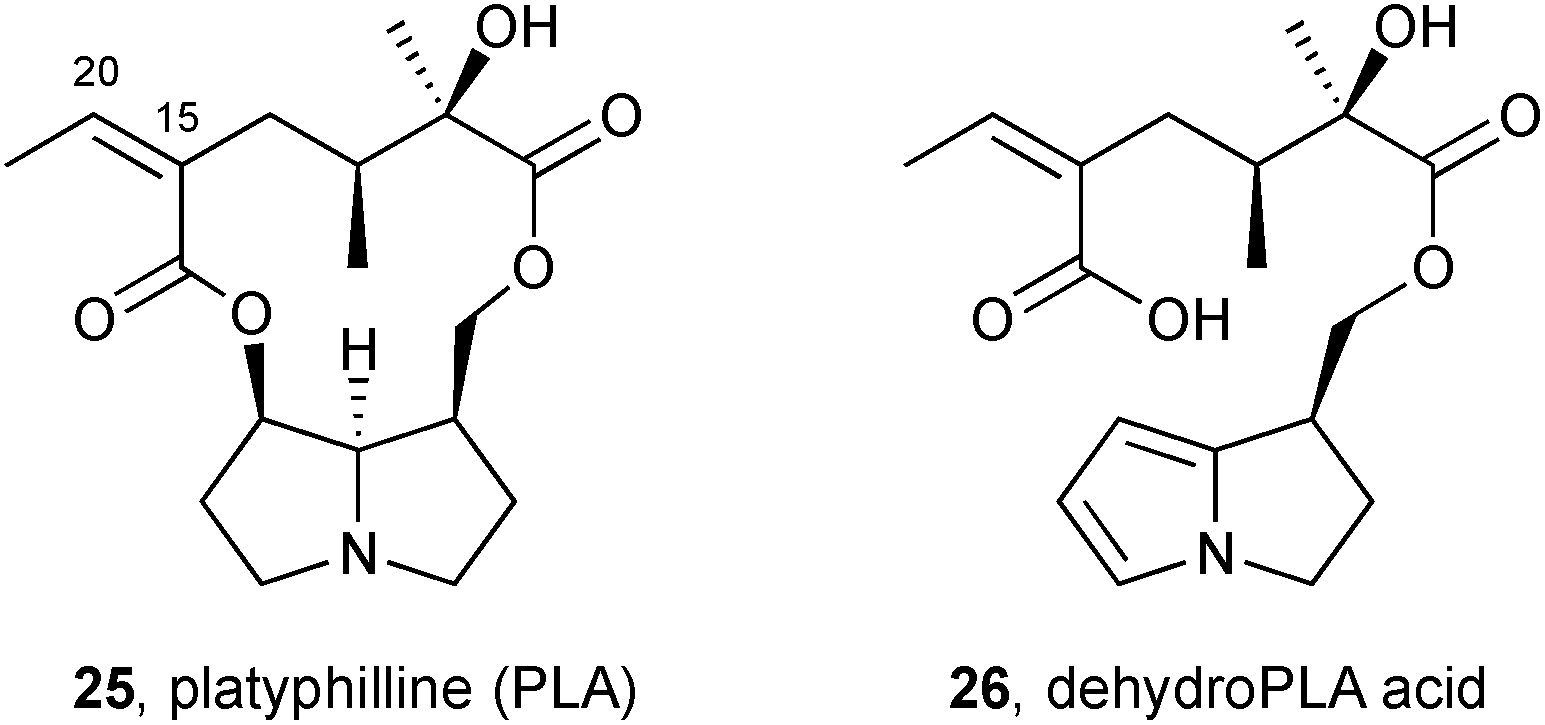
PAs of the platynecine type, lacking C(1–2) unsaturation, are non-toxic. Lin et al. showed that platyphilline 25, a representative alkaloid of this type, is metabolised primarily to the ‘dehydroPLA acid’ 26 shown. Although containing a pyrrole, this metabolite lacks a mechanism for incorporation of a cellular nucleophile via ejection of a suitable leaving group; in addition, the carboxylic acid renders the metabolite relatively water-soluble and readily excreted.13 Minor metabolites included platyphylline N-oxide and the C(15–20) epoxide.
2.2 Estimation in foods etc.
In parallel to trying to understand the toxicity mechanisms of PAs following ingestion, much research has been performed to improve the sensitivity and reliability of analytical techniques that can rapidly quantify the levels of toxic PAs in the complex matrices of foodstuffs, supplements, traditional medicines, and other products with which human or animal populations may come into contact. In addition, there is a developing consensus on the likely dangerous acute and chronic exposure levels. Even within the three-year period under review, many publications have described such studies, with focus on a particular analytical technique, a particular carrier, or a particular location/source. The area has been reviewed14 and the Federal Institute for Risk Assessment (BfR) has issued an informative opinion article with a summary of the existing maximum recommended exposure levels.15For example, reports have described the detection of toxic PAs in honey16–24 and their persistence through fermentation into mead;25 in medicinal or culinary herbs26–30 and herbal or medicinal teas;31–37 in seed oils for cooking, food supplements, and cosmetics;38,39 and in a variety of other sources.40,41 Reports that PAs can persist in contaminated plant-based cattle feed even following ensiling are of potential concern when such PAs pass into milk-producing animals.42–44
The vast majority of analyses are performed by MS and MS/MS protocols usually following pre-processing, derivatisation, or HPLC separation. A comparison of the results provided by 12 analytical laboratories on contaminated animal feed samples showed that an LC-MS/MS method seemed to offer the most consistent results but that there was sufficient variation between the laboratories to point to the need for further development of accurate analytical procedures and tools.45–48 The authors of a separate study tabulated multiple reaction monitoring (MRM) mass spectrometric ion responses of 26 pyrrolizidine ions to highlight the difficulty of quantifying the levels of toxic PA constituents in laboratory samples of, for example, food products and they advocate quantitative NMR as a more reliable means of obtaining meaningful values.49
These studies highlight an emerging recognition of chronic toxicity associated with herbal teas, largely because the consumers are potentially exposed to low toxic PA levels for many years. Wide variations in toxic PA levels were recorded; for example, in a study of herbal teas available to the Swiss market,32 more than one PA was found in 50 of 70 teas studied, of which 24 were at levels above the limit of quantification, and 9 had a Margin of Exposure below 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (an MOE > 10000 is deemed to pose little risk).
000 (an MOE > 10000 is deemed to pose little risk).
In each case, the sources of the contamination vary, but may include occasional (and variable) co-harvesting of toxic PA-containing species, mis-identification of the herb or plant material, contamination during processing, storage, or transport, or insufficient separation of toxic PAs in the case of seed oils.
2.3 Useful bioactivity
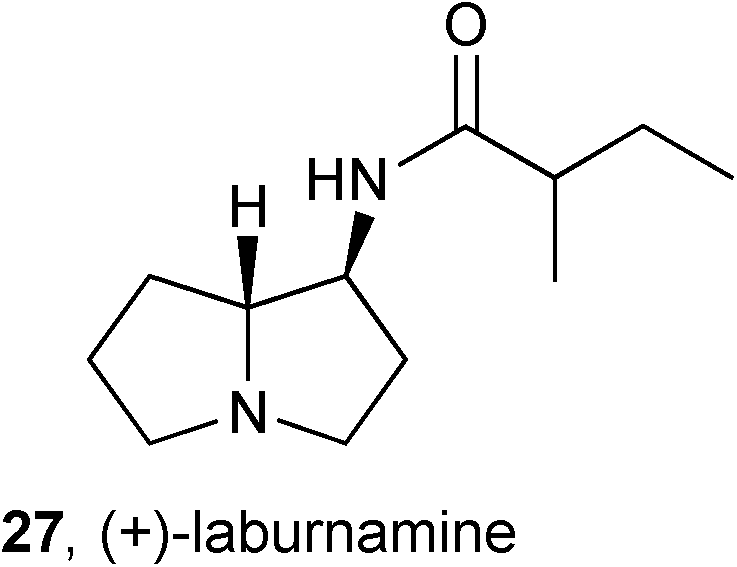
PAs are not all toxic; many are non-toxic, for reasons alluded to above, and some exhibit potentially useful biological activity. Reports of such activity are relatively scarce but researchers will find only what thay assay for; therefore, the few reported ‘hits’ may represent the tip of the iceberg of potential activity that would be revealed by assays against a wider-ranging variety of targets and cell types.During a study of the activity of potential nicotinic ligands prepared from cytisine obtained from Laburnum anagyroides (Fabaceae), (+)-laburnamine 27 was isolated in sufficient quantity to enable a preliminary pharmacological evaluation.50 The authors found that this alkaloid is a selective ligand for the rat cortical α4/β2 neuronal nicotinic acetylcholine receptor subtype (Ki = 0.293 μM) relative to human transfected α3/β4 (Ki = 37 μM) and rat hippocampus α7 (Ki = 40 μM) subtypes. A further study, on the ability of 27 to induce dopamine release relative to nicotine, indicated that (+)-laburnamine acts as a partial agonist.
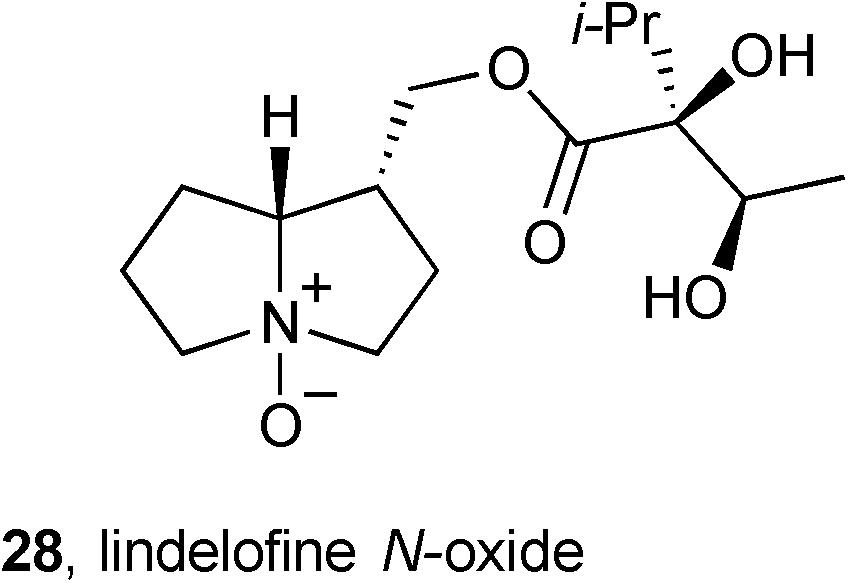
Ten known pyrrolizidines were isolated from Rindera umbellata (Boraginaceae). The distribution of these ten alkaloids varied substantially depending on harvest date (June 2007, May 2008, July 2009) and plant part (aerial parts, roots, seeds).51 The most abundant pyrrolizidine, lindelofine N-oxide 28, was evaluated for its ability to promote tubulin polymerisation; the obtained IC50 = 91 μM compares to 2.4 μM for paclitaxel. The authors suggest that this is the first report of the effect of a PA on tubulin polymerisation.
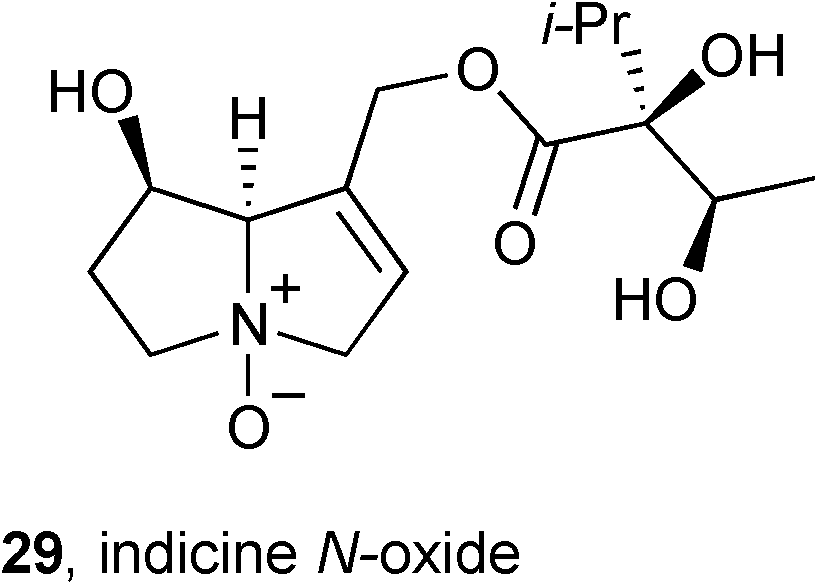
Indicine N-oxide 29 (INO) is cytotoxic against a variety of tumour cell lines and has been evaluated in the clinic for the treatment of leukaemia but was withdrawn from trials due to its severe toxicity. Rathinasamy reported52 on the mechanism underlying this alkaloid's toxicity and found that: (1) INO blocks the cell cycle at mitosis; (2) INO causes spindle abnormalities at the IC50 (∼100 μM) while at 300 μM it depolymerises both interphase and spindle mictrotubules; (3) INO appears to interact with a single binding site on tubulin and this site is not the colchicine binding site; (4) INO leads to DNA cleavage following (computationally predicted) binding at the minor groove.
Monocrotaline is cytoxic towards HepG2 cells (IC50 = 25 μg mL−1) and genotoxic at ∼50 μg mL−1.53
Among fourteen iminosugars isolated from Castanospermum australe (Fabaceae), five tetrahydroxylated pyrrolizidines (australine and epimers) were identified.54 Four of these, along with other iminosugars, were evaluated for glycosidase inhibition and only australine showed significant activity (IC50 26–665 μM against seven different glycosidases).
2.4 Novel PAs and biosynthetic aspects

Investigations of the alkaloidal principal components of plant species following toxicity events in cattle has led to the identification of new pyrrolizidines cryptanthine 32 and echiuplatine 33.56 Thus, samples of Cryptantha inequata and C. utahensis (Boraginaceae) were collected opportunistically following a hepatotoxicity incident in the Kingman area of Arizona, USA, and alkaloid profiles established by HPLC-ESI-MS. For the C. utahensis extract, the major peak (corresponding to cryptanthine, 0.55 ± 0.04 mg g−1 dry weight of plant) was not correlated with known alkaloids but structural elucidation revealed it to be 32, present in the plant primarily as the N-oxide. No cryptanthine was observed in the C. inequata specimen; alongside known PAs, echiuplatine 33 was identified (as its methyl ester) with comparable abundance (0.13 ± 0.001 mg g−1 dry weight of plant) to the known echimidine and its O-acetyl derivative.
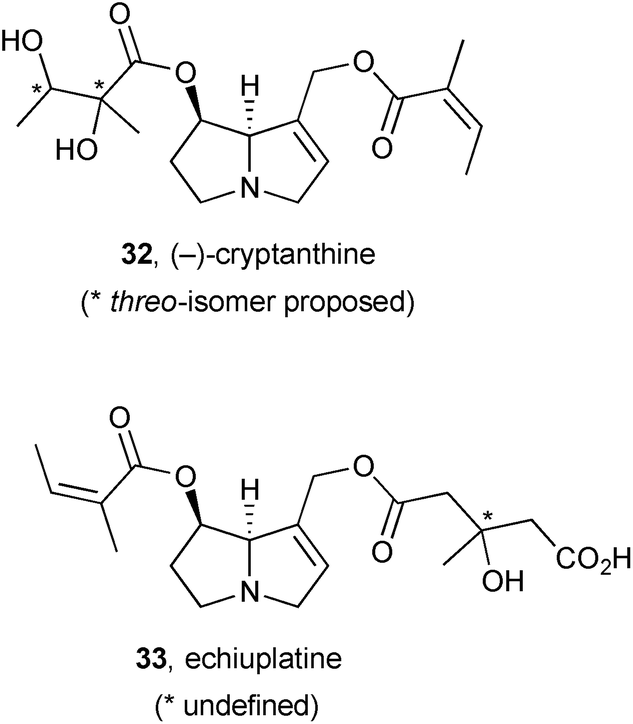
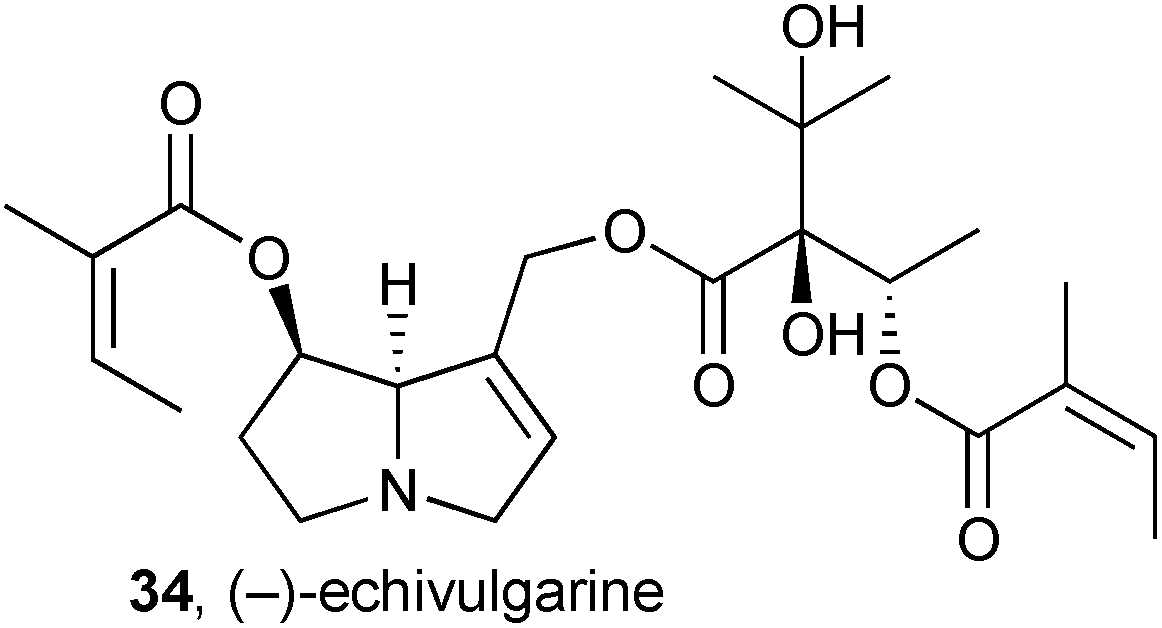
A combination of spectroscopy and computation was used to make a full stereochemical assignment of (−)-echivulgarine 34, obtained from bee pollen granules presumed to have been collected from Echium vulgare (Boraginaceae), commonly occurring in the area.49 From 280 g of the granules, 11 mg of the alkaloid were eventually obtained. Full 1H, 13C, and 15N NMR spectra were compared with predicted spectra based on Boltzmann-weighted conformational distributions for candidate stereoisomers. The absolute stereochemical assignment was supported by a comparison of experimental and theoretical circular dichroism (CD) spectra.
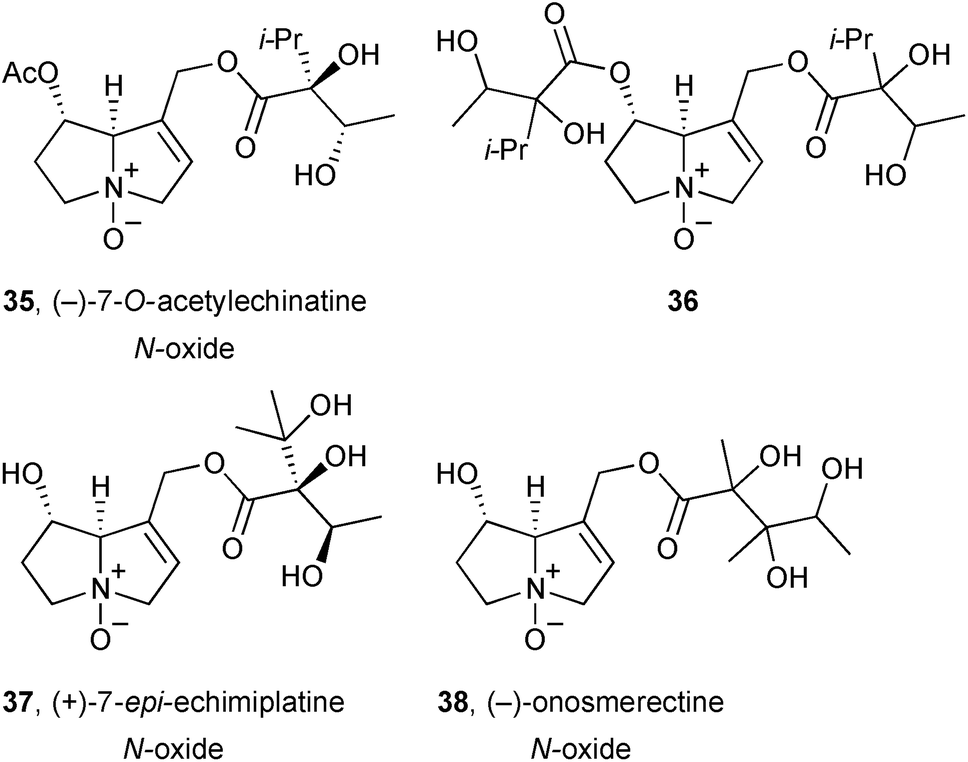
The aerial parts of Onosma erecta (Boraginaceae) yielded four new pyrrolizidines 35–38, with structures assigned primarily on the basis of NMR data.57 Only limited quantities of 36 and 38 were available and stereochemical assignment of the necic acids was not achieved.
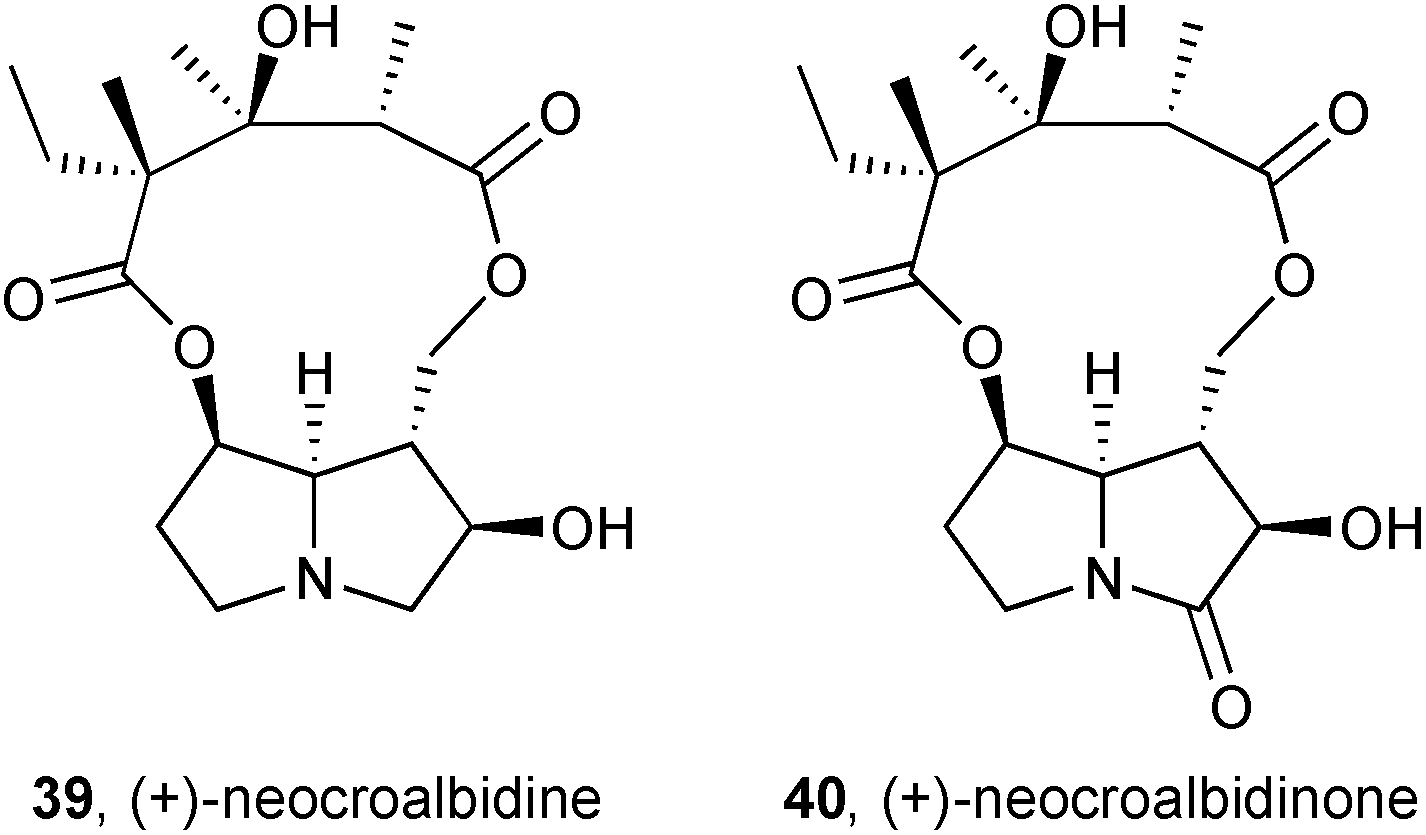
Two new pyrrolizidines, neocroalbidine 39 and neocroalbidinone 40, were isolated from the herb Crotalaria albida (Fabaceae).58 Single crystal X-ray diffraction provided structural confirmation of both alkaloids, including their absolute stereochemistry.
Six new [(+)-nervosines I–VI] and two known [(+)-paludosine, (−)-auriculine] PAs were isolated from Liparis nervosa (Orchidaceae).59 All are nervogenic acid (41) esters of simple 1-hydroxymethyl pyrrolizidines. Six of these are depicted as esters of (−)-isoretronecanol and two of (−)-trachelanthamidine but the text describes these as esters of their enantiomers, (+)-lindelofidine and (+)-laburnine, respectively. Based on reported structures for the known alkaloids, the structures depicted in the paper show the incorrect enantiomer of the necine base. All eight alkaloids showed no cytotoxicity against human tumour cell lines MCF-7, A549, and HepG2, and most were non-toxic to RAW264.7 macrophages; however, all inhibited lipopolysaccharide- (LPS) induced NO production in the same cell line with IC50 = 2.16–38.3 μM.
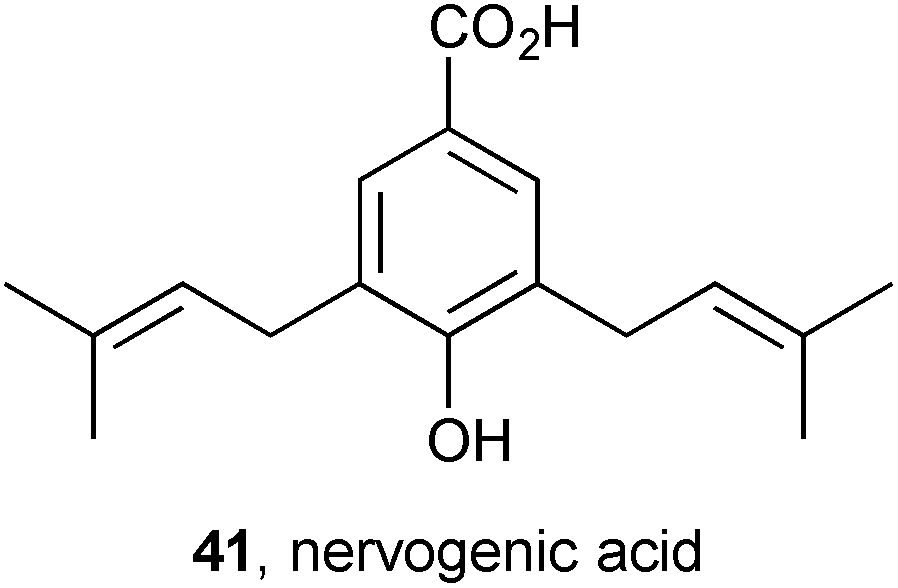
Complete NMR assignments were reported for PAs 42–45 obtained from the roots of Senecio polypodioides (Asteraceae), including the novel pyrrolizidine neosarracine N-oxide 45.60
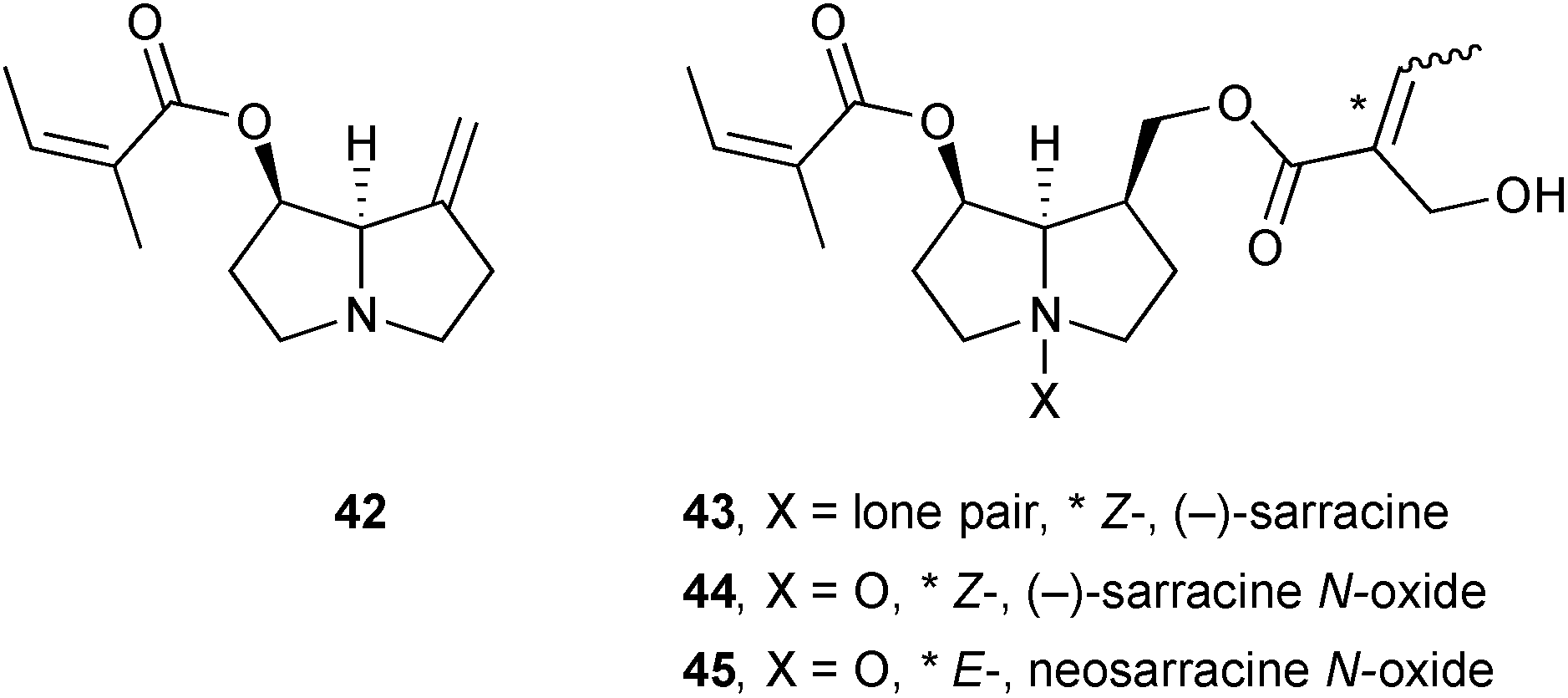
New metabolites were characterised in extracts prepared from transformed root cultures of the plant Bethencourtia hermosae (Asteraceae) from La Gomera (Canary Islands).61 In addition to the known pyrrolizidines senecionine, seneciphylline, and senkirkine, a new pyrrolizidine hermosine 46 was also isolated and characterised through NMR analysis; the stereochemistry around the γ-lactone remained undetermined. Along with many of the other metabolites isolated from this plant, hermosine exhibited some antifeedant activity against aphids.
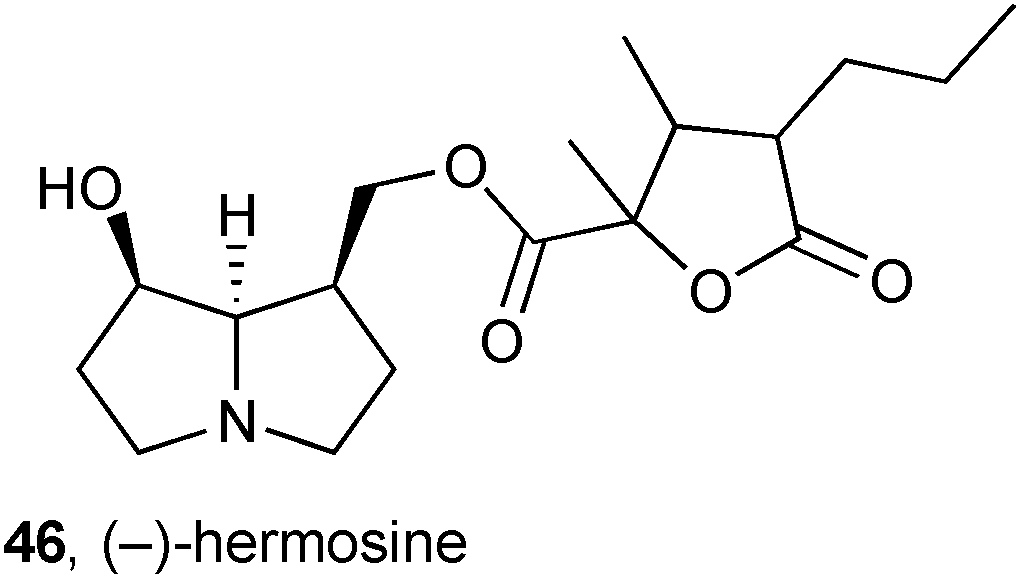
It had been observed that American Monarch butterflies, known to be PA pharmacophagous, are attracted to the freshwater aquatic plant Gymnocoronis spilanthoides (Asteraceae) and on this basis Colegate studied the plant's chemical constituents with the expectation that PAs would be found.62 Methanol extraction of cultivated whole-plant material and gravimetric analysis of the isolated alkaloidal fractions indicated that approximately 0.08% of the fresh weight of the plant comprised pyrrolizidines. Further HPLC MS/MS analysis revealed at least twenty pyrrolizidines to be present. Along with some known alkaloids (e.g. lycopsamine and intermedine), and a number of unidentified components, two new alkaloids were tentatively identified from MS/MS data as spilanthine 47 and gymnocoronine 48. This study raised questions on the relative prevalence of toxic PA content in wild vs. cultivated G. spilanthoides, implications for human health through potential leaching of these compounds into water supplies and the plant's proposed use as a nicotine-free tobacco substitute, and the possibility that this vigorous plant could be used as a sustainable bulk source of PAs.
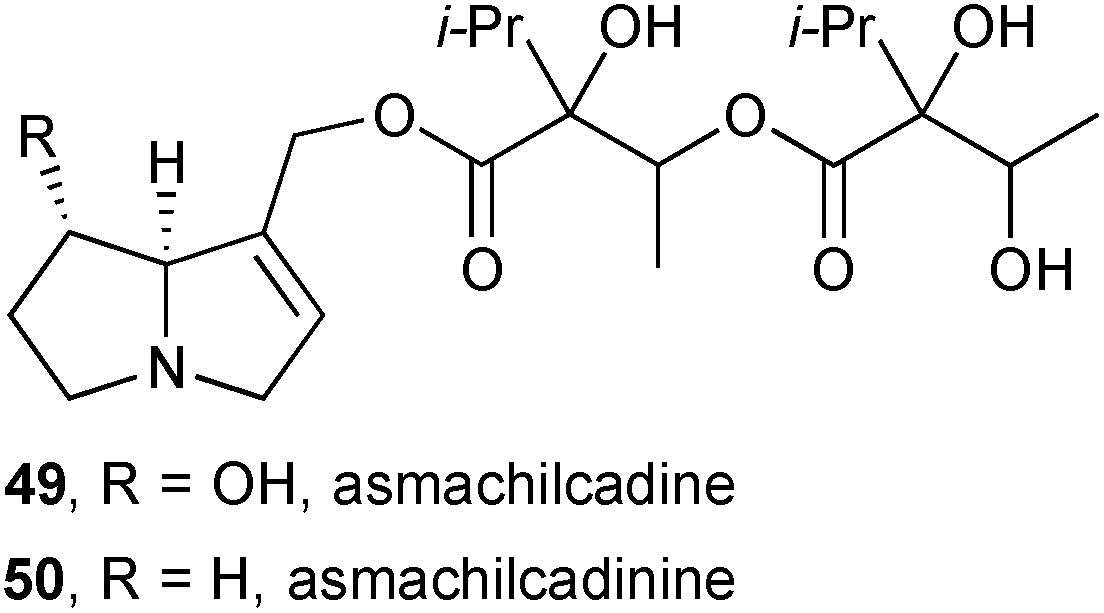
In a second study guided by the observation of insect attraction to plant material, Colegate's group investigated the components of dried leaves and seed heads of ‘asmachilca’, a Peruvian botanical medicine derived, in principle but often not in practice, from Aristeguietia gayana (Asteraceae) and taken traditionally directly or as a tea, and used as a poultice.63 Again, the study was guided by a concern that toxic pyrrolizidines in these traditional preparations could present a significant chronic threat to human health. Six asmachilca samples were analysed; while there were differences in the HPLC profiles between the samples, and at least two different plant species were present, all contained significant quantities of unsaturated PAs (0.4–0.9 weight/dry weight%). Within the 16 identified pyrrolizidines, two new structures were proposed: asmachilcadine 49, a heliotridine ester, and asmachilcadinine 50, the supinidine analogue; the stereochemistry in the necic acids was not confirmed due to inconsistencies with other constituent metabolites present in the extracts. The N-oxides were also observed (by MS) in the extracts as confirmed by oxidation of the parents 49 and 50. Steeping asmachilca samples in boiling water led to alkaloid levels in the tisane that reached a maximum within 3–5 minutes. This work indicates clearly that these preparations present a significant risk of exposure to toxic pyrrolizidines, but variation in the plant source, harvesting, storage, and preparation mean that it is difficult to quantify this risk. Further work is needed to identify the beneficial components of asmachilca preparations with a view to providing a standardised, less toxic preparation.
Three lindelofidine [(+)-isoretronecanol] esters, one novel, were isolated from the Vietnamese medicinal herb Madhuca pasquieri (Sapotaceae).64 The structure of the novel pyrrolizidine, (−)-madhumidine 51, was assigned by NMR experiments; all three alkaloids showed only weak cytotoxicity (IC50 > 100 μM) against three cancer cell lines.

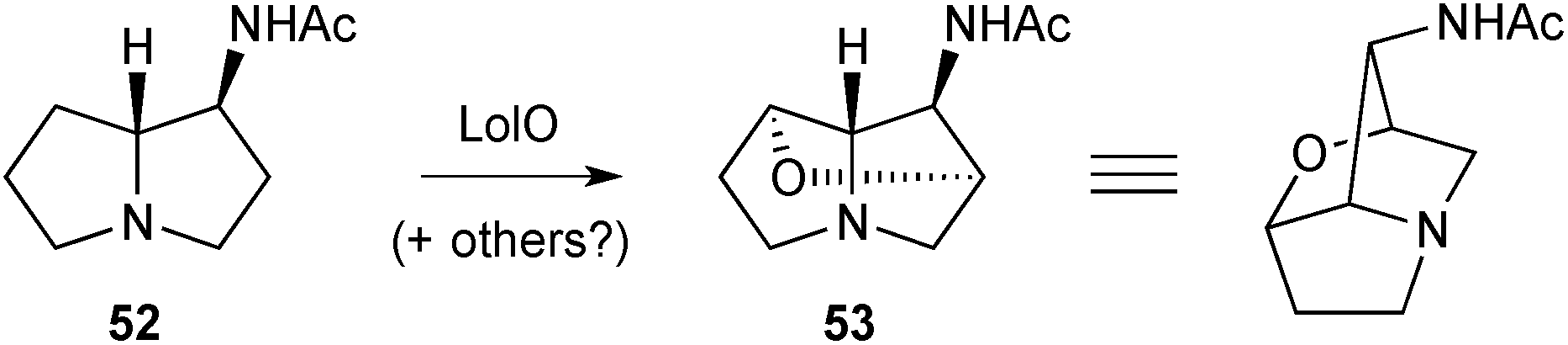 | ||
| Scheme 5 Biosynthetic incorporation of the C(2)–C(7) ether in the lolines. | ||
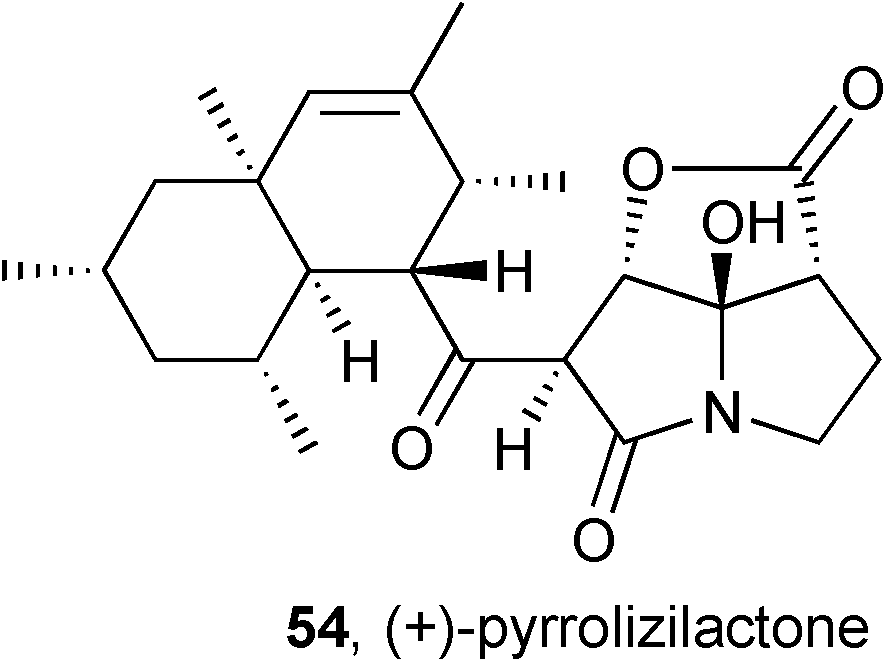
Screening of a fungal fraction library led to the isolation and identification of pyrrolizilactone 54, closely related to cytotoxic antibiotics CJ-16264 and UCS1025A.66 This metabolite was cytotoxic against HL-60 and HeLa cells with IC50 = 1.1 and 3.1 μg mL−1, respectively, but showed no antibacterial activity (vs. E. coli up to 30 μg mL−1).
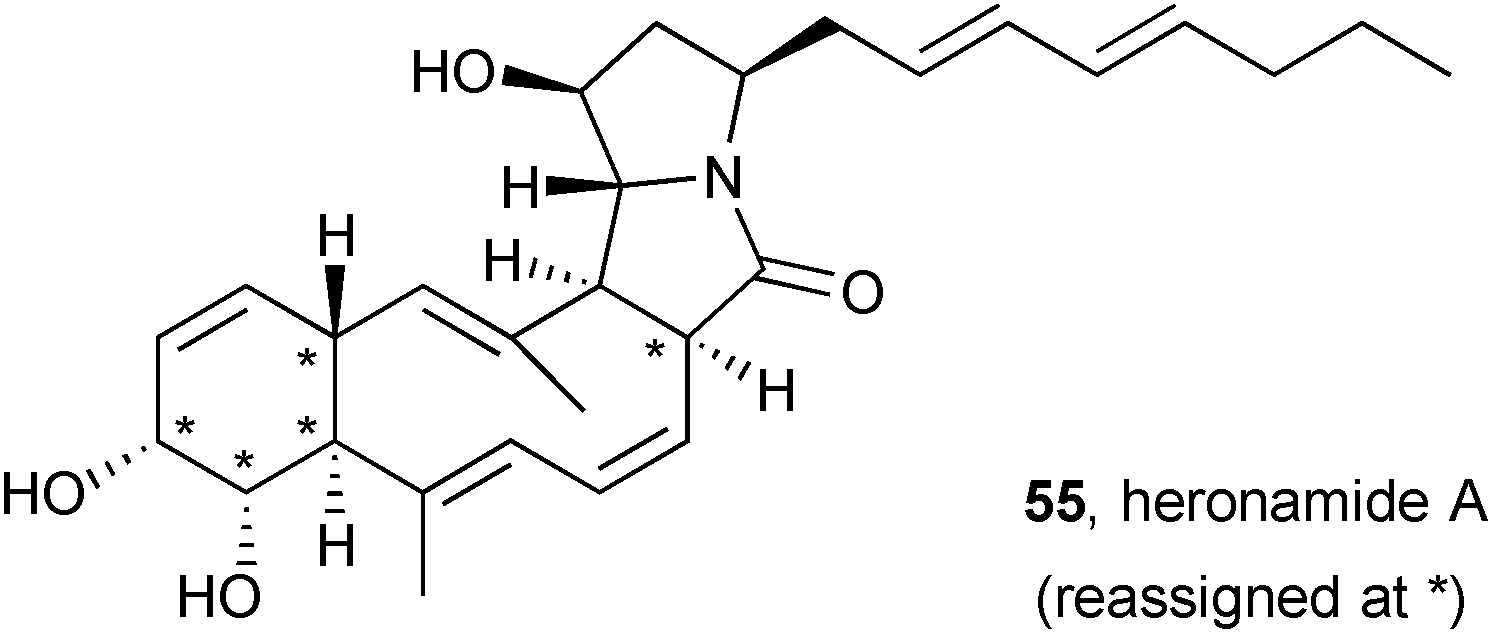
Following a detailed NMR spectroscopic analysis of heronamide A and derivatives, the stereochemistry in this macrocyclic polyketide pyrrolizidinone was reassigned at C(2), C(7–9), and C(12) to that shown (55).67
Houk's group reported DFT calculations to support a transannular [6+4] cycloaddition in the formation of heronamide A from heronamide C.68 Using a side-chain truncated model 56 (Scheme 6) as the basis for calculations, an ambimodal transition state was located that leads to both the [6+4] adduct 58 and an intermediate intramolecular Diels–Alder adduct 57 that then undergoes rapid [3,3]-sigmatropic shift (Cope rearrangement) to produce the more stable [6+4] adduct. The results have implications for broader applications of [6+4] cycloadditions in synthesis.
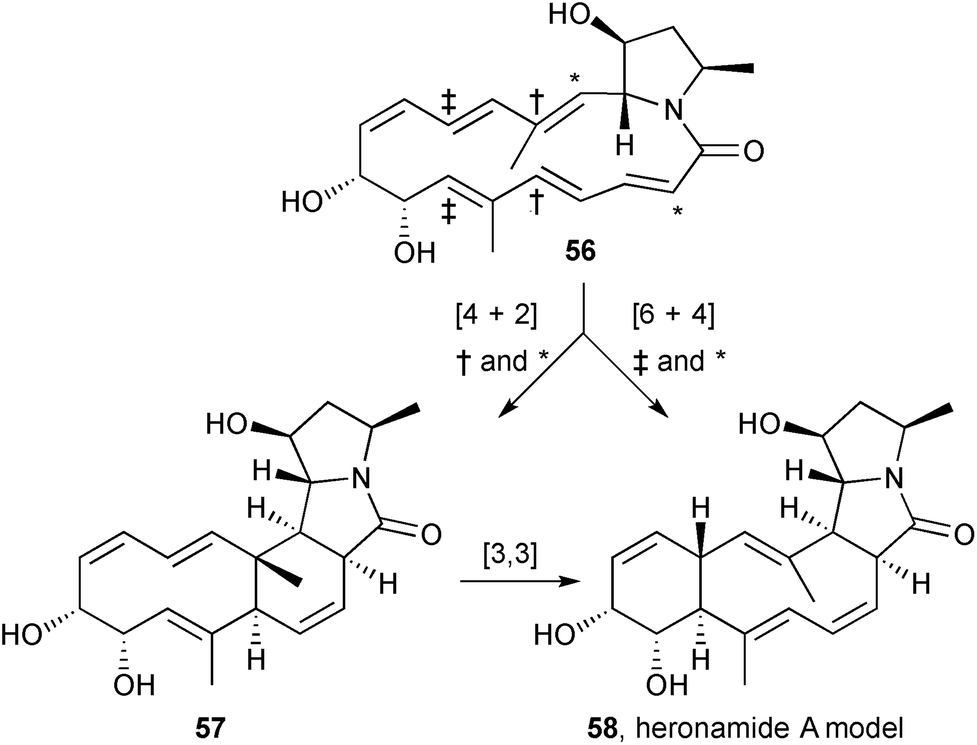 | ||
| Scheme 6 Parallel one-step and two-step mechanistic pathways connect heronamide A and heronamide C. | ||
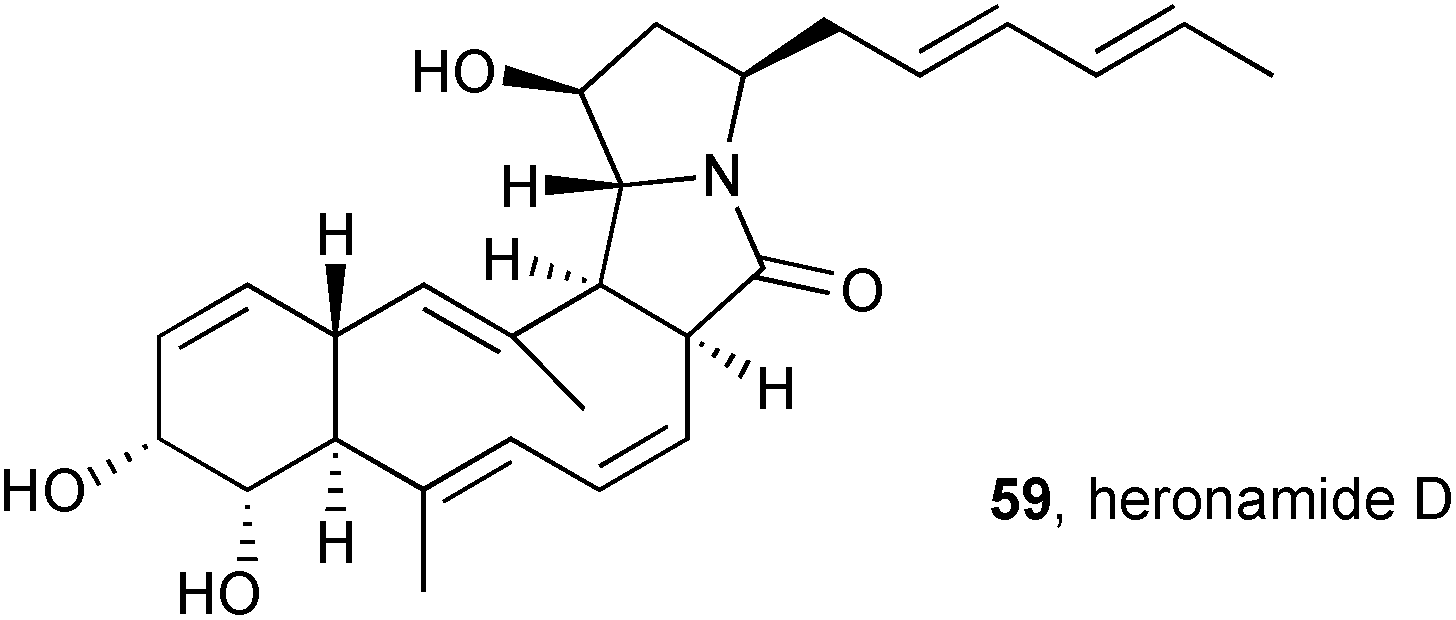
Culturing Streptomyces sp. SCSIO 03032 in a variety of media resulted in the production and isolation of three new macrolactams, heronamides D–F.69 The stereochemistry in heronamide D 59, assigned by extensive 3JHH and NOESY NMR spectroscopic analysis, was found to be identical to that in the recently-revised structure for heronamide A (see above); indeed heronamide D differs from heronamide A simply by virtue of a terminal methyl in place of propyl on the dienyl side chain. The three new heronamides showed no antimicrobial activity (against four bacteria and a fungus), no antioxidant activity (DPPH radical scavenging assay), but did exhibit growth inhibition for three of seven cancer cell lines (IC50 = 15.4–56.4 μM).
Nocardiopsis sp. FU40 ΔApoS is an engineered bacterium in which the ApoS8 gene, encoding the terminal polyketide synthase, is replaced in order to deactivate the production of apoptolidins. Co-culturing this bacterium with competing bacterial strains activates latent metabolic pathways, leading to the production of secondary metabolites unobserved in monoculture.70 A metabolomic response-mapping and comparison approach led to the isolation and identification of a new macrolactam, ciromicin A 60 (Scheme 7), and its isomer ciromicin B 61, a pyrrolizidinone closely resembling heronamide A 55 and D 59. The structural elucidation was achieved by a combination of NMR methods once the molecular formula had been established by HRMS. A sample of pure ciromicin A was converted into ciromicin B, in an overall [6+6] cycloaddition, by exposure to sunlight; ciromicin B was the major product at 400 nm and, although the conversion to ciromicin B reached a maximum at 300 nm, other ciromicin isomers were also produced at this wavelength. The authors propose an outline biosynthesis based on a series of polyketide synthases, then ciromicin-specific enzymes that effect macrolactam formation, closure of the pyrrolidine ring (in circomicin A), and glycosylation. These new metabolites are structurally and biosynthetically related to cytotoxic macrolactams such as vicenistatin; therefore, their in vitro activity was tested against the MV-4-11 human leukaemia cell line and IC50 values of 8.1 and 9.3 μM were found for ciromicin A and B, respectively; there was no antibacterial or antifungal activity found in assays with Bacillus, E. coli, or Saccharomyces species.
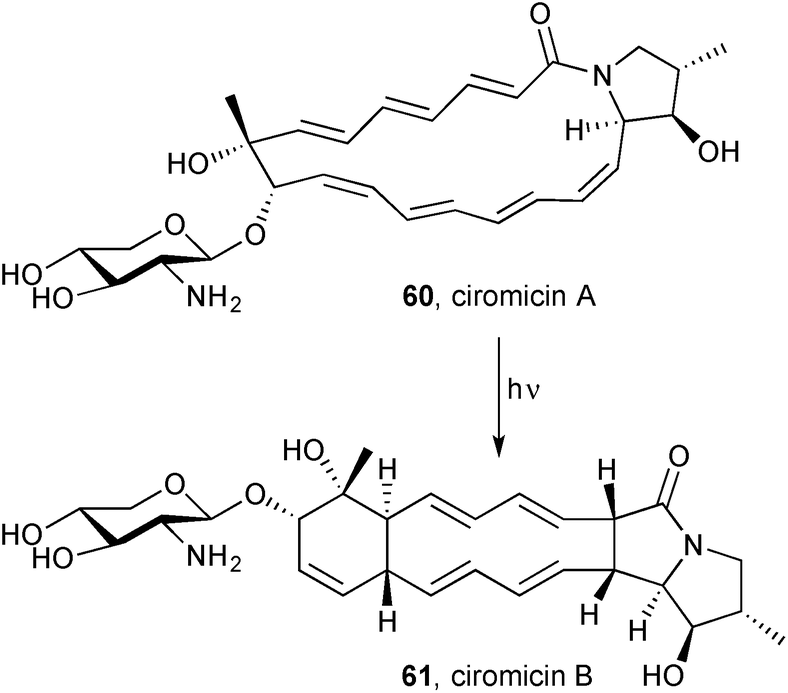 | ||
| Scheme 7 Ciromicin B is formed by photochemical formal [6+6] cycloaddition within ciromicin A. | ||
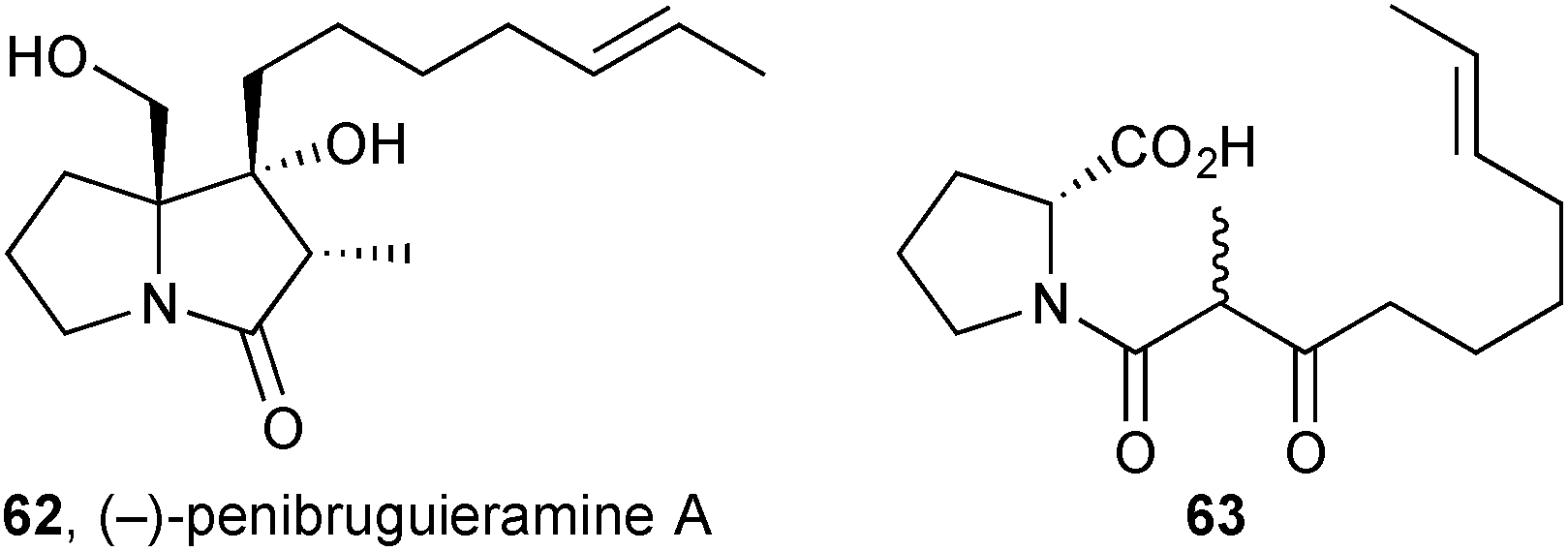
The observation of potent anitibacterial activity in a crude extract of Penicillium sp. strain GD6, isolated from the Chinese mangrove Bruguiera gymnorrhiza (Rhizophoraceae), led to the discovery of a new pyrrolizidine, penibruguieramine A 62.71 This metabolite is proposed to be biosynthesised from acid 63 which has previously been suggested as a precursor to scalusamide A, a simple fatty acid prolinol amide.
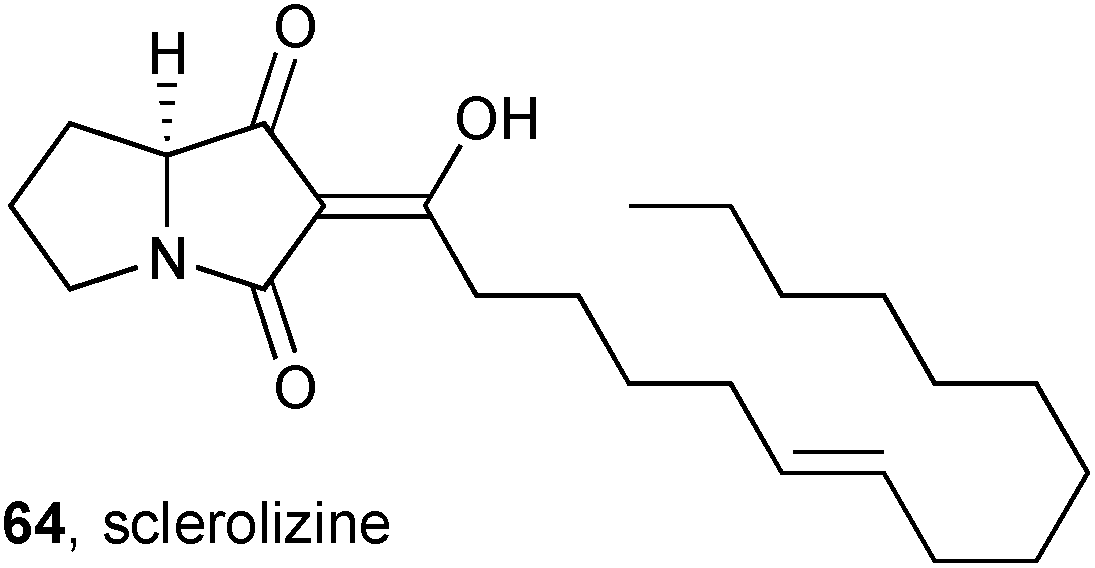
Conceptually related to the induced production of the ciromicins, discussed above, Larsen showed that the growth of the fungus Aspergillus sclerotiicarbonarius in conditions that trigger sclerotium production leads to a greatly altered metabolic profile, with four new compounds identified.72 One of these compounds, sclerolizine 64, is an oxidised pyrrolizidine; the enol stereochemistry was not assigned and the absolute configuration is proposed based solely on a proposed biosynthetic derivation from (S)-proline. The four new metabolites were evaluated as antifungal agents against Candida albicans, and sclerolizine was found to be the most potent with IC50 = 8.5 ± 2.0 μM.
Three papers in quick succession relate to the discovery of bacterial pyrrolizidines of the vinylogous urea type. The first73 reports the isolation, structure, and biosynthesis of legonmycin A 68 and B 69 (Scheme 8), from Streptomyces sp. MA37. The structures were elucidated by a combination of spectroscopic and computational techniques and the major metabolite, legonmycin A, obtained as the racemate. From the draft genome for the MA37 strain, the authors were able to identify a gene cluster (lgn) encoding four key enzymes: LgnA, a thioesterase; LgnC, a flavin dependent monooxygenase; and LgnB and LgnD, two multidomain non-ribosomal peptide synthetases. A precise role for LgnA was not established but enzymes LgnB and LgnD were shown to assemble legonindolizidine A (65, n = 1) and B (65, n = 2). LgnC, along with co-factors FAD+, O2, and NADPH, then effects a four-step transformation into the legonmycins comprising: (1) a Baeyer–Villiger type oxidative ring-expansion; (2) hydrolysis of the so-formed cyclic carbamate (cf.71 below); (3) decarboxylation then condensation to produce the vinylogous urea functionality (→66 and 67); then (4) hydroxylation at C(7a).
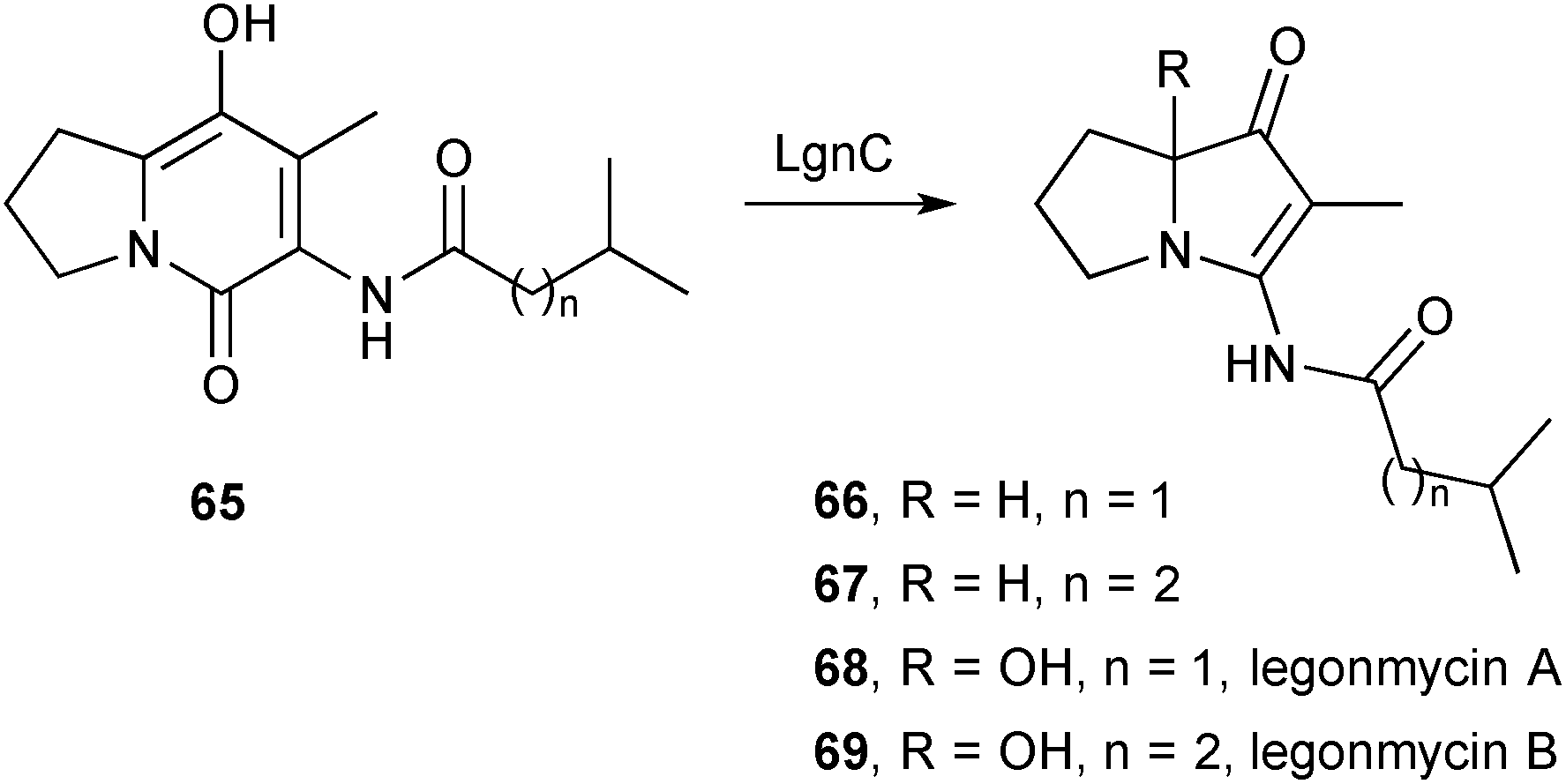 | ||
| Scheme 8 The monooxygenase LgnC effects overall decarbonylation and hydoxylation in the biosynthesis of the legonmycins. | ||
In results that closely parallel those reported by Deng's group in the context of the legonmycins, expression in E. coli of an unknown gene cluster from Xenorhabdus stockiae and differential analysis by 2D NMR spectroscopy (DANS) led to the isolation and characterisation by NMR and MS methods of pyrrolizixenamides A–C 72–74 (Scheme 9).74 The assigned structures were confirmed by total synthesis based on Snider's synthesis of the jenamidines (see previous review). The authors identified a gene cluster pxaAB encoding for PxaA, responsible for producing the pyridone intermediates 70 [R = n-pentyl, n-hexyl, n-heptyl], and PxaB that effects a ring-expansion, hydrolysis, and decarboxylative condensation process. The authors also found that more than 90 bacterial strains from 23 species contain pxaAB homologues suggesting that bacterial pyrrolizidines of this type should occur widely. As an example, when the pyrrolizidine gene cluster in X. szentirmaii was activated by a promoter exchange method, the branched variant pyrrolizixenamide D 75 was produced.
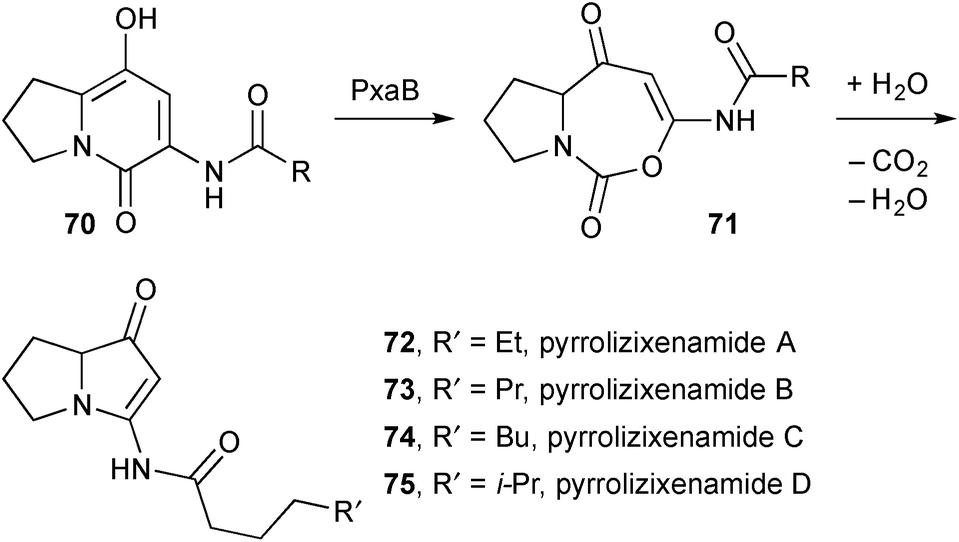 | ||
| Scheme 9 Key steps in the biosynthesis of the pyrrolizixenamides. | ||
Extracts from the Streptomyces spinoverrucosus strain SNB-048 were shown to contain the new pyrrolizidine (+)-spithioneine A 76 and its sulfoxide, spithioneine B 77 (Scheme 10).75 In addition to spectroscopic characterisation, the assigned structures were supported by RANEY® Ni desulfurisation (not shown) of spithioneine A that yielded known components bohemamine C and (S)-hercynine. Additionally, L-ergothioneine and bohemamine 78 were combined under basic conditions to achieve a semi-synthesis of spithioneine A, and this was oxidised to give spithioneine B (the sulfoxide stereochemistry was not established). The authors proposed a plausible biosynthesis for these pyrrolizidines initiating with L-ornithine or L-arginine, polyketide extension, then consecutive N-cyclisations and dehydrations to give bohemamine via bohemamine B. The spithioneines were shown to have no cytotoxicity against four lung cancer cell lines and no antibacterial activity against Pseudomonas aeruginosa and Bacillus subtilus.
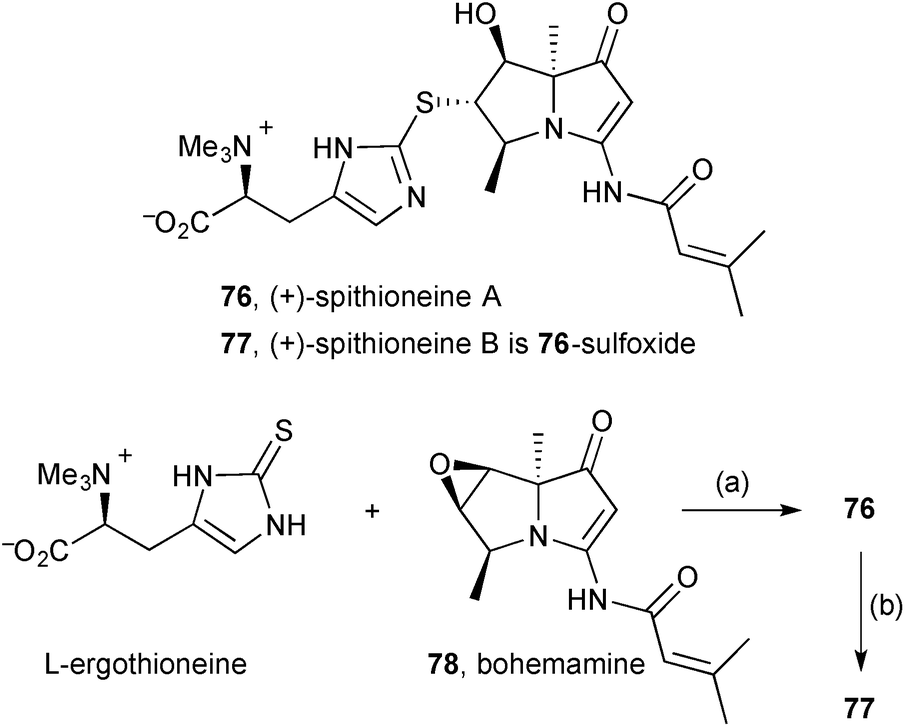 | ||
| Scheme 10 Reagents and conditions: (a) aq. Na2CO3; (b) Oxone®, aq. THF, 0 °C. | ||
Two unusual pyrrolidinyl-oxazinones were isolated from Streptomyces sp. KMF-004 extracted from a sea-water salt-making pool in Korea.76 Salinazinone B 80 (Scheme 11) and its hydroxylated counterpart salinazinone A 81 were characterised spectroscopically and the absolute configuration assigned by comparison of the experimental and calculated electronic CD spectra. A novel pyrrolizidine, bohemamine D 79 and known bohemamine B were found in the same bacterial strain and the authors suggest that the salinazinones derive from them biosynthetically. In essence, the authors' mechanism proceeds via C→O acyl transfer (dotted arrow) then oxidation; however, were oxidation to occur first (either on the external nitrogen, the endo-double bond or, as shown in Scheme 12, via a Baeyer–Villigerase) then an electronically and sterically more reasonable route arises.
 | ||
| Scheme 11 Originally-proposed order-of-events in the biosynthesis of salinazinone B from bohemamine D. | ||
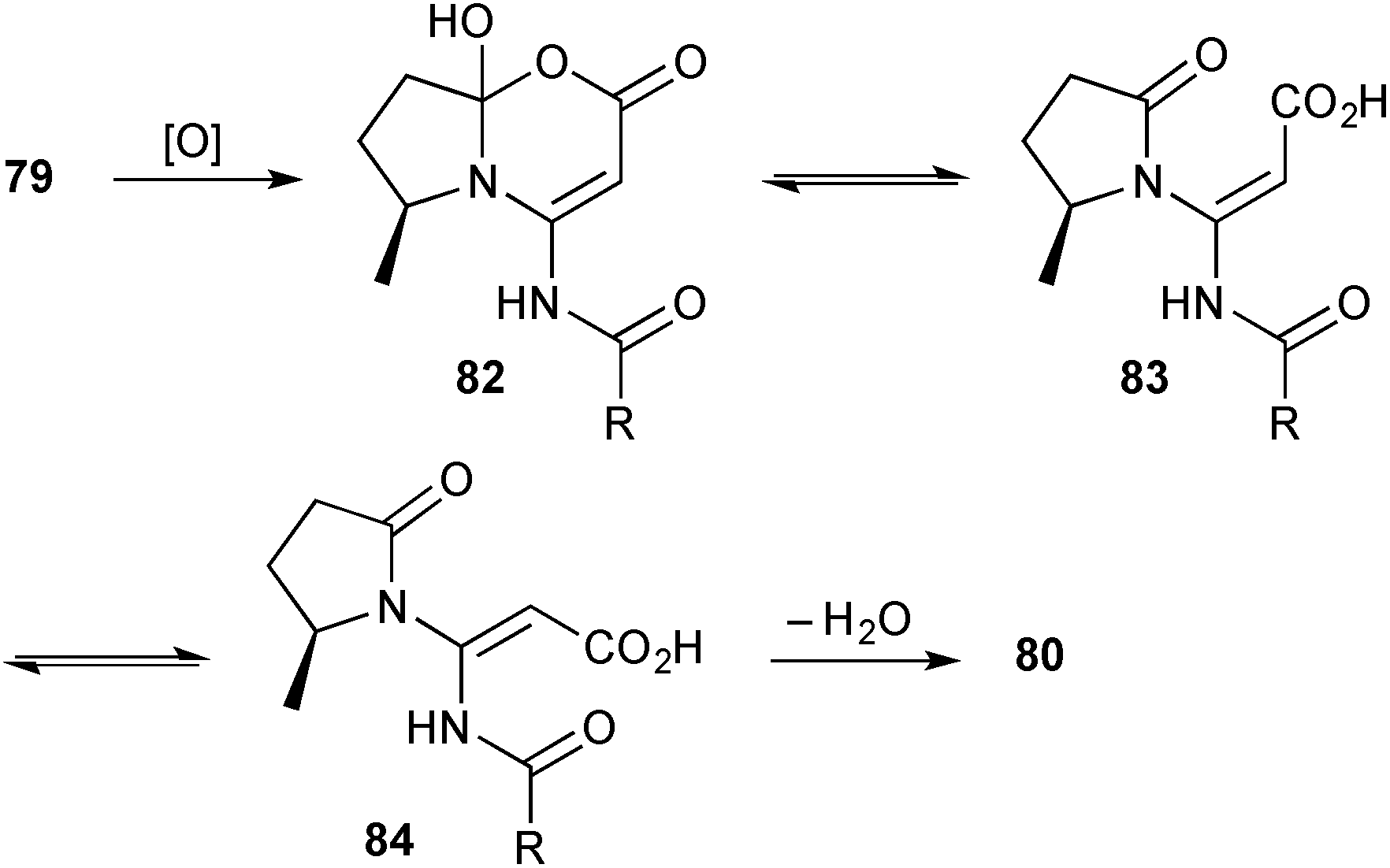 | ||
| Scheme 12 A hypothesis for the biosynthesis of salinazinone B from bohemamine D. | ||
3 Synthetic approaches
3.1 Isoretronecanol and related molecules77
The biosynthetic intramolecular Mannich reaction that introduces the C(1)–C(7a) bond in 1-hydroxymethyl pyrrolizidines inspired a formal synthesis of racemic isoretronecanol (lindelofidine) and trachelanthamidine (laburnine).78 The cyclisation of hydroxylactam 85 (Scheme 13), prepared in two steps from succinic anhydride, was evaluated under a range of acid/solvent/temperature combinations to give varying ratios of diastereomers 86 and 87 following reduction of the intermediate aldehyde. Good to high yields were obtained with a full equivalent or more of TsOH. Under most conditions, with acetonitrile as solvent, the endo-hydroxymethyl diastereomer 86 predominated (up to 9:1 at 15 °C for 3 h) but this ratio was inverted (1:9) in toluene at 45 °C, presumably reflecting kinetic vs. thermodynamic control, respectively. Lactams 86 and 87 have been converted previously into (±)-isoretronecanol and (±)-trachelanthamidine, respectively.
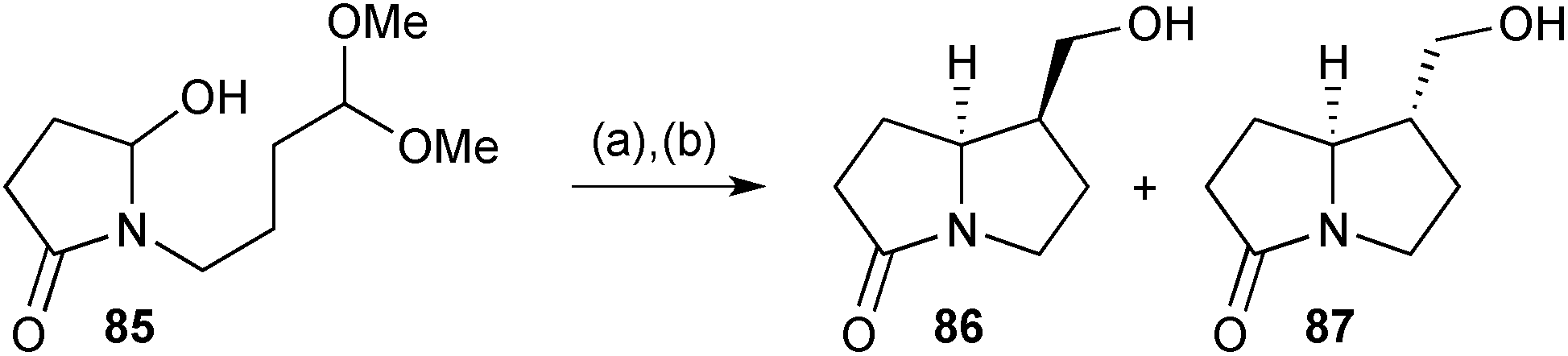 | ||
| Scheme 13 Reagents and conditions: (a) TsOH, CH3CN, 15 °C or PhCH3, 45 °C; (b) NaBH4, MeOH, 0 °C. [86/87 dr = 9:1 (CH3CN), 1:9 (PhCH3)]. | ||
The diene (not shown) formed by diastereoselective 1,4-addition of a chiral ammonia equivalent to enoate 88 (Scheme 14) and enolate allylation in situ, was converted into cycloheptylamine derivative 89 by ring-closing metathesis.79 The so-formed cis-1,2-aminoester was converted efficiently into the trans-isomer 90 by reversible enolisation under basic conditions. Alkene dihydroxylation was moderately selective for the face unencumbered by the amino substituent (dr = 80:20) but this was of no consequence since the next step cleaved this diol (91) to generate a dialdehyde. After amine deprotection, double reductive amination and ester reduction provided (−)-isoretronecanol 92. Alternatively, alkene epoxidation in diastereomeric substrate 89 gave hydroxylactone 93, with the initial epoxidation stereochemistry presumed to result from steric control. Reductive lactone cleavage followed by a parallel end-sequence to that used for (−)-isoretronecanol afforded (−)-trachelanthamidine 94. A variant of each route was also applied to the opposite ester 89/90 diastereomer to provide a second synthesis of both alkaloids.
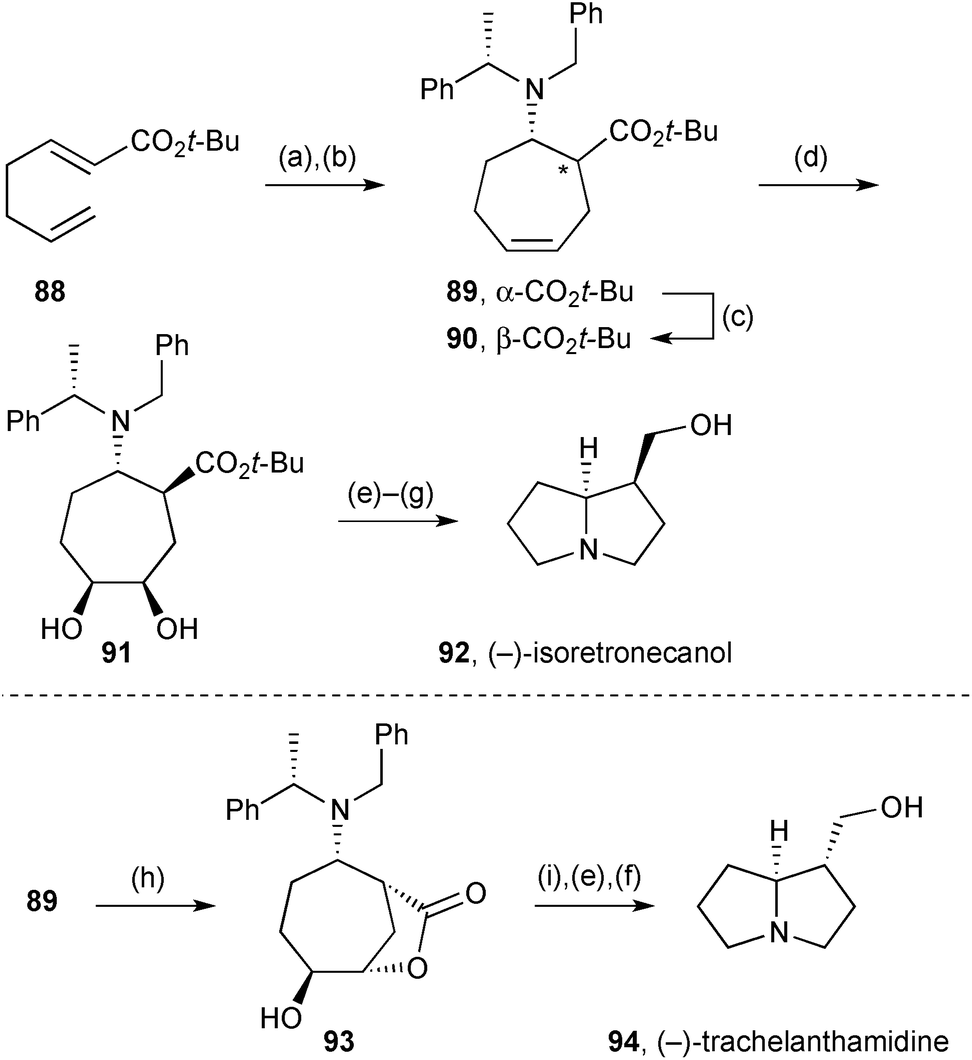 | ||
| Scheme 14 Reagents and conditions: (a) lithium (S)-N-benzyl-N-(α-methylbenzyl)amide, THF, −78 °C then allyl bromide; (b) Grubbs I, CH2Cl2, 30 °C; (c) KHMDS, t-BuOH, THF; (d) OsO4, TMEDA, CH2Cl2, −78 °C then P(CH2OH)3, Et3N, SiO2; (e) NaIO4, MeOH; (f) H2, Pd(OH)2/C, AcOH, MeOH; (g) DIBAL, THF, 0 °C; (h) HBF4, MCPBA, CH2Cl2; (i) LiAlH4, THF, 0 °C. | ||
Methodology developed for the stereoselective synthesis of γ- and δ-lactams was applied to (±)-isoretronecanol (lindelofidine).80 Proton transfer from sulfonyl anhydride 96 (Scheme 15) to imine 95, followed by Mannich-type addition, gave intermediate 97 with excellent diastereoselectivity (dr > 95:5). O- to N-Acyl transfer proceeded under the reaction conditions and esterification in situ gave lactam 98. The second ring was introduced by ring-closing metathesis, and reductive steps completed the route.
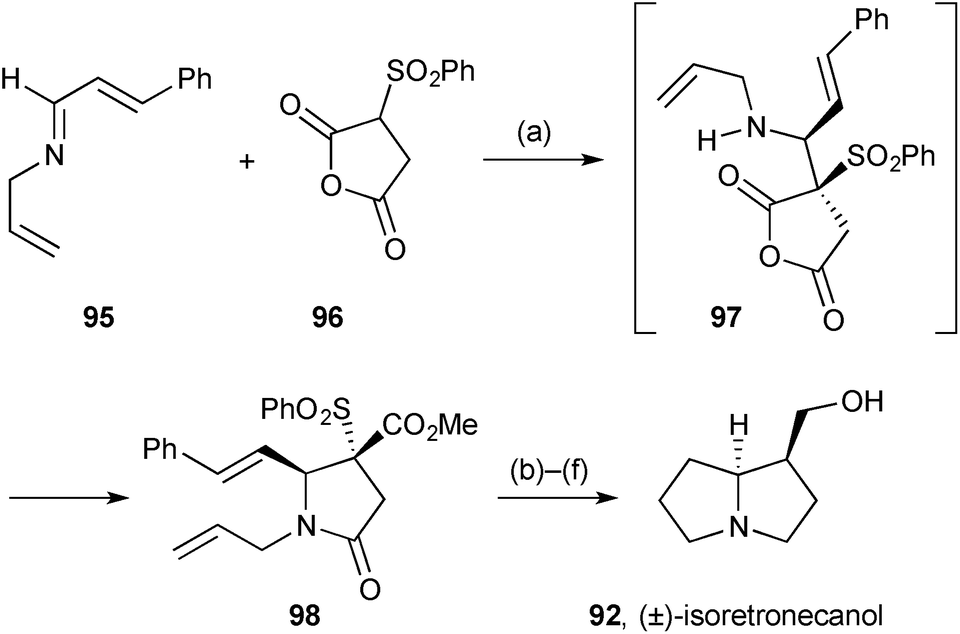 | ||
| Scheme 15 Reagents and conditions: (a) THF then Me3SiCHN2, MeOH, PhCH3, 0 °C; (b) Grubbs II, C6H6, reflux; (c) H2, Pd/C, MeOH; (d) DBU, CHCl3; (e) H2, Pd/C, MeOH; (f) LiAlH4. | ||
Davies' group described a synthesis of the (+)-enantiomer of isoretronecanol (that is, lindelofidine), along with indolizidine and quinolizidine analogues, again based on their chiral ammonia methodology.81 1,4-Addition of the lithiated chiral amine (step a, Scheme 16) to enoate 99 gave the adduct as a single diastereomer. This was not isolated but subjected to immediate Finkelstein reaction; the so-formed iodide cyclised under the reaction conditions with loss of the N-α-methyl-PMB group providing pyrrolidine 100 in 63% overall yield from 99. Ester enolate alkylation with a protected 2-hydroxyethyl electrophile proceeded with high diastereoselectivity, in accordance with the group's previous work, and from 101 completion of the synthesis required just three straightforward steps.
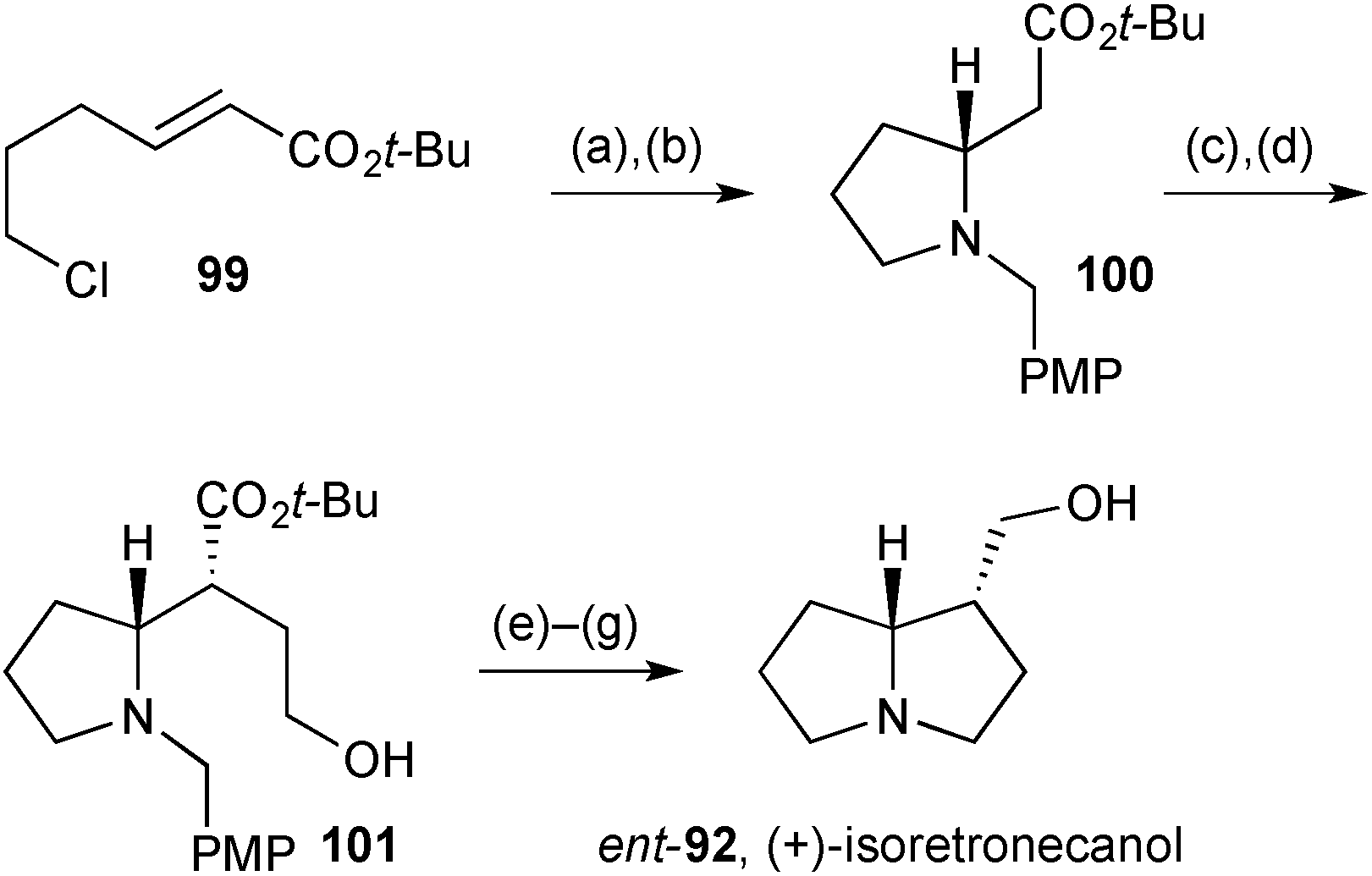 | ||
| Scheme 16 Reagents and conditions: (a) lithium (R)-N-p-methoxybenzyl-N-(α-methyl-p-methoxybenzyl)amide, THF, −78 °C; (b) NaI, CH3CN, reflux; (c) LiHMDS, THF, −78 °C then TBSO(CH2)2I, −78 °C → rt; (d) PPTS, MeOH, CH2Cl2, 50 °C; (e) I2, polymer-supported PPh3, CH3CN, PhCH3; (f) H2, Pd(OH)2/C, MeOH; (g) LiAlH4, THF, reflux. | ||
General methodology for the synthesis of 2,3-cis-disubstituted pyrrolidines was applied to the synthesis of racemic isoretronecanol.82 Ag(I)-catalysed azomethine ylid cycloaddition of iminonitrile 102 (Scheme 17) with methyl acrylate gave 2-cyanopyrrolidine 103 as the single endo-diastereomer shown. A novel procedure for reductive decyanation was developed that the authors proposed to proceed via borohydride mediated E2-type elimination of HCN and then reduction of the so-formed borane-complexed pyrroline with NaBH3CN generated in situ. The route was completed by lactamisation and reduction of both carbonyl groups.
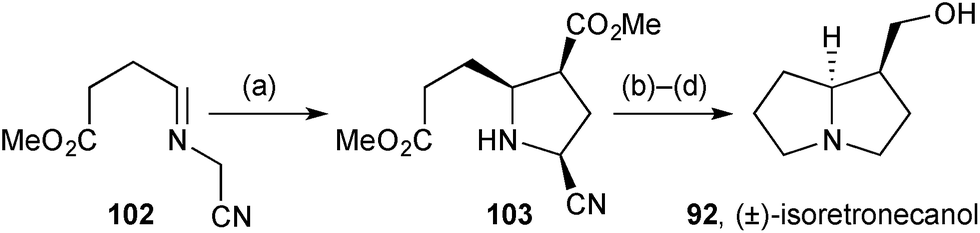 | ||
| Scheme 17 Reagents and conditions: (a) methyl acrylate, AgOAc, DBU, PhCH3, 0 °C; (b) NaBH4, BH3·THF, THF; (c) PhCH3, reflux; (d) LiAlH4, THF, reflux. | ||
Gavhane achieved a formal synthesis of (−)-isoretronecanol and (−)-trachelanthamidine based on Claisen rearrangement.83 Wittig reaction of N-Boc-(S)-prolinal 104 (Scheme 18) gave a mixture of diastereomers 105. These diastereomers were taken on into the thermal rearrangement step and a 7:1 ratio of aldehyde epimers (not shown) was obtained. The authors made no comment on the origin of this preferred stereochemistry nor its relation to the ∼5:1 E-/Z-ratio of enol ether diastereomers. Reduction to alcohol 106 and its epimer completed a formal synthesis that connects with Knight's 1997 route that employed a related, but poorly-stereoselective Ireland–Claisen rearrangement (dr = 1–2:1) as the key step.84
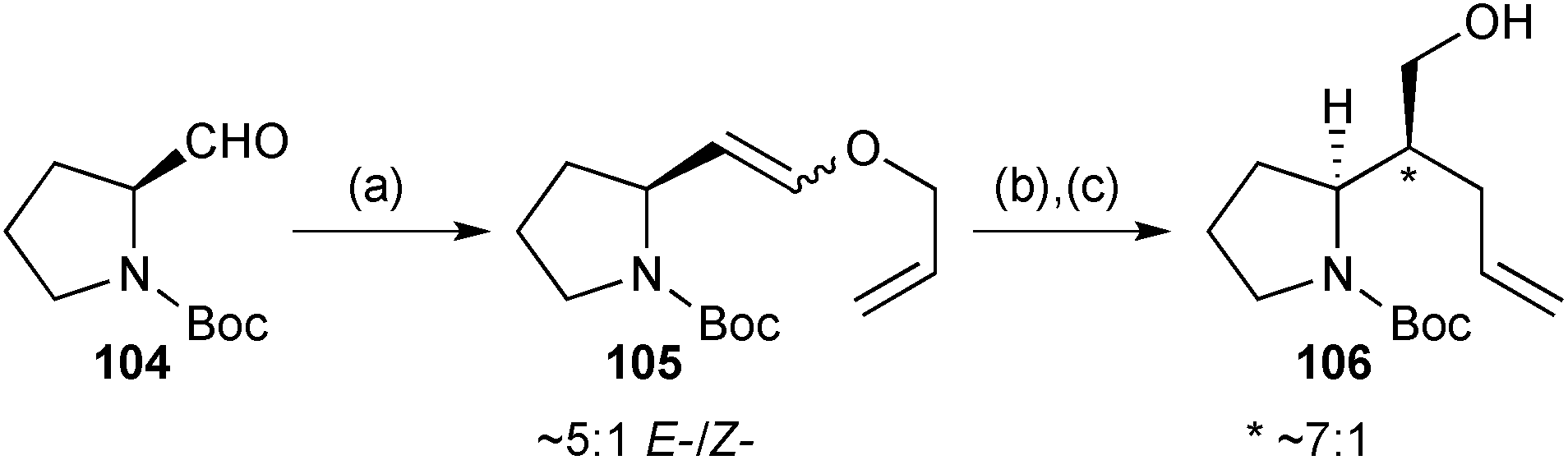 | ||
Scheme 18 Reagents and conditions: (a) Ph3P+CH2OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 Cl−, t-BuOK, PhCH3, 0 °C; (b) C6H6, 80 °C; (c) NaBH4, MeOH, 0 °C. CH2 Cl−, t-BuOK, PhCH3, 0 °C; (b) C6H6, 80 °C; (c) NaBH4, MeOH, 0 °C. | ||
Njardarson reported85 an asymmetric variant of methodology for 2-arylpyrroline synthesis originally described by Steel.86 In one application, addition of the dienolate derived from ethyl bromocrotonate to chiral sulfinylimine 107 (Scheme 19) afforded the (2S)-alkylpyrroline 108 as a single diastereomer. Acidic hydrolysis of the N-sulfinyl group and cyclisation of the resulting free amine completed the pyrrolizidine core. Reduction of the ester gave (−)-supinidine 109, six steps overall from butane-1,4-diol and the shortest asymmetric synthesis of this alkaloid; exo-face hydrogenation of the alkene gave (−)-isoretronecanol 92.
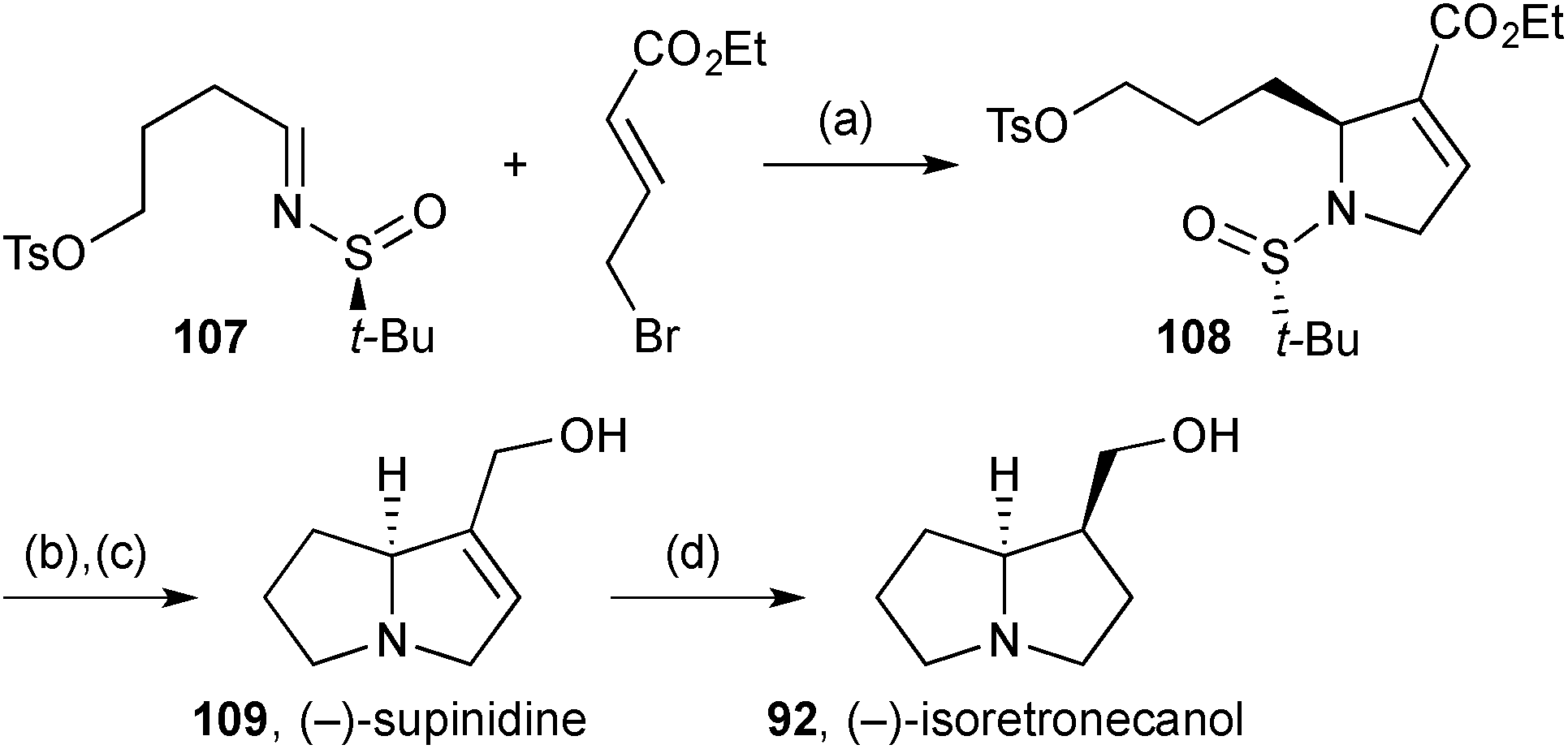 | ||
| Scheme 19 Reagents and conditions: (a) LDA, THF, −78 °C; (b) TMSCl, aq. MeOH then Et3N, CH2Cl2; (c) DIBAL, CH2Cl2, 0 °C; (d) H2, Pd/C, CH2Cl2. | ||
Access to the necine base core of (+)-petasinine 114 (Scheme 20), petasinecine [(2R)-hydroxy-(−)-isoretronecanol], was achieved in an enantiospecific route from (S)-proline.87 The key step in the sequence, aza-Claisen rearrangement of ketene adduct 111, gave amide 112 as a single stereoisomer. Boc deprotection, lactamisation, and ozonolysis of the vinyl side chain gave O-phenyl petasinecine 113.
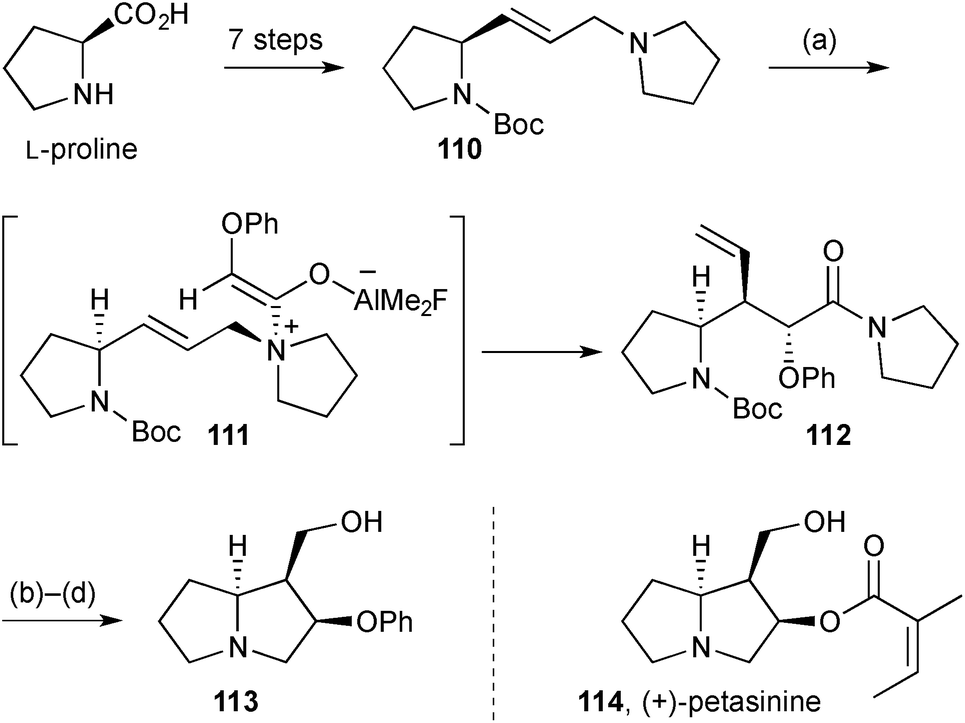 | ||
| Scheme 20 Reagents and conditions: (a) α-phenoxyacetyl fluoride, AlMe3, Na2CO3, CH2Cl2, 0 °C → rt; (b) SOCl2, MeOH, reflux; (c) O3, MeOH, −78 °C then NaBH4, MeOH, −78 °C; (d) BH3·SMe2, THF, 0 °C → rt. | ||
3.2 Simple hydroxypyrrolizidines
The epoxyaldehyde (R,R)-115 (Scheme 21), derived in three steps from cis-but-2-en-1,4-diol, was the starting point for enantioselective syntheses of (1R,7aS)-1-hydroxypyrrolizidine 118 and (1S,2S,7aS)-1,2-dihydroxypyrrolizidine 120.88 The 1,7a-relative stereochemistry in both pyrrolizidines was established by diastereoselective allylation of the benzylimine derived from aldehyde 115; cyclisation then followed under standard Appel halogenation conditions. From intermediate 116, the routes diverged, with epoxide reduction (step e) leading, ultimately, to the monohydroxylated product 118. The trans-diol motif in 120 was established by Lewis acid mediated epoxide alcoholysis (→119), with straightforward deprotection and cyclisation steps completing the route. In both routes, regioselective epoxide-opening may be considered to proceed via SN2-like delivery of hydride or benzyl alcohol at the less sterically encumbered 4-position, which also results in the least reorganisation of the pyrrolidine ring conformation.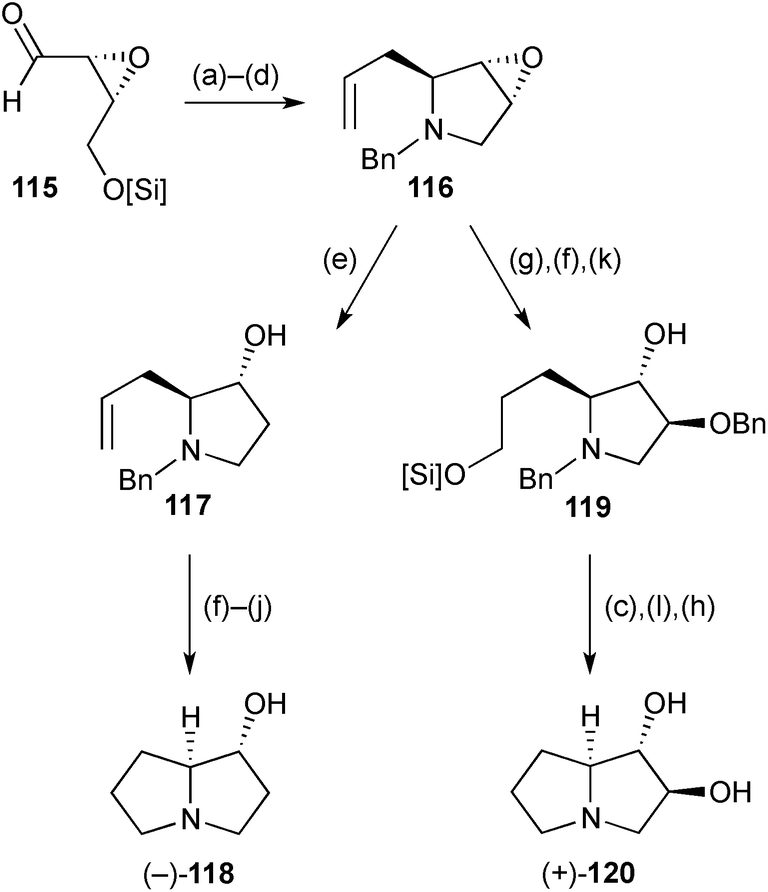 | ||
| Scheme 21 Reagents and conditions: (a) BnNH2, Et2O; (b) CH2CHCH2MgBr, BF3·OEt2, Et2O, −78 °C; (c) NH4F·HF, MeOH; (d) PPh3, CCl4, Et3N, DMF; (e) LiAlH4, Et2O; (f) TBDPSCl, imidazole, DMF; (g) 9-BBN, THF then NaOH, aq. H2O2; (h) NH4+HCO2−, Pd/C, MeOH, reflux; (i) PPh3, CCl4, Et3N, DMF; (j) TBAF, THF; (k) BnOH, Yb(OTf)3, dioxane, 80 °C; (l) TsCl, pyridine, CH2Cl2. [Si] = TBDPS. | ||
Two further syntheses of the lower homologue of lentiginosine, (1S*,2S*,7aS*)-dihydroxypyrrolizidine 120, were reported during the review period. The first, of the (+)-enantiomer (Scheme 22), began with imide formation from L-(+)-tartaric acid, O-silylation, and sulfide oxidation to give tartarimide derivative 122.89 α-Sulfinyl anion addition anti-to the adjacent TBSO-substituent afforded a mixture of inseparable C(7)- and S-stereoisomers 123. Following reductive cleavage of the sulfinyl group, treatment with LiAlH4 achieved stereoselective reduction of the C(7a)-hydroxy group, reduction of the lactam, and desilylation of the hydroxy groups. The stereoselectivity of the reduction at C(7a) was proposed to arise from pseudoaxial delivery of hydride to the N-acyl iminium in a conformation with pseudoequatorial TBSO-substituents in the pyrrolidine ring.
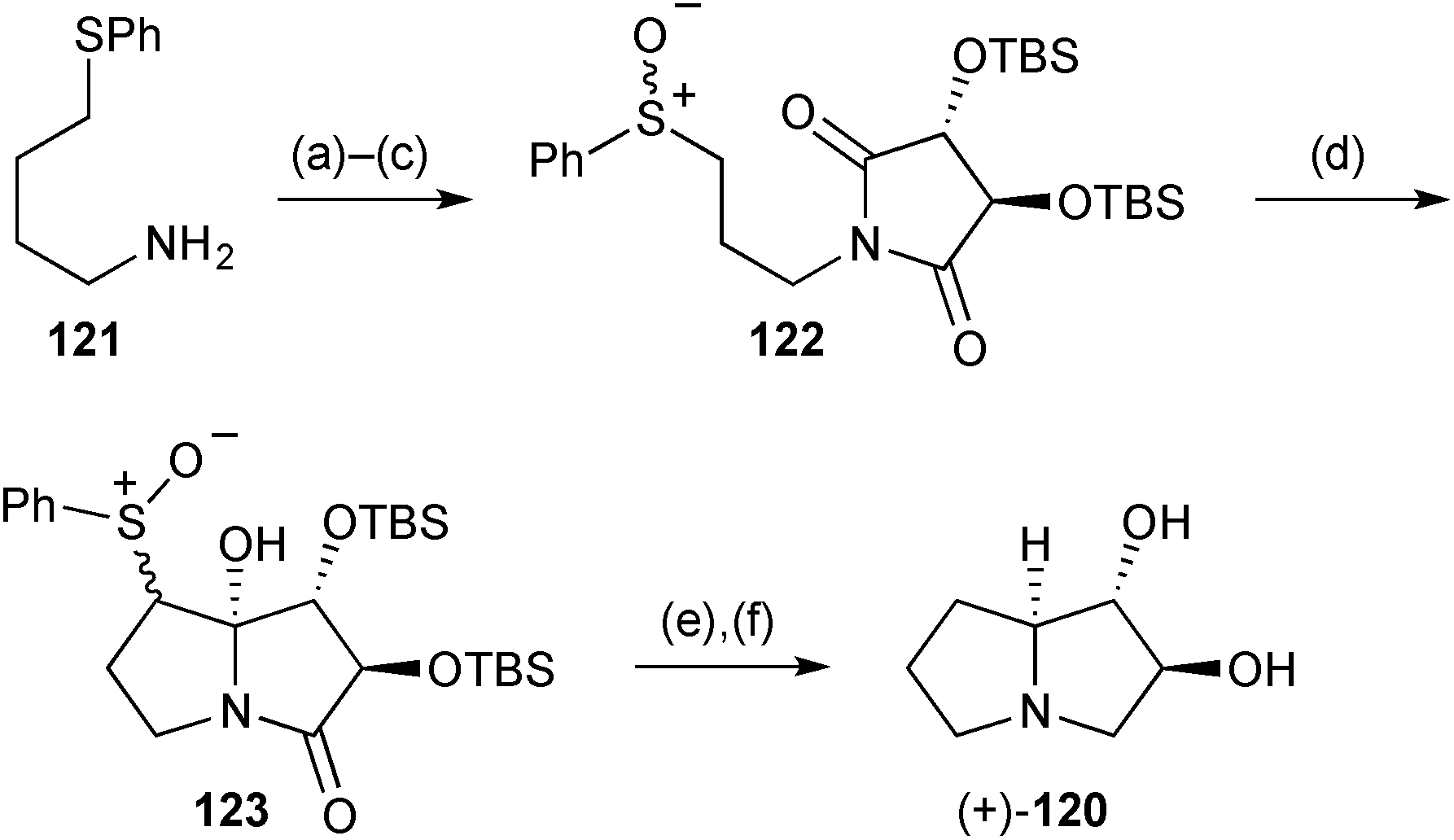 | ||
| Scheme 22 Reagents and conditions: (a) L-(+)-tartaric acid, xylene, reflux; (b) TBSCl, imidazole, DMF, 0 °C → rt; (c) NaIO4, aq. MeOH, 0 °C → rt; (d) LiHMDS, THF, −78 °C; (e) NiCl2·6H2O, NaBH4, aq. MeOH; (f) LiAlH4, THF, reflux. | ||
Kumar's synthesis of the (−)-enantiomer (Scheme 23)90 was elaborated from the hydrazide (not shown) formed by organocatalytic amination of aldehyde 124. Enoate 125 was obtained with ‘94% enantioselectivity’ after HWE olefination in situ using Masamune–Roush conditions. The final stereocentres were then introduced by Sharpless asymmetric dihydroxylation although there is some disparity between the synthetic scheme in the paper, where (DHQD)2PHAL is cited as ligand system, and the text which refers to (DHQD)2AQN. Regardless, the relative stereochemical outcome in this step (dr = 25:75 in favour of that shown in 126) is counter to the inherent 83:17 ratio (in favour of the syn,syn-isomer) obtained for a homologue of 125. Sulfonylation of the 1°-hydroxyls, and hydrogenolysis to reveal the free amine, led to double cyclisation to complete the synthesis.
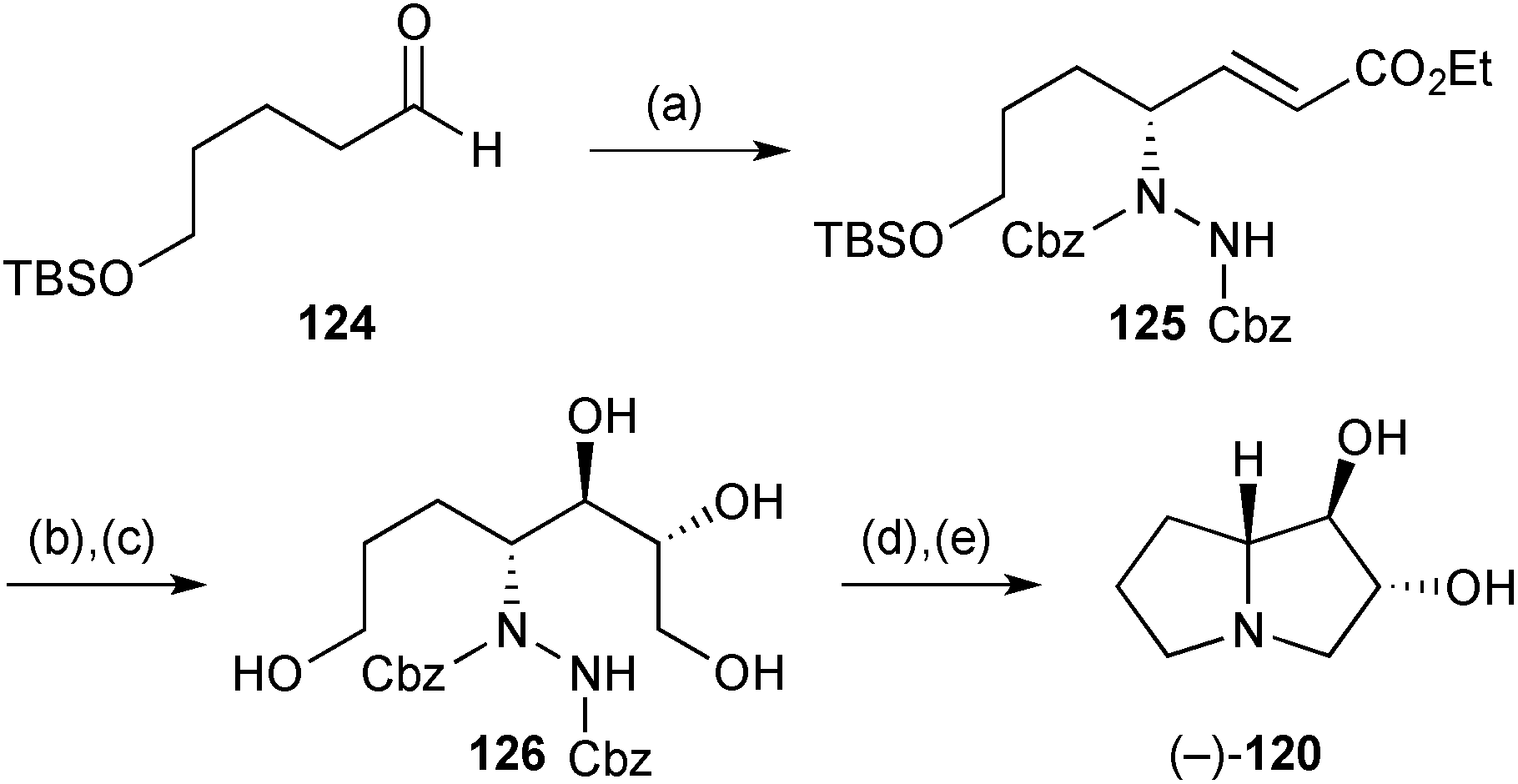 | ||
| Scheme 23 Reagents and conditions: (a) dibenzyl azodicarboxylate, (S)-proline (8 mol%), CH3CN, 0 °C → 10 °C then add LiCl, (EtO)2POCH2CO2Et, DBU, 5 °C; (b) OsO4, K3Fe(CN)6, K2CO3, (DHQD)2AQN, MsNH2, aq. t-BuOH, 0 °C; (c) LiBH4, THF, 0 °C; (d) TsCl, Et3N, CH2Cl2, 0 °C → rt; (e) H2, RANEY® Ni, MeOH then EtOH, 55 °C. | ||
Both enantiomers of a 6-hydroxy derivative of the lentiginosine homologues described above were prepared in short sequences from commercially available 4-hydroxyproline.91 Thus, 4-hydroxyprolinal 127 (Scheme 24) was prepared in three steps from the (R,R)-enantiomer and subjected to Morita–Baylis–Hillman addition of methyl acrylate. The almost complete stereoselectivity in this reaction (step a) was explained by a Felkin–Anh approach to the aldehyde in a conformation stabilised by H-bonding to the hydroxy group. Release of the free amino group, lactamisation, and ozonolysis with a reductive work-up gave the trans-1,2-diol stereochemistry present in the final product (+)-129. Lactam reduction completed the route and the sequence was repeated from (4S)-hydroxy-(S)-proline to give (−)-129.
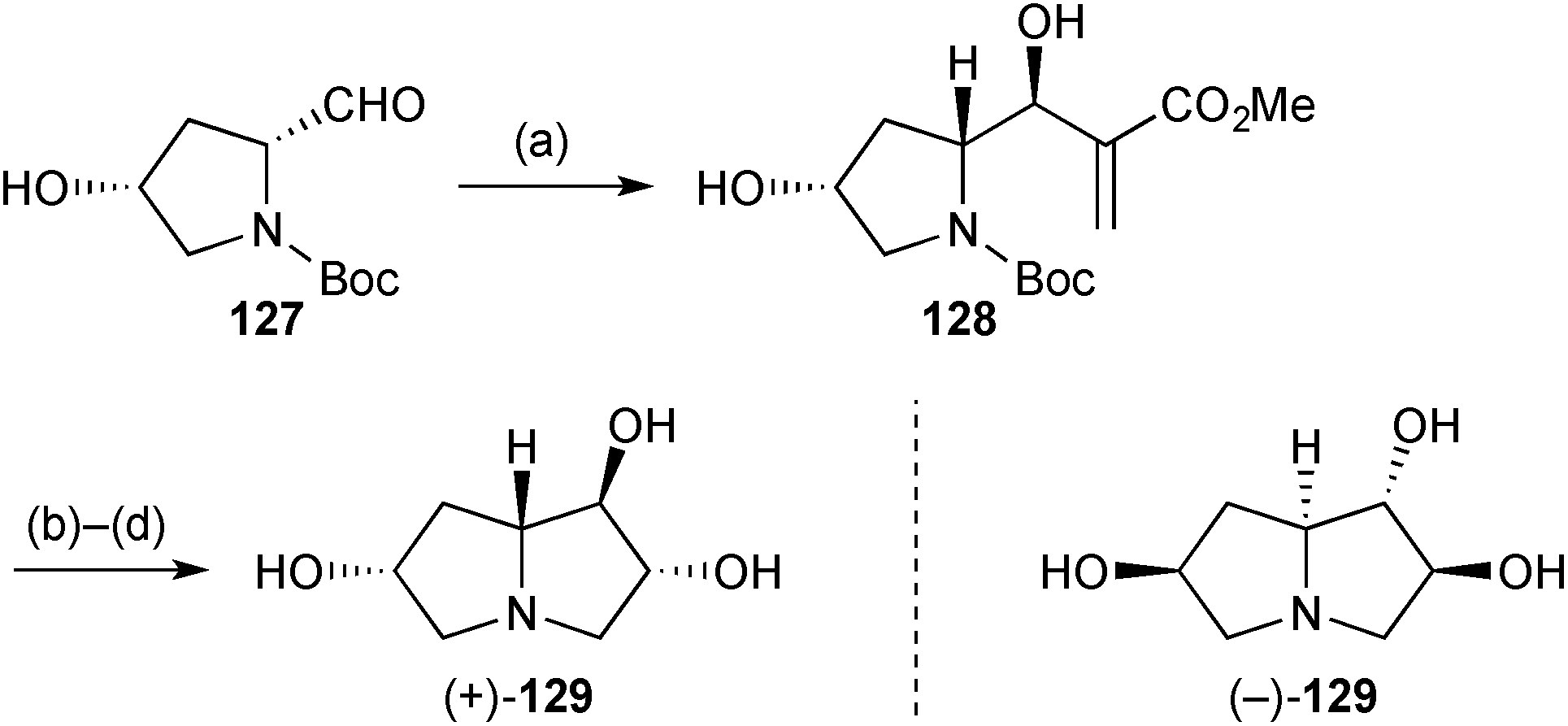 | ||
| Scheme 24 Reagents and conditions: (a) methyl acrylate, DABCO, sonication; (b) aq. HCl, PhCH3, 0 °C then aq. NaOH, 0 °C; (c) O3, MeOH, CH2Cl2, −78 °C then NaBH4, −78 °C → rt; (d) LiAlH4, AlCl3, THF, reflux. | ||
Two syntheses of simple tetrahydroxy pyrrolizidines have been described during the review period, the first being an enantiospecific route from L-erythrose.92 α-Lithio-1-[2-(trimethylsilyl)ethoxy]allene added to the exo-face of nitrone 130 (Scheme 25) then (presumably) acid-catalysed 6-endo-trig O-cyclisation followed via intermediate 131. The resulting cyclic enol ether 132 was hydroborated on the less hindered face and oxidised to afford alcohol 133 with all stereocentres set. Formation of the pyrrolizidine ring system was achieved by protection of the free hydroxy group, N–O reduction with Sm(II), then N-cyclisation via the mesylate. The final product 134 had been shown previously to be an inhibitor of amyloglucosidase from Rhizopus sp.
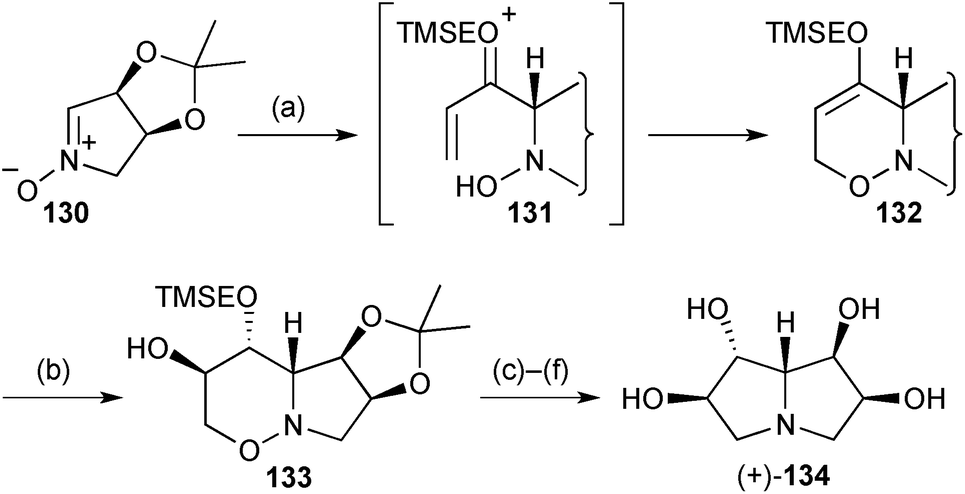 | ||
| Scheme 25 Reagents and conditions: (a) 2-[(trimethylsilyl)ethoxy]allene, BuLi, THF, −78 °C then MgSO4; (b) BH3·THF, THF, −30 °C → rt then aq. NaOH, H2O2, −10 °C → rt; (c) TBDPSCl, imidazole, DMAP, CH2Cl2, 0 °C → rt; (d) SmI2, THF; (e) MsCl, pyridine, 0 °C → rt; (f) Dowex-50, EtOH, 65 °C. | ||
In the second synthesis, the 6,7-di-epi-diastereomer of pyrrolizidine 134 was prepared in ∼20 steps from L-ascorbic acid (Scheme 26).93 Allylic trichloroacetimidate 136 was prepared by routine redox transformations and protecting group steps. This, the corresponding O-benzyl ether, and the free alcohol were studied as substrates for both thermal and Pd(II)-catalysed Overman rearrangement to establish the 7a-stereogenic centre. Among these, only the TBS derivative 136 gave a single 7a-diastereomer and the yield was much improved in the Lewis acid catalysed reaction at room temperature (81%) compared with heating at reflux in xylene (23%). From allylic amine 137 the first ring was constructed by RCM then, after dihydroxylation and acetonide protection, the pyrrolizidine was completed by regioselective O-sulfonylation then global deprotection. Purification via the tetracetate gave the target pyrrolizidine with data in accordance with those obtained by Robina's group as described in the previous review.
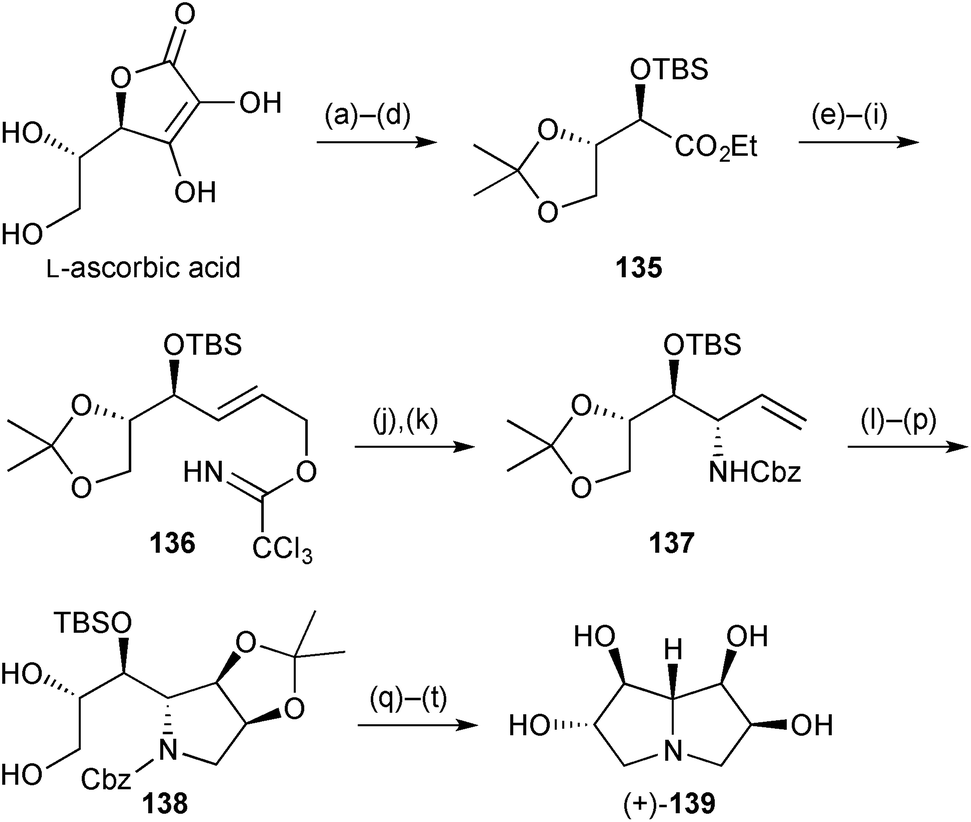 | ||
| Scheme 26 Reagents and conditions: (a) CuSO4, acetone; (b) aq. H2O2, K2CO3; (c) EtI, CH3CN, reflux; (d) TBSCl, imidazole, DMF; (e) DIBAL, CH2Cl2, −10 °C; (f) IBX, DMSO; (g) Ph3PCHCO2Et, CH2Cl2, reflux; (h) DIBAL, CH2Cl2, −30 °C; (i) Cl3CCN, DBU, 0 °C; (j) PdCl2(CH3CN)2, p-benzoquinone, PhCH3; (k) NaOH, aq. THF, 65 °C then CbzCl, RT; (l) NaH, allyl bromide, TBAI, DMF; (m) Grubbs I, CH2Cl2; (n) OsO4, NMO, t-BuOH, aq. acetone; (o) Me2C(OMe)2, TsOH, CH2Cl2; (p) Zn(NO3)2·6H2O, CH3CN, 50 °C; (q) Bu2SnO, TsCl, Et3N, CH2Cl2; (r) H2, Pd(OH)2/C, MeOH then aq. HCl; (s) Ac2O, pyridine; (t) aq. NH3, MeOH. | ||
3.3 Rosmarinecines
Chakraborty's synthesis of 2-epi-(−)-rosmarinecine 145 (Scheme 27) featured Nugent–Rajanbabu–Gansäuer epoxide reductive radical cyclisation of vinylogous carbamate 141.94 This reaction gave trisubstituted pyrrolidine 142 apparently as a single diastereomer. The authors rationalise the outcome as resulting from a Beckwith–Houk transition state assembly with a pseudoaxial (bulky) silyloxy substituent. The C(7)-hydroxy group stereochemistry was set by diastereoselective allylation with allyltributylstannane and Lewis acid activation of the aldehyde (step m); no diastereoselectivity was observed using allylmagnesium bromide. Despite the efficient construction of the first ring (step h), the overall route is long (∼27 steps from L-ascorbic acid), in part due to the >10 protecting group manipulations throughout the synthesis.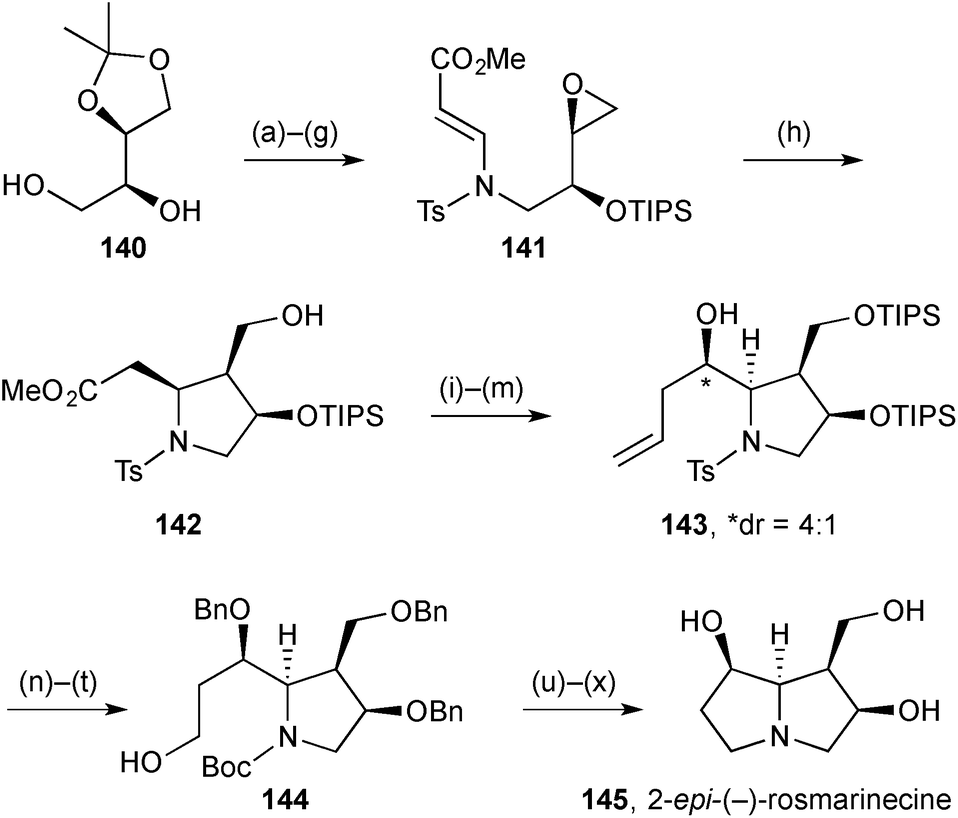 | ||
| Scheme 27 Reagents and conditions: (a) Bu2SnO, Et3N, TsCl, CH2Cl2, 0 °C; (b) TIPSOTf, Et3N, CH2Cl2, 0 °C; (c) TsNH2, KOH, DMSO, 80 °C; (d) methyl propiolate, NMM, CH2Cl2; (e) aq. AcOH, 100 °C; (f) Bu2SnO, Et3N, TsCl, CH2Cl2, 0 °C → rt; (g) NaH, DMF, 0 °C; (h) Cp2TiCl2, Zn, ZnCl2, THF, −20 °C → rt; (i) TIPSOTf, Et3N, CH2Cl2, 0 °C; (j) DIBAL, CH2Cl2, −78 °C → 0 °C; (k) TBSCl, DBU, CH2Cl2; (l) O3, pyridine, MeOH, CH2Cl2, −78 °C then PPh3; (m) allyl-SnBu3, BF3·OEt2, CH2Cl2, −78 °C; (n) Na, naphthalene, THF, −78 °C; (o) Boc2O, Et3N, CH2Cl2; (p) TBAF, THF; (q) BnBr, Bu4N+I−, DMF, THF; (r) OsO4, NMO, PhCH3, aq. acetone; (s) NaIO4, aq. THF, 0 °C; (t) NaBH4, MeOH, 0 °C; (u) TsCl, Et3N, DMAP, CH2Cl2; (v) TFA, CH2Cl2, 0 °C; (w) K2CO3, EtOH, reflux; (x) H2, Pd(OH)2, MeOH. | ||
3.4 Hyacinthacines and their analogues
The more heavily hydroxylated PAs, notably the hyacinthacines, casuarines, and australines, continue to attract attention as targets to highlight stereoselective synthetic methodology and, in combination with their analogues, for biological screening, especially as glycosidase inhibitors.
D-Ribose was used as starting material for a formal synthesis of (+)-3,7a-di-epi-hyacinthacine A1146 [= (+)-2-epi-hyacinthacine A2].95 Davies provided full details96 of syntheses of (−)-hyacinthacine A1, (−)-7a-epi-hyacinthacine A1, (−)-hyacinthacine A2, and (−)-1-epi-alexine 147–150 that were covered in the previous review. Delair and Greene applied their approach from Stericol® as a chiral auxiliary, as reviewed previously, to (+)-hyacinthacine B1151 and (+)-hyacinthacine B215297 and, later, to (+)-hyacinthacine A6153 and (+)-hyacinthacine A7154.98
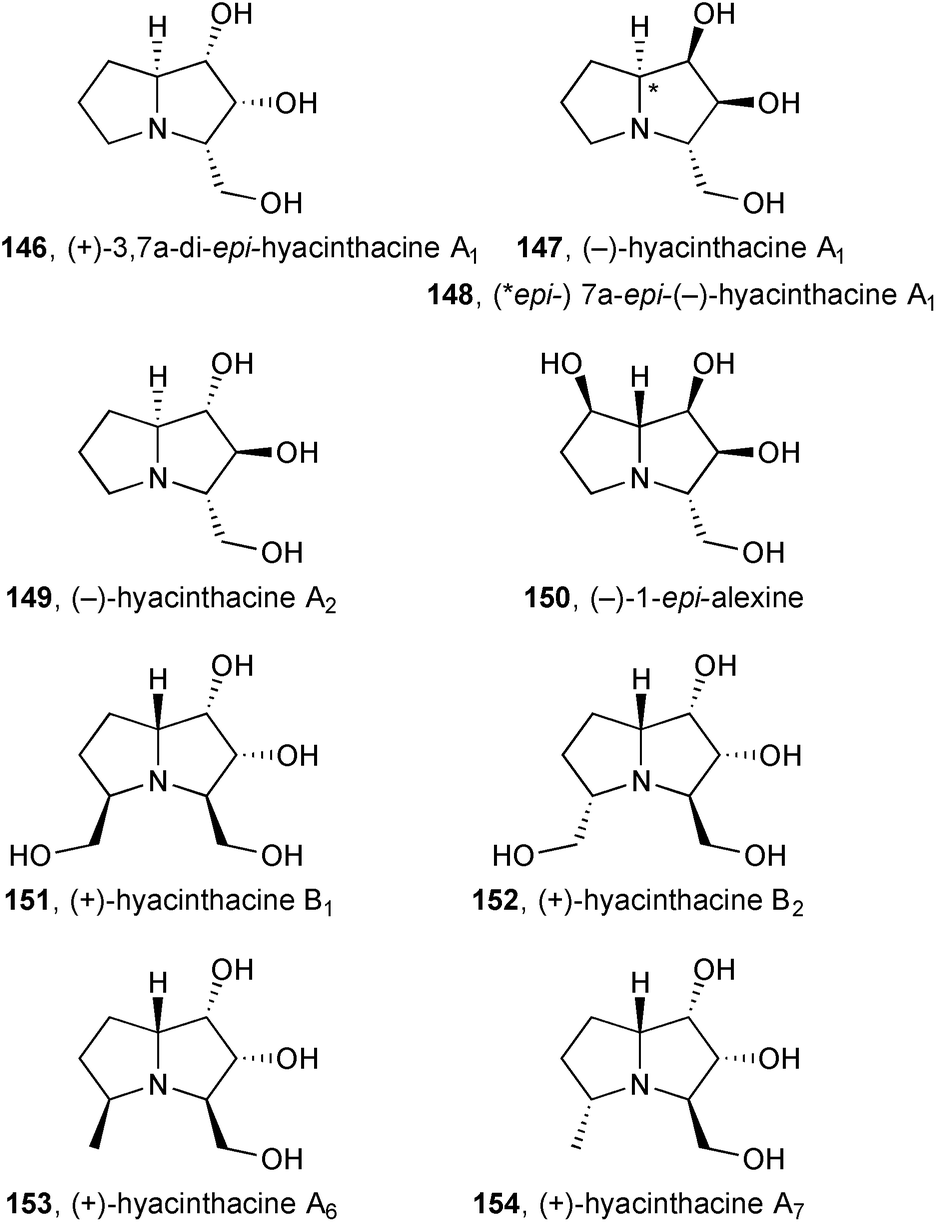
Subsequently, the group reported the synthesis of (+)-hyacinthacine A2ent-149 (Scheme 28).99 Pyrrolizidinone 155, that was an intermediate for the synthesis of (+)-hyacinthacine A1ent-147 (summarised in the previous review), was epoxidised and hydrolysed to give the 1,2-trans-diol functionality. Fleming–Tamao oxidation of the silyl substituent and lactam reduction completed the route.
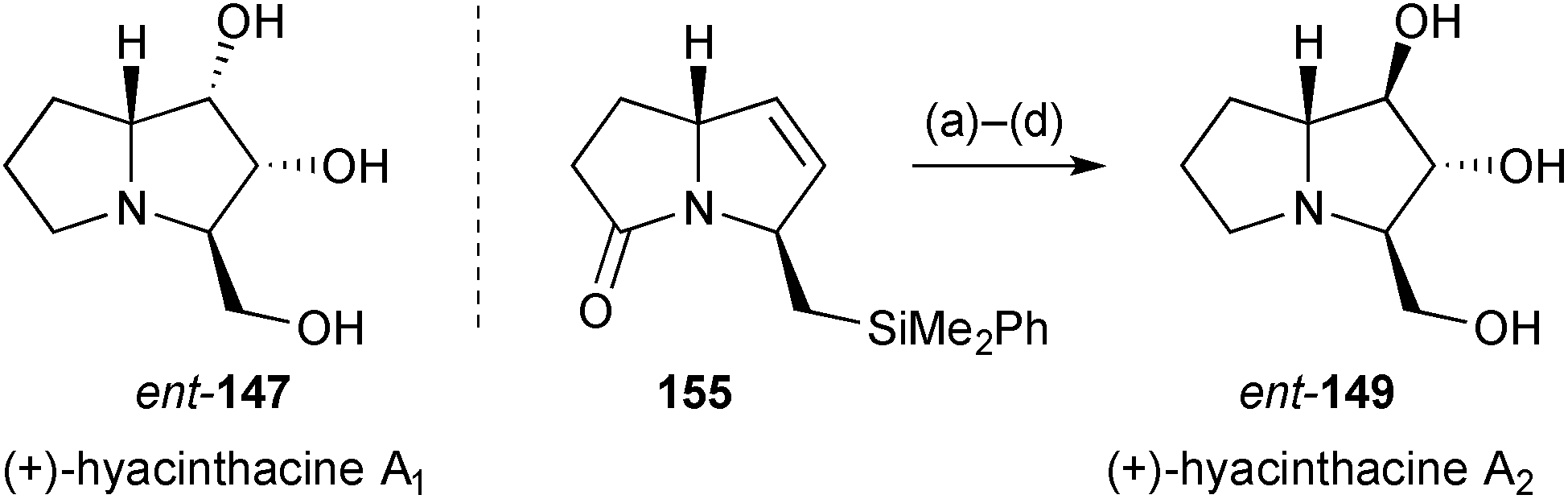 | ||
| Scheme 28 Reagents and conditions: (a) CF3COCH3, Oxone®, EDTA, Na2CO3, CH3CN; (b) CF3CO2H, aq. THF, 70 °C; (c) HBF4·OMe2, CH2Cl2 then KF, MCPBA, DMF; (d) BH3·SMe2, THF. | ||
Concise syntheses of (+)-2-epi-hyacinthacine A2146 and (−)-3-epi-hyacinthacine A1159 (Scheme 29) were achieved from (S)-glutamic acid via aldehyde 156.100 Reagent controlled organocatalytic aldol addition of 2,2-dimethyl-1,3-dioxan-5-one to this aldehyde with the (S)- and (R)-enantiomers of the proline catalyst resulted in diastereomers 157 and 158, respectively, the former existing predominantly in the hemiaminal form shown. These two intermediates were then taken separately through short sequences of deprotection and reductive amination to provide the target molecules.
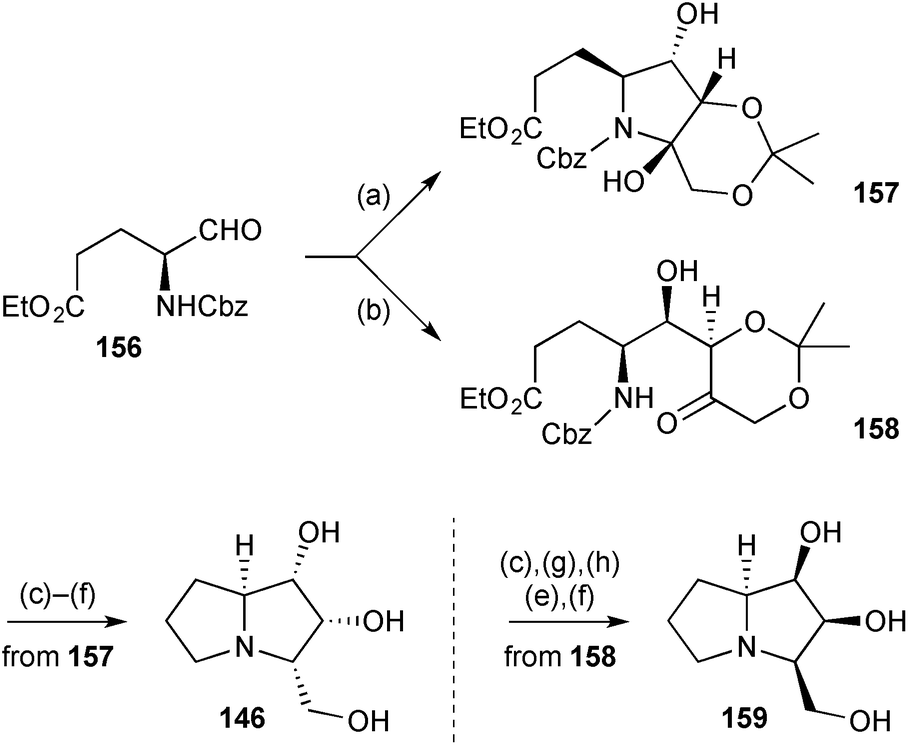 | ||
| Scheme 29 Reagents and conditions: (a) 2,2-dimethyl-1,3-dioxan-5-one, (S)-proline, DMF [dr = 6:1]; (b) 2,2-dimethyl-1,3-dioxan-5-one, (R)-proline, DMF [dr = 10:1]; (c) H2, Pd/C, EtOH; (d) K2CO3, EtOH; (e) LiAlH4, THF, reflux; (f) aq. HCl, MeOH; (g) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C; (h) TBAF, THF. | ||
A synthesis of 2-epi-hyacinthacine A1160 (Scheme 30) was achieved from D-arabinose with the C(5)–C(7) carbons being introduced by iminium allylation and subsequent hydroboration.101
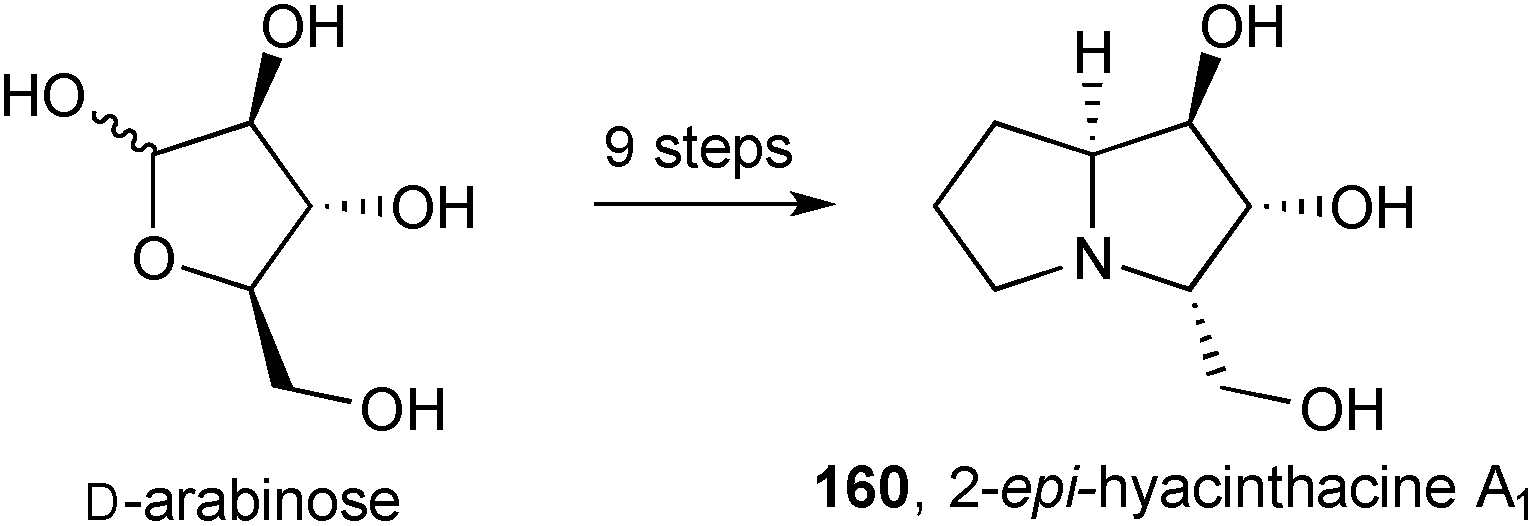 | ||
| Scheme 30 Enantiospecific synthesis of 2-epi-hyacinthacine A1. | ||
Pyne described an improved method for the preparation of key intermediate 162 (Scheme 31) that has been used for the synthesis of several pyrrolizidines. The new route dispensed with the need for a vinyl sulfone starting material by switching from (DHQD)2PHAL to (DHQD)2PYR as ligand system for the dihydroxylation reaction (step a).102 This intermediate was then employed in the synthesis of six hyacinthacine isomers using routes analogous to those detailed in the previous review. The authors concluded that the structures of hyacinthacines B3166, B4170, and B5169 are correctly reported in the earlier literature but hyacinthacine B7168 is incorrectly assigned; they further suggest that natural hyacinthacines B5 and B7 are the same compound, although this proposal could not be proven.
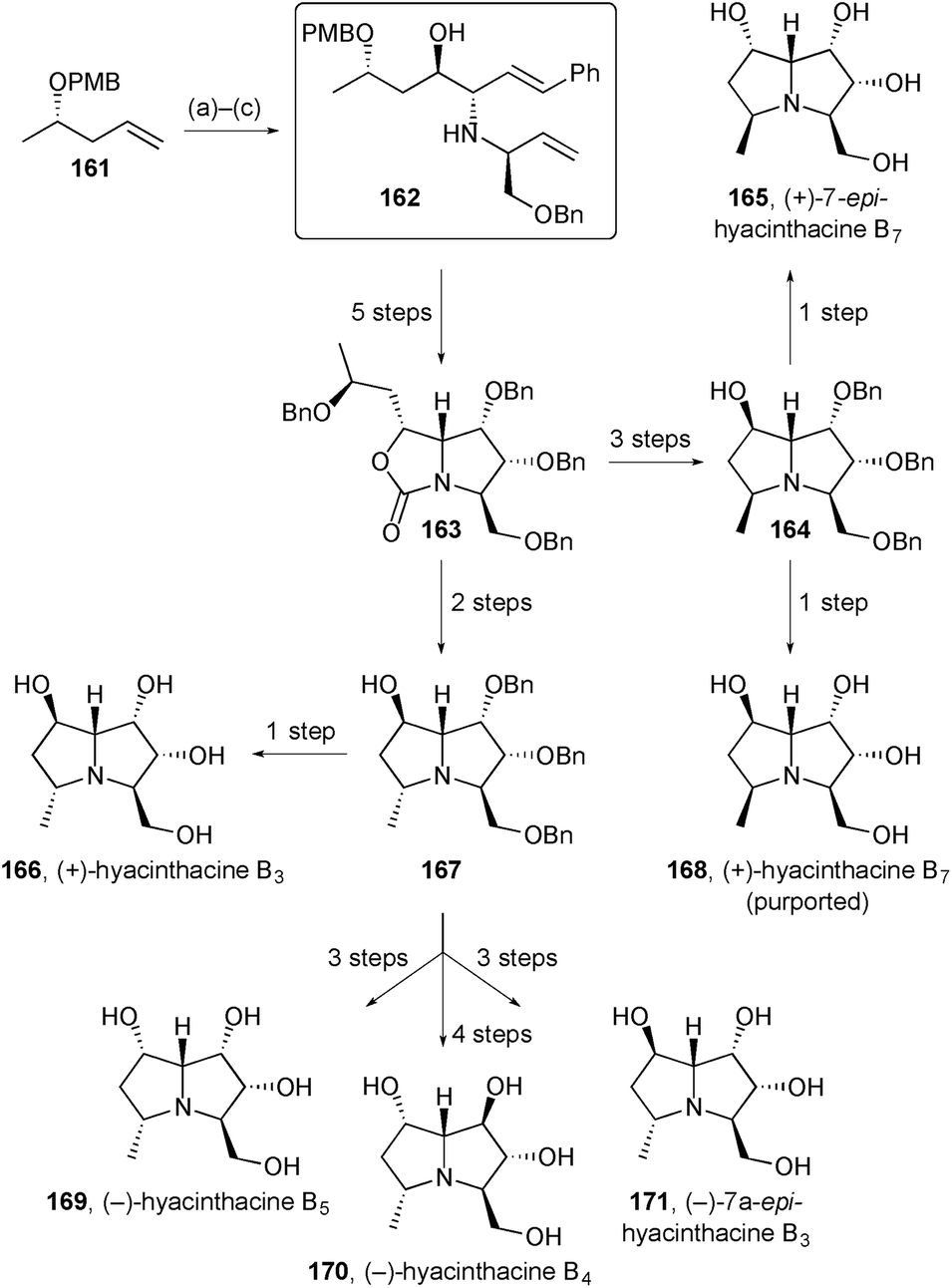 | ||
| Scheme 31 Reagents and conditions: (a) K2OsO4, K3Fe(CN)6, MsNH2, (DHQD)2PYR, aq. t-BuOH; (b) TEMPO, NaOCl, aq. NaHCO3, CH2Cl2; (c) styryl boronic acid, (S)-1-(benzyloxy)but-3-en-2-amine, CH2Cl2. | ||
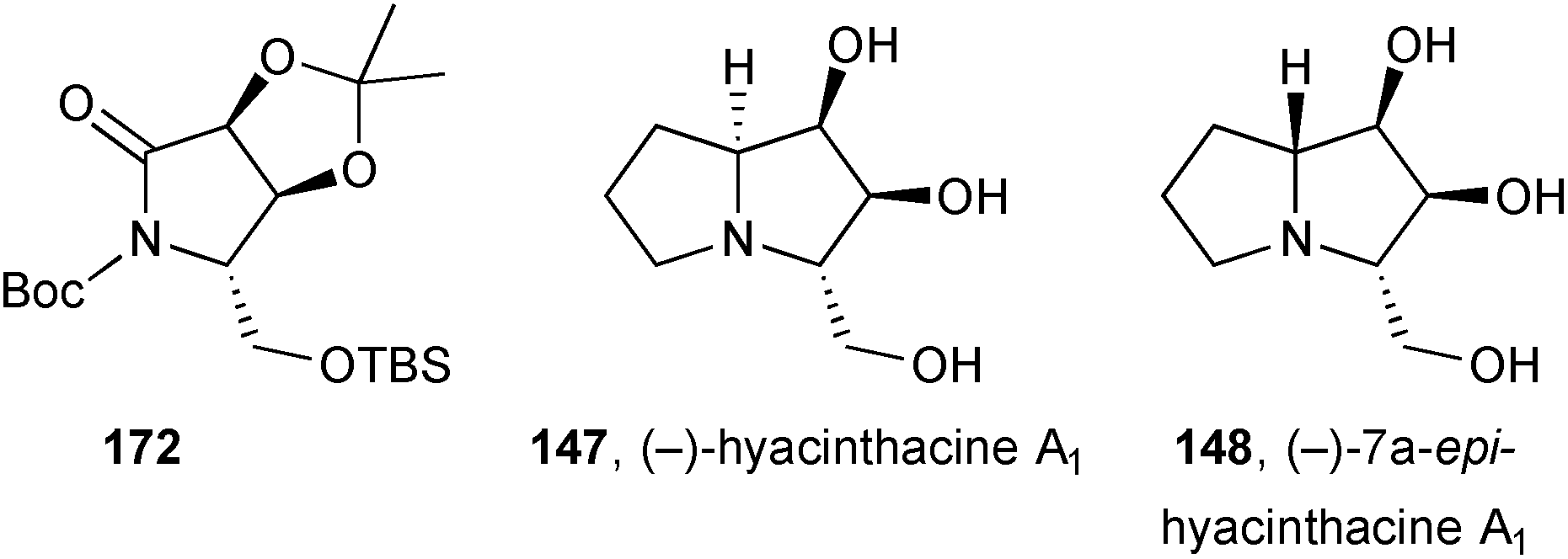
(−)-Hyacinthacine A1147 and its 7a-epimer 148 were prepared from lactam 172 obtained from D-glutamic acid.103 Allyl addition to an iminium ion at pro-C(7a) set the stereochemistry for 7a-epi-hyacinthacine A1. For hyacinthacine A1 itself, lactam 172 was allylated directly giving a ketone which was then reduced and the desired alcohol cyclised via the mesylate.
Britton reported a general approach to the asymmetric synthesis of iminosugars based on tandem proline-catalysed α-chlorination then aldol reaction with dihydroxyacetone derivative 174 to establish a 2,3-dihydroxy-4-chlorocarbonyl stereotriad.104 Two pyrrolizidines were prepared in short routes (Scheme 32). The first, from aldehyde 173, gave ammonium salt 176 in ∼98% ee, via amine 175. Hydrogenolysis and acetonide hydrolysis completed the five step route to (+)-7a-epi-hyacinthacine A1 with 43% overall yield. Alternatively, starting with protected ketoaldehyde 177 and effecting the second cyclisation by reductive amination, (+)-3-epi-hyacinthacine A5 was obtained in 35% overall yield, again with ∼98% ee.
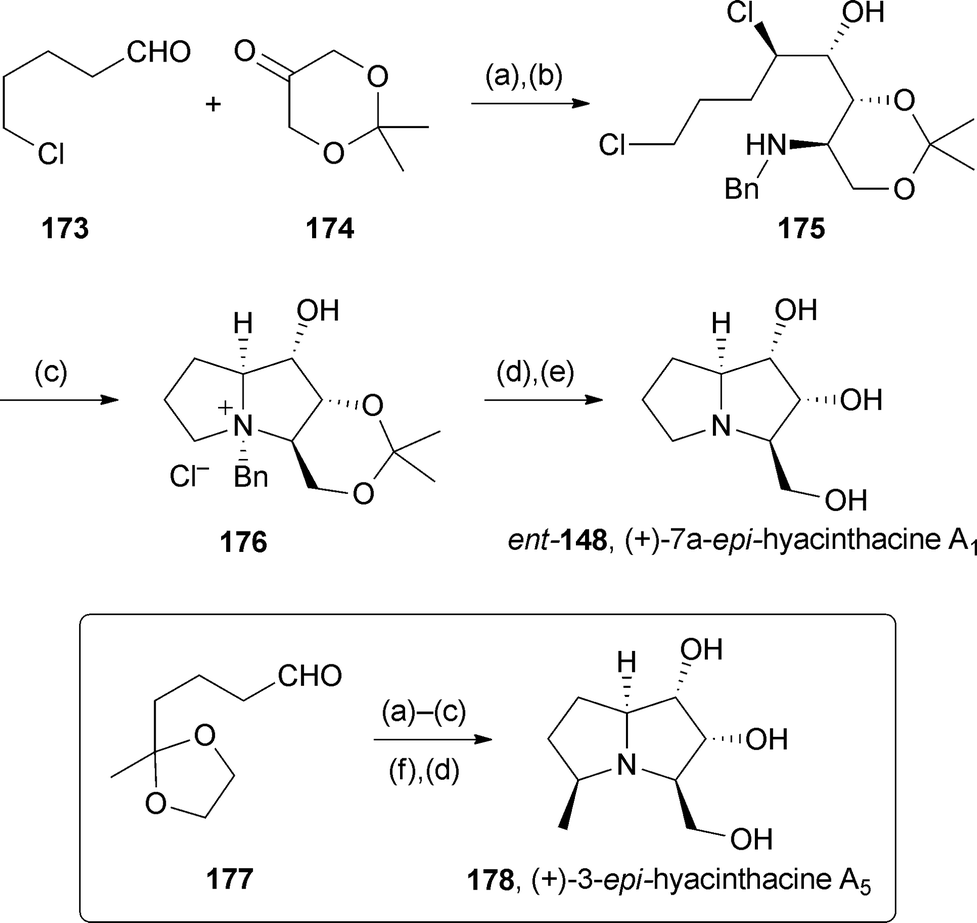 | ||
| Scheme 32 Reagents and conditions: (a) NCS, (S)-proline, CH2Cl2; (b) BnNH2, AcOH, 4 Å MS, THF then NaBH3CN; (c) NaHCO3, PhCH3, 105 °C; (d) H2, MeOH, 60 °C (H-Cube) [then for 178 Dowex 1X8-100 (HO− form)]; (e) NaHCO3, MeOH, 80 °C then PPTS, aq. MeOH, 100 °C then Dowex 1X8-100 (HO− form); (f) PPTS, aq. MeOH, 100 °C. | ||
An enantiospecific synthesis of (+)-hyacinthacine A2ent-149 (Scheme 33) began with N- and O-protection of D-serine then addition of a 2-(chloromethyl)vinyl anion equivalent to the derived Weinreb amide.105 Felkin–Anh selective ketone reduction and silylation of the so-formed 2°-alcohol was followed by cyclisation by Pd(0)-mediated O-allylation to give the anti,syn-oxazine diastereomer 179. This sequence (steps a–g) had been reported and exploited previously by the authors. In order to introduce the C(7a) stereogenic centre, Grignard addition to the aldehyde obtained by alkene ozonolysis was tested under a variety of conditions. In the absence of external additives, low syn-/anti-ratios were obtained but in the presence of a slight excess of ZnCl2 the addition of butenyl magnesium bromide formed essentially one diastereomer, consistent with reaction at the less hindered aldehyde face in a conformation fixed by chelation to the oxazine ring oxygen. After mesylation and N-protection, intermediate 180 was cyclised by N-alkylation and reductive amination steps to give the target alkaloid in fifteen steps overall.
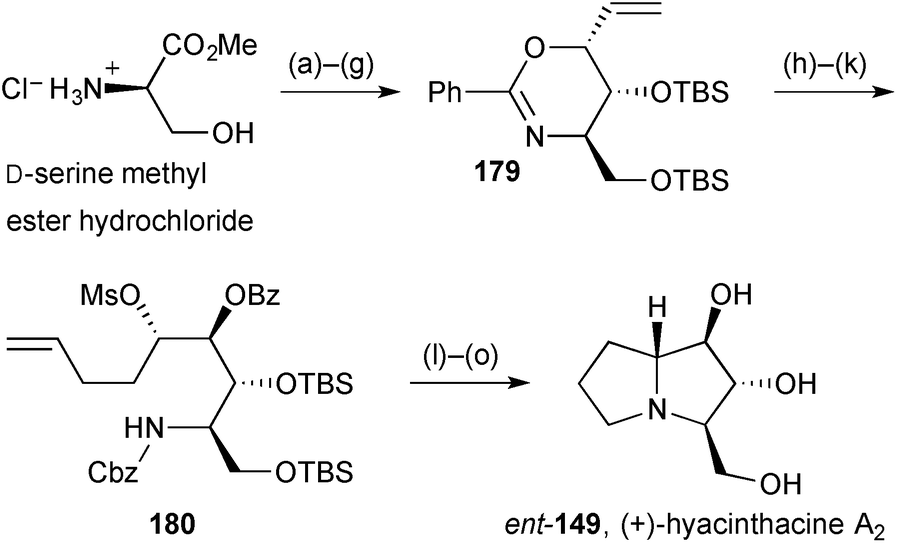 | ||
| Scheme 33 Reagents and conditions: (a) PhCOCl, Et3N, CH2Cl2; (b) TBSCl, imidazole, DMF; (c) (MeO)NHMe·HCl, Me3Al, CH2Cl2; (d) (E)-Bu3SnCHCHCH2Cl, MeLi, THF, −78 °C; (e) LiAlH(Ot-Bu)3, EtOH, −78 °C; (f) TBSCl, imidazole, DMF; (g) NaH, Pd(PPh3)4, TBAI, THF, 0 °C; (h) O3, MeOH, −78 °C then Me2S; (i) but-3-en-1-yl-MgBr, ZnCl2, THF, −78 °C; (j) MsCl, Et3N, CH2Cl2, 0 °C; (k) CbzCl, NaHCO3, aq. CH2Cl2; (l) NaH, THF; (m) O3, MeOH, −78 °C then Me2S; (n) H2, Pd(OH)2, MeOH; (o) HCl, aq. MeOH, reflux [→ent-149·HCl] then Dowex 40W X8. | ||
Goti reported an application of the group's well-developed nitrone cycloaddition/reductive amination strategy to the hyacinthacine B analogue 182 from 181, derived from L-xylose or D-arabinose.106 This compound showed 95% inhibition of Aspergillus niger amyloglucosidase at 1 mM (IC50 = 39 μM). Nitrone cycloaddition followed by reductive amination was also used to give four C(5)-methyl hyacinthacine analogues 183–186.107
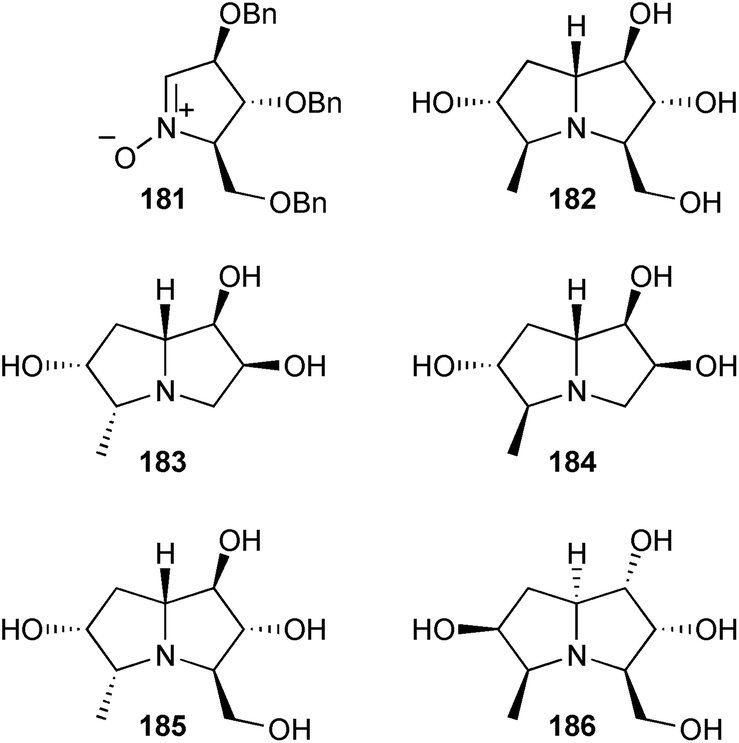
Goti and Cardona also used this strategy to access (6S)-azidohyacinthacine A2.108 This azide was then combined with a variety of symmetrical branched polyalkynes by multiple click cycloadditions to give ‘multivalent pyrrolizidine’ analogues 187–190. These molecules were tested for inhibitory activity against eight different glycosidases. All showed effective inhibition of Aspergillus niger amyloglucosidase at 1 mM (IC50 = 0.7–1.6 μM). Lower level selective inhibition of bovine kidney α-L-fucosidase, coffee bean α-galactosidase, yeast α-glucosidase, and jack bean α-mannosidase was also found.
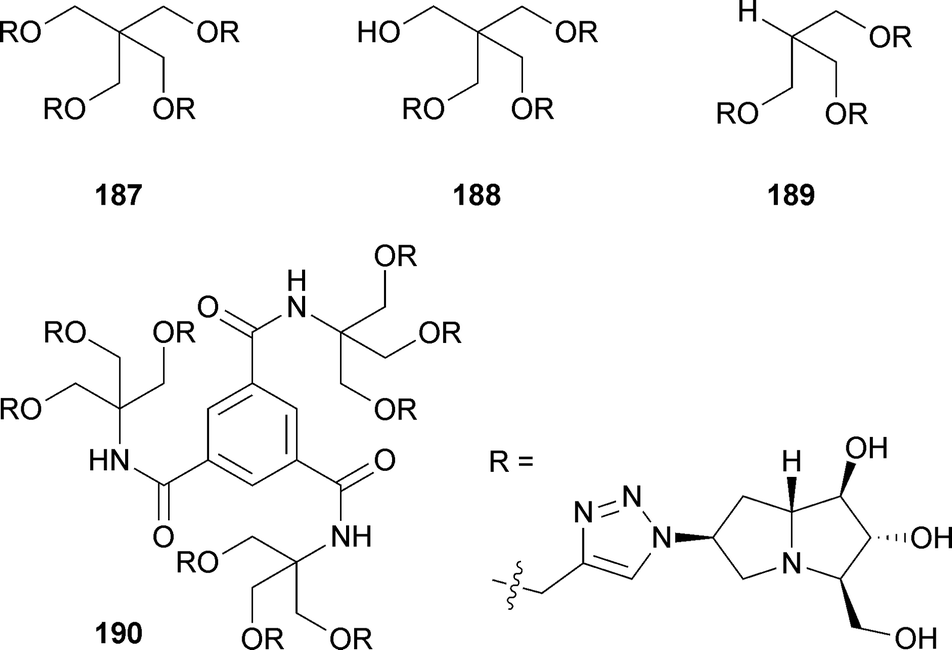
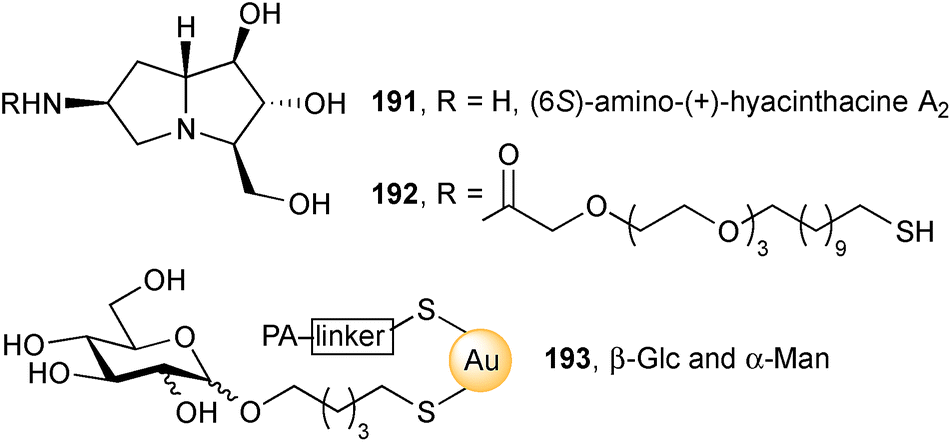
From the same key azide, the group prepared and acylated amine 191, producing thiol 192.109 This was then immobilised at two different ‘densities’ (20% and 40%) onto gold glyconanoparticles (Au-GNPs) adorned with either β-glucosyl or α-mannosyl chains 193. The four iminosugar Au-GNPs exhibited low micromolar inhibition of the amyloglucosidase from Aspergillus niger with the higher density pair having a higher IC50. The authors' work supports the view that increasing the multivalency of iminosugars does not necessarily result in increased bioactivity.
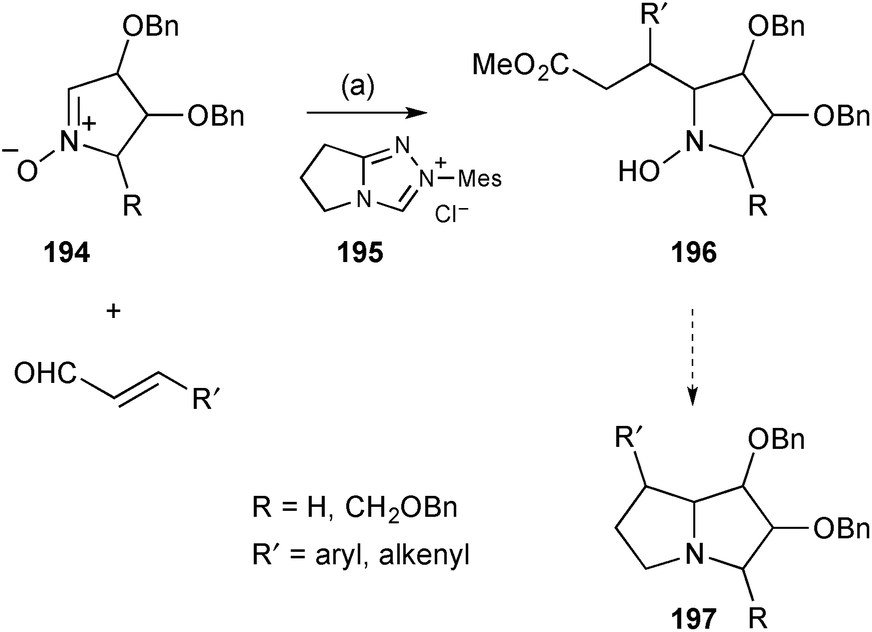
Kato and Yu reported the organocatalysed reaction of sugar-derived nitrones with 3-alkenyl or 3-aryl enals to give functionalised pyrrolidines of the form 196 (Scheme 34).110 Mediated by the pre-catalyst 195 the aldehydes connect at C(3) to the nitrone 194 pro-C(7a) position from the face anti-to the adjacent benzyloxy substituent. During the process, internal proton transfer results in overall oxidation of the aldehyde carbon so that ejection of the catalyst by the nitrone oxygen affords a lactone which is cleaved by methanolysis at the end of the reaction. These intermediates were then reduced and cyclised to give a variety of C(7)-alk(en)yl/aryl substituted PAs 197. The authors reported 16 relevant examples with one of these converted into (7R)-phenylhyacinthacine A2;110a a parallel Chinese patent lists many more examples.110b
 | ||
| Scheme 34 Reagents and conditions: (a) DBU, CH2Cl2, 0 °C → rt then NaOMe, MeOH. | ||
In a further elaboration of sugar-derived nitrones, Fischer's group developed a synthesis of a hyacinthacine C2 analogue 202 (Scheme 35) in which the C(7) hydroxy group in the natural product is moved to C(6) and the C(3) hydroxymethyl substituent is homologated.111 The adduct 199 between vinyl acetate and nitrone 198, from mannose, undergoes loss of acetate in the presence of TMSOTf as Lewis acid, and trapping follows from the exo-face of the so-formed oxonium ion by a glyoxal equivalent (step b). The resulting protected α-ketoester 200 was then taken through a series of reduction and protecting group manipulation steps to generate the PA homologue 202 in a concise overall sequence.
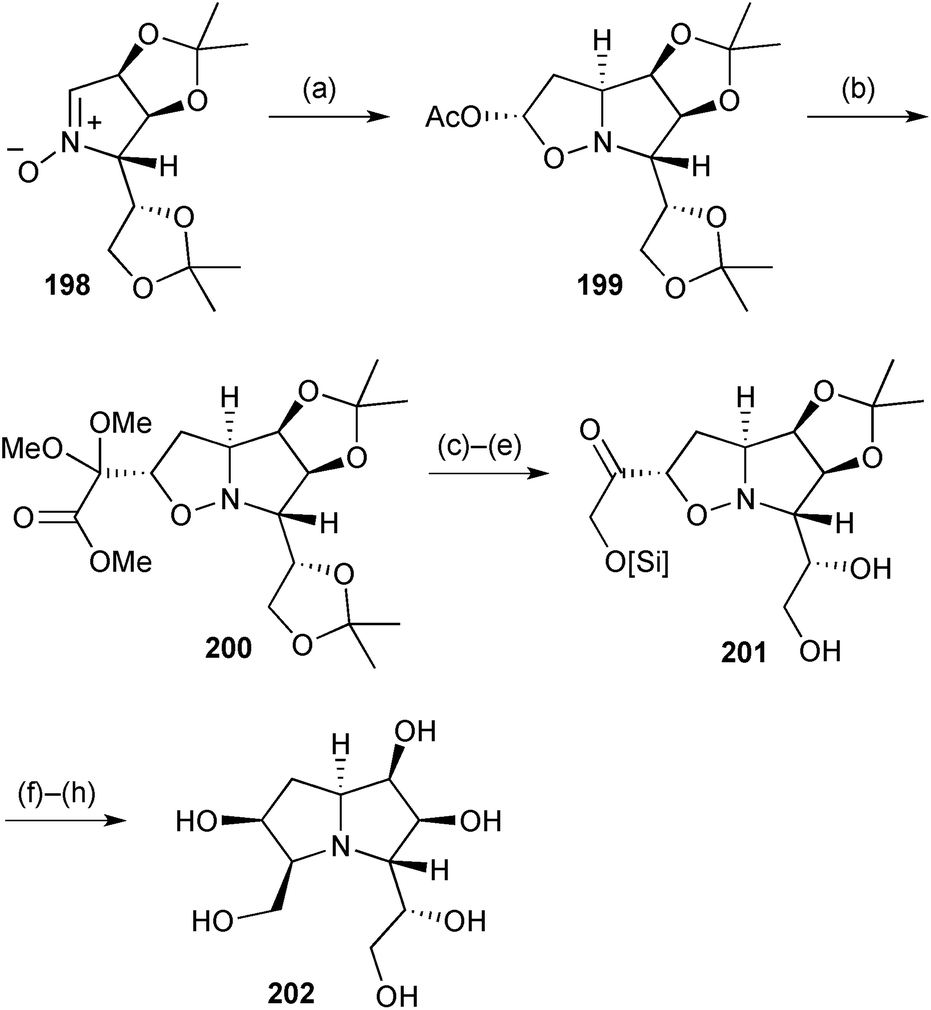 | ||
| Scheme 35 Reagents and conditions: (a) vinyl acetate, 75 °C [dr = 87:13]; (b) (MeO)2CC(OTMS)OMe, TMSOTf, CH2Cl2, −80 °C; (c) LiAlH4, THF, 0 °C; (d) TBDPSCl, imidazole, CH2Cl2; (e) aq. AcOH, 60 °C; (f) H2, Pd/C, EtOH; (g) TBAF, THF; (h) aq. TFA then Dowex (H+ form). [Si] = TBDPS. | ||
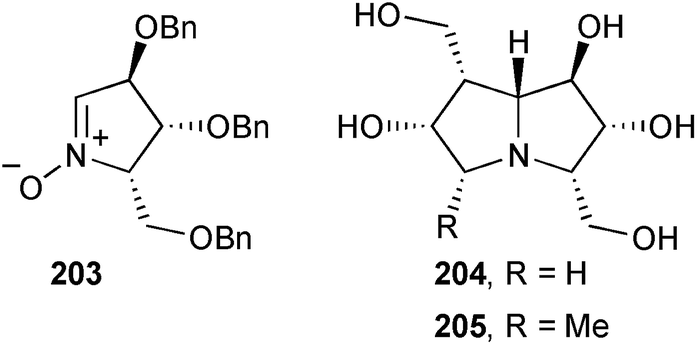
Carbohydrate-derived nitrone 203 was the starting point in routes to the two non-natural pyrrolizidines 204 and 205 that were evaluated for their ability to inhibit a panel of glycosidases.112 The 5-methyl-containing compound 205 was active only against coffee bean α-galactosidase (IC50 = 68.0 μM) but the less substituted compound 204 was both more active for this enzyme (IC50 = 5.4 μM) and showed activity against bovine liver β-galactosidase (IC50 = 82.9 μM).
Based on the occurrence in Scilla species of hydroxylated pyrrolizidines bearing extended side chains at C(5), Toyooka's group developed routes to non-natural hyacinthacine analogues with extended C(3) substituents and tested their glycosidase inhibition.113 The monoacetate (−)-(2S,5R)-207 (Scheme 36) was prepared from N-Boc pyrrole 206 by double carboxylation, two reductive steps, then enzymatic acylation with CAL-B/vinyl acetate. Protecting group and redox manipulation steps were followed by HWE reaction, dihydroxylation, and a second HWE olefination to provide intermediate 208. Removal of the Boc protecting group initiated aza-Michael addition, thought to proceed kinetically via a conformation that avoids steric clashing between the CH2O[Si] and developing CH2CO2Et substituents. Ester reduction and cleavage of the benzyl protecting groups generated analogue 213; alternatively, partial ester reduction then Wittig reaction and reduction produced the three C(3)-alkyl analogues 210–212. These four pyrrolizidines, plus their enantiomers, were evaluated for inhibition of seven glycosidases. Hydroxyethyl analogue 213 showed moderate, selective inhibition of α-L-fucosidase from bovine kidney; most analogues, in both series, inhibited β-galactosidase from bovine liver.
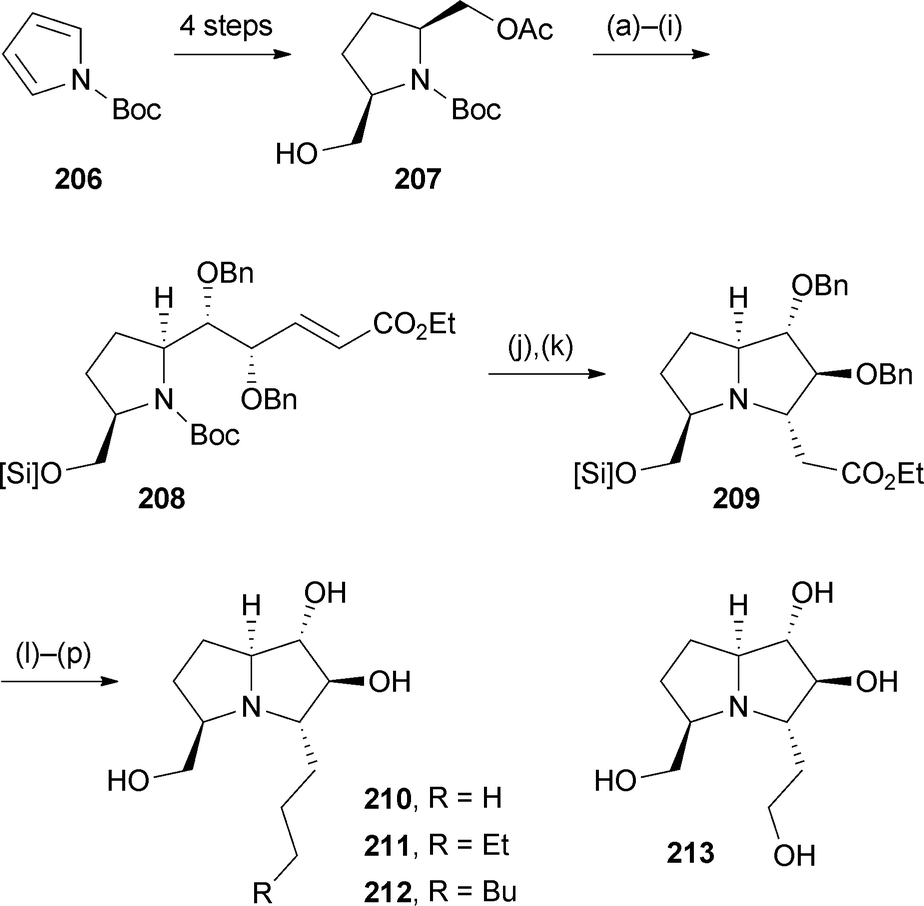 | ||
| Scheme 36 Reagents and conditions: (a) TBDPSCl, imidazole, CH2Cl2; (b) K2CO3, MeOH; (c) SO3·pyridine, Et3N, DMSO; (d) (EtO)2PO·CH2CO2Et, NaH, THF; (e) OsO4, NMO, aq. acetone [dr ∼ 2:1]; (f) NaH, BnBr, DMF; (g) LiBH4, THF; (h) Dess–Martin periodinane, CH2Cl2; (i) (EtO)2PO·CH2CO2Et, NaH, THF; (j) CF3CO2H, CH2Cl2; (k) K2CO3, CH2Cl2; (l) DIBAL, CH2Cl2, −78 °C; (m) Ph3P+CH2R X−, t-BuOK, THF; (n) H2, Pd/C, EtOAc; (o) TBAF, THF; (p) BCl3, THF. [Si] = TBDPS. | ||
3.5 Casuarines and australines
Prior to the publication of the synthesis of a simple tetrahydroxy pyrrolizidine (Scheme 25), Reissig and Goti had disclosed the strategy in the context of the preparation of three PAs related by the same stereochemistry at C(1–3) and C(7a).114 Here, nitrone 181 gave, via adduct 214 (Scheme 37), cyclisation product 215, a common intermediate for the synthesis of (+)-casuarine 216, (+)-australine 217 and its epimer at C(7) 218. Thus, hydroboration and oxidation produced the C(6)–C(7) trans-diol motif present in (+)-casuarine 216, the synthesis being completed by N–O reduction, cyclisation via the mesylate, and deprotection. Alternatively, from 215, performing these last steps first led to the australine core bearing a C(7)-carbonyl; reduction of this ketone afforded (+)-australine 217 with complete stereoselectivity resulting from hydride delivery from the exo-face. A modification of the sequence to casuarine, including C(6)-deoxygenation by vigorous DIBAL reduction of the mesylate, gave access to (−)-7-epi-australine 218. This last PA showed 95% inhibition of Aspergillus niger amyloglucosidase at 1 mM (IC50 = 3.5 μM).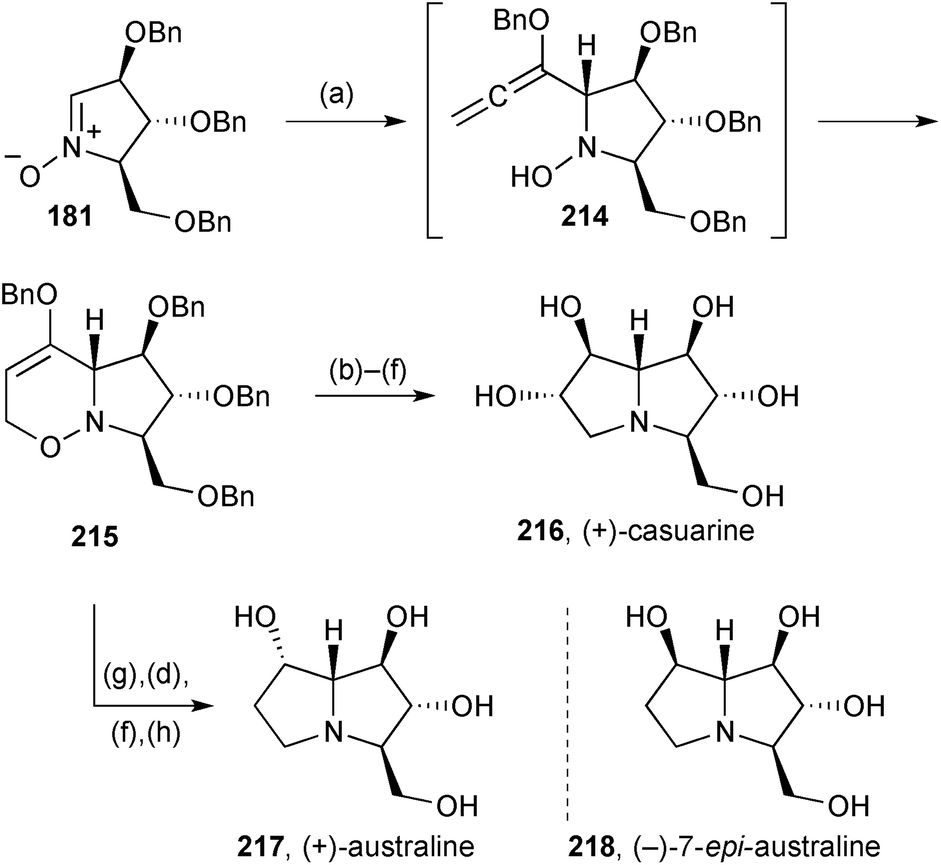 | ||
| Scheme 37 Reagents and conditions: (a) (benzyloxy)allene, BuLi, THF, −78 °C then Na2SO4, CH2Cl2, rt; (b) BH3·THF, THF then H2O2, aq. NaOH; (c) Zn, aq. AcOH, 65 °C or SmI2, THF; (d) MsCl, Et3N, CH2Cl2; (e) LiAlH4, THF, reflux; (f) H2, Pd/C, aq. HCl, MeOH; (g) Mo(CO)6, NaBH4, aq. CH3CN, reflux; (h) NaBH4, MeOH. | ||
Nitrone 181 (Scheme 38) was converted into a mixture of pyrrolizidine diastereomers 220, epimeric at C(7) with the β-isomer being major.115 Hydrogenolysis of the benzyl ethers gave (−)-7-epi-australine 218 and (+)-australine 217. DAST-mediated fluorination of either of the diastereomers of 220 resulted, after deprotection, in the production of the same β-configured 7-fluoro-7-deoxyaustraline derivative 221; the authors speculate that neighbouring group participation by the nitrogen atom intervenes, at least in the case of β-220 where substitution proceeds with clean retention of configuration. Treating the ketone derived from alcohol 220 with DAST led to the 7,7-difluorinated australine analogue 222 after benzyl hydrogenolysis; in the difluorination step a tricyclic compound (not shown) was produced in ∼40% yield by cyclisation of the CH2OBn oxygen at C(3) onto C(7). The same four end products were prepared in the enantiomeric series from ent-181, derived from D-xylose. Evaluation of the eight australine variants’ ability to inhibit a range of glycosidases was also undertaken. The ent-217, 218, 221, 222 variants were essentially inactive in all assays; (+)-australine 217, its 7-epimer 218, and the C(7)-difluoro analogue 222 were effective inhibitors of Aspergillus niger α-glucosidase and amyloglucosidase; the C(7)-monofluoro compound 221 was ∼10 times as effective as (+)-australine in its inhibition of A. niger α-glucosidase (IC50 = 0.63 μM) and also showed reasonable activity against porcine kidney trehalase.
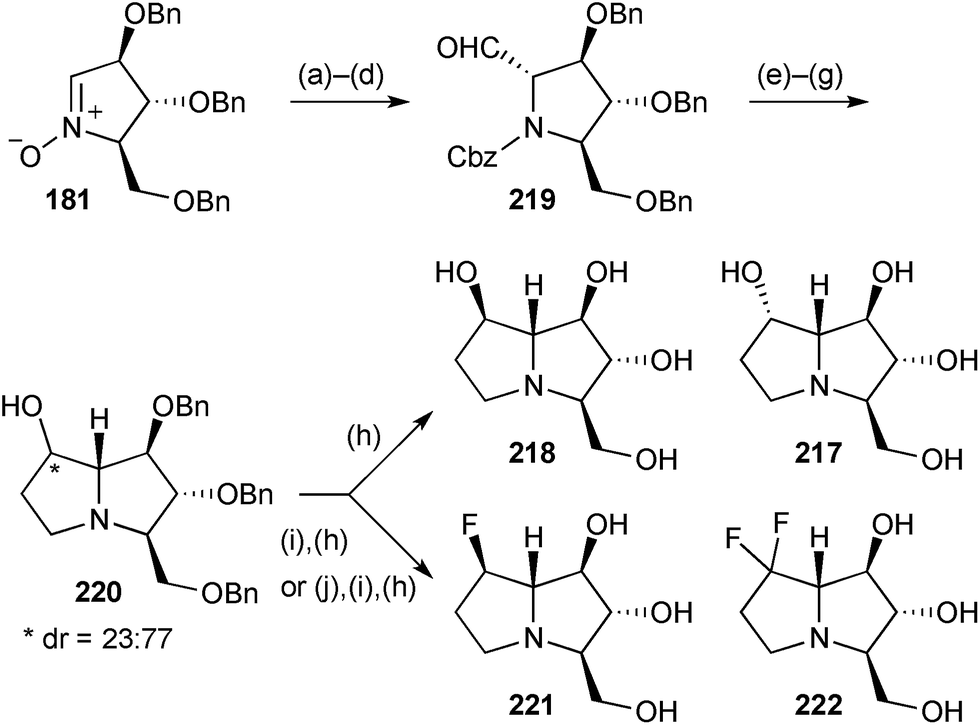 | ||
| Scheme 38 Reagents and conditions: (a) H2CCHMgBr, THF, 0 °C; (b) Zn, AcOH; (c) CbzCl, NaHCO3, aq. THF; (d) ozonolysis; (e) Zn, allyl bromide, NH4Cl, aq. THF; (f) O3, MeOH, −60 °C then Me2S; (g) H2, Pd/C, AcOH, MeOH; (h) H2, Pd/C, HCl, aq. MeOH; (i) DAST, pyridine, CH2Cl2, 0 °C (→221) or rt (→222); (j) (COCl)2, DMSO, CH2Cl2, −40 °C then Et3N. | ||
Pyrrolizidinone 223 (Scheme 39), whose preparation from nitrone 181 was described in the previous review, was inverted at C(6) then reduced to give (+)-dexoyuniflorine A 224.116 This, and three further pyrrolizidines [(−)-uniflorine A, 7-deoxycasuarine, and 7-deoxy-6-(α-glucopyranosyl)casuarine] showed little inhibition of α-amylase (from human saliva), little or comparatively weak activity against porcine trehalase, but moderate to high nanomolar inhibition of insect trehalases from Chironomous riparius, and Spodoptera littoralis. The authors concluded that such selective activity holds promise for the future development of insecticides.
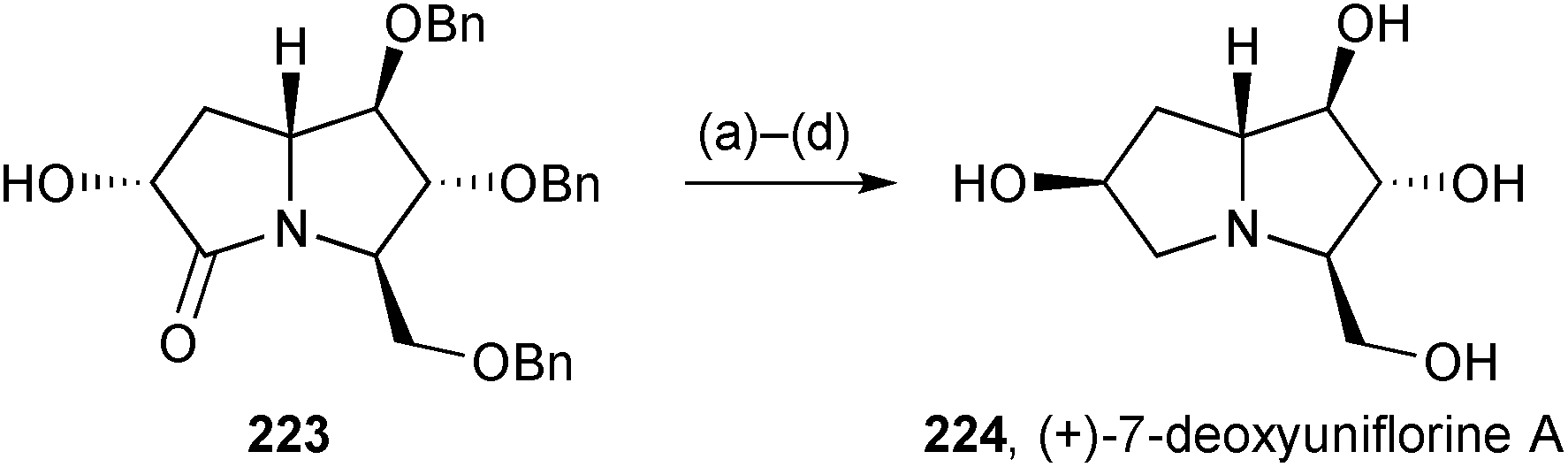 | ||
| Scheme 39 Reagents and conditions: (a) p-NO2C6H4CO2H, DIAD, PPh3, THF; (b) Ambersep 900 OH, MeOH; (c) LiAlH4, THF, reflux; (d) H2, Pd/C, HCl, MeOH then Dowex 50WX8-200. | ||
Further applications of Clapés' chemoenzymatic synthesis of highly oxygenated PAs from dihydroxyacetone 225 are summarised in Scheme 40 which shows stereodivergent routes to ent-casuarine 216, its 3-epimer 229, and both 2-epi- and 2,3-di-epi-casuarine, 231 and 232, respectively.117 These alkaloids were screened for glycosidase activity against bakers yeast α-glucosidase, rice α-glucosidase, and Penicillium decumbens α-rhamnosidase. The most promising compound (229) strongly inhibited rice α-glucosidase (IC50 = 7.9 ± 5.2 μM) and, in follow up, also showed activity against rat intestinal sucrase (IC50 = 3.5 ± 0.6 μM) and rat intestinal maltase (IC50 = 39 ± 13 μM).
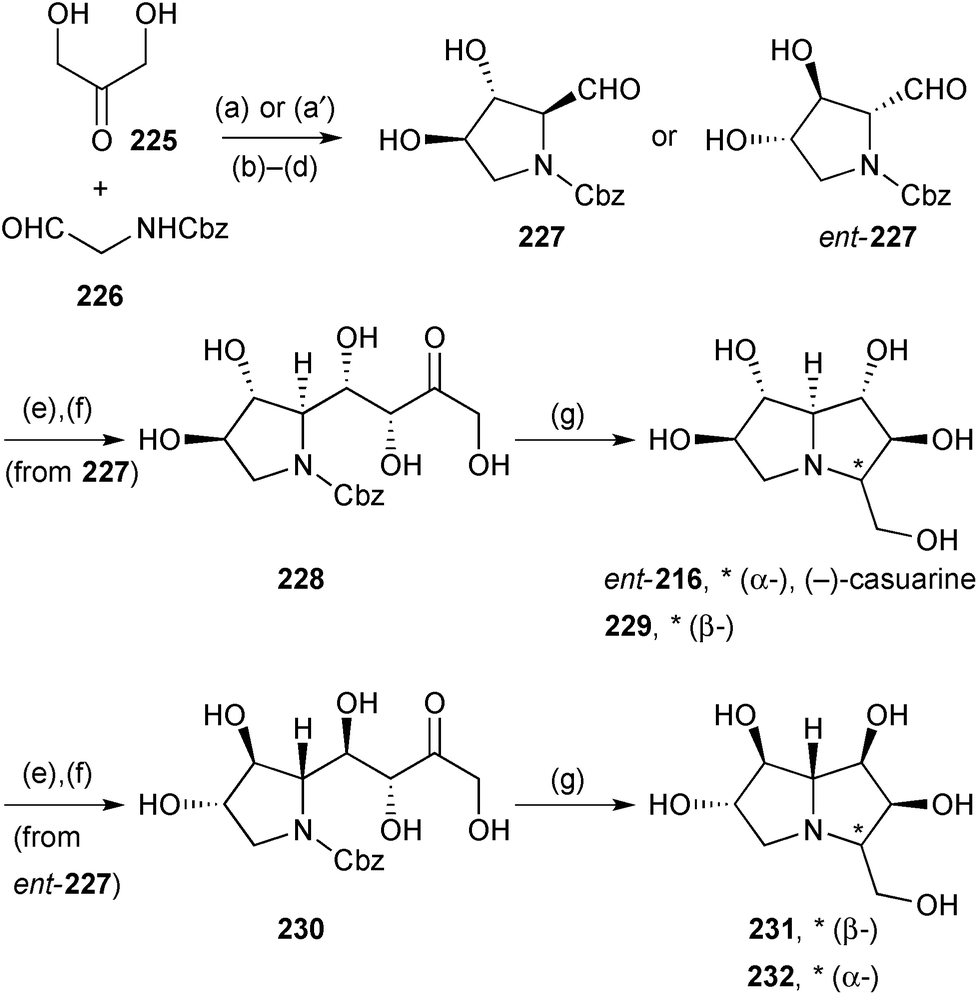 | ||
| Scheme 40 Reagents and conditions: (a) FSAA129S/A165G; (a′) RhuA; (b) H2, Pd/C; (c) Cbz-OSu, aq. dioxane; (d) IBX, EtOAc, reflux; (e) DHAP, FucAF131A, aq. DMF; (f) acid phosphatase, pH 5; (g) H2, Pd/C, aq. MeOH then ion exchange chromatography (CM-Sepharose-NH4+). | ||
A separate paper reports the synthesis of a variety of 5,6-annulated (benzo-, cyclohexano-) hydroxy-PA derivatives.118
More recently, the group described the use of L-rhamnulose-1-phosphate aldolase from Thermotoga maritima (Rhu1PATm) to prepare four hyacinthacine isomers ent-146, 147, 149, and 235 (Scheme 41). There is some ambiguity in the stereochemistry of these pyrrolizidines; the depicted structure for 147 reflects the name provided in the experimental section, rather than that presented in Scheme 4 of the paper (which is the 1,3-diepimer).119
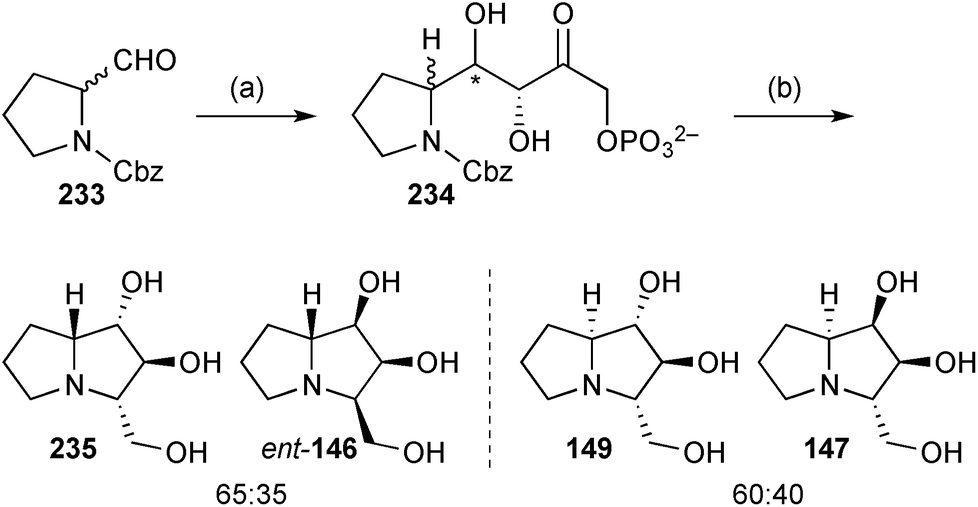 | ||
| Scheme 41 Reagents and conditions: (a) DHAP, Rhu1PATm, aq. DMF then potato acid phosphatase, aq. HCl; (b) H2, Pd/C, aq. MeOH. | ||
Pyrrolizidines ent-235 and 236–240 were prepared by a similar two step process.120

3.6 Aminopyrrolizidines
Davies' synthesis121 of the non-natural (−)-enantiomer of absouline 243 (Scheme 42) is strategically similar to that reported by Scheerer's group, summarised in the previous review. The advance in Davies' version is the use of the chiral ammonia equivalent lithium (S)-N-benzyl-N-(α-methylbenzyl)amide to control the stereochemistry at C(1) (step d). From adduct 242, hydrogenolysis of all benzylic linkages and subsequent lactam formation and reduction led to (1R,7aS)-1-aminopyrrolizidine; acylation with p-methoxycinnamic acid completed the synthesis, in eight steps overall and 20% yield.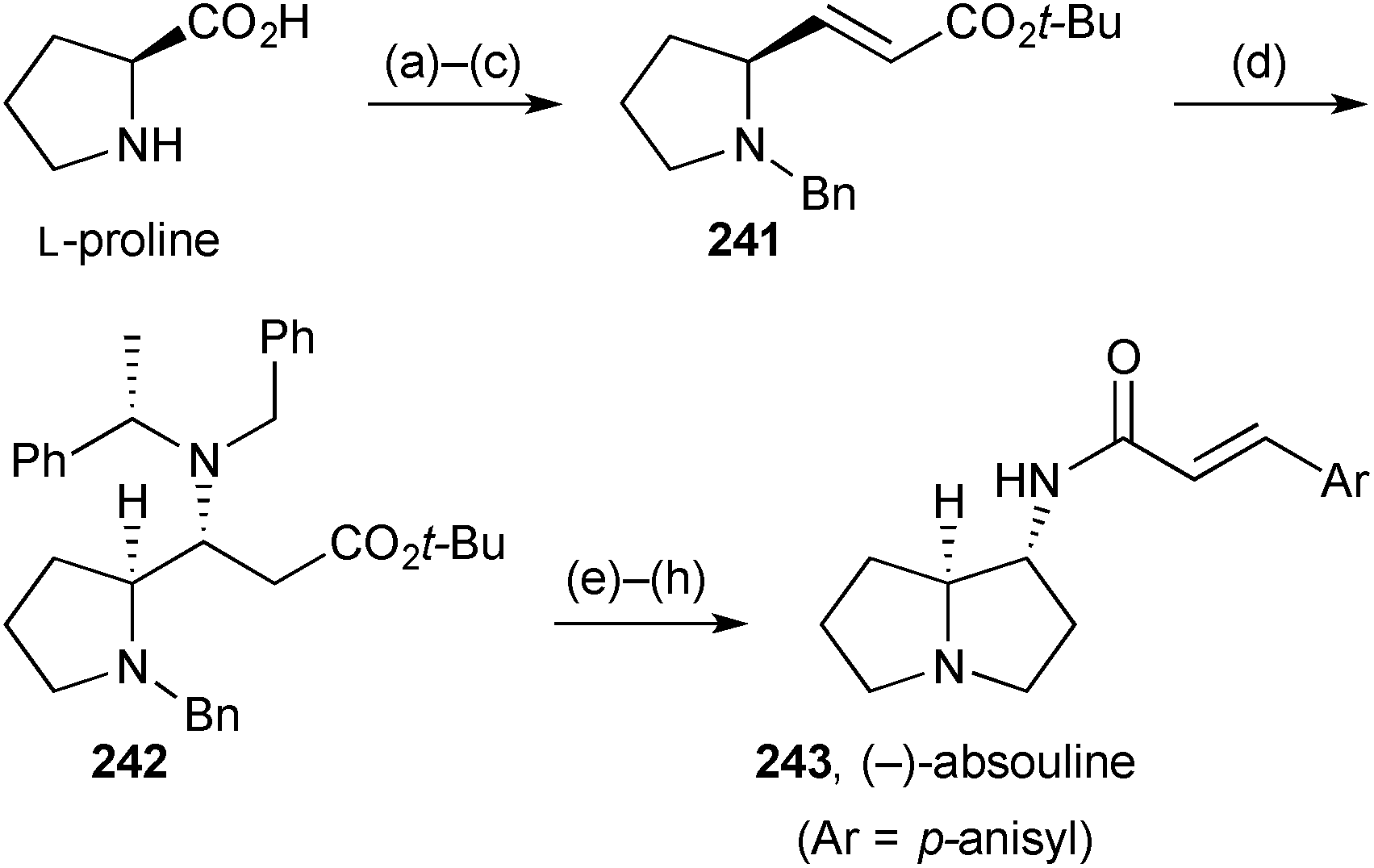 | ||
| Scheme 42 Reagents and conditions: (a) PhCOCl, aq. NaOH, 0 °C; (b) LiAlH4, THF, reflux; (c) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C then Ph3PCHCO2t-Bu, CH2Cl2; (d) lithium (S)-N-benzyl-N-(α-methylbenzyl)amide, THF, −78 °C; (e) H2, Pd(OH)2/C, aq. HCl, MeOH; (f) aq. HCl, 90 °C; (g) DIBAL, THF, 0 °C → rt; (h) trans-ArCHCHCO2H, DCC, DMAP, CH2Cl2, 0 °C → rt. | ||
Brière's group developed a three component synthesis of isoxazolidinones from an aldehyde, an N-alkylhydroxylamine or alkyl hydroxycarbamate, and Meldrum's acid.122 With aldehydes bearing an α-heteroatom, syn-diastereoselectivity was observed. This methodology was applied to a short synthesis of (S,S)-1-aminopyrrolizidinone 245 (Scheme 43). Thus, the syn-1,2-diamino functionality in intermediate 244 was established by condensation with N-Boc (S)-prolinal 104 and aza-Michael addition of benzyl hydroxycarbamate (via246 and 247). Hydrogenolysis of the N–O bond and CBz group in isoxazolidinone 244, followed by acid treatment, gave lactam 245, a known precursor to 1-aminopyrrolizidines.
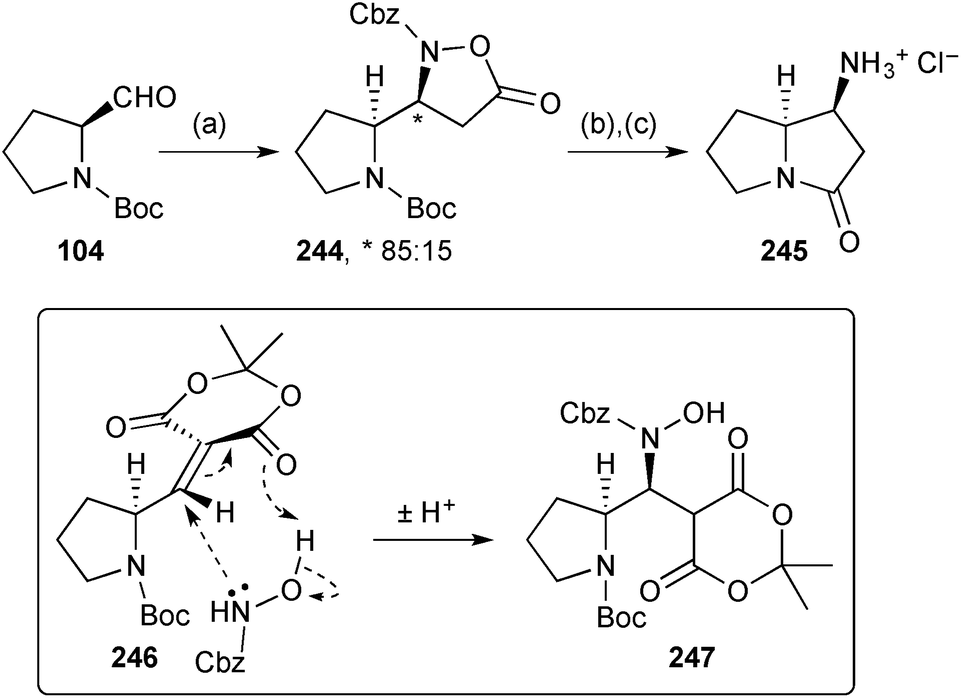 | ||
| Scheme 43 Reagents and conditions: (a) Meldrum's acid, benzyl hydroxycarbamate, DABCO, pyrrolidine, EtOAc; (b) H2, Pd/C, i-PrOH, 60 °C; (c) aq. HCl, 90 °C. | ||
Takahashi reported a synthesis of the structure 252 proposed for (+)-pochonicine (Scheme 44).123 The tetrasubstituted N-Boc-pyrrolidine derivative 249 was prepared from N-acetylglucosamine and allylated to give a roughly 3:1 ratio of diastereomers 250 and 251. The major isomer 250 was taken on to the proposed (+)-pochonicine. During the route, alkene dihydroxylation was not fully selective; therefore, cyclisation via the mesylate provided both C(3) epimers (252 and 253). Neither diastereomer showed spectroscopic data matching the literature values for the natural product. From the minor allylated isomer 251, the same sequence generated C(3) epimers 254 and 255. The NMR spectroscopic data for epimer 255 matched those reported for the natural product but the specific rotation was of opposite sign; accordingly, the structure of (+)-pochonicine is revised to ent-255.
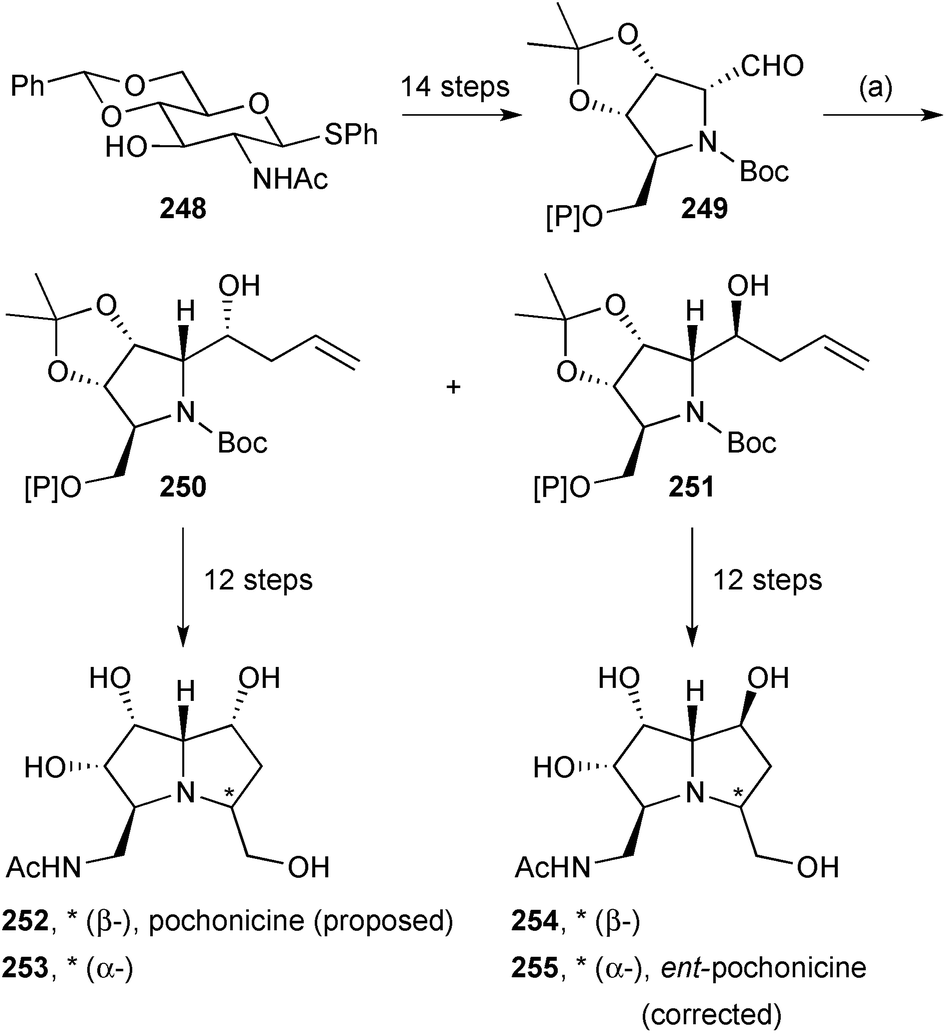 | ||
| Scheme 44 Reagents and conditions: (a) allyl-MgCl, ZnCl2, CH2Cl2, THF, −78 °C [dr = 77:23]. [Si] = TBDPS. | ||
This structural reassignment was subsequently confirmed by Kato and Yu who prepared the same set of diastereomers plus their enantiomers from D-ribose or L-ribose, in an overall more concise route.124 In essence, the synthesis was strategically the same as Takahashi's, with stereochemical branching points at the allylation (→C(1)-epimers 256/257, Scheme 45) and dihydroxylation (→C(3)-epimers, 258/259) stages.
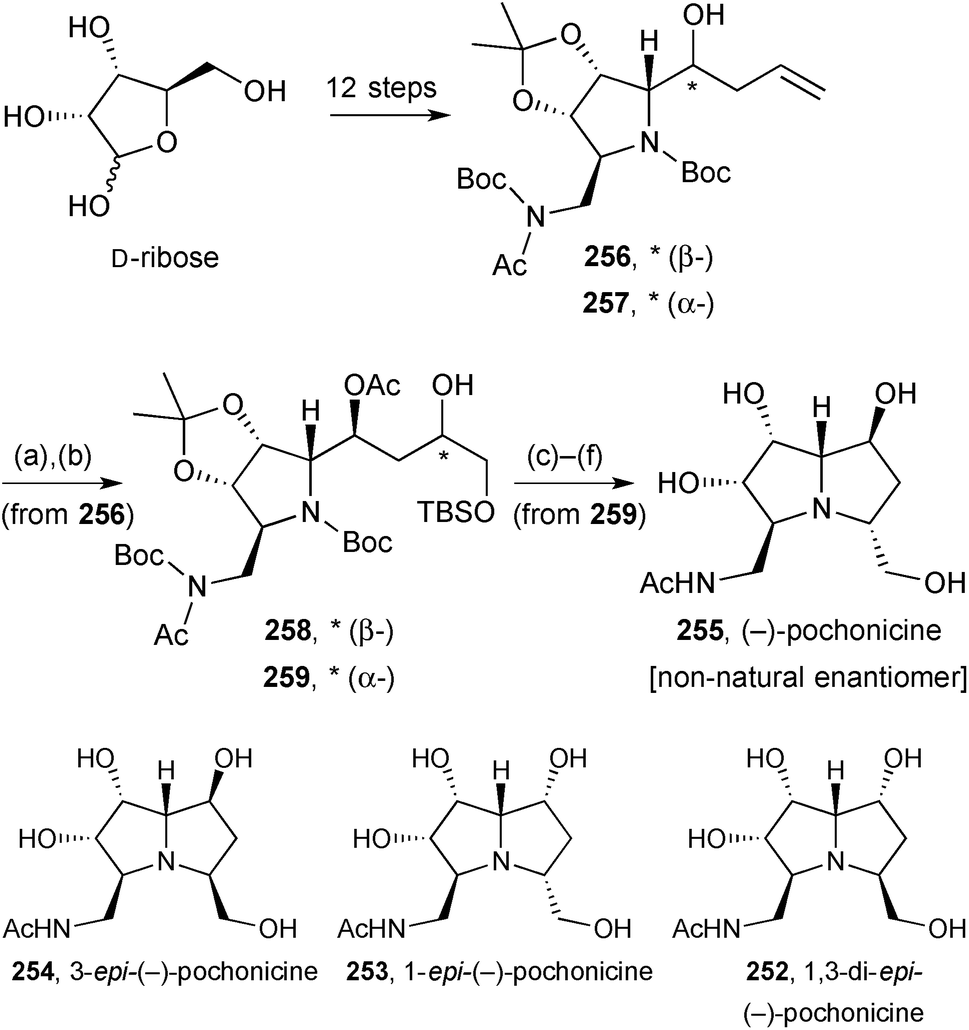 | ||
| Scheme 45 Reagents and conditions: (a) OsO4, NMO, aq. acetone [dr = 1.2:1]; (b) TBSCl, Et3N, DMAP, CH2Cl2; (c) MsCl, Et3N, CH2Cl2; (d) Zn, Br2, p-cresol; (e) K2CO3, MeOH; (f) aq. HCl, MeOH. | ||
In both the Takahashi and Kato–Yu syntheses, the glycosidase inhibitory properties of the various pochonicine isomers were evaluated. (−)-Pochonicine (the non-natural enantiomer) was found to be around 10000 times weaker than the (+)-enantiomer in inhibiting jack bean N-acetylglucosaminidase.123 Whilst pochonicine showed significant inhibition of a panel of glycosidases124 the IC50 values were somewhat less impressive than those reported in the original isolation paper.
In a research programme aimed at understanding the role of lacto-N-biosidase (LNBase), an enzyme that releases lacto-N-biose from human milk oligosaccharides, Stubbs prepared a series of amino- and iminosugars and glycosylated-izidines as potential LNBase inhibitors.125 One of these 265 (Scheme 46) was prepared from castanospermine starting with a series of protecting group manipulations that left a single free hydroxy group in derivative 260. A 2,3,4,6-tetra-O-acetyl-β-galactosyl group was introduced relatively early on in the sequence via trichloroacetimidate 266 (step e). Azide displacement of the mesylate derived from alcohol 262 gave mainly the castanospermine derivative 263 along with a significant amount of the ring-contracted 5-azidomethylpyrrolizidine 264, structurally related to pochonicine (Scheme 45). Deprotection and purification steps afforded the potential inhibitor 265. This small molecule inhibited the LNBase from Bifidobacterium bifidum (Ki = 52 ± 2 μM) but it was the weakest of the six compounds tested and about 10 times less potent than the castanospermine variant derived from 263.
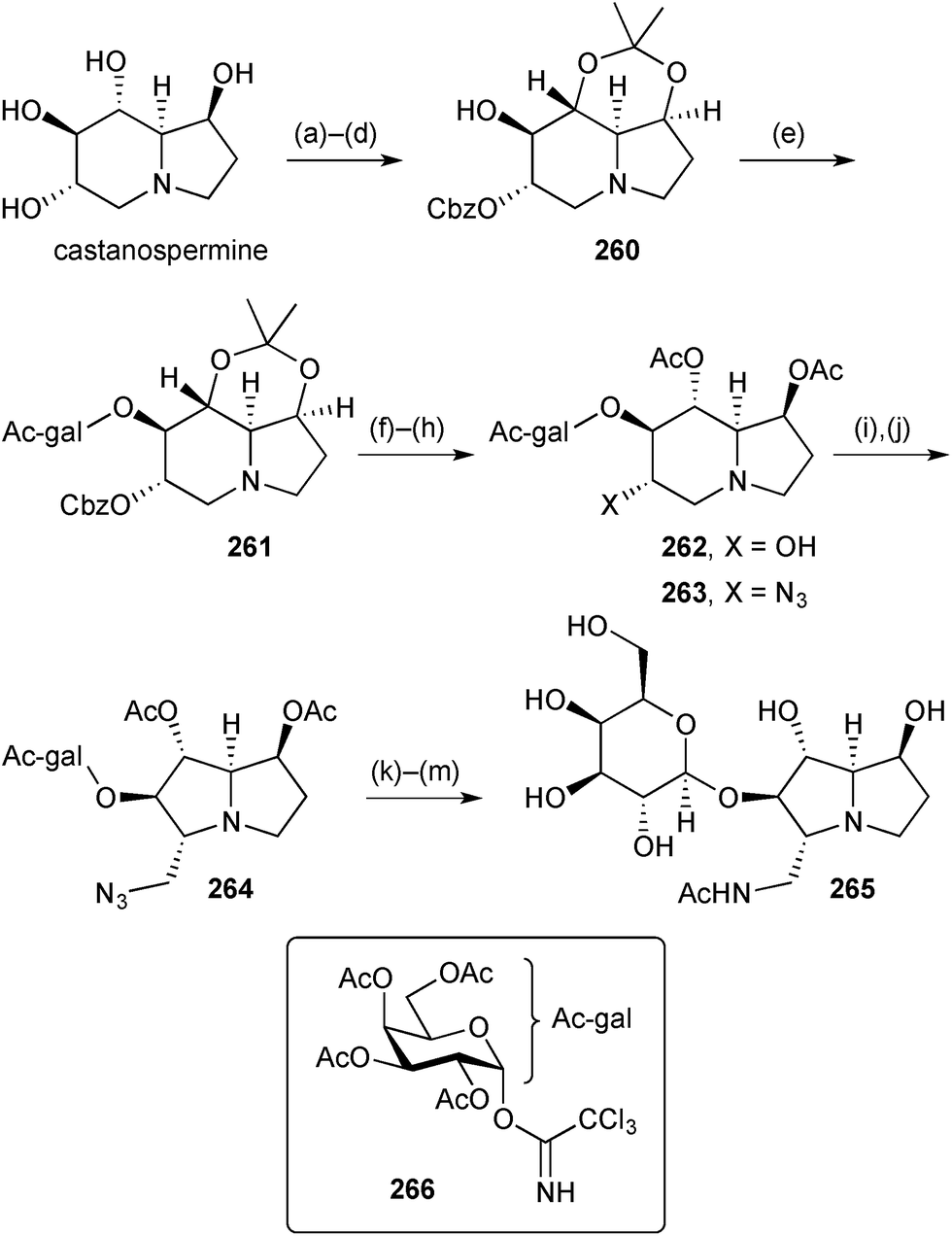 | ||
| Scheme 46 Reagents and conditions: (a) PhCOCl, pyridine; (b) 2-methoxypropene, TsOH·H2O, DME, 55 °C; (c) NaOMe, MeOH; (d) CbzCl, Et3N, THF; (e) 266, TMSOTf, 4 Å MS, CH2Cl2, −30 °C to rt; (f) aq. AcOH, 70 °C; (g) Ac2O, pyridine; (h) H2, Pd/C, MeOH; (i) MsCl, pyridine, 0 °C to rt; (j) NaN3, DMSO [→263, 27% and 264, 15%]; (k) H2, Pd/C, MeOH; (l) Ac2O, pyridine; (m) NaOMe, MeOH then AG50W-X4 (H+), H2O. | ||
Scheerer reported a second generation route to the loline alkaloid system (Scheme 47).126 There are strategic similarities with the first generation route (described in the previous review) but the more recent route provides enantiopure material from L-glutamic acid and access to the Z-alkenyl side chain is achieved more directly by Petasis boronic acid Mannich reaction (step g → 269) then RCM (step i). In this new route, the tethered aminohydroxylation was also optimised and the pentafluorobenzyloxy carbamate, in combination with an enoate (step n → 271), shown to be much more efficient than the original simple carbamate coupled with an allylic alcohol. The route then followed an analogous sequence to that developed earlier for the racemate. Here, ester reduction, double mesylation, and Boc-activation of the oxazinanone enabled cleavage of the imide to be effected exclusively at the endocyclic carbonyl. Cyclisation of the so-formed 2°-hydroxy group gave bicyclic intermediate 272 that cyclised to the norloline derivative 273 upon hydrogenolysis of the Cbz group. Overall, the synthesis is a similar length to the first generation route but is significantly more efficient and provides multi-milligram quantities for further study.
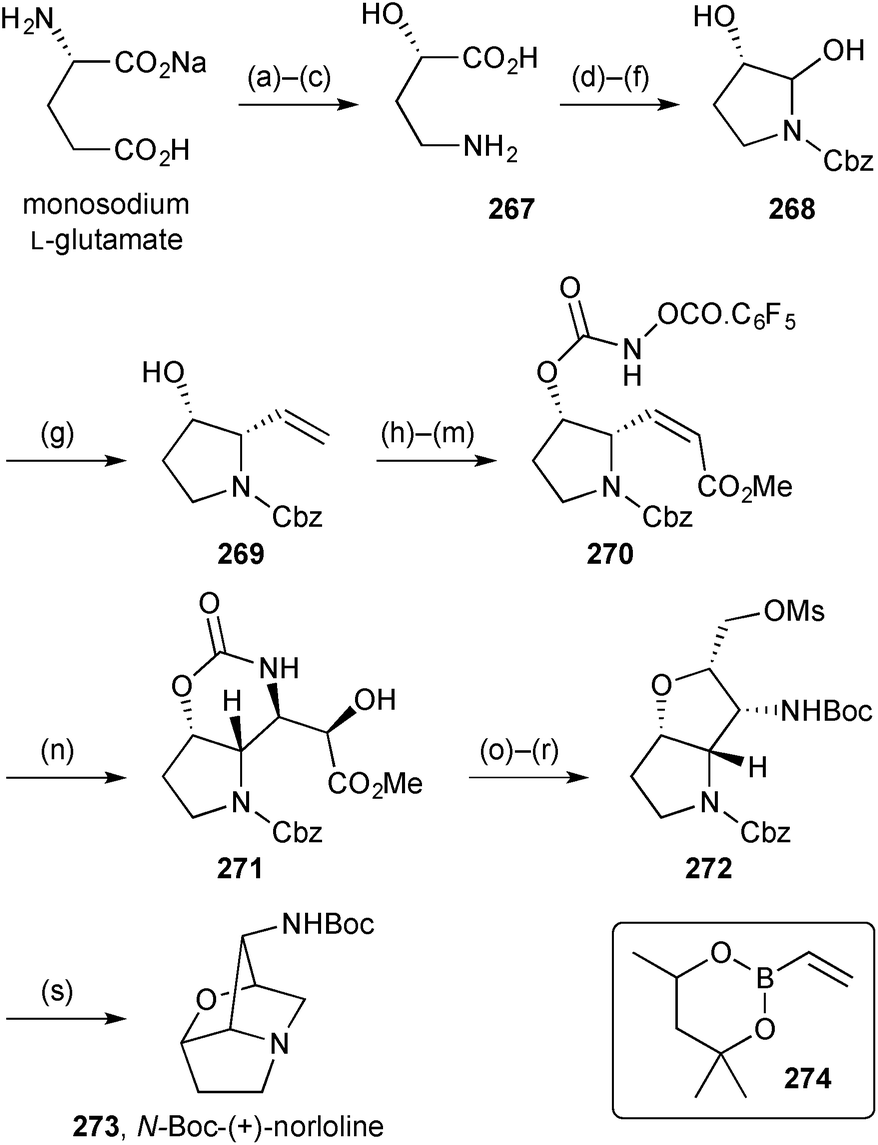 | ||
| Scheme 47 Reagents and conditions: (a) NaNO2, aq. HCl, 0 °C to 10 °C; (b) NH3, EtOH; (c) Cl2, aq. NaOH, 50 °C; (d) HMDS, TMSCl, PhCH3, reflux then EtOH; (e) LHMDS, CbzCl, THF, −78 °C then aq. HCl; (f) NaBH4, MeOH, 0 °C; (g) 274, BF3·OEt2, CH2Cl2, −78 °C to rt; (h) acryloyl chloride, i-Pr2NEt, DMAP, CH2Cl2, −78 °C to rt; (i) Hoveyda–Grubbs II (Hoveyda–Blechert catalyst), DCE, reflux; (j) LiOH·H2O, aq. THF; (k) MeI, K2CO3, DMF; (l) NH2OH·HCl, CDI, pyridine, 0 °C to rt; (m) C6F5COCl, Et3N, CH2Cl2, 0 °C; (n) K2OsO4·H2O, aq. t-BuOH; (o) LiBH4, THF, 0 °C; (p) MsCl, pyridine; (q) Boc2O, DMAP, THF; (r) Cs2CO3, MeOH; (s) H2, Pd(OH)2, MeOH. | ||
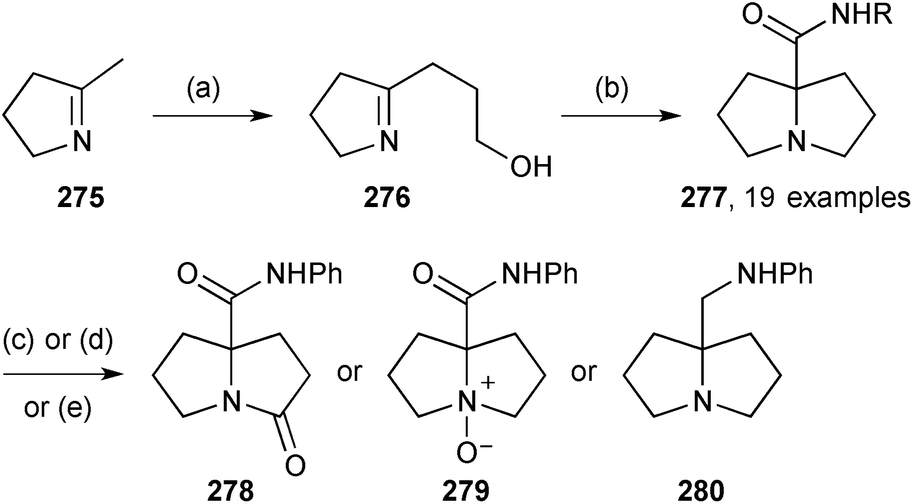
In closing this section, two short syntheses of non-natural 7a-aminoalkyl pyrrolizidines have been reported. The first involves an interesting transformation of 2-hydroxypropyl pyrroline 276 (Scheme 48).127 Presumably, Ritter-type addition of the isocyanide to the protonated pyrroline is followed by capture of the nitrilium ion by the tethered hydroxy group; evolution of the so-formed cyclic imidate to the pyrrolizidine system may be mediated by the chloride ion present in the reaction which can act as a nucleophilic catalyst in a ring-opening/ring-closure sequence. Nineteen varied amides 277 were produced by this methodology and the N-phenyl amide (R = Ph) used to exmplify three redox transformations to give 278–280. Separately, a process for preparing a precursor 281 to homologues of amines of the form 280 was disclosed.128
 | ||
| Scheme 48 Reagents and conditions: (a) LDA, ethylene oxide, THF, −78 °C; (b) RNC, Et3N·HCl, PhCH3, reflux; (c) BnNEt3+MnO4−, CH2Cl2 (→278); (d) MCPBA, CH2Cl2, 0 °C (→279); (e) Red-Al, PhCH3, reflux (→280). | ||
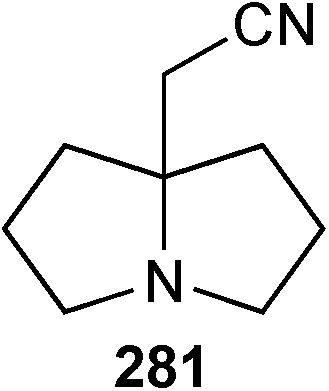
3.7 Pyrrolams, hydroxydanaidone
The product of organocatalytic amination of 5-(tert-butyldimethylsilyl)oxypentanal 125 (Scheme 23) was taken through a similar sequence to provide (−)-(R)-pyrrolam A 282 and pyrrolizidine (hexahydro-1H-pyrrolizine, not shown).129A short RCM route from N-Boc-(2S)-vinylpyrrolidine (from (S)-proline in three steps) and 2-fluoroacryloyl chloride gave access to fluoropyrrolam A derivative 283.130
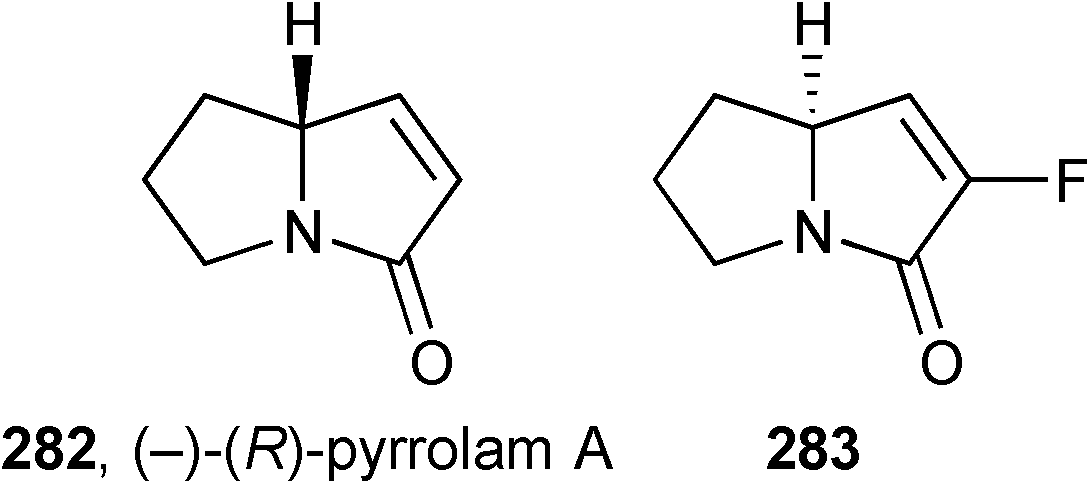
The second synthesis of (−)-(R)-pyrrolam A during the review period employed a chiral equivalent of iminium ion 287 (Scheme 49).131 The strategy was based on protection of the enone double bond of maleimide as a Diels–Alder adduct with anthracene which also served to maintain the absolute stereochemistry during the production and trapping of the iminium centre. In the forward direction, asymmetric borohydride reduction catalysed by oxazaborolidine 288 provided methoxylactam 285 (99% ee) after formation of the aminal from the intermediate hemiaminal. Iminium formation and allylation then N-deprotection afforded exo-allyllactam 286 as a single diastereomer. From this point the synthesis followed conventional lines with flash vacuum pyrolysis releasing the target molecule in the final step; this compound is known to racemise readily but just a slight erosion in enantiomeric purity was found in this instance (282, 94% ee).
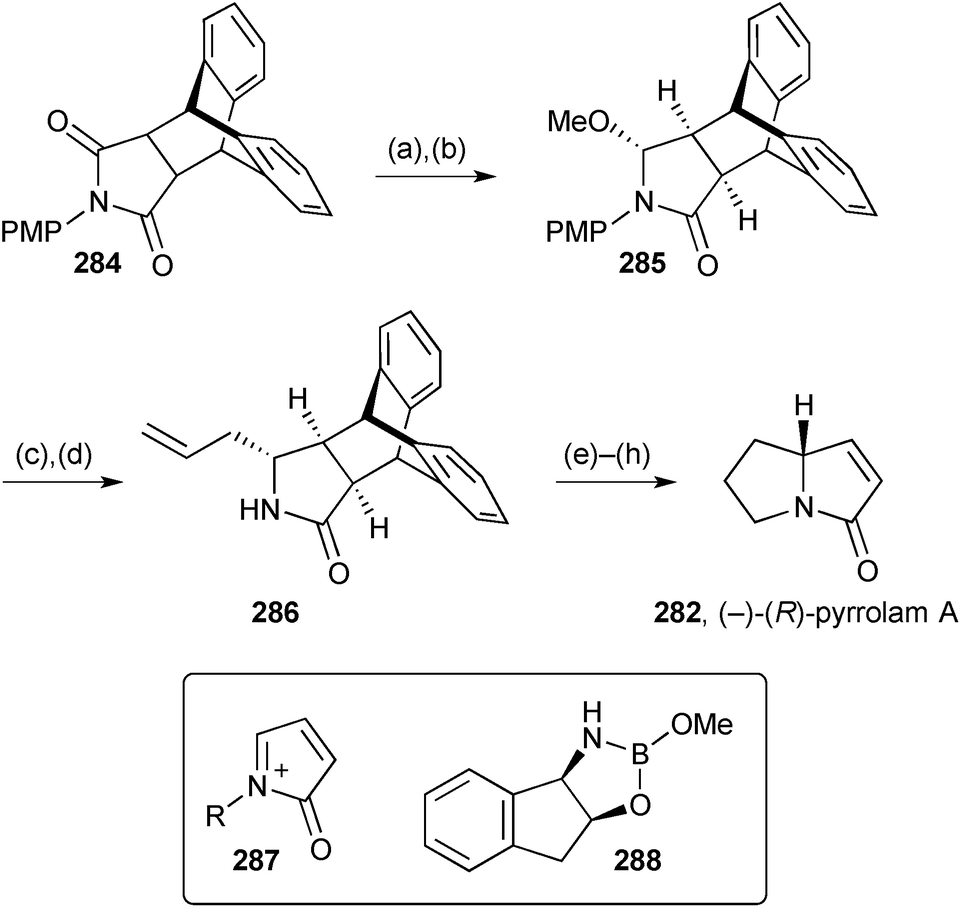 | ||
| Scheme 49 Reagents and conditions: (a) 288 (10 mol%), BH3·THF, THF [ee = 99%]; (b) TsOH, MeOH, 60 °C; (c) allyl-SiMe3, BF3·OEt2, CH2Cl2, −78 °C → rt; (d) CAN, aq. CH3CN, 0 °C → rt; (e) BH3·THF, THF, 0 °C → rt then H2O2, aq. NaOH; (f) MsCl, Et3N, DMAP, CH2Cl2, −10 °C; (g) DBU, EtOH, reflux; (h) 490 °C (FVP). | ||
Shibata has developed the use of an N-(2-pyridyl) group to direct enantioselective C–H activation and alkylation with alkenes. Scheme 50 illustrates an application of this to the formal synthesis of (−)-(R)-pyrrolam A.132 The initial alkylation (step a) was slow, taking one week in boiling dioxane to reach completion, but the product 290 was produced in high yield with a 90% ee. This level of enantiomeric purity was retained during the following four steps that comprised removal of the 2-pyridyl group, ester reduction and tosylation, then base-mediated cyclisation. In the paper, the product 291 is mis-drawn but may be inferred to be that shown here based on retention of stereochemistry from lactam 290. The route stopped at this point but the final steps (f and g) are known.
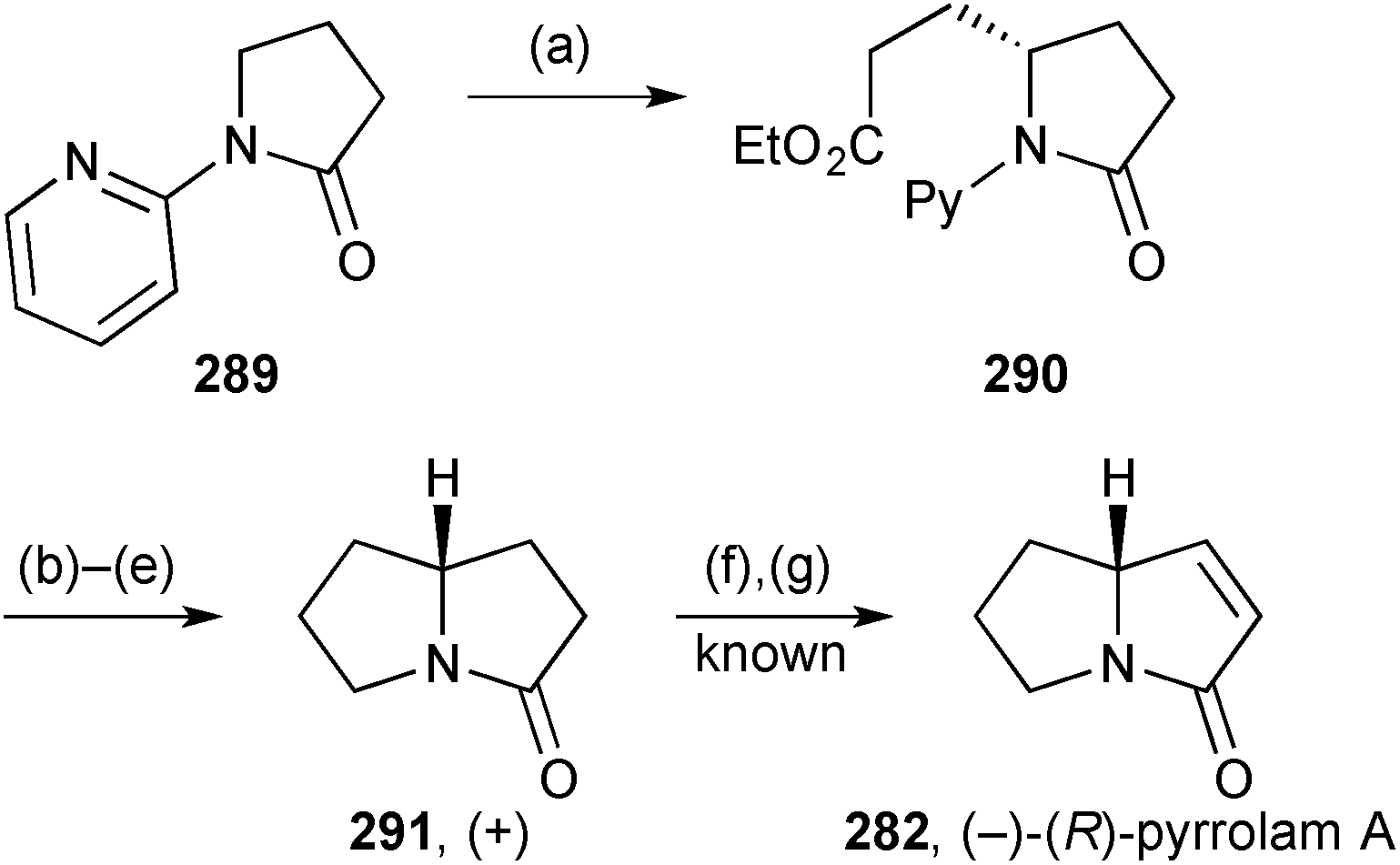 | ||
| Scheme 50 Reagents and conditions: (a) ethyl acrylate, [Ir(cod)2]BF4, (R)-tolBINAP, dioxane, reflux; (b) H2, Pd(OH)2/C, HCl, EtOH; (c) LiAlH4, MeOH; (d) TsCl, Et3N, DMAP, CH2Cl2; (e) NaH, THF; (f) LDA, PhSeCl, THF, −78 °C; (g) H2O2, aq. NaOH. | ||
Glasnov published an efficient microwave mediated hydrolysis of monocrotaline to produce retronecine which was oxidised selectively to give hydroxydanaidal 292 and its O-acetyl derivative 293.133 Hydrogenation of retronecine under various pressures of H2 under continuous flow conditions gave mixtures of desoxyretronecine, retronecanol, and platynecine.
3.8 Miscellaneous134
Trauner described progress towards the total synthesis of the unusual polyketide (−)-PF1018 294 isolated from a fungal Humicola sp. strain, that contains a pyrrolizidine-1,3-dione side chain.135 The synthetic chemistry focused on the tricyclic hydrocarbon moiety, prepared by an elegant 8π-electrocyclisation and Diels–Alder cascade.
Stockman prepared racemic xenovenine 297 (from the ant Solenopsis xenovenum), alkaloid cis-223B 299 (originally from the toad Melanophyniscus stelzneri) and its dipropyl analogue 298 (Scheme 51) by a ‘bioinspired’ triple reductive amination of appropriate tricarbonyl precursors.136 Under the optimised conditions shown (step e), the alkaloids were produced as single diastereomers, an outcome originally envisaged by the authors as resulting from thermodynamic control.
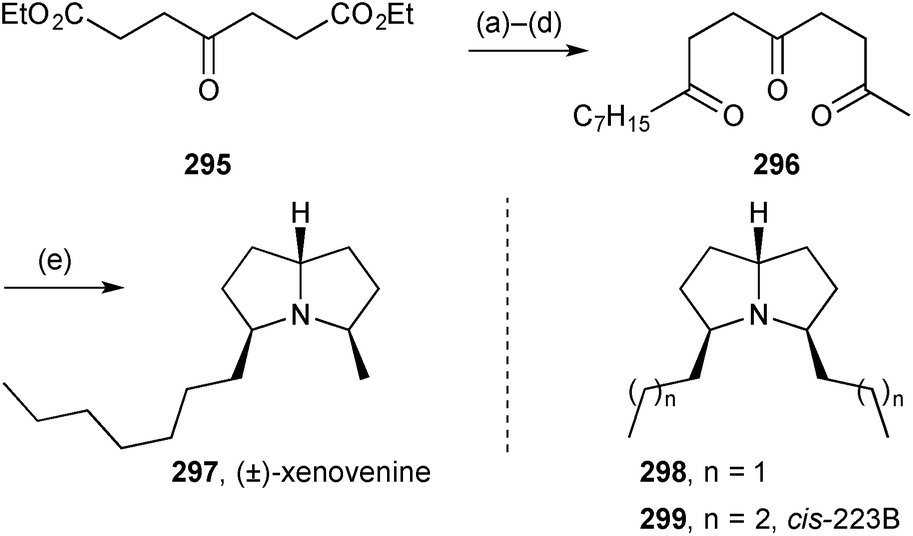 | ||
| Scheme 51 Reagents and conditions: (a) ethylene glycol, PPTS, PhCH3, reflux; (b) NH(OMe)Me, i-PrMgCl, THF, −15 °C → rt; (c) C7H15MgBr then MeMgBr, THF, −60 °C → rt; (d) aq. HCl; (e) NH4OAc, NaBH3CN, MeOH. | ||
Nicolaou conducted a synthesis of candidate stereoisomers of the antibiotic CJ-16,264 because its close relatives (pyrrolizilactone, UCS1025A, and UCS1025B) have different stereochemistry, particularly in the pyrrolizidinone part of the molecule, which is enantiomeric.137 This work resulted in a correction to the relative stereochemistry from 300 (Scheme 52) and assigned the absolute stereochemistry for the (+)-enantiomer as that shown in 303, the differing stereogenic centres being circled in the revised structure. Diastereomers of aldehyde 301, prepared from either (R)- or (S)-citronellol, were coupled with racemic iodopyrrolizidinone 302 to generate, after desilylation and 2°-alcohol oxidation, six candidate structures. By comparison of spectroscopic and specific rotation data, the correct structure could be assigned with confidence.
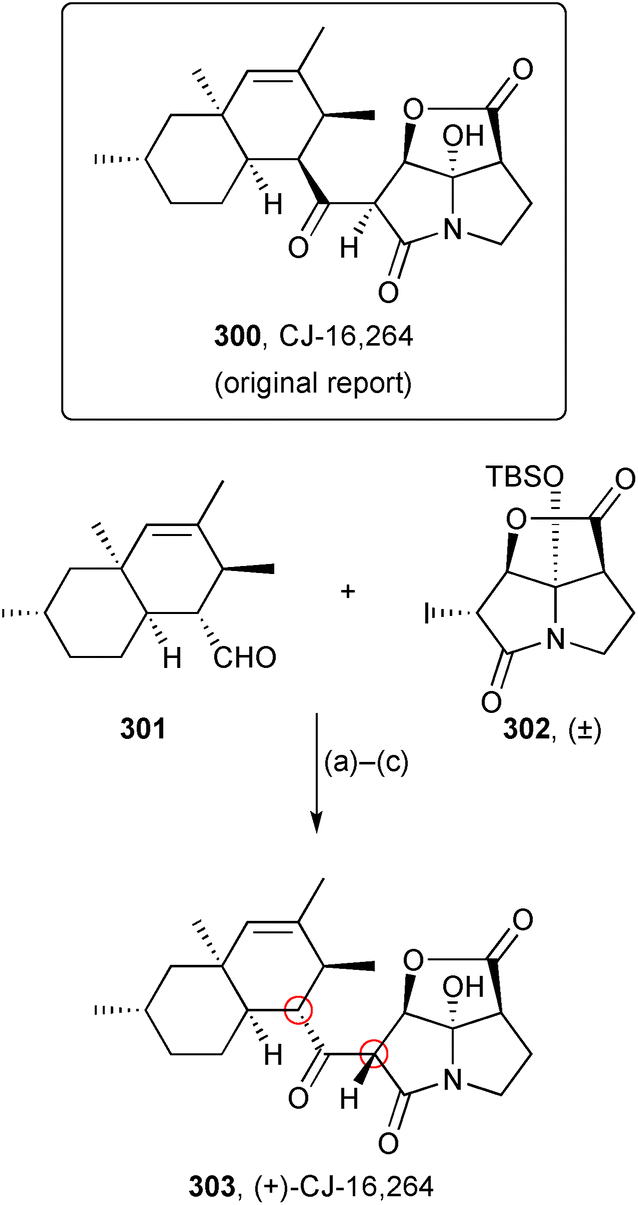 | ||
| Scheme 52 Reagents and conditions: (a) Et3B, PhCH3, −78 °C; (b) tris(dimethylamino)sulfoniumdifluoro trimethylsilicate, THF, 0 °C; (c) Dess–Martin periodinane, CH2Cl2. | ||
(−)-Penibruguieramine A, recently isolated as described earlier,71 was synthesised in a route based on its proposed biosynthesis from ketoamide 63.138 The tert-butyl ester of the enantiomer of this ketoamide 304 (Scheme 53) was prepared in five steps from E-hex-4-en-1-ol via enolate displacement of the derived 1°-alkyl bromide then coupling with (S)-proline as its tert-butyl ester. The key step in this route (step f) involved intramolecular aldol cyclisation which afforded pyrrolizidinone 305 as a single diastereomer in high yield, remarkable given the weak base and protic solvent employed for this transformation; see below for a mechanistic discussion. Reduction of the tert-butyl ester gave material spectroscopically identical to the natural product with an ee in excess of 99%; the structure (of 305) was further confirmed by X-ray crystallography.
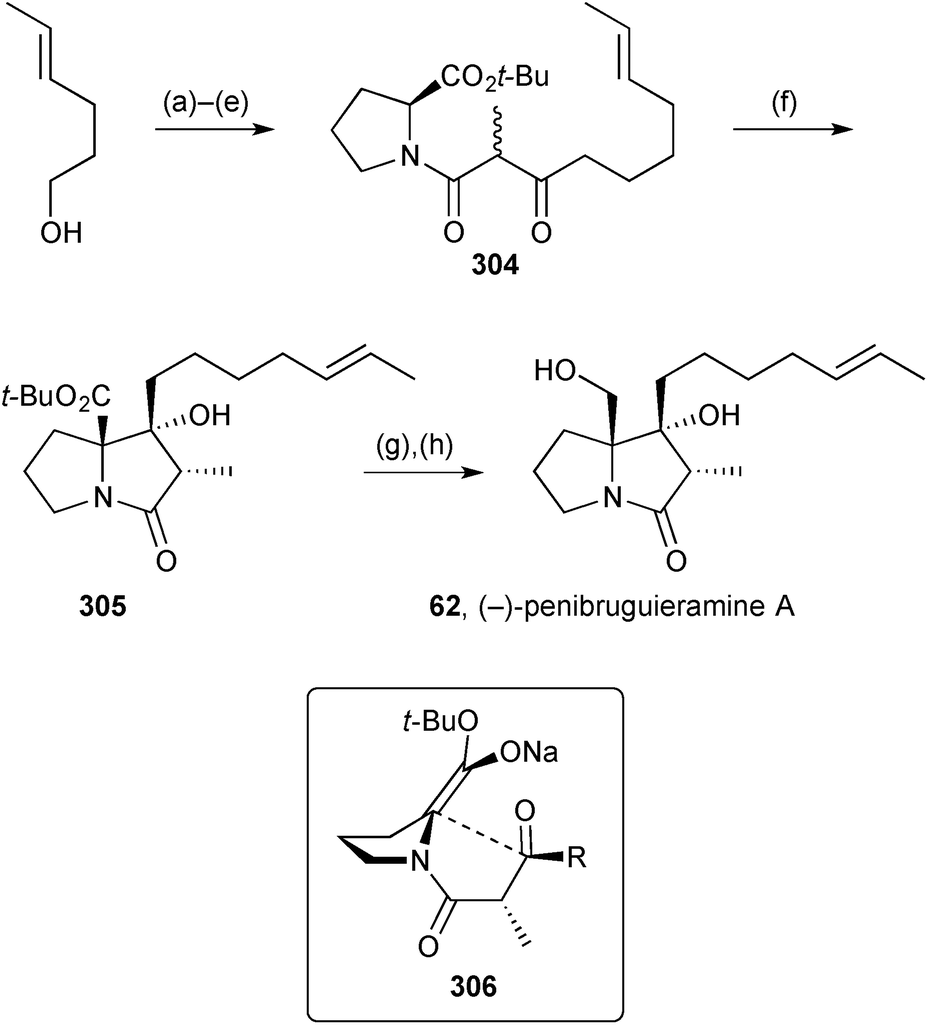 | ||
| Scheme 53 Reagents and conditions: (a) MsCl, Et3N, THF; (b) LiBr, THF, reflux; (c) ethyl 2-methylacetoacetate, NaH, BuLi; (d) KOH, aq. MeOH; (e) (S)-proline tert-butyl ester, DCC, DMAP, CH2Cl2; (f) NaOEt, EtOH; (g) CF3CO2H, CH2Cl2; (h) benzotriazolyloxy-tris(dimethylamino)phosphonium hexafluorophosphate, i-Pr2NEt, THF then NaBH4. | ||
A series of mechanistic investigations shed light on the cyclisation, in particular its stereochemical course: (1) performing the reaction to partial completion in EtOD showed incorporation of deuterium at all acidic positions both in the product and the recovered starting material; (2) subjecting the separated epimers of 304 (at the methyl-bearing ketoamide centre) to the cyclisation conditions showed equilibration to a 1.4:1 mixture of methyl-α and methyl-β isomers (with reference to the structure in Scheme 53) within 10 minutes, then conversion to the cyclised product 305 took place slowly, over ∼10 hours. On the basis of these observations, the authors concluded that all acidic sites are rapidly and reversibly deprotonated including the stereogenic proline α-centre and cyclisation takes place through a conformation 306 in which allylic strain is minimised in the minor epimer at equilibrium. The cyclisation step, therefore, employs both memory of chirality and dynamic kinetic resolution; the product was also shown, by computation, to be the most stable stereoisomer.
4 Outlook
Since their first discovery and characterisation as toxic components in plants the PAs have fascinated researchers across diverse fields of study and continue to do so. The published research summarised herein represents a mere fraction of the activity being undertaken in hundreds of laboratories and field studies worldwide with new developments appearing weekly. In looking back over the recent literature, a few highlights emerge: (1) the importance of the bacterial pyrrolizidines as a class will no doubt increase rapidly now that the genes responsible for their biosynthesis have been characterised and observed in diverse species. (2) A broader approach to assaying PAs for biological activity should be rewarded with new starting points for drug development. (3) A growing awareness of the presence of toxic PAs in herbal preparations means that steps can be taken to minimise chronic exposure and reduce illness whose cause is currently potentially unknown. (4) Synthetic chemists should be inspired by some of the very concise and efficient routes that are becoming more frequent, realise that chiral pool starting points may well offer tempting ‘free’ stereochemistry but often lead to lengthy sequences, and continue to consider PAs as valid targets for stimulating creative new strategies.5 References
- J. Robertson and K. Stevens, Nat. Prod. Rep., 2014, 31, 1721 RSC.
- M. M. Fashe, R. O. Juvonen, A. Petsalo, J. Vepsäläinen, M. Pasanen and M. Rahnasto-Rilla, Chem. Res. Toxicol., 2015, 28, 702 CrossRef CAS PubMed.
- M. M. Fashe, R. O. Juvonen, A. Petsalo, J. Räsänen and M. Pasanen, Chem. Res. Toxicol., 2015, 28, 2034 CrossRef CAS PubMed.
- See ref. 1, p. 1732.
- Q. Xia, Y. Zhao, L. S. Von Tungeln, D. R. Doerge, G. Lin, L. Cai and P. P. Fu, Chem. Res. Toxicol., 2013, 26, 1384 CrossRef CAS PubMed.
- Y. Zhao, S. Wang, Q. Xia, G. G. da Costa, D. R. Doerge, L. Cai and P. P. Fu, Chem. Res. Toxicol., 2014, 27, 1720 CrossRef CAS PubMed.
- X. Jiang, S. Wang, Y. Zhao, Q. Xia, L. Cai, X. Sun and P. P. Fu, J. Food Drug Anal., 2015, 23, 318 CrossRef CAS.
- Q. Xia, L. Ma, X. He, L. Cai and P. P. Fu, Chem. Res. Toxicol., 2015, 28, 615 CrossRef CAS PubMed.
- M. M. Fashe, R. O. Juvonen, A. Petsalo, M. Rahnasto-Rilla, S. Auriola, P. Soininen, J. Vepsäläinen and M. Pasanen, Chem. Res. Toxicol., 2014, 27, 1950 CrossRef CAS PubMed.
- R. A. Field, B. L. Stegelmeier, S. M. Colegate, A. W. Brown and B. T. Green, Toxicon, 2015, 97, 36 CrossRef CAS PubMed.
- A. Xiong, F. Yang, L. Fang, L. Yang, Y. He, Y. Y.-J. Wan, Y. Xu, M. Qi, X. Wang, K. Yu, K. W.-K. Tsim and Z. Wang, Chem. Res. Toxicol., 2014, 27, 775 CrossRef CAS PubMed.
- J. Tang, Z. Wang, T. Akao and M. Hattori, Nat. Prod. Commun., 2013, 8, 1545 CAS.
- J. Ruan, C. Liao, Y. Ye and G. Lin, Chem. Res. Toxicol., 2014, 27, 7 CrossRef CAS PubMed.
- T. Beuerle, I. Mädge and L. Cramer, Dtsch. Lebensm.-Rundsch., 2014, 110, 241 CAS.
- http://www.bfr.bund.de/cm/349/pyrrolizidine-alkaloids-in-herbal-teas-and-teas.pdf [accessed July 2016].
- M. Oplatowska, C. T. Elliott, A.-C. Huet, M. McCarthy, P. P. J. Mulder, C. von Holst, P. Delahaut, H. P. Van Egmond and K. Campbell, Anal. Bioanal. Chem., 2014, 406, 757 CrossRef CAS PubMed.
- C. T. Griffin, M. Danaher, C. T. Elliott, D. G. Kennedy and A. Furey, Food Chem., 2013, 136, 1577 CrossRef CAS PubMed.
- M. Martinello, C. Cristofoli, A. Gallina and F. Mutinelli, Food Control, 2014, 37, 146 CrossRef CAS.
- L. C. Bretanha, M. Piovezan, A. F. V. Sako, M. G. Pizzolatti and G. A. Micke, Am. J. Anal. Chem., 2014, 5, 681 CrossRef.
- F. J. Orantes-Bermejo, J. S. Bonvehí, A. Gómez-Pajuelo, M. Megías and C. Torres, Food Addit. Contam., Part A, 2013, 30, 1799 CrossRef CAS PubMed.
- C. Lu, T. Ding, X. Ma, G. Chen, F. Yang, B. Wu, C. Shen, R. Zhang, X. Fei, X. Zhang, L. Chen and L. Li, Chin. J. Chromatogr., 2013, 31, 1046 CrossRef CAS.
- E. M. Mudge, A. Maxwell, P. Jones and P. N. Brown, Food Addit. Contam., Part A, 2015, 32, 2068 CAS.
- T. Griffin, S. M. Mitrovic, M. Danaher and A. Furey, Food Addit. Contam., Part A, 2015, 32, 214 CrossRef PubMed.
- H. Neumann and A. Huckauf, J. Verbraucherschutz Lebensmittelsicherh., 2016, 11, 105 CrossRef CAS.
- Y. Cao, S. M. Colegate and J. A. Edgar, J. Food Compos. Anal., 2013, 29, 106 CrossRef CAS.
- L. Cramer, H.-M. Schiebel, L. Ernst and T. Beurle, J. Agric. Food Chem., 2013, 61, 11382 CrossRef CAS PubMed.
- L. Zhu, J.-Q. Ruan, N. Li, P. P. Fu, Y. Ye and G. Lin, Food Chem., 2016, 194, 1320 CrossRef CAS PubMed.
- C. Allgaier and S. Franz, Regul. Toxicol. Pharmacol., 2015, 73, 494 CrossRef CAS PubMed.
- B. Avula, S. Sagi, Y.-H. Wang, J. Zweigenbaum, M. Wang and I. A. Khan, Food Chem., 2015, 178, 136 CrossRef CAS PubMed.
- C.-H. Hsieh, H.-W. Chen, C.-C. Lee, B.-J. He and Y.-C. Yang, J. Food Compos. Anal., 2015, 42, 1 CrossRef CAS.
- C. F. Bosi, D. W. Rosa, R. Grougnet, N. Lemonakis, M. Halabalaki, A. L. Skaltsounis and M. W. Biavatti, Rev. Bras. Farmacogn., 2013, 23, 425 CrossRef CAS.
- C. Mathon, P. Edder, S. Bieri and P. Christen, Anal. Bioanal. Chem., 2014, 406, 7345 CrossRef CAS PubMed.
- T. Desta, M. Afework, C. R. Unnithan and H. Alay, Int. J. Pharm. Technol., 2014, 6, 6281 Search PubMed.
- C. T. Griffin, F. Gosetto, M. Danaher, S. Sabatini and A. Furey, Food Addit. Contam., Part A, 2014, 31, 940 CrossRef CAS PubMed.
- M. Schulz, J. Meins, S. Diemert, P. Zagermann-Muncke, R. Goebel, D. Schrenk, M. Schubert-Zsilavecz and M. Abdel-Tawab, Phytomedicine, 2015, 22, 648 CrossRef CAS PubMed.
- I. Mädge, L. Cramer, I. Rahaus, G. Jerz, P. Winterhalter and T. Beuerle, Food Chem., 2015, 187, 491 CrossRef PubMed.
- A. Shimshoni, A. Duebecke, P. P. J. Mulder, O. Cuneah and S. Barel, Food Addit. Contam., Part A, 2015, 32, 2058 Search PubMed.
- L. Cramer, G. Fleck, G. Horn and T. Beurle, J. Am. Oil Chem. Soc., 2014, 91, 721 CrossRef CAS.
- G. Vacillotto, D. Favretto, R. Seraglia, R. Pagiotti, P. Traldi and L. Mattoli, J. Mass Spectrom., 2013, 48, 1078 CrossRef CAS PubMed.
- D. Bodi, S. Ronczka, C. Gottschalk, N. Behr, A. Skibba, M. Wagner, M. Lahrssen-Wiederholt, A. Preiss-Weigert and A. These, Food Addit. Contam., Part A, 2014, 31, 1886 CrossRef CAS PubMed.
- B. Huybrechts and A. Callebaut, Food Addit. Contam., Part A, 2015, 32, 1939 CrossRef CAS PubMed.
- J. Becerra-Jiminez, M. Kuschak, E. Roeder and H. Wiedenfeld, Pharmazie, 2013, 68, 636 CAS.
- C. Gottschalk, S. Ronczka, A. Preiß-Weigert, J. Ostertag, H. Klaffke, H. Schafft and M. Lahrssen-Wiederholt, Anim. Feed Sci. Technol., 2015, 207, 253 CrossRef CAS.
- M. Bolechová, J. Čáslavsky, M. Pospíchalová and P. Kosubová, Food Chem., 2015, 170, 265 CrossRef PubMed.
- M. de Nijs, I. J. W. Elbers and P. P. J. Mulder, Food Addit. Contam., Part A, 2014, 31, 288 CrossRef CAS PubMed.
- S. H. Yoon, M.-S. Kim, S. H. Kim, H. M. Park, H. Pyo, Y. M. Leed, K.-T. Lee and J. Hong, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 992, 56 CrossRef CAS PubMed.
- B. M. Mandić, M. D. Vlajić, S. S. Trifunović, M. R. Simić, L. V. Vujisić, I. M. VuČković, M. M. Novaković, S. D. Nikolić-Mandić, V. V. Tešević, V. V. Vajs and S. M. Milosavljević, Nat. Prod. Res., 2015, 29, 887 CrossRef PubMed.
- A. Schenk, B. Siewert, S. Toff and J. Drewe, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 997, 23 CrossRef CAS PubMed.
- E. Cairns, M. A. Hashmi, A. J. Singh, G. Eakins, M. Lein and R. Keyzers, J. Agric. Food Chem., 2015, 63, 7421 CrossRef CAS PubMed.
- B. Tasso, F. Novelli, F. Sparatore, F. Fasoli and C. Gotti, J. Nat. Prod., 2013, 76, 727 CrossRef CAS PubMed.
- B. M. Mandić, M. R. Simić, I. M. Vučković, L. V. Vujisić, M. M. Novaković, S. S. Trifunović, S. D. Nikolić-Mandić, V. V. Tešević, V. V. Vajs and S. M. Milosavljević, Molecules, 2013, 18, 10694 CrossRef PubMed.
- P. Appadurai and K. Rathinasamy, Toxicol. Lett., 2014, 225, 66 CrossRef CAS PubMed.
- S. S. Kusuma, K. Tanneeru, S. Didla, B. N. Devendra and P. Kiranmayi, Anti-Cancer Agents Med. Chem., 2014, 14, 1237 CrossRef CAS PubMed.
- A. Kato, Y. Hirokami, K. Kinami, Y. Tsuji, S. Miyawaki, I. Adachi, J. Hollinshead, R. J. Nash, J. L. Kiappes, N. Zitzmann, J. K. Cha, R. J. Molyneux, G. W. J. Fleet and N. Asano, Phytochemistry, 2015, 111, 124 CrossRef CAS PubMed.
- D. A. C. Bezerra, J. F. Tavares, P. F. dos Santos, M. V. S. C. Branco, M. de Fátima Agra, F. L. Subrinho, R. Braz-Filho and M. S. da Silva, Magn. Reson. Chem., 2013, 51, 497 CrossRef PubMed.
- S. M. Colegate, D. R. Gardner, T. Z. Davis, J. M. Betz and K. E. Panter, Phytochem. Anal., 2013, 24, 201 CrossRef CAS PubMed.
- H. Damianakos, G. Sotiroudis and I. Chinou, J. Nat. Prod., 2013, 76, 1829 CrossRef CAS PubMed.
- Q.-H. Sun, J.-J. Yang, X.-H. Wei, H. Xu and G.-X. Chou, Phytochem. Lett., 2013, 6, 449 CrossRef CAS.
- S. Huang, X.-l. Zhou, C.-j. Wang, Y.-s. Wang, F. Xiao, L.-h. Shan, Z.-y. Guo and J. Weng, Phytochemistry, 2013, 93, 154 CrossRef CAS PubMed.
- C. Villanueva-Cañongo, N. Pérez-Hernández, B. Hernández-Carlos, E. Cedillo-Portugal, P. Joseph-Nathan and E. Burgueño-Tapia, Magn. Reson. Chem., 2014, 52, 251 CrossRef PubMed.
- B. M. Fraga, C. E. Díaz, L. J. Amador, M. Reina, O. Santana and A. González-Coloma, Phytochemistry, 2014, 108, 220 CrossRef CAS PubMed.
- M. Boppré and S. M. Colegate, Phytochem. Anal., 2015, 26, 215 CrossRef PubMed.
- S. M. Colegate, M. Boppré, J. Monzón and J. M. Betz, J. Ethnopharmacol., 2015, 172, 179 CrossRef CAS PubMed.
- L. S. Hoang, M. H. Tran, J. S. Lee, D. C. To, V. T. Nguyen, J. A. Kim, J. H. Lee, M. H. Woo and B. S. Min, Chem. Pharm. Bull., 2015, 63, 481 CrossRef CAS PubMed.
- J. Pan, M. Bhardwaj, J. R. Faulkner, P. Nagabhyru, N. D. Charlton, R. M. Higashi, A.-F. Miller, C. A. Young, R. B. Grossman and C. L. Schardl, Phytochemistry, 2014, 98, 60 CrossRef CAS PubMed.
- T. Nogawa, M. Kawatani, M. Uramoto, A. Okano, H. Aono, Y. Futamura, H. Koshino, S. Takahashi and H. Osada, J. Antibiot., 2013, 66, 621 CrossRef CAS PubMed.
- R. Sugiyama, S. Nishimura and H. Kakeya, Tetrahedron Lett., 2013, 54, 1531 CrossRef CAS.
- P. Yu, A. Patel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 13518 CrossRef CAS PubMed.
- W. Zhang, S. Li, Y. Zhu, Y. Chen, Y. Chen, H. Zhang, G. Zhang, X. Tian, Y. Pan, S. Zhang, W. Zhang and C. Zhang, J. Nat. Prod., 2014, 77, 388 CrossRef CAS PubMed.
- D. K. Derewacz, B. C. Covington, J. A. McLean and B. O. Bachmann, ACS Chem. Biol., 2015, 10, 1998 CrossRef CAS PubMed.
- Z.-F. Zhou, T. Kurtán, X.-H. Yang, A. Mándi, M.-Y. Geng, B.-P. Ye, O. Taglialatela-Scafati and Y.-W. Guo, Org. Lett., 2014, 16, 1390 CrossRef CAS PubMed.
- L. M. Petersen, J. C. Frisvad, P. B. Knudsen, M. Rohlfs, C. H. Gotfredsen and T. O. Larsen, J. Antibiot., 2015, 68, 603 CrossRef CAS PubMed.
- S. Huang, J. Tabudravu, S. S. Elsayed, J. Travert, D. Peace, M. H. Tong, K. Kyeremeh, S. M. Kelly, L. Trembleau, R. Ebel, M. Jaspars, Y. Yu and H. Deng, Angew. Chem., Int. Ed., 2015, 54, 12697 CrossRef CAS PubMed.
- O. Schimming, V. L. Challinor, N. J. Tobias, H. Adihou, P. Grün, L. Pöschel, C. Richter, H. Schwalbe and H. B. Bode, Angew. Chem., Int. Ed., 2015, 54, 12702 CrossRef CAS PubMed.
- P. Fu and J. B. MacMillan, Org. Lett., 2015, 17, 3046 CrossRef CAS PubMed.
- M. C. Kim, J. H. Lee, B. Shin, L. Subedi, J. W. Cha, J.-S. Park, D.-C. Oh, S. Y. Kim and H. C. Kwon, Org. Lett., 2015, 17, 5024 CrossRef CAS PubMed.
- Review: M. Brambilla, S. G. Davies, A. M. Fletcher and J. E. Thomson, Tetrahedron: Asymmetry, 2014, 25, 387 CrossRef CAS.
- D. Koley, K. Srinivas, Y. Krishna and A. Gupta, RSC Adv., 2014, 4, 3934 RSC.
- M. Brambilla, S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Tetrahedron, 2014, 70, 204 CrossRef CAS.
- N. A. Sorto, M. J. Di Maso, M. A. Muñoz, R. J. Dougherty, J. C. Fettinger and J. T. Shaw, J. Org. Chem., 2014, 79, 2601 CrossRef CAS PubMed.
- S. G. Davies, A. M. Fletcher, E. M. Foster, I. T. T. Houlsby, P. M. Roberts, T. M. Schofield and J. E. Thomson, Org. Biomol. Chem., 2014, 12, 9223 CAS.
- J. Li, H. Zhao, X. Jiang, X. Wang, H. Hu, L. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6306 CrossRef CAS PubMed.
- K. B. Gavhane, S. B. Bhorkade and M. G. Kulkarni, Tetrahedron: Asymmetry, 2015, 26, 746 CrossRef CAS.
- D. W. Knight, A. C. Share and P. T. Gallagher, J. Chem. Soc., Perkin Trans. 1, 1997, 2089 RSC.
- I. Chogii and J. T. Njardarson, Angew. Chem., Int. Ed., 2015, 54, 13706 CrossRef CAS PubMed.
- D. J. Aldous, A. S. Batsanov, D. S. Yufit, A. J. Dalençon, W. M. Dutton and P. G. Steel, Org. Biomol. Chem., 2006, 4, 2912 CAS.
- C. Heescher, D. Schollmeyer and U. Nubbemeyer, Eur. J. Org. Chem., 2013, 4399 CrossRef CAS.
- E. Zapata-Machin, T. Castellan, C. Baudoin-Dehoux and Y. Génisson, Synth. Commun., 2015, 45, 635 CrossRef CAS.
- S. Du-a-man, D. Soorukram, C. Kuhakarn, P. Tuchinda, V. Reutrakul and M. Pohmakotr, Eur. J. Org. Chem., 2014, 1708 CrossRef CAS.
- S. V. Kauloorkar, V. Jha, G. Jogdand and P. Kumar, Org. Biomol. Chem., 2014, 12, 4454 CAS.
- K. R. Luna-Freire, J. P. S. Scaramal, J. A. L. C. Resende, C. F. Tormena, F. L. Oliveira, R. Aparicio and F. Coelho, Tetrahedron, 2014, 70, 3319 CrossRef CAS.
- M. Jasinski, E. Moreno-Clavijo and H.-U. Reissig, Eur. J. Org. Chem., 2014, 442 CrossRef CAS.
- P. Rajasekaran, A. A. Ansari and Y. D. Vankar, Eur. J. Org. Chem., 2015, 2902 CrossRef CAS.
- S. Basu, P. S. Kandiyal, R. S. Ampapathi and T. K. Chakraborty, RSC Adv., 2013, 3, 13630 RSC.
- A. Rajender, J. P. Rao and B. V. Rao, Eur. J. Org. Chem., 2013, 1749 CrossRef CAS.
- E. A. Brock, S. G. Davies, J. A. Lee, P. M. Roberts and J. E. Thomson, Org. Biomol. Chem., 2013, 11, 3187 CAS.
- P. V. Reddy, J. Smith, A. Kamath, H. Jamet, A. Veyron, P. Koos, C. Philouze, A. E. Greene and P. Delair, J. Org. Chem., 2013, 78, 4840 CrossRef CAS PubMed.
- J. Smith, A. Kamath, A. E. Greene and P. Delair, Synlett, 2014, 25, 209 CAS.
- A. Veyron, P. V. Reddy, P. Koos, A. Bayle, A. E. Greene and P. Delair, Tetrahedron: Asymmetry, 2015, 26, 85 CrossRef CAS.
- J. Marjanovic, V. Divjakovic, R. Matovic, Z. Ferjancic and R. N. Saicic, Eur. J. Org. Chem., 2013, 5555 CrossRef CAS.
- R. Lahiri, Y. S. Reddy, S. A. Kulkarni and Y. D. Vankar, RSC Adv., 2013, 3, 23242 RSC.
- K. Savaspun, C. W. G. Au and S. G. Pyne, J. Org. Chem., 2014, 79, 4569 CrossRef CAS PubMed.
- C.-M. Si, Z.-Y. Mao, R.-G. Ren, Z.-T. Du and B.-G. Wei, Tetrahedron, 2014, 70, 7936 CrossRef CAS.
- M. Bergeron-Brlek, M. Meanwell and R. Britton, Nat. Commun., 2015, 6, 6903 CrossRef CAS PubMed.
- S.-H. Park, X. Jin, J.-C. Kang, C. Jung, S.-S. Kim, S.-S. Kim, K.-Y. Leeb and W.-H. Ham, Org. Biomol. Chem., 2015, 13, 4539 CAS.
- D. Martella, F. Cardona, C. Parmeggiani, F. Franco, J. A. Tamayo, I. Robina, E. Moreno-Clavijo, A. J. Moreno-Vargas and A. Goti, Eur. J. Org. Chem., 2013, 4047 CrossRef CAS.
- P. Gkizis, N. G. Argyropoulos and E. Coutouli-Argyropoulou, Tetrahedron, 2013, 69, 8921 CrossRef CAS.
- G. D'Adamio, C. Parmeggiani, A. Goti, A. J. Moreno-Vargas, E. Moreno-Clavijo, I. Robina and F. Cardona, Org. Biomol. Chem., 2014, 12, 6250 Search PubMed.
- C. Matassini, M. Marradi, F. Cardona, C. Parmeggiani, I. Robina, A. J. Moreno-Vargas, S. Penadés and A. Goti, RSC Adv., 2015, 5, 95817 RSC.
- (a) W.-Y. Xu, R. Iwaki, Y.-M. Jia, W. Zhang, A. Kato and C.-Y. Yu, Org. Biomol. Chem., 2013, 11, 4622 RSC; (b) C. Yu, W. Xu, Y. Jia, W. Zhang and Z. She, CN 103467470 A, 2013.
- D. Benadiková, M. Medvecký, A. Filipová, J. Moncoľ, M. Gembický, N. Prónayová and R. Fischer, Synlett, 2014, 25, 1616 CrossRef.
- R. Lahiri, A. Palanivel, S. A. Kulkarni and Y. D. Vankar, J. Org. Chem., 2014, 79, 10786 CrossRef CAS PubMed.
- D. Minehira, T. Okada, R. Iwaki, A. Kato, I. Adachi and N. Toyooka, Tetrahedron Lett., 2015, 56, 331 CrossRef CAS.
- C. Parmeggiani, F. Cardona, L. Giusti, H.-U. Reissig and A. Goti, Chem.–Eur. J., 2013, 19, 10595 CrossRef CAS PubMed.
- Y.-X. Li, Y. Shimada, K. Sato, A. Kato, W. Zhang, Y.-M. Jia, G. W. J. Fleet, M. Xiao and C.-Y. Yu, Org. Lett., 2015, 17, 716 CrossRef CAS PubMed.
- G. D'Adamio, A. Sgambato, M. Forcella, S. Caccia, C. Parmeggiani, M. Casartelli, P. Parenti, D. Bini, L. Cipolla, P. Fusi and F. Cardona, Org. Biomol. Chem., 2015, 13, 886 Search PubMed.
- A. L. Concia, L. Gómez, T. Parella, J. Joglar and P. Clapés, J. Org. Chem., 2014, 79, 5386 CrossRef CAS PubMed.
- P. Laborda, F. J. Sayago, C. Cativiela, T. Parella, J. Joglar and P. Clapés, Org. Lett., 2014, 16, 1422 CrossRef CAS PubMed.
- I. Oroz-Guinea, K. Hernández, F. C. Bres, C. Guérard-Hélaine, M. Lemaire, P. Clapés and E. García-Junceda, Adv. Synth. Catal., 2015, 357, 1951 CrossRef CAS.
- A. Soler, M. L. Gutiérrez, J. Bujons, T. Parella, C. Minguillon, J. Joglar and P. Clapés, Adv. Synth. Catal., 2015, 357, 1787 CrossRef CAS.
- S. G. Davies, A. M. Fletcher, C. Lebée, P. M. Roberts, J. E. Thomson and J. Yin, Tetrahedron, 2013, 69, 1369 CrossRef CAS.
- C. Berini, M. Sebban, H. Oulyadi, M. Sanselme, V. Levacher and J.-F. Brière, Org. Lett., 2015, 17, 5408 CrossRef CAS PubMed.
- Y. Kitamura, H. Koshino, T. Nakamura, A. Tsuchida, T. Nitoda, H. Kanzaki, K. Matsuoka and S. Takahashi, Tetrahedron Lett., 2013, 54, 1456 CrossRef CAS.
- J.-S. Zhu, S. Nakagawa, W. Chen, I. Adachi, Y.-M. Jia, X.-G. Hu, G. W. J. Fleet, F. X. Wilson, T. Nitoda, G. Horne, R. van Well, A. Kato and C.-Y. Yu, J. Org. Chem., 2013, 78, 10298 CrossRef CAS PubMed.
- M. Hattie, T. Ito, A. W. Debowski, T. Arakawa, T. Katayama, K. Yamamoto, S. Fushinobud and K. A. Stubbs, Chem. Commun., 2015, 51, 15008 RSC.
- K. E. Miller, A. J. Wright, M. K. Olesen, M. T. Hovey and J. R. Scheerer, J. Org. Chem., 2015, 80, 1569 CrossRef CAS PubMed.
- I. P. Kerschgens and K. Gademann, Synthesis, 2015, 47, 3153 CrossRef CAS.
- C. Lu, P. Zeng, M. Sun and X. Gu, CN 104311561, 2015.
- S. V. Kauloorkar, V. Jha and P. Kumar, RSC Adv., 2013, 3, 18288 RSC.
- M. Marhold, C. Stillig, R. Fröhlich and G. Haufe, Eur. J. Org. Chem., 2014, 5777 CrossRef CAS.
- B. J. Marsh, H. Adams, M. D. Barker, I. U. Kutama and S. Jones, Org. Lett., 2014, 16, 3780 CrossRef CAS PubMed.
- Y.-K. Tahara, M. Michino, M. Ito, K. S. Kanyivab and T. Shibata, Chem. Commun., 2015, 51, 16660 RSC.
- S. T. Martinez, A. C. Pinto, T. Glasnov and C. O. Kappe, Tetrahedron Lett., 2014, 55, 4181 CrossRef CAS.
- Review of the synthesis of a variety of -izidine alkaloids from proline: C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405 RSC.
- R. Webster, B. Gaspar, P. Mayer and D. Trauner, Org. Lett., 2013, 15, 1866 CrossRef CAS PubMed.
- A. Barthelme, D. Richards, I. R. Mellor and R. A. Stockman, Chem. Commun., 2013, 49, 10507 RSC.
- K. C. Nicolaou, A. A. Shah, H. Korman, T. Khan, L. Shi, W. Worawalai and E. A. Theodorakis, Angew. Chem., Int. Ed., 2015, 54, 9203 CrossRef CAS PubMed.
- J. H. Kim, S. Lee and S. Kim, Angew. Chem., Int. Ed., 2015, 54, 10875 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |