
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient route for design of luminescent composite materials based on polyethylene containing europium dibenzoylmethanate†
Dmitrii M.
Roitershtein
 *ab,
Lada N.
Puntus
ac,
Konstantin A.
Lyssenko
d,
Ilya V.
Taidakov
de,
Evgenia A.
Varaksina
ae,
Mikhail E.
Minyaev
a,
Victor A.
Gerasin
a,
Maria A.
Guseva
a,
Alexey A.
Vinogradov
ab,
Maria S.
Savchenko
f and
Ilya E.
Nifant'ev
af
*ab,
Lada N.
Puntus
ac,
Konstantin A.
Lyssenko
d,
Ilya V.
Taidakov
de,
Evgenia A.
Varaksina
ae,
Mikhail E.
Minyaev
a,
Victor A.
Gerasin
a,
Maria A.
Guseva
a,
Alexey A.
Vinogradov
ab,
Maria S.
Savchenko
f and
Ilya E.
Nifant'ev
af
aA.V. Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences, 29 Leninsky Prospect, 119991, Moscow, Russia. E-mail: roiter@yandex.ru
bN.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky Prospect, 119991, Moscow, Russia
cV.A. Kotel'nikov Institute of Radioengineering and Electronics, Russian Academy of Sciences, 11-7 Mokhovaya Str., 125009, Moscow, Russia
dA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilova Str., 119991, Moscow, Russia
eP.N. Lebedev Physical Institute, Russian Academy of Sciences, 53 Leninsky Prospect, Moscow 119991, Russia
fM.V. Lomonosov Moscow State University, Chemistry Department, 1 Leninskie Gory Str., Building 3, 119991, Moscow, Russia
First published on 3rd October 2017
Abstract
Luminescent composite materials based on linear polyethylene doped by a well dispersed tetrakis dibenzoylmethanate europium complex have been developed. The anion of the latter serves as an efficient light harvesting antenna and possesses the desired photophysical properties. The targeted modification of the Eu complex, namely introduction of a long hydrocarbon chain into the tetraalkylammonium cation [RNEt3]+, has ensured the compatibility of the complex with the polyethylene matrix. The [RNEt3]+ cation has been obtained by using long-chained 1-iodoalkanes synthesized via the Nd-catalyzed ethylene oligomerization process. The photophysical properties and the homogeneity of the obtained composites have been controlled by optical spectroscopy, luminescence intensity mapping as well as scanning electron microscopy.
Introduction
The use of lanthanide complexes in the development of new optical materials has markedly increased in the last few decades. Due to the unique photophysical properties,1–3 coordination and organometallic compounds of f-elements are important for the design of new luminescent materials for optical amplifiers for telecommunication networks,4 light-emitting devices,5 lasers,6 luminescent films,7 and organic light emitting diodes.8 Unlike organic compounds possessing similar characteristics, lanthanide complexes have low extinction coefficients, which ensure transparency in the visible region. Owing to high quantum yields, monochromatic emission (the line width does not exceed 10 nm), and long luminescence lifetimes, lanthanide complexes with organic ligands can serve in the development of luminescent labels9 and anti-counterfeiting taggants.10Lanthanide complexes with organic ligands providing an “antenna effect”9 have a great Stokes shift, which allows one to effectively convert UV radiation (200–400 nm) to visible light. Therefore, such complexes can be successfully used to prepare optical materials.11 Radiation converters that transform the UV part of the solar spectrum to visible radiation are of considerable interest. In particular, increasing the proportion of red light in the spectra of light sources used in agriculture can intensify green plant growth. In this case, Eu(III) chelate complexes with the emission maximum at 610–620 nm are appropriate for the purpose.12
Of particular interest are luminescent composite materials based on various polymers. When homogeneous distribution of luminescent materials in the polymer matrix is achieved, the intensity and efficiency of luminescence can be substantially increased.13,14 Moreover, the composite materials based on polymers doped by the lanthanide complexes are very attractive for different applications since a high luminance of the composite can be achieved by relatively low concentrations of complexes that remarkably decreases the cost of such materials.
The most intriguing challenge is to obtain luminescent composite materials based on polyethylene, which possesses the excellent photostability under UV/blue irradiation required from modern luminescent materials.13 Besides, obtaining a homogenous distribution of a luminescent complex in the polyethylene matrix is associated with well-known difficulties.8,15–17
The known luminescent polymer composites fabricated, for example, using polyvinylpyrrolidone,18 polyvinylpyridine and the polyethylene methacrylate copolymer,19 and silicone rubber20 either do not utilize polyethylene, or use a polyethylene as a surface coating for luminescent materials rather than as a matrix in which they are distributed.21 There is only one reported example of formation of polyethylene composites based on europium complexes, including hexanoic, octanoic, lauric, and stearic acid salts.22 The composites thus obtained showed the typical Eu luminescence; however, the authors did not elucidate the degree of homogeneity of the europium distribution in the polyethylene matrix.
Compatibility of the matrix materials and lanthanide complex is the key problem arising in the design of hybrid materials composed of polymers doped by the lanthanide complexes. In particular, the well known brightly luminescent complex Na[Eu(tta)4], (Htta is 2-thenoyltrifluoroacetone) is sparingly soluble in low-polarity polymers like polystyrene, and is well compatible only with more polar polyesters (e.g. PMMA).23 The presence of perfluorinated substituents may induce the aggregation and phase separation of the complex in the matrix.
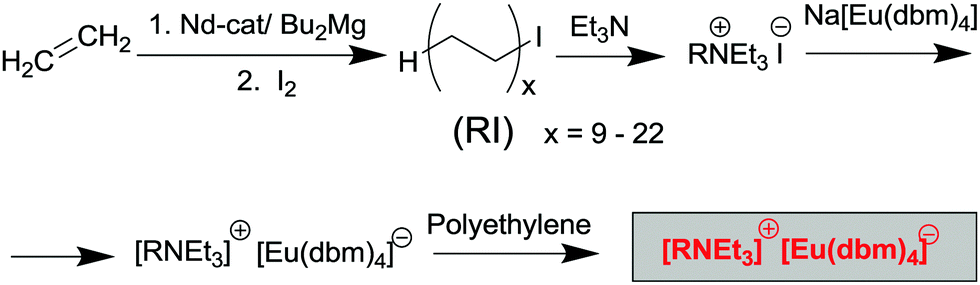
We here report a straightforward and efficient route for design of luminescent composite materials based on linear polyethylene (PE) (Scheme 1), including the synthesis of three tetrakis-Eu complexes containing dibenzoylmethane (1,3diphenylpropane-1,3-dione (Hdbm)): Na[Eu(dbm)4] (1),24 [(C12H25NMe3)]2[Eu(dbm)4]I (2), and [(RNEt3)] [Eu(dbm)4] (3).
 | ||
| Scheme 1 | ||
Dibenzoylmethane is known as the ligand serving as the efficient light harvesting antenna for the Eu ion. Also, the dibenzoylmethanate complexes of europium exhibit both photoluminescent and triboluminescent properties.25–27 The anionic tetrakis-type of complex was determined by the general assumption that the use of a luminescent complex containing two parts, namely the luminescent anion and cation with a long hydrocarbon chain can prevent the separation of the polymer and complex and ensure homogeneity of the composite. The introduction of a long chain cation into the tetrakis complexes 2 and 3 should lead to better distribution of these complexes into PE. The photophysical properties of both the tetrakis complexes and composites containing these complexes were studied systematically by optical spectroscopy. The homogeneity of the distribution of the luminescent centres in the polymer matrix was controlled by luminescence intensity mapping and scanning electron microscopy.
Results and discussion
The tetraalkylammonium salt, (RNEt3)I, was prepared by the reaction of RI with triethylamine.28 Hereinafter, R designates a set of CnH2n+1 oligomers with n = 18–44, where n is an even number (with the weight-average molecular weight corresponding to n = 30–32). The sample of RI 1-iodoalkanes used to prepare the ammonium salt was synthesized by catalytic oligomerization of ethylene followed by treating the reaction mixture with iodine28 (Scheme 2). | ||
| Scheme 2 | ||
Our preliminary experiments demonstrated excellent compatibility of polyethylene (PE) with the tetraalkylammonium salt (RNEt3)+I. The [(RNEt3)]+[Eu(dbm)4]− complex, consisting of the tetraalkylammonium cation compatible with the polyethylene matrix and the [Eu(dbm)4]− anion was expected to provide high luminescence.
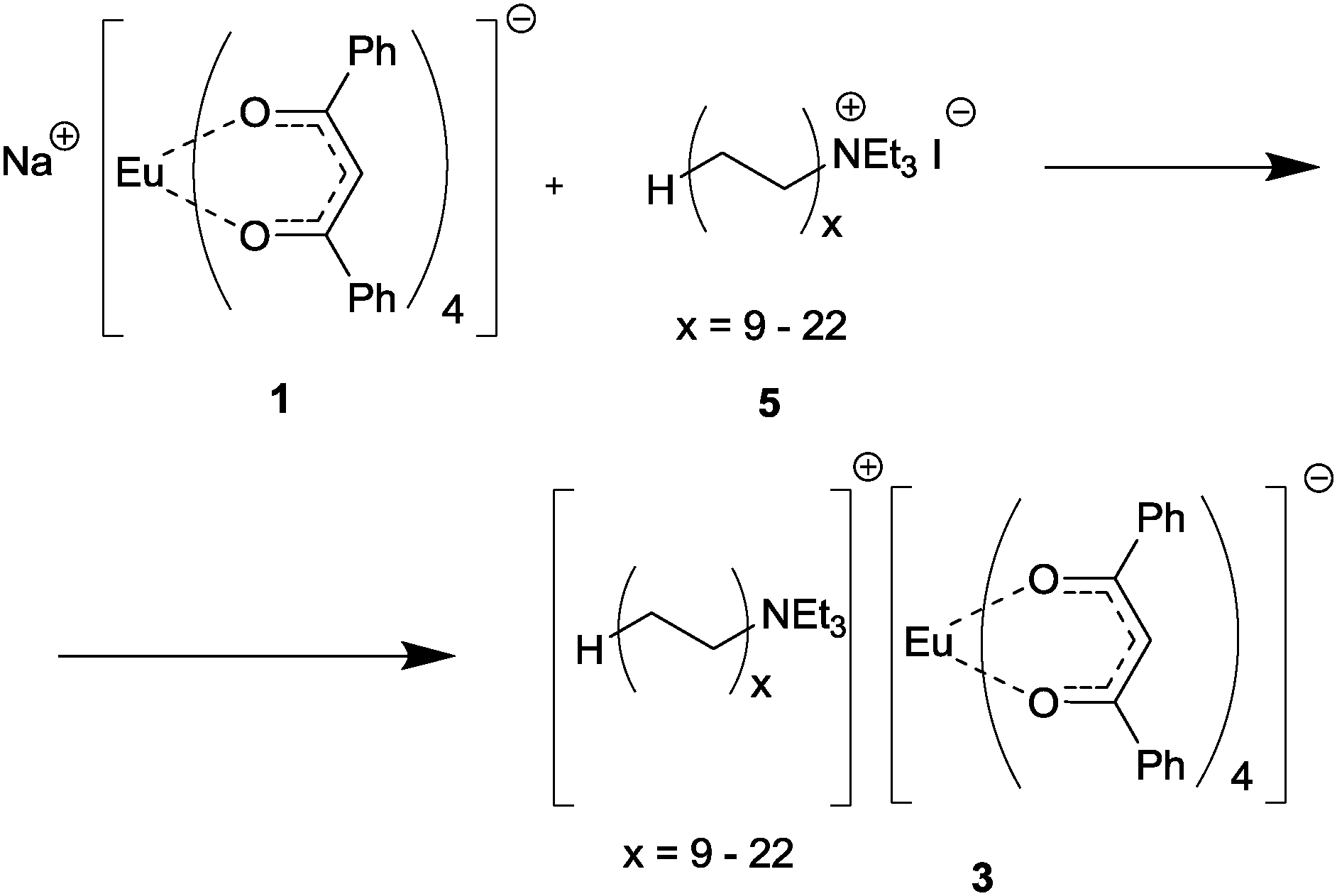
In order to evaluate the advantages and the scope of the method applicability, we used three sorts of luminescent complexes for composite preparation, namely, three europium tetrakis-β-diketonate complexes: Na[Eu(dbm)4] (1),24 [(C12H25NMe3)]2[Eu(dbm)4]I (2), and [(RNEt3)] [Eu(dbm)4] (3). Complex 1, which served as a precursor for compounds 2 and 3, was used for comparison as a standard europium complex with ligands that provide an “antenna effect”. Complex 2 was an individual compound, intended to be used as a model of 3 for verification and subsequent optimization of all procedures en route from the europium complex to the polyethylene-based composite. Compound 3 was not a single compound, but a mixture of europium dibenzoylmethanate complexes with the [(RNEt3)]+ cations (R = CnH2n+1, n = 18–44).
Synthesis of europium complexes
Complex 1 was prepared by a known procedure,24 compounds 2 and 3 were synthesized by the reactions of 1 with the ammonium salts (C12H25NMe3)+I− (4) and (RNEt3)+I− (5), which were prepared by the reaction of dimethyldodecylamine with CH3I and by the reaction of the oligomeric RI mixture with triethylamine, respectively (Scheme 2). The structure of (C12H25NMe3)+I− was established by X-ray diffraction (see structural studies for details).It was found that the reaction of the oligomeric iodoalkane RI mixture does not proceed to 100% conversion under typical conditions of tertiary amine quaternization with alkyl halides29 even upon considerable increase in the reaction time and with a large excess of Et3N (8 to 20 equivalents). Therefore, samples of the (RNEt3)+I− salt contained higher iodoalkanes RI as an irremovable impurity.
Compounds 2 and 3 were synthesized by ion exchange between complex 1 and the corresponding ammonium salts upon refluxing in THF. The reaction of 1 with the (C12H25NMe3)+I− salt did not give the expected compound, [(C12H25NMe3)][Eu(dbm)4], but yielded its ate-complex with the initial ammonium salt, [(C12H25NMe3)]2[Eu(dbm)4]I (2) instead (according to elemental analysis and 1H NMR data) (Scheme 3). The NMR spectrum of the resulting ate-complex was quite informative, despite the presence of the paramagnetic Eu3+ cation. It is interesting that the [Eu(dbm)4]− anion behaves as a shift-reagent relative to the (C12H25NMe3)+ cation, inducing only a slight line broadening and a small downfield signal shift. As compared with the initial ammonium salt, (C12H25NMe3)+I−, the (CH3)3N– signals in the Eu complex 2 have more pronounced downfield shifts (4.81 ppm vs. 3.47 ppm) than the (CH3)3N–CH2– signals (4.44 ppm vs. 3.63 ppm). The presence of iodide ions was confirmed by HRMS data (negative ion m/z = 126.9051). A twofold increase in the reaction time (from 40 to 80 hours) did not change the composition of the reaction products.
 | ||
| Scheme 3 | ||
The reaction of (RNEt3)I (5) with complex 1 (Scheme 4) was also carried out by refluxing in THF. The RI impurity present in the (RNEt3)+I− salt can also react with complex 1 (see S2, ESI†). To control the completeness of reaction between 5 and 1, the reaction time was varied from 50 to 100 hours. After 50 hours of refluxing, the composition of the reaction products remained unchanged, according to the chemical analysis data.
 | ||
| Scheme 4 | ||
Structural studies
Although all attempts to obtain single crystals of 2 were unsuccessful we were able to analyze the crystal structures of 1 (Na+(EtOH)2[Eu(dbm)4]−) (Fig. 1) and 4 (C12H25NMe3)+I−. | ||
| Fig. 1 The general view of 1 in the representation of atoms by thermal ellipsoids (p = 50%). The sodium and ethanol molecules with occupancies equal to 0.32 are omitted for clarity. | ||
The europium cation in 1 is eight-coordinated and adopts the distorted square antiprism polyhedron. Its distortion and variation of Eu–O bond lengths 2.346(2)–2.401(2) Å is clearly the consequence of the cation⋯anion interactions in crystal. Indeed, salt 1 crystallizes as the contact ion pair in which the coordination number of the sodium cation is equal to five and its sphere is composed of three oxygen atoms of the dbm ligand that correspond to the same base of the europium antiprism polyhedron (the Eu(1)⋯Na(1) distance is 3.381(2) Å), and of two oxygen atoms of ethanol molecules. The Na⋯O distances for oxygen atoms of dbm ligands and ethanol vary in the ranges 2.339(3)–2.620(4) and 2.246(5)–2.251(6) Å, respectively.
It should be noted that for oxygen atoms of dbm that are involved in additional coordination with sodium, the Eu–O distances are systematically longer (av. 2.40 Å) than for those which are not (av. 2.37 Å).
The above contact ion pairs are assembled in the crystal into infinite chains by means of O–H⋯O hydrogen bonds with the O⋯O separation being equal to 2.806(5) and 2.831(6) Å. (Fig. 2).
 | ||
| Fig. 2 The general view of the H-bonded chain directed along crystallographic axis b in the crystal of 1. | ||
In the crystal of 4 (Fig. 3), cations form a typical packing, namely, chains in which the neighboring cations are arranged in a head-to-tail pattern and are assembled by means of weak C–H⋯I contacts with H⋯I distances of ca. 3.0 Å. The C12-chain is characterized by almost all trans-conformations, although some of the C–C–C–C torsion angles are as small as 130–140°.
 | ||
| Fig. 3 The cation⋯anion interactions in the crystal of 4. | ||
To some extent, the crystal data for 1 and 4 shed some light on the possible structure of the ate-complex 2. For example, in both salts, contact ion pairs are formed via the Na⋯O and C–H⋯I interactions. Analysis of the CSD30,31 demonstrates that ammonium salts of lanthanide-containing anions form similar contact ion pairs, in which the alkyl substituents at nitrogen are involved in C–H⋯O contacts with oxygens of β-diketonate ligands, CSD refcodes: (FIDNIP01),32 (FIDNOV),33 (LOLWEQ), (LOLWIU), (LOLWAM)27 and (GUVWAW).34 The iodide anion in complex 2 can also (by analogy with salt 4) be involved in the additional cation binding, which should considerably change the charge distribution in the lanthanide coordination sphere and, hence, should affect the luminescent characteristics of the lanthanide ion.
Preparation of composite films
For the preparation of Eu-containing composites, samples 1–3 were mixed with PE in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio (w/w) by the melt mixing technique using a laboratory micro-extruder. A 0.3 mm-thick film was formed from the composite materials obtained. The subsequent measurements were performed with film samples.
:10 ratio (w/w) by the melt mixing technique using a laboratory micro-extruder. A 0.3 mm-thick film was formed from the composite materials obtained. The subsequent measurements were performed with film samples.
Initially, samples of compounds 2 and 3 were investigated by TGA and DSC for their thermal stability. According to TGA, compound 3 is stable up to 200 °C and undergoes reversible phase transition in the temperature range of 45–90 °C (Fig. S8, ESI†). Compound 2, which was isolated as an ate-complex [(C12H25NMe3)]2[Eu(dbm)4]I−, starts to decompose at 138 °C (Fig. S9, ESI†). Since the film is formed at a higher temperature, evidently, a substantial part of complex 2 may decompose and contaminate the composite with the products of thermolysis. Therefore, compound 2, which was initially intended for the investigation of the behavior of europium complexes in the polymer matrix, is inapplicable for modeling of the composite luminescence base, because of low thermal stability. However, the obtained results are suitable for evaluation of the limitations of this approach.
For the luminescent compound introduced into the matrix, we chose the complex [(RNEt3)]+[Eu(dbm)4]−, consisting of the tetraalkylammonium cation, which meets the compatibility requirement with the polyethylene matrix, and the [Eu(dbm)4]− anion, which is expected to ensure high luminescence.
Metal-centered luminescence of tetrakis complexes
The luminescence spectra of the europium complexes 1–3 were recorded from both ligand-centered excitation at 380 nm and metal-based excitation at 465 nm, which gives rise to similar characteristic Eu3+ emission peaks (Fig. 4). These spectra show the typical narrow bands corresponding to the Eu3+ 5D0 → 7FJ (J = 0–4) transitions. Generally the intensities and Stark splittings of the 5D0 → 7FJ transitions are influenced by the strength and symmetry of the ligand field. A forbidden 5D0 → 7F0 transition (region 570–585 nm) of the Eu3+ ion is presented by a weak symmetric single line in all cases which indicates the presence of only one type of Eu environment. Moreover all the luminescence decays of the excited 5D0 level of the Eu ion were treated as a single exponential function and the corresponding lifetimes confirm the presence of only one type of luminescence center for every complex as well (Table 1 and Fig. 5).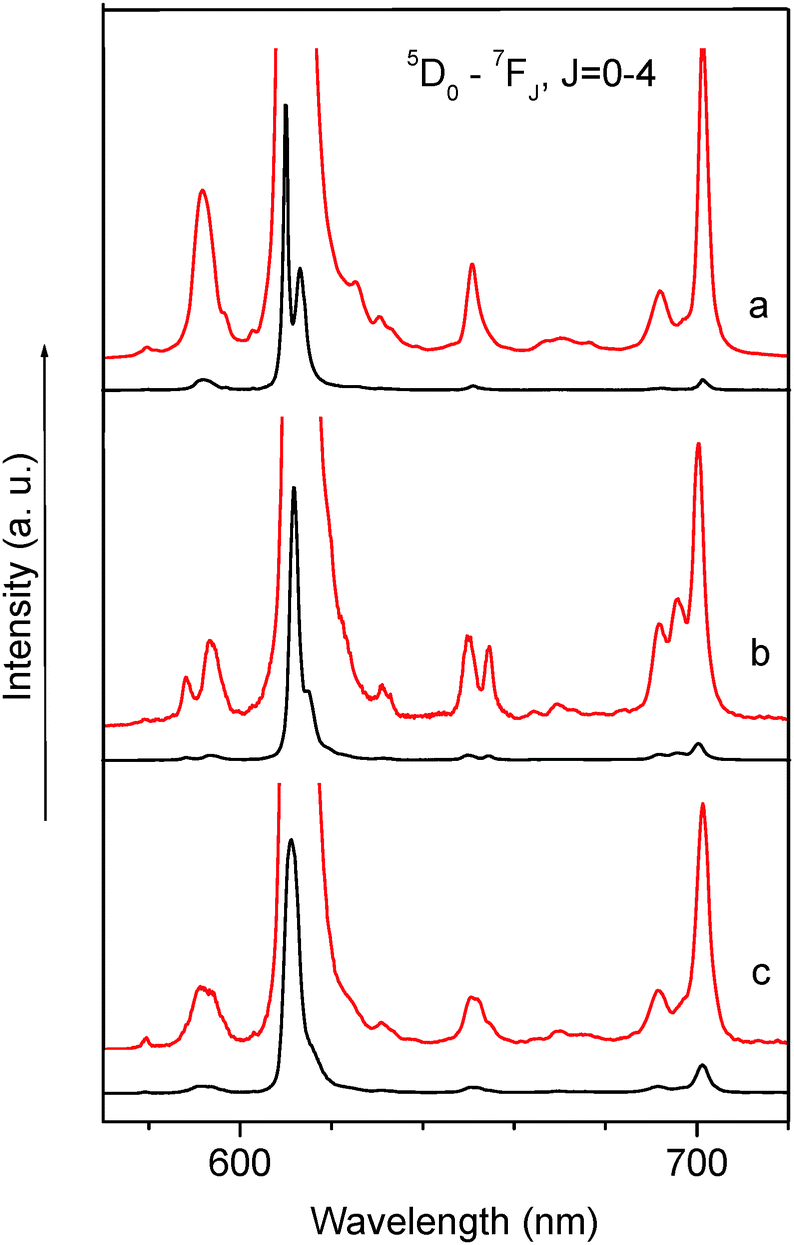 | ||
| Fig. 4 Luminescence spectra of complexes 1 (a), 2 (b) and 3 (c) at 300 K, λexc = 380 nm. | ||
| Complex | 5D0–7F0 | 5D0–7F2 | 5D0–7F4 | I tot/IMD,0 | τ obs (ms−1) | Q EuEu ± 2% | Q LigEu ± 5% |
|---|---|---|---|---|---|---|---|
| 1 | 0.04 | 15 | 1 | 18 | 0.54 ± 0.02 | 49 | 26 |
| 2 | 0.1 | 28 | 3 | 33 | 0.26 ± 0.01 | 42 | 2.5 |
| 3 | 0.04 | 21 | 3 | 26 | 0.49 ± 0.02 | 63 | 10 |
 | ||
| Fig. 5 Photoluminescence decay curves for complexes 1, 2, 3 (λem = 615 nm, λex = 380 nm, T = 300 K). | ||
The integrated intensity of this transition is the maximum in the case of complex 2 (Table 1 and Fig. 5). The electric dipole 5D0 → 7F2 transition (region 600–620 nm) is extremely sensitive to the symmetry of the europium surroundings and is called hypersensitive and so the ratio of the integrated intensity of the 5D0 → 7F2 transition to the one of the 5D0 → 7F1 transition is a measure of the symmetry of the coordination sphere. In a centrosymmetric environment the magnetic dipole 5D0 → 7F1 transition is predominant and above the ratio <1 while the distortion of the symmetry around the ion causes an intensity enhancement of the 5D0 → 7F2 transition. In these complexes this ratio equals 15, 28 and 21 for complexes 1–3, respectively, and indicates relatively strong deviation of the Eu3+ site symmetry from the inversion center as well as a highly polarizable chemical environment around the ion. The maximum value obtained for complex 2 (28) along with the maximum value of integrated intensity discussed for the 5D0 → 7F0 transition evidences the strongest deviation of site symmetry of the Eu ion from the inversion center. The comparison performed on integrated intensities together with analysis of the fine structure of the luminescence spectra as well as the lifetime of the 5D0 level of the europium ion point out a similar site symmetry of the Eu3+ ion in complexes 1 and 3. The difference observed for all these parameters for complex 2 can be explained by the fact that this complex exists as an ate-complex [(C12H25NMe3)]2[Eu(dbm)4]I (2) and contains two [RNR3′]+, cations and additional anion (I−). As was discussed above, the ammonium salts of lanthanide anions can form tight ion pairs, in which the alkyl substituents linked with nitrogen atom form C–H⋯O short contacts with oxygen atoms of the β-diketonate. Previously we have shown that noncoordinated anions (chlorides and triflates) can effect Stark splittings of the electronic transitions of the Eu ion as well as the formation charge transfer states which are involved in Eu luminescence sensitization.35–37
The high intensity of the first Stark component of the 5D0 → 7F2 transition at 300 K potentially can be successfully used for obtaining a relatively high color purity (for example in complex 1 the line at 610 nm, ∼45% of total integrated intensity, fwhm (full width-at-half-maximum) = 34 cm−1).
The efficiency observed of the energy transfer processes in the tetrakis complexes has been estimated by the calculation of the intrinsic quantum yield of the europium centered emission, QEuEu, by means of Werts' formula.38 In the considered europium complexes 1–3 the value of the intrinsic quantum yield is quite similar and equal to 49%, 42% and 63%, respectively (Table 1) which indicates the intense dependence on nonradiative deactivation processes as well as on structural peculiarities as was discussed above. The highest value of the intrinsic quantum yield observed in 3 is also in line with the lowest site symmetry of the europium surroundings. The overall absolute quantum yield QLLn upon ligand excitation was measured and for the most luminescent complex 1 it amounts to 26 ± 0.3% and so the efficacy with which electromagnetic energy is transferred from the surroundings onto the Eu ion (ηsens) amounts to ∼55%.
The excitation spectra of the europium complexes 1–3 measured at 300 K display in addition to the narrow f–f transitions 5D4,2,1,0 ← 7F0, (Fig. 6), a broad band expanded in the range 250–500 nm with a broadened vibronic structure (a case of medium electron–phonon interaction). The longest wavelength maximum of this band in the case of complexes 1 and 3 is centered at 400 nm and assigned to the S1 state of the dbm ligand. Vibronic satellites appearing in the excitation spectra of complexes 1 and 3 are assigned to the symmetric vibration of the β-diketonate chelating ring with a large contribution of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching with a frequency of 1470 cm−1. The longest wavelength band is slightly blue shifted in the case of complex 2 up to 390 nm. It is worth mentioning that a broad pedestal is found in the region 420–500 nm, which is most clearly observed in the excitation spectrum of complex 3. This covered weak band can be tentatively assigned to the charge transfer state. Generally the energy of the charge transfer state is determined by such parameters as the optical electronegativity of the ligand and so the higher the energy of the charge transfer state the higher the value of this parameter. Taking into account the structural peculiarities of the complexes one can conclude that the presence of two ammonium cations due to C–H⋯O interactions and the steric repulsion in complex 2 promotes the elongation of the Eu–Ligand bonds. As a result the optical electronegativity of dbm is the highest in complex 2.37,39,40 The relatively low energy of this charge-transfer band (its long wavelength edge is ∼20500 cm−1) can be the reason for the not very high value of the overall absolute quantum yield QLLn.
O bond stretching with a frequency of 1470 cm−1. The longest wavelength band is slightly blue shifted in the case of complex 2 up to 390 nm. It is worth mentioning that a broad pedestal is found in the region 420–500 nm, which is most clearly observed in the excitation spectrum of complex 3. This covered weak band can be tentatively assigned to the charge transfer state. Generally the energy of the charge transfer state is determined by such parameters as the optical electronegativity of the ligand and so the higher the energy of the charge transfer state the higher the value of this parameter. Taking into account the structural peculiarities of the complexes one can conclude that the presence of two ammonium cations due to C–H⋯O interactions and the steric repulsion in complex 2 promotes the elongation of the Eu–Ligand bonds. As a result the optical electronegativity of dbm is the highest in complex 2.37,39,40 The relatively low energy of this charge-transfer band (its long wavelength edge is ∼20500 cm−1) can be the reason for the not very high value of the overall absolute quantum yield QLLn.
 | ||
| Fig. 6 Luminescence excitation spectra of complexes 1 (a), 3 (b) and 2 (c) at 300 K, λreg = 612 nm. | ||
All prepared polyethylene films exhibit a characteristic luminescence of the Eu3+ ion with a maximum at 610 nm (Fig. 7).
 | ||
| Fig. 7 Luminescence spectra of PE films doped by 10% of complexes 2 (a), 3 (b) and 1 (c) at 300 K. | ||
The luminescence spectra of these polyethylene films differ from the spectra of initial complexes. The observed spectra suggest that the composite formation has induced some changes in the coordination sphere of the Eu ion but the nature of the changes will be the subject of further studies.
Of most interest was to elucidate the homogeneity of the europium distribution in the polymer matrix. Luminescence intensity mapping experiments (Fig. 8a) demonstrated a homogeneous distribution of the luminescence centers in the PE matrix.
 | ||
| Fig. 8 Images of luminescence intensity mapping (λem = 620 nm, λex = 375 nm), sample sizes 80 × 80 μm. Composite films: (a) PE – 3 (10%), (b) PE – 3 (1%), (c) PE – 1 (10%), (d) PE – 2 (10%). | ||
Comparison of the luminescence intensity mapping results (Fig. 8) for films made of the three composite materials (PE–3 (a), PE–1 (c), PE–2 (d)) indicates that the most homogeneous distribution of the luminescent dopant in PE has been attained for the composite based on 3 (Fig. 8(a)). The use of inorganic complex 1 (Fig. 8(c)) gives rise to a considerable film inhomogeneity, while the use of 2 (Fig. 8(d)) results in the formation of aggregates in which the luminescent dopant is concentrated. The latter may be attributable to the formation of micelle-like aggregates in the composite containing 2. In the composite doped with 3, the formation of such aggregates is prevented by the inhomogeneity of the [RNEt3]+ cations, which differ in the chain length of the organic group R, or because their hydrocarbon chain is (on average) much longer. One more possible reason for the inhomogeneous distribution of 2 is its noticeable thermal decomposition during the film formation caused by the low thermal stability of the complex. The film with a lower europium content (1% of 3) obtained by dilution of the PE–10% 3 was also characterized by a homogeneous distribution of luminescence centers over the film area (Fig. 8(b)).
The SEM images of the composite film surfaces (Fig. 9) also show a marked inhomogeneity of the composites prepared using 1 or 2, as compared with the film made of the composite doped with 3.
 | ||
| Fig. 9 SEM images of composite films: (a) PE – 3 (10%), (b) PE – 1 (10%), (c) PE – 2 (10%). | ||
Conclusions and outlooks
To find a straightforward route to the PE-compatible complexes of rare earths, the reaction of alkylammonium iodides (RNR′3)+I, (R = CnH2n+1, n = 18–44) with Na[Eu(dbm)4] was applied and led to the exchange of sodium with the organic cation, containing a long linear hydrocarbon chain.The chosen approach allows one to introduce a long linear hydrocarbon chain into the lanthanide complex, possessing the desired photophysical properties. This finding provided a high compatibility with polyethylene and allowed us to obtain polyethylene based optical composite materials with pre-determined properties.
A high degree of homogeneity in the distribution of the luminescent centers in polyethylene has been achieved by the proposed method. The developed procedure opens up the route to obtain composite materials emitted in different regions of the spectrum from visible (Tb, Sm, Dy ions) to the NIR part (Nd, Yb, Er ions) by using different lanthanide ions. Moreover, the unlimited row of existed lanthanide complexes also allows optimizing the optical properties of forthcoming luminescent composite materials based on polyethylene and that is very attractive for practical applications, these materials containing a small amount of lanthanide complexes (10 mol%) can be highly luminescent.
To improve the photophysical characteristics of the composite within the proposed methodology, it is necessary to optimize the synthesis of the alkylammonium salt used, and to test other lanthanide complexes concerning the thermostability of the luminescent compound.
Experimental
General details
Tetrahydrofuran and diethyl ether were predried over NaOH and distilled from potassium/benzophenone ketyl. Hexane was distilled from the Na/K alloy. Na[Eu(dbm)4](C2H5OH)2 (1),24 was prepared according to literature procedures, synthesis of the oligomer mixture “RI” was performed according to the published procedure28 with some modifications, as described in the ESI† (S1). The matrix polymer used to prepare the composite was PE, Mw = 3 × 105, Mn = 1 × 105, d = 0.940 g cm−3, Mp. = 126 °C. Metal analyses were performed by direct complexometric titrations with EDTA-disodium salt, using a Xylenol Orange indicator. TGA analyses were conducted using the TGA/DSC1 Mettler Toledo system at a rate of 10 K min−1 under an argon flow of 10 mL min−1 in the range from 303 K to 673 K. Standard differential scanning calorimetry (DSC) thermograms were recorded on a DSC823 Mettler Toledo system at a rate of 10 K min−1 under an argon atmosphere in the range from 293 K to 553 K. The 1H NMR spectra were recorded with Bruker AV400 (400 MHz) or Bruker AV600 (600 MHz) instruments in CDCl3. MALDI mass spectra were recorded with a Bruker autoflex speed instrument in positive ion reflectron mode using a solid-state UV-laser (λ = 355 nm). ESI mass spectra were recorded with a MALDI-TOF Bruker ultraflex III.X-ray crystallography
X-ray diffraction data were collected on a APEX II DUO CCD diffractometer using molybdenum radiation [λ(MoKα) = 0.71072 Å, ω-scans] for 1 and 4. The substantial redundancy in data allowed empirical absorption correction to be applied with SADABS by multiple measurements of equivalent reflections. The structures were solved by direct methods and refined by the full-matrix least-squares technique against F2 in the anisotropic-isotropic approximation. Assuming the proximity of the β angle in 1 to 90° upon the refinement it was treated as the merohedral twin (monoclinic cell emulating the orthorhombic one). The usage of the TWIN −1 0 0 0 −1 0 0 0 1 instruction led to the BASF parameter being equal to 0.39832 and to a significant decrease of the R1 value. In the case of 4 despite the pseudosymmetry (P21/m space group) we have solved and refined it in P21. The choice of the latter space group was due to the significant deviation from planarity for the C12-chain (see torsion angles) as well as due to the unrealistic anisotropic displacement parameters of carbon atoms in the case of the P21/m space group.The positional and anisotropic displacement parameters of the disordered Na(HOC2H5)2 fragment in 1 (see Fig. S1 in ESI†) were refined with constraints on the O–C and C–C bond length (DFIX) and anisotropic displacement parameters (EADP).
The occupancies for Na(1) and Na(1′) atoms were equal to 0.680(3) and 0.320(3) respectively. C–H hydrogen atoms in all structures were placed in calculated positions and refined within the riding model. Hydroxy H-atoms in 1 were located from the Fourier density synthesis. All calculations were performed with the SHELXTL software package.41 Crystal data and structure refinement parameters are listed in Table 2. Crystallographic data for the structures reported in this paper have been deposited in the Cambridge Crystallographic Data Centre as CCDC 1567021 (for 1) and 1567020 (for 4).†
| 1 | 4 | |
|---|---|---|
| Brutto formula | C64H56EuNaO10 | C15H34IN |
| Formula weight | 1160.03 | 355.33 |
| T/K | 120 | 120 |
| Space group | P21/c | P21 |
| Z(Z′) | 4(1) | 2(1) |
| a/Å | 25.835(2) | 5.7367(6) |
| b/Å | 8.3357(8) | 7.4012(8) |
| c/Å | 25.370(2) | 21.041(2) |
| β/° | 90.1060(19) | 91.012(2) |
| Volume/Å3 | 5463.3(9) | 893.25(17) |
| ρ calc/g cm−3 | 1.410 | 1.321 |
| μ/cm−1 | 12.16 | 17.79 |
| F(000) | 2376 | 368 |
| 2θmax, ° | 58 | 17.79 |
| Reflections collected (Rint) | 55473 (0.0554) | 14807 (0.0378) |
| Independent reflections | 14499 | 4748 |
| Reflections with I > 2σ(I) | 12081 | 4266 |
| Parameters | 710 | 160 |
| R 1 [I > 2σ(I)] | 0.0381 | 0.0276 |
| wR2 | 0.0824 | 0.0622 |
| GOF | 0.977 | 1.040 |
| Residual electron density, e Å−3 (ρmin/ρmax) | −1.001/1.336 | −0.460/0.752 |
Syntheses
MS MALDI-TOF [m/z (I/I0)]: found for C15H34N+: 228.26 (100%), 229.27 (22%), 230.28 (2%). Calcd.: 228.27 (100.0%), 229.27 (17%), 230.28 (1%). NMR 1H (600 MHz, CDCl3), δ: 0.88 (t, 3H, –CH2CH3), 1.26 (br. s, 14H, –NC3H6 –C8H16–CH3), 1.28 (m, 2H, –NC2H4–CH2–C8H17), 1.77 (m, 2H, –NCH2CH2–) 3.47 (s, 9H, –N(CH3)), 3.63 (m, 2H, –NCH2–).
MALDI-TOF detected ions Et3N+(CH2–CH2)nH with n = 9–22 (with the highest peaks for n = 13–16). Mn = 526, Mw = 536. (Fig. S10, ESI†) NMR 1H (400 MHz, CDCl3), δ: 0.85 (t. –CH2CH3), 1.24 (br. s –(CH2)n–CH3), 1.39 (t, –N(CH2–CH3)3), 1.70 (m, –NCH2–CH2–), 1.80 (m., 4H, I–CH2–CH2–), 3.17 (t, I–CH2–), 3.25 (br. m, –N–CH2–), 3.50 (q., –N(CH2–CH3)3).
The product may contain an admixture of linear low molecular weight polyethylene, which is difficult to trace due to overlapping of signals at the NMR-spectrum.
ESI-MS [m/z (I/I0)]: (+ve) 228.268 (100%) (C15H34N+), 338.341(6%), 449.127 (7%)
ESI-MS [m/z (I/I0)]: (−ve) 126.905 (100%) (I−), 223.076 (13%), 255.233 (15%), 281.248 (10%), 355.095 (11%).
1H NMR (500 MHz, CDCl3), δ: 0.92 (br. s, 6H, –CH2CH3), 1.31 (br. s, 28H, –NC4H8–C7H14–CH3), 1.43 (br. s, 4H, –NC3H6–CH2–C8H17), 1.61 (br. s, 4H, –NC2H4–CH2–C9H19), 2.19 (br. s, 4H, –NCH2–CH2–), 4.44 (br. m, 4H, –NCH2–), 4.81 (br. s, 18 H, –N(CH3)), 7.00–7.20 (m. 20H), 7.35–7.60, (m. 20H); 13C NMR (150.9 MHz, CDCl3), δ: 13.7, 22.2, 23.3, 26.1, 28.9, 29.2, 31.5, 54.8, 67.6, 125.5, 125.9, 126.7, 128.2, 130.2, 181.3, 185.3.
ESI MS detected ions Et3N+(CH2–CH2)nH with n = 12–20 (the highest peaks for n = 15–16), negative range: ESI-MS [m/z (I/I0)]: (−ve) 126.905 (100%) (I−), 223.076 (13%), 255.233 (15%), 281.248 (10%), ESI-MS and NMR spectra are presented in Fig. S13, S6 and S7 (ESI†), respectively. Elemental analysis: Found: C, 63.70; H, 6.98; N, 0.56; Eu, 9.48, the difference in the results of the analysis between samples from two independent synthetic experiments did not exceed 0.5%.
Preparation of the composite and composite films
Composites PE-1, PE-2 and PE-3 were prepared by single-stage mixing in a melt under the action of shear stress. The laboratory twin-screw micro-extruder HAAKE MiniLab Rheomex CTW5 was loaded with PE 4.500 g and 0.500 g of the corresponding europium complexes. The mixing was carried out at a temperature of 155 °C and at a screw speed of 100 rpm for 15 minutes. The composite with a lower content of Eu (1% of 3) was obtained by “diluting” of PE-3 Eu with polyethylene to the required concentration. After unloading from the extruder, the composites were pressed on a press with heating plates at a temperature of 155 °C and a pressure of 5 MPa for 5 minutes, and then were cooled in air. Composite films with a diameter of ca. 5 cm and a thickness of 0.3 mm were obtained.Scanning electron microscopy
A target-oriented approach was utilized for the optimization of the analytic measurements.42 Before measurements the samples were mounted on a 25 mm aluminum specimen stub, fixed by conductive silver paint and coated with a thin film (15 nm) of carbon. The observations were carried out using a Hitachi SU8000 field-emission scanning electron microscope (FE-SEM). Images were acquired in backscattered electron mode at 15 kV accelerating voltage and at a working distance of 10–11 mm. The morphology of the samples was studied taking into account the possible influence of the carbon coating on the surface.Photo-physical study
The luminescence measurements (spectra and lifetimes) were recorded on a Fluorolog FL 3-22 spectrometer from Horiba-Jobin-Yvon-Spex at 300 K. Lifetimes (τ) were measured on samples put into quartz capillaries; they are averages of at least three independent measurements, which were achieved by monitoring the decay at the maxima of the emission spectra. The single or biexponential decays were analyzed with Origin 7.0. Quantum yields (Q) of Eu-centered luminescence were determined by an absolute method with a specially designed integration sphere(G8, GMP SA, Switzerland). The calculation of the intrinsic quantum yield of the europium centered emission, QEuEu, has been performed by means of the Werts' formulawhere AMD,0 is a constant spontaneous emission probability (14.65 s−1 for EuIII), n the refractive index, Itot the total area of the emission spectrum (5D0 → 7FJ, J = 0–6), and IMD,0 the 5D0 → 7F1 band area.38 The refractive index was taken as 1.5 for all cases. Luminescence intensity mapping experiments were carried out with the use of a MicroTime 200 time-correlated single photon counting system (PicoQuant, Germany) coupled to an inverted fluorescent microscope IX71 (Olympus, Japan). The samples of flat films were placed on the microscope stage and excited by a PDL 828 pulsed laser (376 nm emission, 50 ps pulse duration, 100 kHz pulse repetition rate). The emission spectral band was selected by interference band-pass filters (Chroma, USA) with a bandwidth of 10 nm. The filter band was centered at 620 nm. The luminescent signal was acquired by a τ-SPAD photon-counting avalanche photodiode and a PicoHarp 300 correlation system of MicroTime 200. The horizontal positioning accuracy of the piezopositioner mounted into the microscope stage was about 10 nm. Spatial resolution of 300 nm was achieved with the use of the confocal scheme together with a UPlanSApo 100 microscope objective. The maximum size of the region of interest (ROI) was 80 × 80 microns.

Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Russian Science Foundation (Grant No. 17-13-01357). The authors are grateful to the Department of Structural Studies of the Zelinsky Institute of Organic Chemistry, Moscow, for electron microscopy characterization. The X-ray diffraction and optical data were obtained using the equipment of the Center For Molecule Composition Studies of INEOS RAS.References
- S. V. Eliseeva and J.-C. G. Bünzli, New J. Chem., 2011, 35, 1165 RSC.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53 CrossRef PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- S. Moynihan, R. Van Deun, K. Binnemans, J. Krueger, G. von Papen, A. Kewell, G. Crean and G. Redmond, Opt. Mater., 2007, 29, 1798 CrossRef CAS.
- P. P. Sun, J. P. Duan, J. J. Lih and C. H. Cheng, Adv. Funct. Mater., 2003, 13, 683 CrossRef CAS.
- K. Nakamura, Y. Hasegawa, H. Kawai, N. Yasuda, N. Kanehisa, Y. Kai, T. Nagamura, S. Yanagida and Y. Wada, J. Phys. Chem. A, 2007, 111, 3029 CrossRef CAS PubMed.
- C. Yang, Z. F. Sun, L. Liu and L. Q. Zhang, J. Mater. Sci., 2008, 43, 1681 CrossRef CAS.
- J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357 CrossRef CAS PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542 CrossRef CAS PubMed.
- O. Guillou, C. Daiguebonne, G. Calvez and K. Bernot, Acc. Chem. Res., 2016, 49, 844 CrossRef CAS PubMed.
- S. F. H. Correia, V. de Zea Bermudez, S. J. L. Ribeiro, P. S. Andre, R. A. S. Ferreira and L. D. Carlos, J. Mater. Chem. A, 2014, 2, 5580 CAS.
- The Rare Earth Elements: Fundamentals and Applications, ed. A. D. Atwood, Wiley, 2012, p. 575 Search PubMed.
- J. Rocha, L. D. Carlos, F. A. Almeida Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926 RSC.
- L. D. Carlos, R. A. S. Ferreira, V. D. Z. Bermudez, B. Julian-Lopez and P. Escribano, Chem. Soc. Rev., 2011, 40, 536 RSC.
- S. Liu, Y. Gao, T. Chen and Q. Wang, Appl. Mech. Mater., 2013, 467, 86 CrossRef.
- S. S. Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539 CrossRef CAS.
- M. Alexandre and P. Dubois, Mater. Sci. Eng., 2000, 28, 1 Search PubMed.
- Q. Shi, Z. XinyiJin, Y. Shen and Y. Liu, Mater. Lett., 2015, 147, 113 CrossRef CAS.
- C. Liu and B. Yan, Eur. J. Inorg. Chem., 2015, 2797 Search PubMed.
- S. Wen, X. Zhang, L. Yao, M. Xi, L. Zhang, H. Fong and L. Liu, J. Mater. Chem. C, 2013, 1, 1613 RSC.
- J. Zhang, A. Z. Mohammad, S. Rahman, Y. Li, J. Yang, B. Zhao, E. Lu, P. Zhang, X. Cao, R. Yu and B. Wang, Opt. Mater., 2013, 36, 471 CrossRef CAS.
- M. Lin, Q. An, Z. Hu, H. Zeng and Q. Ling, Chin. J. Appl. Chem., 2014, 31, 1164 CAS.
- I. V. Taydakov, S. A. Ambrozevich, E. A. Varaksina, A. G. Vitukhnovsky, A. A. Tyutyunov and O. A. Melnik, J. Russ. Laser Res., 2016, 37, 192 CrossRef CAS.
- W. G. Quirino, C. Legnani, R. M. B. dos Santos, K. C. Teixeira, M. Cremona, M. A. Guedes and H. F. Brito, Thin Solid Films, 2008, 517, 1096 CrossRef CAS.
- H.-Y. Wong, W.-S. Lo, W. T. K. Chan and G.-L. Law, Inorg. Chem., 2017, 56, 5135 CrossRef CAS PubMed.
- R. G. Xiong and X. Z. You, Inorg. Chem. Commun., 2002, 5, 677 CrossRef CAS.
- S. Akerboom, M. S. Meijer, M. A. Siegler, W. T. Fu and E. Bouwman, J. Lumin., 2014, 145, 278 CrossRef CAS.
- M. E. Minyaev, A. A. Vinogradov, D. M. Roitershtein, R. S. Borisov, I. V. Ananyev, A. V. Churakov and I. E. Nifant'ev, J. Organomet. Chem., 2016, 818, 28 CrossRef.
- J. E. Gordon, J. Am. Chem. Soc., 1965, 87, 4347 CrossRef CAS.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, B72, 171 CrossRef PubMed.
- F. A. Cotton, L. M. Daniels and P. Huang, Inorg. Chem. Commun., 2001, 4, 319 CrossRef.
- L. M. Sweeting and A. L. Rheingold, J. Am. Chem. Soc., 1987, 109, 2652 CrossRef CAS.
- M. Milanova, J. Zaharieva, B. Morgenstern, K. Hegetschweiler and D. Todorovsky, Z. Kristallogr. - New Cryst. Struct., 2010, 225, 17 CAS.
- L. Puntus and K. Lyssenko, J. Rare Earths, 2008, 26, 146 CrossRef.
- L. Puntus, K. Lyssenko, I. Pekareva and M. Yu. Antipin, Mol. Phys., 2010, 108, 557 CrossRef CAS.
- Yu. V. Nelyubina, L. N. Puntus and K. A. Lyssenko, Chem. – Eur. J., 2014, 20, 2860 CrossRef CAS PubMed.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- L. N. Puntus, K. A. Lyssenko, I. S. Pekareva and J.-C. G. Bunzli, J. Phys. Chem. B, 2009, 113, 9265 CrossRef CAS PubMed.
- L. N. Puntus, K. A. Lyssenko, M. Yu. Antipin and J.-C. G. Bunzli, Inorg. Chem., 2008, 47, 11095 CrossRef CAS PubMed.
- M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef PubMed.
- V. V. Kachala, L. L. Khemchyan, A. S. Kashin, N. V. Orlov, A. A. Grachev, S. S. Zalesskiy and V. P. Ananikov, Russ. Chem. Rev., 2013, 82, 648 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1567020 and 1567021. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj03055j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |