
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddition of N-nucleophiles to gold(III)-bound isocyanides leading to short-lived gold(III) acyclic diaminocarbene complexes†
Tatyana B.
Anisimova
a,
Mikhail A.
Kinzhalov
b,
M. Fátima C.
Guedes da Silva
 a,
Alexander S.
Novikov
b,
Vadim Yu.
Kukushkin
b,
Armando J. L.
Pombeiro
*a and
Konstantin V.
Luzyanin
*bc
a,
Alexander S.
Novikov
b,
Vadim Yu.
Kukushkin
b,
Armando J. L.
Pombeiro
*a and
Konstantin V.
Luzyanin
*bc
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
bSaint Petersburg State University, 7/9 Universitetskaya Nab., Saint Petersburg 199034, Russian Federation
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: konstantin.luzyanin@liverpool.ac.uk
First published on 6th April 2017
Abstract
Reaction of [AuCl3(CNR1)] (R1 = Xyl, Cy, (S)-CHMePh) with amines unexpectedly proceeds via the redox pathway giving gold(I)–isocyanides and imines, while the addition of benzophenone hydrazone to the isocyanide ligand in [AuCl3(CNR1)] at RT leads to short-lived gold(III) acyclic diaminocarbene complexes [AuCl3{C(NHNCPh2)NHR1}].
In recent years, complexes with N-heterocyclic carbenes (NHCs) and acyclic diaminocarbenes (ADCs) have emerged as valuable alternatives to other traditional catalysts in a number of organic transformations.1 Most common routes to their preparation include (i) a coordination of the pre-formed carbenes (generated in situ from appropriate precursors and a base, or obtained via transmetallation from M-NHCs) to a metal center,1b,e,2 (ii) an oxidative addition of appropriate carbene precursors to electron-rich metal centers,1d,e,2 and (iii) a metal-mediated nucleophilic addition or a dipolar cycloaddition to isocyanides.1a,2,3
A few reported Au(III)–NHC complexes were generated via (i) oxidation of the corresponding Au(I)–NHC species using X2 (X = Cl,4 Br,4b,5 I4b,5a,6), PhICl2,4a,5,6b,7 ICl,5b,6b ICF3,5b or CsBr3;8 (ii) reaction of N-alkyltriazolium salts with H[AuCl4]9 or Na[AuCl4]10 followed by addition of a base, or interaction of lithiated triazole with [AuCl3(THT)] (THT = tetrahydrothiophene) followed by addition of an acid;11 (iii) treatment of Na[AuCl4] with C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N(CH2)nOH leading to a substitution of chloride with the isocyanide with consecutive intramolecular attack of the OH group on the carbon of the coordinated isocyanide;12 (iv) transmetallation of the carbene ligand from the Ag(I), W(I), or Cr(I) to Au(III) center;13 and (v) disproportionation of Au(I)–NHC in the presence of [AuCl(SMe2)] furnishing Au(III)–NHC complexes and metallic gold.14
N(CH2)nOH leading to a substitution of chloride with the isocyanide with consecutive intramolecular attack of the OH group on the carbon of the coordinated isocyanide;12 (iv) transmetallation of the carbene ligand from the Ag(I), W(I), or Cr(I) to Au(III) center;13 and (v) disproportionation of Au(I)–NHC in the presence of [AuCl(SMe2)] furnishing Au(III)–NHC complexes and metallic gold.14
As far as open-chain acyclic diaminocarbenes bound to a gold(III) center are concerned, data available are even more scarce than those for gold(III)–NHC congeners and only a few gold(III)–ADCs were reported. The known gold(III)–ADCs are mostly derived from the oxidative addition of bromine and iodine to the corresponding gold(I)–ADCs.15 Single examples of other approaches include (i) intramolecular attack of the cycloaurated 2-(2-pyridylamino)phenyl ligand on an isocyanide;16 carbene transmetallation from the chromium(I) to the gold(III) center;17 and (ii) amine addition to gold(III)-bound CNRs.18
Although the complexes of gold(I) with ADC ligands1a,15b,19 are commonly assembled via the addition of sp3-N and sp3-O nucleophiles to gold(I)–isocyanides, only two examples of nucleophilic addition to gold(III)–CNRs were reported. Thus, Bartel and Fehlhammer studied12 the reaction of Na[AuCl4] with CN(CH2)nOH leading to the substitution of a chloride with the isocyanide followed by an intramolecular attack of the OH group on the C-atom of the coordinated isocyanide12 giving an NHC–Au(III) complex; the intermediate ADC species were not isolated but postulated as reaction intermediates. In the other study, addition of amines to the isocyanide in [Au(C6F5)3(p-CNC6H4Me)] gave the corresponding gold(III)–ADC species;18 no spectroscopic properties of these complexes were observed.18 Intrigued by a limited number of known gold(III)–ADCs, we aimed to expand their family via the reaction of other gold(III)–isocyanide precursors, viz. [AuCl3(CNR1)], with different NH-nucleophiles. Considering limited spectroscopic data reported for a few known gold(III)–ADCs, we also aimed to extensively characterize new gold(III)–ADC species.
We initiated this study from the preparation of gold(III)–isocyanides via the oxidative addition of chlorine to [AuCl(CNR1)] (Scheme 1). Thus, vigorous bubbling of dry gaseous chlorine through a solution of [AuCl(CNR1)] (R1 = Xyl 1; Cy 2, But3, (S)-CHMePh 4) in dry CH2Cl2 led to the formation of the corresponding complexes [AuCl3(CNR1)] (R1 = Xyl 5; Cy 6, But7, (S)-CHMePh 8). At 20–25 °C, all reactions were completed almost immediately furnishing gold(III)–isocyanide species that were isolated as either yellow crystalline solids (5–7) or an yellow oily residue (8) in 96–99% yield. Preparation of the related [AuBr3(CNR1)] species via the oxidative addition of bromide to [AuBr(CNR1)] was previously described.20
 | ||
| Scheme 1 Chlorination of 1–4 leading to gold(III)–isocyanides. | ||
The prepared isocyanide complexes 5–8 were characterized by ESI+/−-MS, FT-IR, 1H and 13C{1H} NMR spectroscopy, while complexes 6 and 7 were characterized additionally by CHN elemental analyses, and 5 by single-crystal X-ray diffraction (Fig. 1S and 2S in the ESI†).
Amines are sp3-N nucleophiles conventionally used for coupling with coordinated isocyanides, including gold(I)–CNRs,1a therefore, we have selected representative primary and secondary amines as our first choice in the reaction with gold(III)–isocyanides.
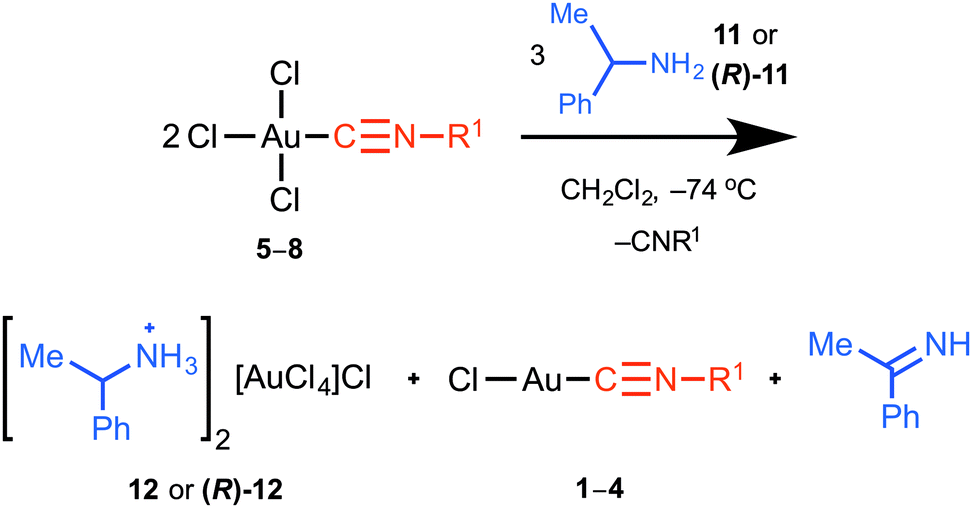
Reaction of [AuCl 3 (CNR 1 )] with amines. Reaction of complexes 5–8 with one or two equivalents of primary (benzylamine 9) or secondary (morpholine 10) amine in CH2Cl2 led to the formation of the corresponding amine hydrochloride and the product of reduction of the starting complex in solution (to 1–4, respectively) along with some yet unidentified species. With the more sterically hindered α-methylbenzylamine (11) as a nucleophile, the reaction proceeds via an unusual reduction pathway (Scheme 2).
 | ||
| Scheme 2 Interplay between α-methylbenzylamine and AuIII–isocyanide complexes. | ||
Thus, the reaction of equimolar amounts of any one of 5–8 with 11 in CH2Cl2 at −74 °C led to the precipitation of complex salt 12 as a bright yellow crystalline solid (ca. 20% yield). The maximum yield of 12 (40–42%) was achieved with a 5–8 to 11 ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. In the solution, the presence of 12·HCl and respective gold(I)–isocyanide complexes 1–4, alongside 1-phenylethan-1-imine and trace amounts of some yet unidentified species, was established using ESI-MS, FT-IR, and 1H NMR spectroscopy. No carbene complexes were detected in this mixture. Similar results were obtained for optically pure α-(R)-methylbenzylamine (R)-11. Complex salts 12 and (R)-12 were isolated and characterized by elemental analyses, ESI-MS, FT-IR and NMR and X-ray diffraction for (R)-12 (see the ESI†).
:3. In the solution, the presence of 12·HCl and respective gold(I)–isocyanide complexes 1–4, alongside 1-phenylethan-1-imine and trace amounts of some yet unidentified species, was established using ESI-MS, FT-IR, and 1H NMR spectroscopy. No carbene complexes were detected in this mixture. Similar results were obtained for optically pure α-(R)-methylbenzylamine (R)-11. Complex salts 12 and (R)-12 were isolated and characterized by elemental analyses, ESI-MS, FT-IR and NMR and X-ray diffraction for (R)-12 (see the ESI†).
We attempted to shed light on the nature of this process. In the early works by Castro et al.,21 oxidation of the amines by iron(III)–porphyrins led to the formation of appropriate imines as oxidation products, corresponding iron(II)–porphyrine complexes as reduction products as well as amine hydrochlorides (eqn (1)). Reduction of gold(III)–NHCs to the corresponding gold(I)-derivatives in the presence of free isocyanide is also known from the studies of Bartel and Fehlhammer.12
2[FeIIIP]Cl + 5R1R2CHNH2 → 2[FeIIP](H2NCHR1R2)2 + R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH + 2R1R2CHNH3Cl NH + 2R1R2CHNH3Cl | (1) |
Taking these data into consideration and on the basis of our experimental evidence (gold(I)–isocyanides 1–4, free isocyanide and 1-phenylethan-1-imine were identified in the reaction mixture) as well as the observed stoichiometry, we assume that the overall reaction of 5–8 with 11 proceeds via the redox pathway depicted in Scheme 2. Herein, one equiv. of methylbenzylamine (11) is oxidized to 1-phenylethan-1-imine by one equiv. of gold(III)–isocyanides (5–8) that are simultaneously reduced to the corresponding gold(I) species (1–4). Two equivalents of protons that are released during this oxidation are accepted by two additional equivalents of free amine furnishing the corresponding quaternary ammonium salt [Ph(Me)CHNH3]Cl. This further reacts with the second equiv. of [AuCl3(CNR1)] giving rise to a complex salt 12 and free isocyanide. To the best of our knowledge, this gold(III)-mediated oxidation of amines was not previously reported. In the earlier study, addition of amines to [Au(C6F5)3(p-CNC6H4Me)] gave the corresponding gold(III)–ADC species, and no oxidation of amine by gold(III) was observed.18 We believe that this can be rationalized by a stronger sigma donation from the coordinated C6F5 in [Au(C6F5)3(p-CNC6H4Me)] (when compared to that from chlorides in 5–8) resulting in a decrease of the oxidation potential of gold(III) below the required threshold.
Insofar as our attempts to prepare gold(III)–ADCs via coupling of gold(III)–CNRs with amines led to the oxidation of the latter, we focused our attention on hydrazines that are stronger nucleophiles than the relevant amines due to the α-effect.22 Among hydrazine derivatives, aldo- and ketohydrazones are recognized as the most stable toward oxidation23 and were selected for further studies.
Reaction of [AuCl 3 (CNR 1 )] with hydrazones. Recently we reported the addition of aldo- and ketohydrazones to an isocyanide ligand in cis-[MCl2(CNR)2] (M = Pd(II), Pt(II)) that result in the corresponding aminocarbene species.24 Extensive experimental and theoretical studies on the properties of hydrazones25 revealed that the hybridization of the NH2 center is intermediate between sp3-N and sp2-N, and it depends strongly on the nature of substituents, approaching sometimes the pure sp2-N state.
To our knowledge, nucleophilic attack of H2N–NCPh2 on gold-bound isocyanides has never been described, presumably because Au(I) does not provide sufficient electrophilic activation to a coordinated isocyanide moiety toward nucleophilic attack. To compare the susceptibility of gold(III)– and gold(I)–isocyanides toward nucleophilic attack, we carried out theoretical DFT calculations [viz., a geometry optimization procedure in the gas phase, calculations of vibrational frequencies, and natural bond orbital (NBO) analysis] using [AuCl3(CNMe)] and [AuCl(CNMe)] as model compounds (see full details in the ESI†). The energy level of the first unoccupied MO bearing π*(CN) orbitals (viz., LUMO−1, Fig. S3, ESI†) in [AuCl3(CNMe)] (−2.07 eV) is significantly lower than that in [AuCl(CNMe)] (−1.47 eV), suggesting that the Au(III) center is a stronger activator of the isocyanide ligands toward the addition of nucleophiles. The computed NBO charges on isocyanide carbon atoms in [AuCl3(CNMe)] and [AuCl(CNMe)] are 0.44 and 0.30, respectively. Finally, the calculated value of the unscaled normal mode frequency ν(CN) in [AuCl3(CNMe)] (2389 cm−1) is greater than that in [AuCl(CNMe)] (2321 cm−1), and this reflects higher electrophilic activation of CNRs bound to the gold(III) center.3a Note that these calculated values of the normal mode frequencies ν(CN) are unscaled, and are expectedly overestimated.26 Recommended multiplication of the calculated ν(CN) values on the scaling factor 0.96 gives 2293 cm−1 and 2228 cm−1, respectively, and it is in perfect agreement with the experimental data.26 Thus, orbital, charge, and frequency arguments reveal that the Au(III) center is a better activator of the CNR ligand toward nucleophilic addition when compared to Au(I). Taking these results into consideration, we studied the reactivity of Au(III)–isocyanide complexes in the coupling with hydrazones.
Reaction of 5, 6, or 8 with benzophenone hydrazone (13, 1 equiv.) in dry CH2Cl2 proceeds at RT for ca. 15 min giving the short-lived complexes [AuCl3{C(NHNCPh2)NHR1}] (R1 = Xyl 14; Cy 15, (S)-CHMePh 16; Scheme 3). No such reaction was observed between 7 and 13, which is apparently because of the low reactive ButNC.27 Reaction of 5–8 with 9-fluorenone hydrazone (17) and salicylaldehyde hydrazone (18) in dry CH2Cl2 or toluene at RT led to the formation of unstable species that almost immediately decompose giving metallic gold and a solution of yet unidentified species.
 | ||
| Scheme 3 Nucleophilic addition of benzophenone hydrazone to AuIII–bound isocyanide. | ||
Compounds 14 and 16 possess low stability in solution and in the solid state, but we could characterize them by FT-IR, ESI-MS, and 1H and 13C{1H} (only for 14) NMR spectroscopy. In addition, the structure of 14, which was stable enough at 150 K when compared to those of 15 and 16, was elucidated by single-crystal X-ray diffraction. Compound 15 rapidly (ca. 10 min) decomposes both in solution and in the solid state, and was characterized solely by solution 1H NMR spectroscopy immediately after its formation. The detailed characterization of 14–16 is provided in the ESI,† and herein we discuss the most important details of the solid-state X-ray structure of complex 14 (Table S1, the ESI† contains crystallographic data and processing parameters for 14).
The asymmetric unit of 14 contains one molecule of the complex and one of chloroform (Fig. 1). The square-planar coordination environment of 14 (τ4 = 0.05)28 is filled by one carbene and three chloride ligands. All bond angles around the Au(III) center are close to 90° varying from 87.21(16)° to 92.63(5)°. The Au–Cl1 (2.3255(12) Å) distance opposite to the carbene fragment is slightly longer than the other two Au–Cl distances (2.2687(14) and 2.3001(14) Å) and that in 5 (2.2687(19) Å) indicating significant ground-state trans influence of the carbene ligand when compared to the parent isocyanide in 5. The Au–C bond distance (2.005(6) Å) is comparable to those observed in 5 (1.954(7) Å) and in the related complexes containing N-heterocyclic carbenes [AuCl3(IMes)] (2.016(7) Å),9 [AuCl3(1,3-dihydro-1-methyl-3-(2,4,6-trimethylphenyl)-2H-imidazol-2-ylidene)] (1.994(4) Å),7 and [AuCl3(IPr)] (2.013(9) Å).4a The carbene moiety is almost planar (C1 deviates from the Au1–N1–N2 plane by 0.017 Å) and the angles around the carbene C1 atom are nearly equal to 120° varying from 118.4(4)° to 121.9(3)°, indicating its sp2 hybridization. Both Ccarbene–N distances are equal within 3σ (1.312(6) and 1.318(5) Å) and their values are typical for delocalized one-and-a-half C–N bond (e.g. 1.337(12) for Car–N in pyridine).29 The carbene plane (built on N1, C1, and N2 atoms) is almost perpendicular to the plane defined by the chloride ligands (angle of 77.66°). The carbene C1–N1 and C1–N2 bonds adopt E- and Z-configurations, respectively. The solid-state structure of 12 exhibits intermolecular T-shaped C−H⋯π interactions involving both the C26−H8 and the C23−H10 bonds of every molecule and the C3−C8 phenyl rings of adjacent ones. Such interactions give rise to infinite chains along the crystallographic a axis (see the ESI† for more details).
 | ||
| Fig. 1 Crystal structure of 14 with the atomic numbering scheme (hydrogen labels and chloroform molecules were omitted for simplicity). Thermal ellipsoids are drawn at the 25% probability level. Selected bond lengths (Å) and angles (°): Au1–Cl1 2.3255(12), Au1–Cl2 2.2687(14), Au1–Cl3 2.3001(14), Au1–C1 2.005(5), C1–N1 1.312(6), C1–N2 1.318(5), C2–N3 1.293(6), N2–N3 1.387(5), C1–Au1–Cl1 178.55(12), C1–Au1–Cl2 87.21(16), C1–Au1–Cl3 89.29(16), Cl1–Au1–Cl2 92.63(5), Cl2–Au1–Cl3 175.16(5), N1–C1–Au1 121.9(3), N1–C1–N2 118.4(4), N2–C1–Au1 119.7(3). | ||
Finally, evaluation of the bonding situation in the dimeric clusters of 5, 14, and (R)-12 was carried out using theoretical DFT calculations and topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM analysis);30 this approach has already been successfully used by us in the studies of non-covalent interactions and properties of coordination bonds in various transition metal complexes.31 QTAIM analysis (Table S2, ESI†) indicates the presence of two bond critical points (3, −1) (BCPs) for covalent bonds Au–Cl and two BCPs for Au⋯Cl non-covalent interactions, and no BCPs for Au⋯Au contacts in all studied dimers. The magnitudes of the electron density, values of the Laplacian and energy density, the −G(r)/V(r) ratio in BCPs for Au–Cl and Au⋯Cl contacts as well as appropriate Wiberg bond indices (WI) are typical for covalent bonds M–L in coordination complexes and for non-covalent electrostatic interactions, respectively. We have defined energies for these contacts according to the procedures proposed by Espinosa et al.32 and Vener et al.33 (Table S2, ESI†), and one can state that the relativistic and non-relativistic approaches give very similar estimates. The results of theoretical calculations led to the conclusion that weak Au⋯Cl non-covalent electrostatic interactions are most likely responsible for the stabilization of dimeric associates 5, 14, and (R)-12 in the solid state.
As a conclusion, direct oxidative addition of chlorine to [AuCl(CNR1)] gives the corresponding AuIII–CNR species in nearly quantitative yields. Attempted nucleophilic addition of amines to gold(III)–isocyanides does not lead to carbene complexes but furnishes imines and gold(I)–isocyanides generated via the redox pathway. At the same time, addition of benzophenone hydrazone to the coordinated isocyanide in [AuCl3(CNR1)] furnishes new types of short-lived gold(III)–ADC species that gradually decompose to give metallic gold. Insofar as the stability of gold(III)–ADCs is concerned, one should consider that although the instability of complexes has a negative impact on their shelf-life, it might turn positive for their catalytic activity. It is argued that many modern organometallic catalytic processes, i.e. cross-coupling, are essentially catalysed by the nano-sized particles formed from the starting molecular compounds during the precatalyst activation step.34 Herein, in situ prepared gold(III)–ADCs can promptly generate gold nanoparticles enabling subsequent catalytic transformations and further studies in this direction are currently underway in our group.
Acknowledgements
This work has been partially supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal (through the PPCDT program – FEDER funded and research project Pest – UID/QUI/00100/2013), the Russian Foundation for Basic Research (grants 15-33-20536, 16-33-60123, and 16-33-60063), and the grant program of the President of the Russian Federation (grant MK-7425.2016.3). The authors are grateful to the Center for Magnetic Resonance, Center for Chemical Analysis and Materials Research (Saint Petersburg State University), and the Portuguese NMR (grant NRNNM2013) and MS (IST Node, grant REM 2013 da FCT) networks. Alexander S. Novikov is thankful to the Saint Petersburg State University and Santander Bank for fellowship (internships of young scientists in Ibero-America universities and scientific organizations).References
- (a) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed; (b) V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef CAS; (c) L. M. Slaughter, ACS Catal., 2012, 2, 1802–1816 CrossRef CAS; (d) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC; (e) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (f) K. V. Luzyanin and A. J. L. Pombeiro, Carbene complexes derived from metal-bound isocyanides, in Isocyanide chemistry, ed. V. Nenajdenko, Wiley-VCH, 2012, ch. 15, pp. 531–550 Search PubMed.
- J. Vignolle, X. Catton and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS PubMed.
- (a) R. A. Michelin, A. J. L. Pombeiro and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2001, 218, 75–112 CrossRef CAS; (b) F. E. Hahn, Angew. Chem., Int. Ed. Engl., 1993, 32, 650–665 CrossRef; (c) M. Tamm and F. E. Hahn, Coord. Chem. Rev., 1999, 182, 175–209 CrossRef; (d) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 3122–3172 CrossRef CAS PubMed; (e) P. G. Edwards and F. E. Hahn, Dalton Trans., 2011, 10278–10288 RSC; (f) M. C. Jahnke and F. E. Hahn, Coord. Chem. Rev., 2015, 293–294, 95–115 CrossRef CAS; (g) F. E. Hahn, V. Langenhahn, T. Lügger, T. Pape and D. L. Van, Angew. Chem., Int. Ed., 2005, 44, 3759–3763 CrossRef CAS PubMed; (h) O. Kaufhold, A. Stasch, T. Pape, A. Hepp, P. G. Edwards, P. D. Newman and F. E. Hahn, J. Am. Chem. Soc., 2009, 131, 306–317 CrossRef CAS PubMed; (i) A. Flores-Figueroa, T. Pape, K.-O. Feldmann and F. E. Hahn, Chem. Commun., 2010, 46, 324–326 RSC; (j) M. Schmidtendorf, T. Pape and F. E. Hahn, Angew. Chem., Int. Ed., 2012, 51, 2195–2198 CrossRef CAS PubMed; (k) A. C. Dumke, T. Pape, J. Kösters, K.-O. Feldmann, C. Schulte to Brinke and F. E. Hahn, Organometallics, 2013, 32, 289–299 CrossRef CAS; (l) J. Ruiz, G. García, M. E. G. Mosquera, B. F. Perandones, M. P. Gonzalo and M. Vivanco, J. Am. Chem. Soc., 2005, 127, 8584–8585 CrossRef CAS PubMed; (m) J. Ruiz, B. F. Perandones, G. García and M. E. G. Mosquera, Organometallics, 2007, 26, 5687–5695 CrossRef CAS; (n) J. Ruiz, L. García, C. Mejuto, M. Vivanco, M. R. Díaz and S. García-Granda, Chem. Commun., 2014, 50, 2129–2132 RSC; (o) J. Ruiz, L. García, D. Sol and M. Vivanco, Angew. Chem., Int. Ed., 2016, 55, 8386–8390 CrossRef CAS PubMed.
- (a) S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394–402 CrossRef CAS; (b) M. Baron, C. Tubaro, M. Basato, M. M. Natile and C. Graiff, J. Organomet. Chem., 2013, 723, 108–114 CrossRef CAS.
- (a) H. V. Huynh, S. Guo and W. Q. Wu, Organometallics, 2013, 32, 4591–4600 CrossRef CAS; (b) J. Gil-Rubio and J. Vicente, Dalton Trans., 2015, 19432–19442 RSC.
- (a) J. Gil-Rubio, V. Cámara, D. Bautista and J. Vicente, Inorg. Chem., 2013, 52, 4071–4083 CrossRef CAS PubMed; (b) M. Blaya, D. Bautista, J. Gil-Rubio and J. Vicente, Organometallics, 2014, 33, 6358–6368 CrossRef CAS.
- M. Pazicky, A. Loos, M. J. Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jakel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448–4458 CrossRef CAS.
- M. Kriechbaum, D. Otte, M. List and U. Monkowius, Dalton Trans., 2014, 8781–8791 RSC.
- S. F. Zhu, R. X. Liang, L. J. Chen, C. Wang, Y. W. Ren and H. F. Jiang, Tetrahedron Lett., 2012, 53, 815–818 CrossRef CAS.
- M. Deetlefs, H. G. Raubenheimer and M. W. Esterhuysen, Catal. Today, 2002, 72, 29–41 CrossRef CAS.
- H. G. Raubenheimer, P. J. Olivier, L. Lindeque, M. Desmet, J. Hrusak and G. J. Kruger, J. Organomet. Chem., 1997, 544, 91–100 CrossRef CAS.
- K. Bartel and W. P. Fehlhammer, Angew. Chem., Int. Ed. Engl., 1974, 13, 599–600 CrossRef.
- (a) H. G. Raubenheimer and S. Cronje, Chem. Soc. Rev., 2008, 37, 1998–2011 RSC; (b) T. T. Zou, C. T. Lum, S. S. Y. Chui and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS PubMed.
- (a) J. Dinda, T. Samanta, A. Nandy, K. Das Saha, S. K. Seth, S. K. Chattopadhyay and C. W. Bielawskie, New J. Chem., 2014, 38, 1218–1224 RSC; (b) J. Dinda, S. Das Adhikary, S. K. Seth and A. Mahapatra, New J. Chem., 2013, 37, 431–438 RSC.
- (a) R. Usón, A. Laguna, J. Vicente, J. García, B. Bergareche and P. Brun, Inorg. Chim. Acta, 1978, 28, 237–243 CrossRef; (b) R. Usón, A. Laguna, J. Vicente, J. García and B. Bergareche, J. Organomet. Chem., 1979, 173, 349–355 CrossRef; (c) J. E. Parks and A. L. Balch, J. Organomet. Chem., 1974, 71, 453–463 CrossRef CAS; (d) G. Minghetti and F. Bonati, J. Organomet. Chem., 1973, 54, C62–C63 CrossRef CAS; (e) L. Manojlović-Muir, J. Organomet. Chem., 1974, 73, C45–C46 CrossRef; (f) A. L. Bandini, G. Banditelli, G. Minghetti, B. Pelli and P. Traldi, Organometallics, 1989, 8, 590–593 CrossRef CAS.
- O. Crespo, M. C. Gimeno, A. Laguna, S. Montanel-Pérez and M. D. Villacampa, Organometallics, 2012, 31, 5520–5526 CrossRef CAS.
- E. O. Fischer and M. Bock, J. Organomet. Chem., 1985, 287, 279–285 CrossRef CAS.
- R. Usón, A. Laguna, M. Laguna, E. Fernandez, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1982, 1971–1976 RSC.
- (a) J. Vicente, M. T. Chicote, M. D. Abrisqueta and P. G. Jones, Organometallics, 1997, 16, 5628–5636 CrossRef CAS; (b) J. Vicente, M. T. Chicote, M. D. Abrisqueta, M. C. Ramírez de Arellano, P. G. Jones, M. G. Humphrey, M. P. Cifuentes, M. Samoc and B. Luther-Davies, Organometallics, 2000, 19, 2968–2974 CrossRef CAS; (c) J. Vicente, M. T. Chicote, M. M. Alvarez-Falcón, M. D. Abrisqueta, F. J. Hernández and P. G. Jones, Inorg. Chim. Acta, 2003, 347, 67–74 CrossRef CAS; (d) J. Vicente, M. T. Chicote, M. D. Abrisqueta, M. M. Alvarez-Falcón, F. J. Hernández, M. C. Ramírez de Arellano and P. G. Jones, Organometallics, 2003, 22, 4327–4333 CrossRef CAS.
- D. Schneider, O. Schuster and H. Schmidbaur, Organometallics, 2005, 25, 3547–3551 CrossRef.
- C. E. Castro, M. Jamin, W. Yokoyama and R. Wade, J. Am. Chem. Soc., 1986, 108, 4179–4187 CrossRef CAS.
- (a) J. O. Edwards and R. G. Pearson, J. Am. Chem. Soc., 1962, 84, 16–24 CrossRef CAS; (b) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry. Part A: Structure and Mechanisms, Springer, Berlin, 5th edn, 2007 Search PubMed.
- (a) J. B. Aylward, J. Chem. Soc. C, 1969, 1663–1665 RSC; (b) R. I. J. Amos, B. S. Gourlay, C. H. Schiesser, J. A. Smith and B. F. Yates, Chem. Commun., 2008, 1695–1697 RSC.
- (a) E. A. Valishina, T. M. Buslaeva and K. V. Luzyanin, Russ. Chem. Bull., 2013, 62, 1361–1365 CrossRef CAS; (b) B. G. M. Rocha, E. A. Valishina, R. S. Chay, M. F. C. Guedes da Silva, T. M. Buslaeva, A. J. L. Pombeiro, V. Y. Kukushkin and K. V. Luzyanin, J. Catal., 2014, 309, 79–86 CrossRef CAS; (c) K. V. Luzyanin, M. F. C. Guedes da Silva, V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2009, 362, 833–838 CrossRef CAS; (d) E. A. Valishina, M. F. C. Guedes da Silva, M. A. Kinzhalov, S. A. Timofeeva, T. M. Buslaeva, M. Haukka, A. J. L. Pombeiro, V. P. Boyarskiy, V. Y. Kukushkin and K. V. Luzyanin, J. Mol. Catal. A: Chem., 2014, 395, 162–171 CrossRef CAS.
- (a) Y. P. Kitaev and B. I. Buzykin, Usp. Khim., 1972, 41, 995–1037 CrossRef CAS; (b) V. V. Zverev and Y. P. Kitaev, Zh. Strukt. Khim., 1974, 15, 128–132 CAS.
- M. L. Laury, M. J. Carlson and A. K. Wilson, J. Comput. Chem., 2012, 33, 2380–2387 CrossRef CAS PubMed.
- M. A. Kinzhalov, V. P. Boyarskiy, K. V. Luzyanin, F. M. Dolgushin and V. Y. Kukushkin, Dalton Trans., 2013, 42, 10394–10397 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- H. F. Allen, O. Kennard, D. G. Watson, L. Brammer, A. Guy Orpen and T. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- (a) A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, D. V. Dar'in, G. L. Starova and V. Y. Kukushkin, J. Am. Chem. Soc., 2016, 138, 14129–14137 CrossRef CAS PubMed; (b) D. M. Ivanov, M. A. Kinzhalov, A. S. Novikov, I. V. Ananyev, A. A. Romanova, V. P. Boyarskiy, M. Haukka and V. Y. Kukushkin, Cryst. Growth Des., 2017, 17, 1353–1362 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1988, 285, 170–173 CrossRef.
- M. V. Vener, A. N. Egorova, A. V. Churakov and V. G. Tsirelson, J. Comput. Chem., 2012, 33, 2303–2309 CrossRef CAS PubMed.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595–1604 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of complexes, theoretical evaluation of the bonding situation and Cartesian atomic coordinates. CCDC 1471327–1471329. See DOI: 10.1039/c7nj00529f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |