
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, X-ray structure and electrochemical properties of hybrid binuclear metallophthalocyaninate-capped tris-pyridineoximates†
Semyon V.
Dudkin
a,
Alexander S.
Belov
a,
Yulia V.
Nelyubina
a,
Anastasia V.
Savchuk
b,
Alexander A.
Pavlov
a,
Valentin V.
Novikov
a and
Yan Z.
Voloshin
 *ac
*ac
aNesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, 119991, Moscow, Russia. E-mail: voloshin@ineos.ac.ru; Tel: +7 499 135 9344
bVernadskii Institute of General and Inorganic Chemistry of the National Academy of Sciences of Ukraine, 03680 Kiev, Ukraine
cGubkin Russian State University of Oil and Gas, 119991 Moscow, Russia
First published on 10th March 2017
Abstract
New antimony-capped iron and nickel(II) tris-pyridineoximates with a labile triethylantimony cross-linking group were obtained by template condensation of 2-acetylpyridinoxime with triethylantimony(V) dibromide on the corresponding metal ion as a matrix. They easily undergo transmetallation (capping group exchange) with Lewis-acidic zirconium and hafnium(IV) phthalocyaninates (Pc) to give binuclear MPc-capped tris-pyridineoximates. The obtained hybrid complexes and their precursors were thoroughly characterized (among others, by single-crystal X-ray diffraction), and their redox properties were studied by cyclic and differential pulse voltammetry.
Introduction
Macrobicyclic cage complexes with an encapsulated metal ion (clathrochelates)1 are a special case of coordination compounds with unusual physicochemical properties.2 Owing to this, these cage complexes have found use as electrocatalysts for hydrogen evolution1b,3 and as prospective materials for biomedical applications,4 including topological drugs,5 anti-fibrillogenic agents,6 and selective cytotoxic compounds.7 Paramagnetic cobalt(II) clathrochelates have also been recognized as single-molecule magnets (SMMs),8i.e. molecules able to retain their magnetization in the absence of an applied magnetic field,9 and they are expected to revolutionize the field of high-density information storage or quantum computing. Among various building blocks for the design of SMMs, porphyrins and phthalocyanines (Pcs) have been widely used.10 Therefore, hybrid molecular systems that combine a transition metal (pseudo)clathrochelate and a porphyrin or a phthalocyanine complex seem to be even more prospective candidates for manufacturing new types of SMMs.In the present paper, we report a two-step approach to the first binuclear metallophthalocyaninato-capped metal(II) tris-pyridineoximates, representatives of a new type of hybrid polytopic and multicentered molecular system, and their antimony-capped precursors. As obtaining these complexes with a cobalt ion is still a real challenge, the proposed approach is tested here on iron and nickel(II) as the encapsulated ions. Such complexes were obtained by a transmetallation reaction (capping group exchange), which has been successfully used for the synthesis of various polyazomethine phthalocyaninato- and porphyrinoclathrochelates (Scheme 1).11
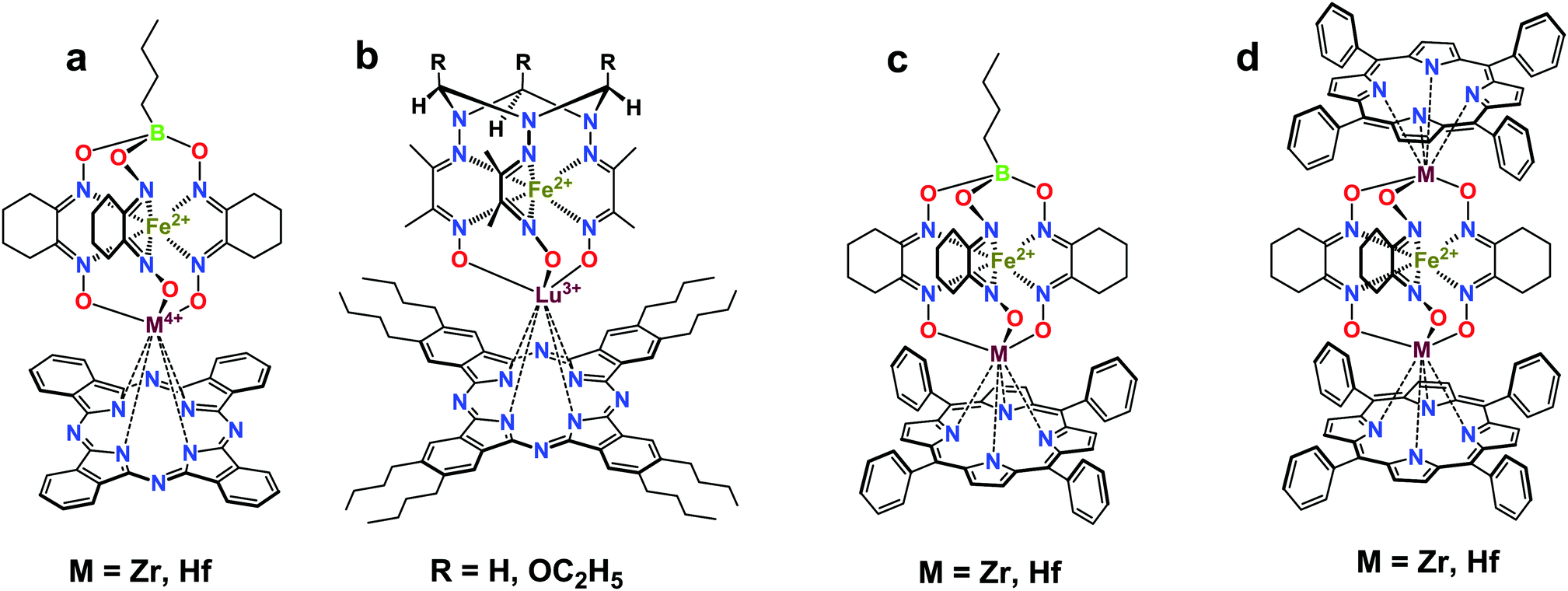 | ||
| Scheme 1 Hybrid iron(II) phthalocyaninato- (a and b) and porphyrinoclathrochelates (c and d). | ||
Results and discussion
While antimony-capped metal tris-pyridineoximates are rare,12 tris-dioximates of iron(II) and cobalt(III) are easy to obtain13 in moderate yields via template condensation with triethylantimony dibromide.Here, antimony-capped iron and nickel(II) tris-pyridineoximates ([Fe(AcPyOx)3(Sb(C2H5)3)](ClO4) and [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4)) were prepared in high yields (85 and 90%, respectively) via the template self-assembly of a metal(II) salt, 2-acetypyridineoxime and triethylantimony(V) dibromide in an ethanol medium, with NaHCO3 as a base (Scheme 2). We also attempted to use triethylamine as a base; however, isolation of the target products was more challenging.
 | ||
| Scheme 2 Synthesis of antimony-capped iron and nickel(II) tris-pyridineoximates and their binuclear MIVPc-capped hybrid derivatives. | ||
The binuclear MPc-capped hybrid derivatives of the compounds ([Fe(AcPyOx)3(ZrPc)](ClO4), [Fe(AcPyOx)3(HfPc)](ClO4), [Ni(AcPyOx)3(ZrPc)](ClO4) and [Ni(AcPyOx)3(HfPc)](ClO4)) were synthesized in relatively high yields (65–79%) by transmetallation of the above antimony(V)-capped precursors under mild reaction conditions (with a methanol–dichloromethane mixture as a solvent and at ambient temperature) using equimolar amounts of Lewis-acidic zirconium and hafnium(IV) phthalocyaninates (Scheme 2).
The formation of these new metallophthalocyaninato-capped tris-pyridineoximates was confirmed by elemental analysis, 1D (1H and 13C{1H}) and 2D NMR, MALDI-TOF MS, UV-vis spectra and single-crystal X-ray diffraction; their redox properties were studied by cyclic (CV) and differential pulse (DPV) voltammetry.
Introduction of the Pc fragment into [Fe(AcPyOx)3(SbEt3)]ClO4 affects the 1H and 13C{1H} NMR spectra of the resulting phthalocyaninatoclathrochelates [Fe(AcPyOx)3(HfPc)] and [Fe(AcPyOx)3(ZrPc)]: the signals of the protons of the methyl group and those of the pyridine ring shift to higher values by 2.5 and 1.0 ppm, respectively, as these nuclei are located in the shielding region of the π-electron current in the conjugated macrocyclic system. The signals of the nuclei of all the carbon atoms in [Fe(AcPyOx)3(HfPc)] and [Fe(AcPyOx)3(ZrPc)] are shifted towards the low-field region by ∼1 ppm for the same reason (see the ESI,† Fig. S1, S2, and S5–S8). As the chemical properties of zirconium and hafnium are similar, the values of the chemical shifts for [Fe(AcPyOx)3(HfPc)] and [Fe(AcPyOx)3(ZrPc)], and [Ni(AcPyOx)3(HfPc)] and [Ni(AcPyOx)3(ZrPc)] differ only slightly. A similar behavior has been observed earlier for other zirconium- and hafnium(IV)-capped phthalocyaninatoclathrochelates.11a,c
The antimony-cross-linked nickel(II) complex [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4) and its MIVPc-capped derivatives [Ni(AcPyOx)3(HfPc)](ClO4) and [Ni(AcPyOx)3(ZrPc)](ClO4) are paramagnetic compounds (s = 1) owing to the d8 high-spin configuration of the nickel(II) ion. Their NMR spectra (see the ESI,† Fig. S3, S4, S9 and S10) are thus dominated by paramagnetic effects: contact shifts that depend on the spin density distribution in a molecule and pseudocontact shifts that are associated with dipole–dipole electron–nucleus interactions. As the paramagnetic shifts reach several hundred ppm, they mask the effect of the π-electron current.
Like the earlier reported phenylboron-capped iron(II) tris-pyrazoloximates with a similar distribution of unpaired electrons,14 the trigonal-prismatic nickel(II) complexes are expected to have a very small pseudocontact contribution due to the orbitally non-degenerate ground state. Therefore, their NMR spectra are mainly determined by the contact shifts, so assignment of the signals in these spectra is nontrivial and was successfully completed only when assisted by DFT calculations of the complexes [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4) and [Ni(AcPyOx)3(ZrPc)](ClO4). For [Ni(AcPyOx)3(ZrPc)](ClO4) and [Ni(AcPyOx)3(HfPc)](ClO4), the paramagnetic broadening of these signals prevented us from obtaining their 13C NMR spectra.
Additionally, the excellent agreement between the experimental paramagnetic shifts and the calculated contact shifts (Fig. 1) confirmed the relatively low importance of the pseudocontact shifts. Note that the paramagnetic nickel(II) ion affects the 1H NMR shifts of the protons of the Pc-fragments only slightly, as the contact shift becomes negligible when the nucleus under question and the paramagnetic center are separated by five or more chemical bonds.
 | ||
| Fig. 1 Experimental vs. calculated paramagnetic shifts for [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4) (a and b) and [Ni(AcPyOx)3(ZrPc)](ClO4) (c). | ||
MALDI-TOF mass spectra of all the complexes obtained contain the intensive peaks of the corresponding cationic species [M − ClO4−]+. UV-vis spectra of [Fe(AcPyOx)3(ZrPc)](ClO4), [Fe(AcPyOx)3(HfPc)](ClO4), [Ni(AcPyOx)3(ZrPc)](ClO4) and [Ni(AcPyOx)3(HfPc)](ClO4) and their antimony-capped precursors ([Fe(AcPyOx)3(Sb(C2H5)3)](ClO4) and [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4)) in dichloromethane are shown in Fig. 2 and Fig. S11–S17 (see the ESI†). A deconvoluted spectrum of the antimony-capped iron(II) tris-pyridineoximate [Fe(AcPyOx)3(Sb(C2H5)3)](ClO4) contains, in the visible range (440–550 nm), four intensive (ε ∼ 2–11 × 103 mol−1 L cm−1) bands assigned to Fed → Lπ* metal-to-ligand charge transfer (MLCT). Intensive (ε ∼ 3–30 × 103 mol−1 L cm−1) bands in the UV range of the spectrum were assigned to π–π* intraligand transitions. The UV-vis spectrum of the nickel(II)-containing analogue [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4) also contains intensive π–π* intraligand transition bands in the UV range. A broad asymmetrical band of low intensity (ε ∼ 25 mol−1 L cm−1) is observed at 800–1000 nm, together with more intensive (ε ∼ 115–260 mol−1 L cm−1) bands with maxima from 400 to 500 nm. These bands were assigned to d–d transitions characteristic of a six-coordinate nickel(II) ion.
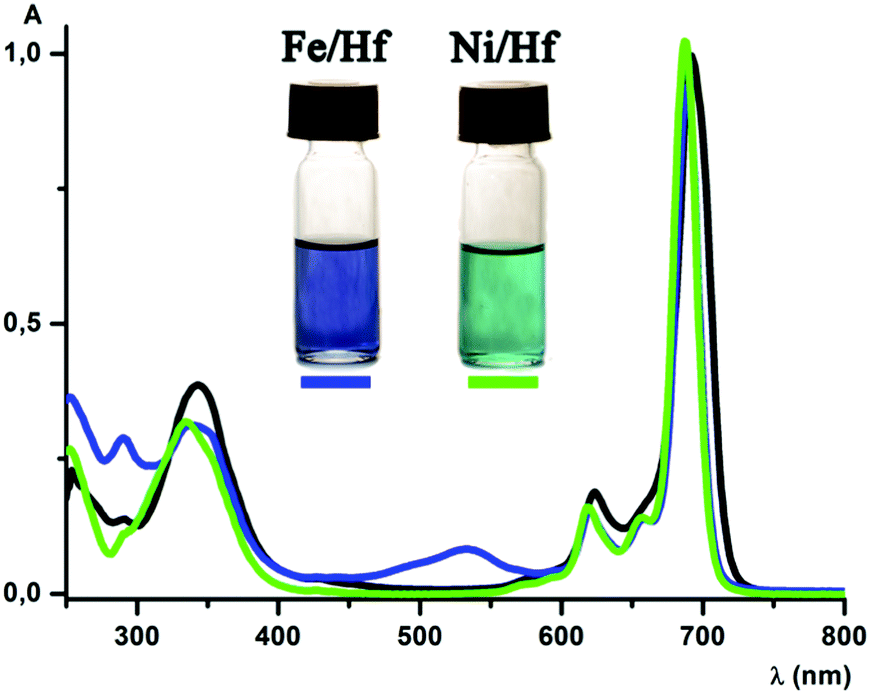 | ||
| Fig. 2 UV-vis spectra of the parent phthalocyaninate Hf(Cl2)Pc in DMSO (in black) and its hybrid derivatives [Fe(AcPyOx)3(HfPc)](ClO4) (in blue) and [Ni(AcPyOx)3(HfPc)](ClO4) (in green) in dichloromethane. | ||
The UV-vis spectra of the MIVPc-capped hybrid tris-pyridineoximates can be superposed on the corresponding spectra of the metal(IV) phthalocyaninate and metal(II) tris-pyridineoximate chromophores. In particular, the spectrum of the hybrid iron(II) complex [Fe(AcPyOx)3(HfPc)](ClO4) (Fig. 2) contains one intensive absorption band in the red and near-IR region at 689 nm (ε ∼ 205 × 103 mol−1 L cm−1), and its two vibronic satellites are found at 660 nm (ε ∼ 28 × 103 mol−1 L cm−1) and 620 nm (ε ∼ 32 × 103 mol−1 L cm−1). In addition, there is also a broad intensive (ε ∼ 65 × 103 mol−1 L cm−1) band, characteristic of the Pc fragment, at 340 nm. A broad band observed at 534 nm (ε ∼ 16 × 103 mol−1 L cm−1) was assigned to the Fed → Lπ* MLCT in the iron(II)-containing tris-pyridineoximate fragment. The UV-vis spectrum of the [Fe(AcPyOx)3(ZrPc)](ClO4) complex is identical to that of its hafnium cross-linked analogue (see the ESI,† Fig. S15).
The spectrum of the hybrid complex [Ni(AcPyOx)3(HfPc)](ClO4) contains one intensive (ε ∼ 243 × 103 mol−1 L cm−1) band in the red and near-IR region at 688 nm and its two vibronic satellites: a shoulder at 656 nm (ε ∼ 34 × 103 mol−1 L cm−1) and a band at 619 nm (ε ∼ 39 × 103 mol−1 L cm−1). A broad B band of the metal(IV)-containing Pc fragment appears as a band at 335 nm (ε ∼ 78 × 103 mol−1 L cm−1) with a shoulder at 352 nm (ε ∼ 64 × 103 mol−1 L cm−1) (Fig. 2). The observed splitting of this B band may be attributed to the contribution of the tris-pyridineoximate fragment. The UV-vis spectrum of the zirconium(IV) Pc-capped complex [Ni(AcPyOx)3(ZrPc)](ClO4) is almost identical to that of the Hf(IV) analogue (see the ESI,† Fig. S17).
The structures of the obtained hybrid complexes [Fe(AcPyOx)3(HfPc)](ClO4), [Fe(AcPyOx)3(ZrPc)](ClO4), [Ni(AcPyOx)3(HfPc)](ClO4), and [Ni(AcPyOx)3(ZrPc)](ClO4) (Fig. 3 and 4) were confirmed by single-crystal X-ray diffraction analysis, which also allowed the structure of one of their precursors, [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4), to be elucidated (Fig. S18, see the ESI†). In the hybrid complexes (Table 1), the MIIN6 polyhedron adopts a geometry that is intermediate between a trigonal prism (TP, the distortion angle φ = 0°) and a trigonal antiprism (TAP, φ = 60°), and may be alternatively described as a truncated trigonal pyramid (for it has bases of a slightly different size due to the very different nature of the apical fragments). The central metal ion MII is located almost in the center of the MN6 polyhedron (Fe–N 1.860(9)–1.978(7) Å; Ni–N 2.010(3)–2.112(4) Å), as is typical of low-spin Fe(II) and high-spin Ni(II) ions, and is only slightly shifted towards the capping metal ion MIV (Zr or Hf) along the line MII–MIV.
 | ||
| Fig. 3 General view of the hybrid tris-pyridineoximate cations [Fe(AcPyOx)3(HfPc)]+ (a) and [Fe(AcPyOx)3(ZrPc)]+ (b) with atoms shown as thermal ellipsoids at p = 30%; hydrogen atoms and perchlorate anions are omitted for clarity. | ||
 | ||
| Fig. 4 General view of the hybrid tris-pyridineoximate cations [Ni(AcPyOx)3(HfPc)]+ (a) and [Ni(AcPyOx)3(ZrPc)]+ (b) with atoms shown as thermal ellipsoids at p = 30%; hydrogen atoms and perchlorate anions are omitted for clarity. | ||
| [Fe(AcPyOx)3(HfPc)](ClO4) | [Fe(AcPyOx)3(ZrPc)](ClO4) | [Ni(AcPyOx)3(HfPc)](ClO4) | [Ni(AcPyOx)3(ZrPc)](ClO4) | |
|---|---|---|---|---|
| MII/MIV | Fe/Hf | Fe/Zr | Ni/Hf | Ni/Zr |
| MII–N1 (Å) | 1.878(8) | 1.869(7) | 2.032(4) | 2.010(3) |
| MII–N2 (Å) | 1.962(8) | 1.944(7) | 2.084(4) | 2.105(4) |
| MII–N3 (Å) | 1.897(8) | 1.860(9) | 2.039(4) | 2.014(3) |
| MII–N4 (Å) | 1.970(7) | 1.978(7) | 2.105(4) | 2.081(4) |
| MII–N5 (Å) | 1.888(8) | 1.848(8) | 2.018(4) | 2.036(4) |
| MII–N6 (Å) | 1.974(7) | 1.968(7) | 2.112(4) | 2.097(4) |
| N–O (Å) |
1.343(9)–1.349(10)
av. 1.345 |
1.375(9)–1.415(10)
av. 1.392 |
1.352(5)–1.369(5)
av. 1.361 |
1.357(4)–1.365(4)
av. 1.361 |
| MIV–O (Å) |
2.096(7)–2.126(6)
av. 2.114 |
2.111(6)–2.170(6)
av. 2.131 |
2.072(3)–2.119(3)
av. 2.100 |
2.114(3)–2.147(3)
av. 2.125 |
| MIV–N (Å) |
2.219(8)–2.241(7)
av. 2.230 |
2.222(7)–2.244(7)
av. 2.233 |
2.239(4)–2.262(4)
av. 2.250 |
2.254(3)–2.248(3)
av. 2.249 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (Å) N (Å) |
1.285(12)–1.307(12)
av. 1.298 |
1.276(10)–1.327(12)
av. 1.299 |
1.282(6)–1.293(6)
av. 1.287 |
1.280(6)–1.292(5)
av. 1.285 |
| C–C (Å) |
1.437(15)–1.461(13)
av. 1.449 |
1.453(11)–1.456(12)
av. 1.454 |
1.466(7)–1.473(7)
av. 1.469 |
1.478(6)–1.492(6)
av. 1.484 |
| NC–CN (°) |
1.1(11)–5.4(13)
av. 3.8 |
0.6(11)–8.1(11)
av. 3.4 |
4.4(7)–8.8(6)
av. 6.4 |
1.2(5)–7.5(5)
av. 4.0 |
| φ (°) | 45.3 | 46.4 | 36.7 | 37.8 |
| α (°) | 80.6 | 80.2 | 77.6 | 77.4 |
| h (Å) | 2.17 | 2.12 | 2.36 | 2.32 |
The degree of TP-to-TAP distortion in the MIIN6 polyhedron depends on MII and only slightly on MIV. It is much closer to TAP in the iron complexes than in their nickel analogues (Table 1); the corresponding distortion angle φ varies from 45.3–46.4° to 36.7–37.8° with a simultaneous increase in the height of the TP–TAP polyhedron from 2.12–2.17 to 2.32–2.36 Å. For comparison, the φ angle of the TP–TAP polyhedron and its height in [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4) are 35.8° and 2.37 Å (see the ESI,† Table S8). These distortions are larger than in the previously reported zirconium/hafnium-capped iron(II) Pc-clathrochelates with two capping fragments (φ ∼ 30°).11c,d The reason for this may be the higher flexibility of the rigid caging framework in the obtained hybrid complexes, as one of their ‘caps’ is a perchlorate anion that is loosely bound to the pyridine moieties via weak C–H⋯OCl contacts (O⋯H distance from ∼2.6 Å). Note that similar to antimony, large zirconium and hafnium(IV) ions are known to give clathrochelates with a more TAP-like geometry around the encapsulated metal ion MII.11c The average bite angle (α; the N–M–N angle) as well as the lengths of the CN, C–C and O–N bonds in the caging ligand are similar in all four hybrid complexes (Table 1).
The capping metal ion MIV coordinates three oxygen atoms of the caging ligand and four nitrogen atoms of the Pc core, with only minor variations in the MIV–O (2.072(3)–2.170(6) Å) and MIV–N (2.219(8)–2.262(4) Å) bonds formed by the similarly-sized heptacoordinate Zr(IV) and Hf(IV) ions (Shannon radii of 0.92 and 0.90 Å, respectively). In all cases, the MIV ion is displaced by approximately 1.1 Å from the plane of the four nitrogen atoms it is bound to in the phthalocyanine macrocycle, which itself has a dome-like geometry. Despite this, the main supramolecular motif adopted by these hybrid complexes is a ‘base-to-base’ dimer produced by stacking interactions between the phthalocyanine macrocycles. The shortest N⋯N distance observed between the pyrrole moieties is close to 3.5 Å, as previously reported for zirconium/hafnium-capped iron(II) phthalocyaninatoclathrochelates with two capping fragments.11c,d
The electrochemical properties of the obtained complexes were studied by cyclic (CV) and differential pulse (DPV) voltammetry in a dichloromethane/TBAP system (Fig. 5 and Fig. S18, ESI†). For all the MPc-capped tris-pyridineoximate complexes, two subsequent reduction waves are observed in the cathodic range (see the ESI,† Fig. S19). These processes are quasi-reversible, as follows from ΔEp being in the range of 60–90 mV and from the current ratio for the direct and reverse processes being close to 1. The first reduction waves show diffusional control, as the peak current depends linearly on the square root of the scan rate. The potentials of these reductions are the same for all four hybrid complexes and are characteristic of Pc-centered reductions.11b Note that the corresponding reduction waves were absent in the CVs of the antimony-capped precursors, which do not have a Pc fragment, thus confirming this assignment.
 | ||
| Fig. 5 DPVs for 0.1 mM dichloromethane solutions of the complexes [Fe(AcPyOx)3(HfPc)](ClO4) (a), [Fe(AcPyOx)3(ZrPc)](ClO4) (b), [Ni(AcPyOx)3(HfPc)](ClO4) (c), and [Ni(AcPyOx)3(ZrPc)](ClO4) (d). | ||
In the anodic range, several other waves of quasi-reversible diffusionally-controlled oxidation processes were detected. Although these waves significantly overlap, DPV allowed four and three oxidation peaks to be observed for the iron and nickel(II) complexes, respectively (Fig. 5). While the very close redox potentials of these processes complicate the assignment of all of the peaks, the potentials of the first oxidation peaks are similar for the iron and nickel complexes and are characteristic of Pc-localized redox processes.11b The second oxidation peak in the case of the iron complexes is probably due to a Fe2+/Fe3+ process, but the next oxidation is also localized on the Pc moiety.11 The nickel complexes, in contrast to the iron(II) complexes, show an irreversible oxidation at approximately 0.82 V (vs. Fc/Fc+), assigned to a Ni2+/3+ process. As expected, the substitution of zirconium for hafnium does not result in any significant changes in the redox properties of the complexes. The irreversible waves, assigned to the destruction of the macrobicyclic moiety, are observed at potentials larger than −1.8 and 1.0 V (vs. Fc/Fc+) in the cathodic and anodic regions, respectively. This is the case for all of the compounds, including the semiclathrochelate precursors.
Experimental section
Materials and physical measurements
The reagents used (FeCl2·4H2O, Ni(ClO4)2·6H2O, NaClO4·H2O, NaHCO3, CaCO3, 2-acetylpyridine, sorbents and solvents) were obtained commercially (SAF). The zirconium and hafnium(IV) phthalocyaninates (Zr(Cl2)Pc and Hf(Cl2)Pc), 2-acetylpyridineoxime and triethylantimony(V) dibromide were prepared as described earlier.15The analytical data for the C, H and N content were obtained with a Carlo Erba model 1106 microanalyzer; the data for the Sb content were obtained using the X-ray fluorescence method.
MALDI-TOF mass spectra were recorded in the positive and negative ranges with a MALDI-TOF-MS Bruker Autoflex II (Bruker Daltonics) mass spectrometer in reflecto-mol mode. Ionization was induced by a UV-laser with the wavelength of 337 nm. The samples were applied to a nickel plate, and 2,5-dihydroxybenzoic acid was used as the matrix. The accuracy of the measurements was 0.1%.
UV-vis spectra of all of the complexes in dichloromethane were recorded in the range of 250–800 nm with a Varian Cary 50 spectrophotometer. The individual Gaussian components of these spectra were calculated using the Fityk program.16
1H and 13C NMR spectra were recorded from CD2Cl2 and CDCl3 solutions of these complexes with a Bruker Avance 600 spectrometer. The measurements were done using the residual signals of CD2Cl2 (1H 5.32 ppm, 13C 54.00 ppm) and CDCl3 (1H 7.26 ppm, 13C 77.16 ppm). Data acquisition and processing were performed with Topspin 2.1 and Mestrenova 9.0.0 software, respectively.
Cyclic voltammetry experiments were carried out in acetonitrile or dichloromethane solutions with 0.1 M TBAP as a supporting electrolyte using a Metrohm Autolab PGSTAT128N potentiostat with a conventional one-compartment three-electrode cell (5 mL of solution). The platinum disk electrode (MF-2013, BASi), which was used as a working electrode, was thoroughly polished with 0.05 μm alumina slurry, sonicated for two minutes in deionized water and rinsed before every measurement. A platinum wire counter electrode and standard Ag/AgCl/NaClaq. reference electrode (RE-5B, BASi) were used. To account for drift of the reference electrode, ferrocene was added after the measurements as an internal standard, and all of the potentials are reported relative to the Fc/Fc+ redox couple. The solutions were thoroughly deaerated by passing argon through them before the CV experiments and above them during the measurements.
All quantum chemical calculations were performed using the ORCA program package v. 3.0.3.17 The X-ray structures of the complexes were used as an initial approximation for geometry optimization, which was performed with the non-hybrid PBE functional18 and the def2-TZVP basis set.19 After the geometry optimization, the tensors of hyperfine interactions for hydrogen and carbon nuclei were calculated using the hybrid PBE0 functional20 and the def2-TZVP basis set, with the primitives of a higher order of the exponent added for a better description of the electron density around the nuclei. The paramagnetic shifts for hydrogen and carbon nuclei were calculated using the following equation:21
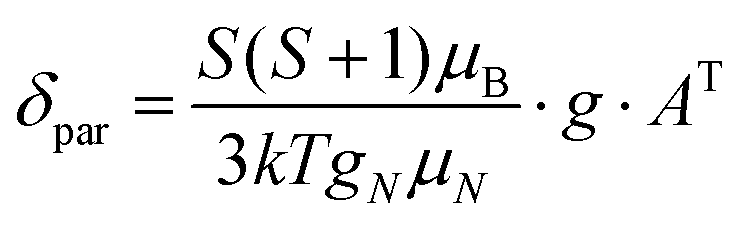
Synthesis
N), 158.62 (s, 2-Py). UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 235(30), 267(2.8), 305(9.4), 306(19), 399(4.0), 445(2.1), 467(1.9), 498(6.7), 551(11.5).
N), 377.65 (br. s, CH3), 415.01 (br. s, 3-Py), 566.40 (br. s, 5-Py). UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 265 (30), 307 (2.4), 319 (24), 345 (6.2), 389 (0.27), 484 (0.12), 771 (0.03), 938 (0.005).
General procedure for preparation of the metal(IV)phthalocyaninate-capped iron and nickel(II) complexes
The corresponding metallophthalocyaninate {Zr(Cl2)Pc or Hf(Cl2)Pc} (0.13 mmol), the appropriate triethylantimony-capped precursor {[Fe(AcPyOx)3(Sb(C2H5)3)](ClO4) or [Ni(AcPyOx)3(SbEt3)](ClO4)} (0.13 mmol), CaCO3 (0.05 g) and silica gel (0.05 g) were dissolved/suspended in a dichloromethane–methanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture (4 mL). The reaction mixture was stirred for 8 h at room temperature and extracted with dichloromethane. The extract was evaporated to approximately 10 mL and separated using column chromatography on silica gel (eluents: dichloromethane and a dichloromethane–acetone (10:1) mixture). The first eluate was thrown out, and the second eluate was collected and evaporated to dryness. The solid residue was washed with hexane and dried in vacuo.
:1). Anal. calcd for C53H37N14O7FeHfCl (%): C, 50.85; H, 2.98; N, 15.67. Found (%): C, 50.92; H, 3.01; N, 15.70. 1H NMR (CD2Cl2): δ (ppm) 1.00 (s, 9H, CH3), 5.80 (d, 3JHH = 5.42 Hz, 3H, 6-Py), 6.67 (m, 3H, 5-Py), 6.97 (d, 3JHH = 8.44 Hz, 3H, 3-Py), 7.41 (m, 3H, 4-Py), 8.24 (m, 8H, β-Pc), 9.34 (m, 8H, α-Pc). 13C NMR (CD2Cl2): δ (ppm) 11.53 (s, CH3), 123.23 (s, 3-Py), 123.76 (s, α-Pc), 124.36 (s, 5-Py), 131.32 (s, β-Pc), 136.87 (s, 4-Py), 136.95 (s, C(Pc)), 151.53 (s, 6-Py), 153.68 (s, CN(Pc)), 153.73 (s, CN(Pc)), 155.67 (s, CN), 157.38 (s, 2-Py). MALDI-TOF MS: calcd for C53H37N14O7FeHfCl 1252.1473; found, 1153.0602 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 341 (68.60), 532 (17.48); 620 (34.41); 656 sh (29.94); 689 (218.09).
:1). Anal. calcd for C53H37N14O7FeZrCl (%): C, 54.67; H, 3.20; N, 16.84. Found (%): C, 54.82; H, 3.06; N, 16.96. 1H NMR (CD2Cl2): δ (ppm) 0.96 (s, 9H, CH3), 5.84 (d, 3JHH = 5.45 Hz, 3H, 6-Py), 6.68 (m, 3H, 5-Py), 6.97 (d, 3JHH = 8.39 Hz, 3H, 3-Py), 7.41 (m, 3H, 4-Py), 8.23 (m, 8H, β-Pc), 9.34 (m, 8H, α-Pc). 13C NMR (CD2Cl2): δ (ppm) 11.55 (s, CH3), 123.27 (s, 3-Py), 123.75 (s, α-Pc), 124.40 (s, 5-Py), 131.27 (s, β-Pc), 136.94 (s, 4-Py), 137.42 (s, C(Pc)), 151.62 (s, 6-Py), 153.94 (s, CN(Pc)), 154.00 (s, CN(Pc)), 155.61 (s, CN), 158.01 (s, 2-Py). MALDI-TOF MS: calcd for C53H37N14O7FeZrCl, 1164.1052; found, 1063.1984 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 341 (77.11), 534 (19.66), 621 (38.81), 658 sh (34.05), 690 (245.85).
:1). Anal. calcd for C53H37N14O7NiHfCl (%): C, 50.74; H, 2.97; N, 15.63. Found (%): C, 50.92; H, 2.92; N, 15.78. 1H NMR (CD2Cl2): δ (ppm) −26.10 (br. s, 9H, CH3), 8.22 (m, 8H, β-Pc), 9.35 (m, 8H, α-Pc), 14.34 (s, 4-Py), 48.64 (br. s, 5-Py), 56.30 (br. s, 3-Py), 141.47 (br. s, 6-Py). MALDI-TOF MS: calcd for C53H37N14O7NiHfCl, 1254.1528; found, 1155.3228 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 335 (77.99), 352 sh (64.41), 619 (39.20), 656 sh (34.29), 688 (243.47).
:1). Anal. calcd for C53H37N14O7NiZrCl (%): C, 54.53; H, 3.19; N, 16.80. Found (%): C, 54.62; H, 3.25; N, 16.65. 1H NMR (CD2Cl2): δ (ppm) −26.02 (br. s, 9H, CH3), 8.15 (m, 8H, β-Pc), 9.23 (m, 8H, α-Pc), 14.39 (s, 4-Py), 48.83 (br. s, 5-Py), 56.46 (br. s, 3-Py), 141.27 (br. s, 6-Py). MALDI-TOF MS: calcd for C53H37N14O7NiZrCl, 1166.1055; found, 1067.3603 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 336 (74.93), 352 sh (59.54), 620 (36.91), 657 sh (32.74), 688 (228.50).
:1) mixture (4 mL). The reaction mixture was stirred for 8 h at room temperature and extracted with dichloromethane. The extract was evaporated to approximately 10 mL and separated using column chromatography on silica gel (eluents: dichloromethane and a dichloromethane–acetone (10:1) mixture). The first eluate was thrown out, and the second eluate was collected and evaporated to dryness. The solid residue was washed with hexane and dried in vacuo.
:1). Anal. calcd for C53H37N14O7FeHfCl (%): C, 50.85; H, 2.98; N, 15.67. Found (%): C, 50.92; H, 3.01; N, 15.70. 1H NMR (CD2Cl2): δ (ppm) 1.00 (s, 9H, CH3), 5.80 (d, 3JHH = 5.42 Hz, 3H, 6-Py), 6.67 (m, 3H, 5-Py), 6.97 (d, 3JHH = 8.44 Hz, 3H, 3-Py), 7.41 (m, 3H, 4-Py), 8.24 (m, 8H, β-Pc), 9.34 (m, 8H, α-Pc). 13C NMR (CD2Cl2): δ (ppm) 11.53 (s, CH3), 123.23 (s, 3-Py), 123.76 (s, α-Pc), 124.36 (s, 5-Py), 131.32 (s, β-Pc), 136.87 (s, 4-Py), 136.95 (s, C(Pc)), 151.53 (s, 6-Py), 153.68 (s, CN(Pc)), 153.73 (s, CN(Pc)), 155.67 (s, CN), 157.38 (s, 2-Py). MALDI-TOF MS: calcd for C53H37N14O7FeHfCl 1252.1473; found, 1153.0602 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 341 (68.60), 532 (17.48); 620 (34.41); 656 sh (29.94); 689 (218.09).
:1). Anal. calcd for C53H37N14O7FeZrCl (%): C, 54.67; H, 3.20; N, 16.84. Found (%): C, 54.82; H, 3.06; N, 16.96. 1H NMR (CD2Cl2): δ (ppm) 0.96 (s, 9H, CH3), 5.84 (d, 3JHH = 5.45 Hz, 3H, 6-Py), 6.68 (m, 3H, 5-Py), 6.97 (d, 3JHH = 8.39 Hz, 3H, 3-Py), 7.41 (m, 3H, 4-Py), 8.23 (m, 8H, β-Pc), 9.34 (m, 8H, α-Pc). 13C NMR (CD2Cl2): δ (ppm) 11.55 (s, CH3), 123.27 (s, 3-Py), 123.75 (s, α-Pc), 124.40 (s, 5-Py), 131.27 (s, β-Pc), 136.94 (s, 4-Py), 137.42 (s, C(Pc)), 151.62 (s, 6-Py), 153.94 (s, CN(Pc)), 154.00 (s, CN(Pc)), 155.61 (s, CN), 158.01 (s, 2-Py). MALDI-TOF MS: calcd for C53H37N14O7FeZrCl, 1164.1052; found, 1063.1984 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 341 (77.11), 534 (19.66), 621 (38.81), 658 sh (34.05), 690 (245.85).
:1). Anal. calcd for C53H37N14O7NiHfCl (%): C, 50.74; H, 2.97; N, 15.63. Found (%): C, 50.92; H, 2.92; N, 15.78. 1H NMR (CD2Cl2): δ (ppm) −26.10 (br. s, 9H, CH3), 8.22 (m, 8H, β-Pc), 9.35 (m, 8H, α-Pc), 14.34 (s, 4-Py), 48.64 (br. s, 5-Py), 56.30 (br. s, 3-Py), 141.47 (br. s, 6-Py). MALDI-TOF MS: calcd for C53H37N14O7NiHfCl, 1254.1528; found, 1155.3228 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 335 (77.99), 352 sh (64.41), 619 (39.20), 656 sh (34.29), 688 (243.47).
:1). Anal. calcd for C53H37N14O7NiZrCl (%): C, 54.53; H, 3.19; N, 16.80. Found (%): C, 54.62; H, 3.25; N, 16.65. 1H NMR (CD2Cl2): δ (ppm) −26.02 (br. s, 9H, CH3), 8.15 (m, 8H, β-Pc), 9.23 (m, 8H, α-Pc), 14.39 (s, 4-Py), 48.83 (br. s, 5-Py), 56.46 (br. s, 3-Py), 141.27 (br. s, 6-Py). MALDI-TOF MS: calcd for C53H37N14O7NiZrCl, 1166.1055; found, 1067.3603 [M − ClO4−]+. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, mol−1 L cm−1): 336 (74.93), 352 sh (59.54), 620 (36.91), 657 sh (32.74), 688 (228.50).
X-ray crystallography
Single crystals of the [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4), [Fe(AcPyOx)3(HfPc)](ClO4), [Fe(AcPyOx)3(ZrPc)](ClO4), [Ni(AcPyOx)3(HfPc)](ClO4), and [Ni(AcPyOx)3(ZrPc)](ClO4) complexes suitable for X-ray diffraction experiments were grown at room temperature from their solutions in a dichloromethane–hexane mixture ([Ni(AcPyOx)3(ZrPc)], [Fe(AcPyOx)3(HfPc)] and [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4)) or in a dichloromethane–benzene mixture ([Fe(AcPyOx)3(ZrPc)] and [Ni(AcPyOx)3(HfPc)]). The intensities of the reflections were measured at 120 K (for [Ni(AcPyOx)3(Sb(C2H5)3)](ClO4), [Fe(AcPyOx)3(HfPc)](ClO4), [Ni(AcPyOx)3(HfPc)](ClO4) and [Ni(AcPyOx)3(ZrPc)](ClO4)) or at 100 K (for [Fe(AcPyOx)3(ZrPc)](ClO4)) with a Bruker Apex II DUO CCD diffractometer using Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined using the full-matrix least-squares technique against F2 in the anisotropic–isotropic approximation. Hydrogen atom positions were calculated, and they were refined in the isotropic approximation within the riding model. The unit cells of [Ni(AcPyOx)3(ZrPc)](ClO4) and [Fe(AcPyOx)3(HfPc)](ClO4) contain disordered solvent species, which were assumed to be dichloromethane and hexane, both used in the mixture for crystallization of these complexes; these species have been treated as a diffuse contribution to the overall scattering without specific atom positions by SQUEEZE/PLATON.22 All the calculations were performed with the SHELXTL program package.23Details of data collection and refinement are given in Table S7 (see the ESI†).
Conclusions
We developed a two-step synthetic pathway for new hybrid Pc-capped metal complexes by transmetallation of antimony-capped iron and nickel(II) tris-pyridineoximates with Lewis-acidic zirconium and hafnium(IV) phthalocyaninates. They combine a transition metal clathrochelate and a phthalocyaninate complex, both of which are now used as ‘building blocks’ in single molecule magnets. The obtained hybrid complexes are therefore an important molecular platform on which new classes of these exciting materials can be built in the future.Acknowledgements
The synthesis of the hybrid complexes was supported by the Russian Science Foundation (project 14-13-00724). Spectral and structural characterization was performed under the financial support from the Russian Foundation for Basic Research (projects 16-03-00368, 16-03-00688, 16-33-00233 and 15-29-01112) and Russian Academy of Science (program P8). Electrochemical measurements were supported by a grant from the President of the Russian Federation (projects MK-6320.2016.3 and 2179.2017.3).Notes and references
- (a) Y. Voloshin, I. Belaya and R. Krämer, The Encapsulation Phenomenon: Synthesis, Reactivity and Applications of Caged Ions and Molecules, Springer International Publishing, Switzerland, 2016 Search PubMed; (b) Y. Z. Voloshin, N. A. Kostromina and R. Krämer, Clathrochelates: Synthesis, Structure and Properties, Elsevier, Amsterdam, 2002 Search PubMed; (c) A. Ingham, M. Rodopoulos, K. Coulter, T. Rodopouls, S. Subramanian and A. McAuley, Coord. Chem. Rev., 2002, 233–234, 255–271 CrossRef CAS.
- (a) O. A. Varzatskii, I. N. Denisenko, A. S. Belov, A. V. Vologzhanina, Y. N. Bubnov, S. V. Volkov and Y. Z. Voloshin, Inorg. Chem. Commun., 2014, 44, 134–138 CrossRef CAS; (b) Y. Z. Voloshin, A. S. Belov, Z. A. Starikova and A. V. Vologzhanina, Russ. Chem. Bull., Int. Ed., 2013, 62, 1858–1865 CrossRef CAS; (c) O. A. Varzatskii, I. N. Denisenko, S. V. Volkov, A. S. Belov, A. V. Dolganov, A. V. Vologzhanina, V. V. Novikov, Y. N. Bubnov and Y. Z. Voloshin, Eur. J. Inorg. Chem., 2013, 3178–3184 CrossRef CAS; (d) O. A. Varzatskii, S. V. Kats (Menkach), L. V. Penkova, S. V. Volkov, A. V. Dolganov, A. V. Vologzhanina, Y. N. Bubnov and Y. Z. Voloshin, Eur. J. Inorg. Chem., 2013, 1987–1992 CrossRef CAS; (e) Y. Z. Voloshin, O. A. Varzatskii, A. V. Belov, Z. A. Starikova, A. V. Dolganov and T. V. Magdesieva, Polyhedron, 2007, 27, 325–334 CrossRef; (f) Y. Z. Voloshin, O. A. Varzatskii, A. S. Belov and Y. N. Bubnov, Russ. Chem. Bull., Int. Ed., 2006, 55, 1119–1125 CrossRef CAS; (g) Y. Z. Voloshin, O. A. Varzatskii, A. S. Belov, A. Y. Lebedev, I. S. Makarov, M. E. Gurskii, M. Y. Antipin, Z. A. Starikova and Y. N. Bubnov, Inorg. Chim. Acta, 2007, 360, 1543–1554 CrossRef CAS.
- (a) A. V. Dolganov, A. S. Belov, V. V. Novikov, A. V. Vologzhanina, G. V. Romanenko, Y. G. Budnikova, G. E. Zelinskii and Y. Z. Voloshin, Dalton Trans., 2015, 44, 2476–2487 RSC; (b) A. V. Dolganov, I. G. Belaya and Y. Z. Voloshin, Electrochim. Acta, 2014, 125, 302–306 CrossRef CAS; (c) L. El Chachtouli, M. Fournier, S. Cherdo, R. Guillot, M.-F. Charlot, E. Anxolabéhère-Mallart, M. Robert and A. Aukauloo, J. Phys. Chem. C, 2013, 117, 17073–17077 CrossRef; (d) I. G. Belaya, S. V. Svidlov, A. V. Dolganov, G. E. Zelinskii, T. V. Potapova, A. V. Vologzhanina, O. A. Varzatskii, Y. N. Bubnov and Y. Z. Voloshin, Dalton Trans., 2013, 42, 13667–13678 RSC; (e) A. V. Dolganov, A. S. Belov, V. V. Novikov, A. V. Vologzhanina, A. Mokhir, Y. N. Bubnov and Y. Z. Voloshin, Dalton Trans., 2013, 42, 4373–7376 RSC; (f) D. Kochubey, V. Kaichev, A. Saraev, S. Tomyn, A. Belov and Y. Voloshin, J. Phys. Chem. C, 2013, 117, 2753–2759 CrossRef CAS; (g) E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 6104–61207 CrossRef PubMed; (h) Y. Z. Voloshin, A. S. Belov, A. V. Vologzhanina, G. G. Aleksandrov, A. V. Dolganov, V. V. Novikov, O. A. Varzatskii and Y. N. Bubnov, Dalton Trans., 2012, 41, 6078–6093 RSC; (i) Y. Z. Voloshin, A. V. Dolganov, O. A. Varzatskii and Y. N. Bubnov, Chem. Commun., 2011, 47, 7737–7739 RSC; (j) M. T. D. Nguen, M.-F. Charlot and A. Aukauloo, J. Phys. Chem. A, 2011, 115, 911–922 CrossRef PubMed; (k) O. Pantani, S. Naskar, R. Guillot, P. Millet, E. Anxolabéhère-Mallart and A. Aukauloo, Angew. Chem., Int. Ed., 2008, 47, 9948–9950 CrossRef CAS PubMed.
- (a) Y. Z. Voloshin, O. A. Varzatskii, K. Y. Zhizhin, N. T. Kuznetsov and Y. N. Bubnov, Russ. Chem. Bull., Int. Ed., 2006, 55, 22–25 CrossRef CAS; (b) Y. Z. Voloshin, O. A. Varzatskii, A. V. Palchik, Z. A. Starikova, M. Y. Antipin, E. G. Lebed and Y. N. Bubnov, Inorg. Chim. Acta, 2006, 359, 553–569 CrossRef CAS; (c) A. B. Burdukov, E. G. Boguslavskii, V. A. Reznikov, N. V. Pervukhina, M. A. Vershinin, Y. Z. Voloshin, O. A. Varzatskii and Y. N. Bubnov, Russ. Chem. Bull., Int. Ed., 2005, 54, 1125–1130 CrossRef CAS; (d) Y. Z. Voloshin, A. S. Belov, A. Y. Lebedev, O. A. Varzatskii, M. Y. Antipin, Z. A. Starikova and T. E. Kron, Russ. Chem. Bull., Int. Ed., 2004, 53, 1218–1222 CrossRef CAS; (e) Y. Z. Voloshin, O. A. Varzatskii, Z. A. Starikova, M. Y. Antipin, A. Y. Lebedev and A. S. Belov, Russ. Chem. Bull., Int. Ed., 2004, 53, 1496–1502 CrossRef CAS; (f) Y. Z. Voloshin, O. A. Varzatskii, T. E. Kron, V. K. Belsky, V. E. Zavodnik and A. V. Palchik, Inorg. Chem., 2000, 39, 1907–1918 CrossRef CAS PubMed; (g) Y. Z. Voloshin, O. A. Varzatskii, A. V. Palchik, A. I. Stash and V. K. Belsky, New J. Chem., 1999, 23, 355–358 RSC.
- (a) Y. Z. Voloshin, V. V. Novikov and Y. V. Nelyubina, RSC Adv., 2015, 5, 72621–72637 RSC; (b) Y. Z. Voloshin, O. A. Varzatskii and Y. N. Bubnov, Russ. Chem. Bull., Int. Ed., 2007, 56, 577–605 CrossRef CAS.
- V. B. Kovalska, M. Y. Losytskyy, O. A. Varzatskii, V. V. Cherepanov, Y. Z. Voloshin, A. A. Mokhir, S. M. Yarmoluk and S. V. Volkov, Bioorg. Med. Chem., 2014, 22, 1883–1888 CrossRef CAS PubMed.
- J. Blechinger, O. A. Varzatskii, V. Kovalska, G. E. Zelinskii, Y. Z. Voloshin, E. Kinski and A. Mokhir, Bioorg. Med. Chem., 2016, 26, 626–629 CrossRef CAS PubMed.
- (a) A. A. Pavlov, Y. V. Nelubina, S. V. Kats, L. V. Penkova, N. N. Efimov, A. O. Dmitrienko, A. V. Vologzhanina, A. S. Belov, Y. Z. Voloshin and V. V. Novikov, J. Phys. Chem. Lett., 2016, 7, 4111–4116 CrossRef CAS PubMed; V. V. Novikov, A. A. Pavlov, Y. N. Nelyubina, M.-E. Boulon, O. A. Varzatskii, Y. Z. Voloshin and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137, 9792–9795 Search PubMed; (b) V. V. Novikov, I. V. Ananyev, A. A. Pavlov, M. V. Fedin, K. A. Lyssenko and Y. Z. Voloshin, J. Phys. Chem. Lett., 2014, 5, 496–500 CrossRef CAS PubMed; (c) V. V. Novikov, A. A. Pavlov, A. S. Belov, A. V. Vologzhanina, A. Savitsky and Y. Z. Voloshin, J. Phys. Chem. Lett., 2014, 5, 3799–3803 CrossRef CAS PubMed.
- (a) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS; (b) D. Gatteschi, R. Sessoli and J. Villain, Molecular nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed.
- (a) H. Wang, B.-W. Wang, Y. Bian, S. Gao and J. Jiang, Coord. Chem. Rev., 2016, 306, 195–216 CrossRef CAS; (b) K. L. M. Harriman and M. Murugesu, Acc. Chem. Res., 2016, 49, 1158–1167 CrossRef CAS PubMed.
- (a) S. V. Dudkin, N. R. Erickson, A. V. Vologzhanina, V. N. Novikov, H. V. Rhoda, C. D. Holstrom, Y. V. Zatsikha, M. S. Yusubov, Y. Z. Voloshin and V. N. Nemykin, Inorg. Chem., 2016, 55, 11867–11882 CrossRef CAS PubMed; (b) Y. Z. Voloshin, O. A. Varzatskii, L. G. Tomilova, M. O. Breusova, T. V. Magdesieva, Y. N. Bubnov and R. Krämer, Polyhedron, 2007, 26, 2733–2740 CrossRef CAS; (c) Y. Z. Voloshin, O. A. Varzatskii, S. V. Korobko, V. Y. Chernii, S. V. Volkov, L. A. Tomachynski, V. I. Pehn'o, V. Y. Antipin and Z. A. Starikova, Inorg. Chem., 2005, 44, 822–824 CrossRef CAS PubMed; (d) J. R. Sabin, O. A. Varzatskii, Y. Z. Voloshin, Z. A. Starikova, V. V. Novikov and V. N. Nemykin, Inorg. Chem., 2012, 51, 8362–8372 CrossRef CAS PubMed.
- M. Hołyńska, Inorg. Chem. Commun., 2012, 20, 322–325 CrossRef.
- (a) Y. Z. Voloshin, O. A. Varzatskii, S. V. Korobko, M. Y. Antipin, I. I. Vorontsov, K. A. Lyssenko, D. I. Kochubey, S. G. Nikitenko and N. G. Strizhakova, Inorg. Chim. Acta, 2004, 357, 3187–3204 CrossRef CAS; (b) Y. Z. Voloshin, O. A. Varzatskii, A. S. Belov, Z. A. Starikova, A. V. Dolganov and T. V. Magdesieva, Polyhedron, 2008, 27, 325–334 CrossRef CAS.
- O. A. Varzatskii, L. V. Penkova, S. V. Kats (Menkach), A. V. Dolganov, A. V. Vologzhanina, A. A. Pavlov, V. V. Novikov, A. S. Bogomyakov, V. N. Nemykin and Y. Z. Voloshin, Inorg. Chem., 2014, 53, 3062–3071 CrossRef CAS PubMed.
- (a) L. A. Tomachynski, I. N. Tretyakova, V. Y. Chernii, S. V. Volkov, M. Kowalska, J. Legendziewicz, Y. S. Gerasymchuk and S. Radzki, Inorg. Chim. Acta, 2008, 361, 2569–2581 CrossRef CAS; (b) C. B. Aakeröy, A. S. Sinha, K. N. Epa, C. L. Spartz and J. Desper, Chem. Commun., 2012, 48, 11289–11291 RSC; (c) K. Isseib and B. Hamann, Z. Anorg. Allg. Chem., 1965, 339, 289–297 CrossRef.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- S. Moon and S. Patchkovskii, in Calculation of NMR and EPR Parameters. Theory and Applications, ed. M. Kaupp, M. Bühl and V. G. Malkin, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details of analytical and spectroscopic data collection, together with the crystallographic, UV-vis and electrochemical data. CCDC 1503088–1503092. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj00131b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |