
Fabrication of porous magnetic nanocomposites for bone tissue engineering†
Arundhati
Bhowmick
a,
Nilkamal
Pramanik
a,
Tapas
Mitra
a,
Arumugam
Gnanamani
b,
Manas
Das
c and
Patit Paban
Kundu
*ad
aDepartment of Polymer Science and Technology, University of Calcutta, 92 A.P.C. Road, Kolkata-700009, India. E-mail: ppk923@yahoo.com; Tel: +91-33-2352-5106
bMicrobiology Division, CSIR-Central Leather Research Institute, Adyar, Chennai-600020, Tamil Nadu, India
cDepartment of Chemical Engineering, University of Calcutta, 92 A.P.C. Road, Kolkata-700009, India
dDepartment of Chemical Engineering, Indian Institute of Technology, Roorkee-247667, India
First published on 22nd November 2016
Abstract
Here, the fabrication and characterization of porous magnetic nanocomposites was carried out via the blending of chitosan, polyethylene glycol and nano-hydroxyapatite–Fe3O4. Scanning electron microscope images revealed a highly interconnected macro- and micro-porous structure. These nanocomposites showed good water uptake abilities and have good antimicrobial properties. The tensile strengths of these nanocomposites were enhanced significantly compared to previously reported results, after the addition of nano-Fe3O4. Moreover, these nanocomposites could be applied for magnetic therapy as this material exhibited superparamagnetic properties. Finally, these nanocomposites were good supports for human osteoblast-like MG-63 cells’ growth, attachment and proliferation and they showed good cytocompatibility. No negative effect on the MG-63 cells was observed, suggesting that these nanocomposites have great potential to be applied for bone regeneration.
1 Introduction
In recent times, the integration of porous scaffold materials in bone tissue engineering has become an emerging substitute for autografting and allografting,1 as these materials stimulate bone formation from the surrounding tissue and also can act as templates to grow cells for bone tissue regeneration. However, fabrication of a desirable porous material for successful bone regeneration is a great challenge. Moreover, it should have an interconnected porous structure that can support cell penetration, new tissue ingrowths, nutrient diffusion and neovascularisation, and have good mechanical properties and biocompatibility.2–4 Matrices are usually implemented to achieve the requirements for bone regeneration to promote osteoblast proliferation and osteogenesis by providing a suitable microenvironment.5 Since natural bone is composed of inorganic hydroxyapatite (HAP, Ca10(PO4)6(OH)2) and organic collagen fibrils,6,7 a number of HAP–polymer hybrid materials8–16 have been fabricated where the polymer gives flexibility, toughness and bioresorbability, and HAP brings stiffness, strength and osteoconductivity. Among the different polymers used, chitosan (CTS), a natural polysaccharide present in chitin in which glucosamine and N-acetylglucosamine units are connected via β (1–4) linkages, is attractive as it is flexible, resistance to heat, biocompatible and nonimmunogenic.17–19 In spite of the attractive properties of CTS, poor mechanical strength limited its applications in bone tissue engineering.20 Again, the brittleness and poor mechanical stability of pure HAP restrict its use for bone regeneration and tissue engineering applications.21 To address the limitation related to poor mechanical properties associated with CTS, it has been blended with several synthetic polymers (poly[vinyl alcohol], polycaprolactone and poly[acrylamide]).22–24 In addition to this, PEG has become a widely used polymer for application in medical implants. In recent years, various attempts have been made for the fabrication of nanocomposites, wherein nano-HAP particles are embedded in polyethylene glycol (PEG) polymeric matrices,25,26 while a few approaches have been developed for blending CTS with PEG.27,28 These limited approaches demand the fabrication of composites containing CTS and HAP with PEG to obtain a suitable scaffold for bone regeneration.Magnetic therapy has been considered as a promising technique in the treatment of bone diseases. The rationale for designing magnetic scaffolds is to achieve such a structure as will be able to reach very high magnetization values by applying an external magnetic field, and upon removal of the magnetic field it will be magnetically “turned off”. There are several advantages of using a magnetic field as reported previously such as the fact that magnetic fields may stimulate the proliferation of osteoblasts, promote the expression of growth factors, increase osteointegration, enhance bone density and calcium content, expedite the recovery of bone fractures and accelerate new bone formation.29–32 The incorporation of magnetic nanoparticles (MNPs) into polymeric scaffolds is considered to accelerate the rate of osteoblast cell growth and differentiation due to the ability of bone tissue to recognize mechano-electrical conversion, which results in increased cellular proliferation and expression levels of multiple genes related to bone differentiation.33,34 MNPs are also found to significantly improve the hydrophilicity as well as the tensile mechanical properties of the nanocomposite-based polymeric 3D scaffold and increase the degradation rate of the composite.35–37 The improved initial cell adhesion and subsequent penetration through the porous scaffolds is another effect of the MNPs.
Therefore, appropriately designed magnetic iron oxide nanoparticles (IONPs) are used for magnetic therapy because of their biocompatibility and nontoxicity and they can be used in biomedical applications such as magnetic resonance imaging, drug delivery and tissue targeting, and also for in vivo applications.38,39 These IONPs become superparamagnetic at a size below 50 nm.40 Each magnetic domain provides intrinsic magnetic therapy which further enhances its functionality in the presence of an external magnetic field.41,42 However, very few studies have reported the integration of IONPs into bone substitutes to produce magnetic scaffolds.33,43–50 Bock et al. integrated IONPs into a HAP–collagen scaffold and in vitro study showed no negative effect on human bronchial smooth muscle cells.44 Meng et al. fabricated magnetic nanofibrous films composed of polylactic acid, HAP and γ-Fe2O3 and observed that the presence of a static magnetic field (SMF) significantly enhanced the pre-osteoblast MC3T3-E1 cell proliferation rate after the incorporation of superparamagnetic nanoparticles in the scaffold.33 They have recently validated in vivo the osteogenic potential of the magnetic nanofibrous scaffolds coupled with an external SMF.47 After implementation of the scaffolds in the lumbar transverse defect of New Zealand white rabbits, histological observations indicate that osteoblast cells appeared and new bone tissue formed around the scaffold pieces, as well as blood vessels over longer periods. Homogenous and close-to-natural bone morphology was observed using computed tomography images when the animals were exposed to an external magnetic field, compared to animals that were not exposed to the magnetic field. They have also found that the scaffolds degraded faster using an external magnet.
Tampieri et al. directly nucleated superparamagnetic Fe-HA crystals on self-assembling collagen fibers (telopeptides-free type I, from horse tendons) where synthesis temperatures were varied from 25° to 50°.45 Fe-HA/coll25 (synthesized at 25°) provided the best results both in terms of osteoblast-like human cell adhesion, viability and proliferation, independent of the presence of a static magnetic field.45 Wei et al. fabricated magnetic biodegradable fibers in which good cell adhesion and proliferation was observed.50
Further, in orthopedic implants bacterial infection is an issue that leads to implant failure, hospitalization and sometimes mortality of the patient. Hence, an ideal scaffold material should have antimicrobial properties for successful bone regeneration.
Considering all of the above issues, we report porous, magnetic nanocomposites (BPMs) containing CTS/PEG/Fe3O4–HAP. Their physicochemical properties, morphology, mechanical properties, antimicrobial activities, magnetic properties and in vitro cell proliferation behaviour have been investigated.
2 Experimental section
2.1 Materials
The molecular weight of CTS (Acros Organics, 86% deacetylated) was 230 kDa, determined using the Ubbelohde viscometric method. Nano-HAP has been synthesized previously by us.51 The synthesis of Fe3O4 nanoparticles and nano-HAP–Fe3O4 was reported by us recently.52 The molecular weight of polyethylene glycol (PEG) was 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Merck).
000 (Merck).
2.2 Synthesis of chitosan–PEG–nano-HAP–Fe3O4 porous magnetic nanocomposites (BPM I–III)
BPM I–III nanocomposites were fabricated by blending various weight percentages of CTS, PEG and nano-Fe3O4–HAP (Table 1). Initially, an appropriate amount of (55–75 wt%) CTS was dissolved in 15 mL of 85% formic acid and PEG (20–40 wt%) was added to that with continuous stirring. Then, a fixed amount (5 wt%) of HAP was added to that mixture. After stirring overnight, the homogeneous mixture was transferred to petri-dishes to obtain the films, which were dried further at 60 °C for 48 h.| Bone-like porous magnetic-materials (BPMs) | CTS (wt%) | PEG (wt%) | HAP–Fe3O4 (wt%) |
|---|---|---|---|
| BPM I | 55 | 40 | 5 |
| BPM II | 65 | 30 | 5 |
| BPM III | 75 | 20 | 5 |
2.3 Physicochemical properties
The presence of the individual components of BPM I–III was confirmed using Fourier transform infrared (FTIR) absorption spectra analysis (Model Perkin-Elmer-782) within a scanning range of 4000–400 cm−1.X-ray diffraction (XRD) patterns of BPM I–III were obtained using powder XRD (Model Goniometer Miniflex, JAPAN) using Cu Kα radiation operating at a voltage of 30 kV and a current of 15 mA. The samples were scanned from 5° to 70° in 2θ at a speed of 4° min−1.
Study of the water uptake abilities of BPM I–III was performed by taking the dry weights (Wd) of the samples (BPM I–III) and then immersing them in distilled water for up to 24 hours. After removing them from the water, the wet samples were blotted on filter paper and weighed again (Ww). Five replicates were used for each study. The following eqn (1)53 was used to determine the percentage of water absorption (EA) of the samples.
| EA = [(Ww − Wd)/Wd] × 100 | (1) |
2.4 Microstructure of the composites
Scanning electron microscopic (SEM) examination (Model EVO-18, Carl-Zeiss) was performed after coating BPM I–III with gold using a E-1010 Hitachi Ion Sputter.Transmission Electron Microscopic (TEM) images of nano-Fe3O4 were acquired using an FEI Tecnai G2 Spirit Twin. For this experiment, nano-Fe3O4 was dispersed in a 1:1 methanol–water mixture using ultrasonication and loaded onto a Cu grid.
2.5 Porosity and density measurement
The porosity and density of the prepared nanocomposites were determined using the liquid displacement method.54 Ethanol was used as the displacement liquid. At first, the dry weights (W) of the samples (BPM I–III) were measured. A known volume of ethanol was measured and recorded as V1. The samples were then immersed in the ethanol in a cylinder and saturated for 24 hours. Then, the total volume of the ethanol and the ethanol-impregnated samples was denoted V2. Then, the samples were removed from the cylinder and the residual ethanol volume was recorded as V3.The density (d) of the nanocomposites was expressed as
| d = W/V2 − V3 | (2) |
The porosity (μ) of the nanocomposites was calculated based on the following formula:
| μ = (V1 − V3)/(V2 − V3) | (3) |
2.6 Mechanical properties
Tensile testing of BPM I–III and CBPM was performed using a Universal Testing Machine (Model LR01KOLPLUS, England) at a crosshead speed of 5 mm min−1 with a 30 mm gauge length. The samples were 21 mm in diameter and 0.05 mm in thickness. The mechanical properties of BPM I–III and CBPM under hydrated conditions were also measured. For these experiments, BPM I–III and CBPM were immersed in phosphate buffer solution (PBS) at 37 °C for 24 hours before tensile testing.2.7 Antimicrobial activities
For antimicrobial study, Gram-negative bacteria Escherichia coli XL1B strain and Gram-positive strains Lysinibacillus fusiformis and Bacillus cereus were grown in agar media at 37 °C. The activity of BPM I–III against these bacterial strains was observed following the disk diffusion method.522.8 Magnetization analysis
For magnetization analysis, a superconducting quantum interference device-vibrating sample magnetometer (SQUID-VSM) (MPMS, USA) was used and was operated at temperatures 2 K < T < 300 K. For field-dependent magnetization measurements, −10000 to 10000 Oe of magnetic field was applied.
2.9 Cell culturing and maintenance
Human MG-63 cells were procured from NCCS, Pune, India. The cultures were maintained in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% Fetal Bovine Serum (FBS), glutamine (200 mM), sodium bicarbonate (2 mg ml−1) and antibiotic and antimycotic solution (1%). The medium was changed from time to time. The cells were cultured in tissue culture flasks, incubated at 37 °C in a humidified 5% CO2 atmosphere. To detach the cells, trypsin (0.05%) was applied.2.10 In vitro cell proliferation study (MTT assay)
For MTT assay, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was supplied by Sigma Aldrich. MG-63 cells were grown in DMEM supplemented with FBS (10% v/v) and antibiotic (1%) and were incubated at 37 °C in a 5% CO2 humidified atmosphere. 96 well culture polystyrene plates, obtained from Tarson, India, were coated with BPM I and CBPM. After drying and sterilizing the plates, the cells were seeded at a density of 0.5 × 106 per well. After 72 hours of incubation, the supernatant of each well was replaced with MTT, which was diluted in a serum-free medium and again incubated for 4 hours. After removing the MTT solution, the blue crystals were dissolved by adding 0.04 N hydrochloric acid in isopropanol. Finally, the UV absorbance at 570 nm was measured. The experiment was performed at least three times. Sets of three wells were employed for each experimental variable.3 Results and discussion
Initially, we developed porous magnetic nanocomposites (BPM I–III) by blending CTS, PEG and nano-HAP–Fe3O4 in different weight percentages (see the Experimental section). Investigation of the physicochemical properties, morphology, mechanical properties, antimicrobial activities, magnetic properties and in vitro cell proliferation was done and is discussed below.3.1 Physicochemical properties
The formation of BPM I–III was confirmed using FT-IR, powder XRD and SEM studies.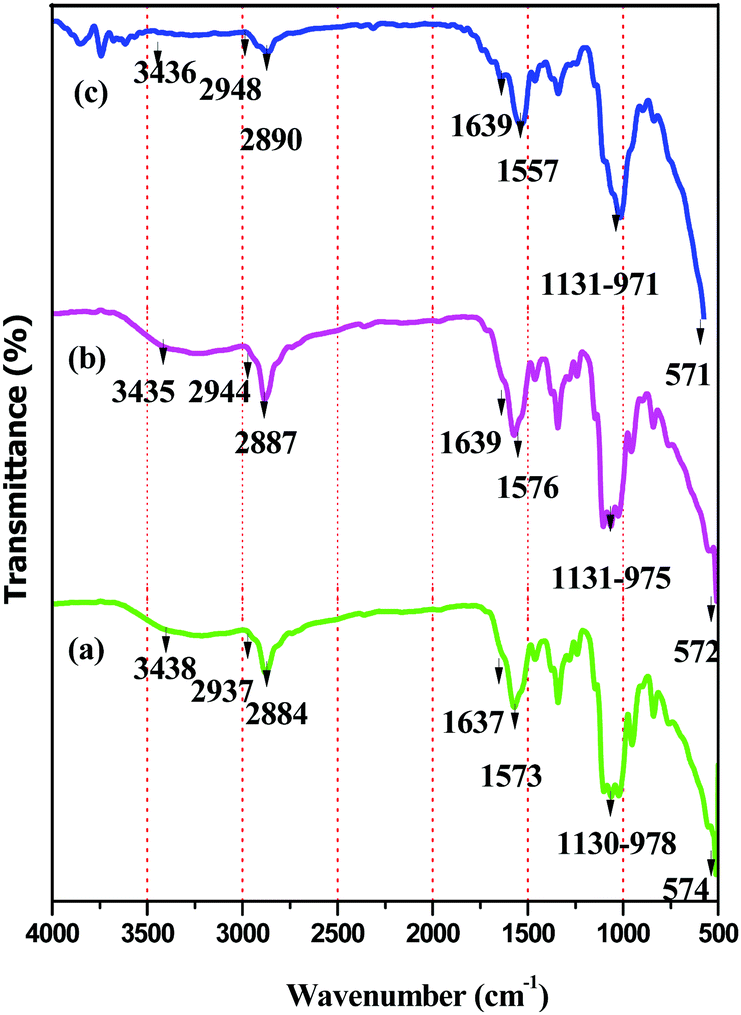 | ||
| Fig. 1 FT-IR spectra of [a] BPM I, [b] BPM II and [c] BPM III. | ||
| BPM I (cm−1) | BPM II (cm−1) | BPM III (cm−1) | Assignments |
|---|---|---|---|
| 3438 | 3435 | 3436 | OH stretching |
| 2937 | 2944 | 2948 | CH aliphatic stretching |
| 2884 | 2887 | 2890 | CH aliphatic stretching |
| 1637 | 1639 | 1639 | CO stretching (amide I) |
| 1573 | 1576 | 1557 | NH bending (amide I) |
| 1130–978 | 1131–975 | 1131–971 | P–O stretching for PO43− |
| 574 | 572 | 571 | Fe–O deformation |
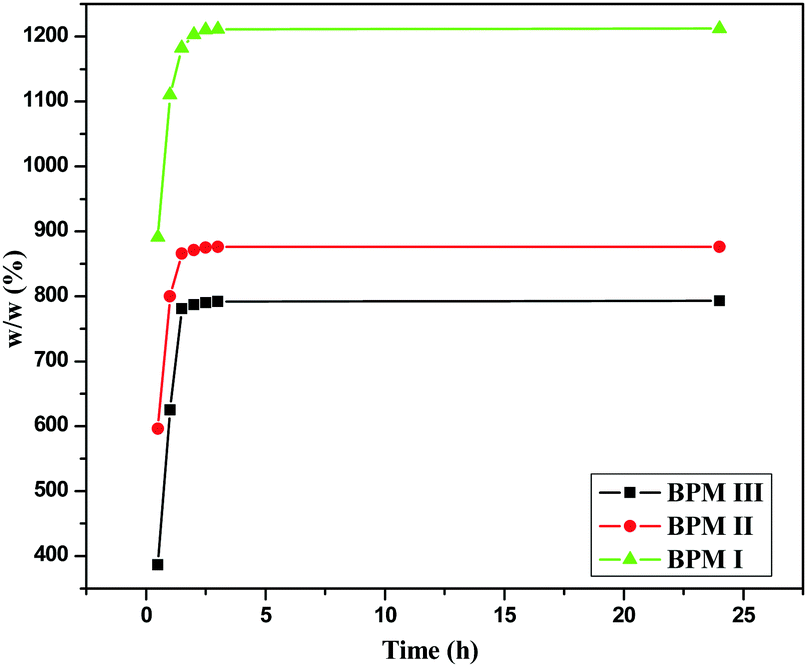 | ||
| Fig. 2 Water-uptake studies of BPM I–III. | ||
3.2 Microstructure of the composites
SEM study revealed that the size of nano-Fe3O4 was ∼20–60 nm (ESI 2†). In addition, the SEM images of BPM I–III (Fig. 3) showed highly porous structures that had two kinds of pores. The macroscopic pores had a size of approximately 20 to 50 μm (Fig. 3a) and were interconnected with each other (Fig. 3b), and many 1–2 μm micropores were also observed (Fig. 3b, d and f). It was previously reported that the ability to generate pore sizes of between about 20–50 μm is advantageous as this more closely mimics the natural cellular environment.62 The presence of Fe3O4 nanoparticles was confirmed by white spots that were found in the SEM micrographs (Fig. 3c and f). Thus, these macropores should be able to promote the formation of internal mineralized bone, and the micropores should be able to support nutrient delivery.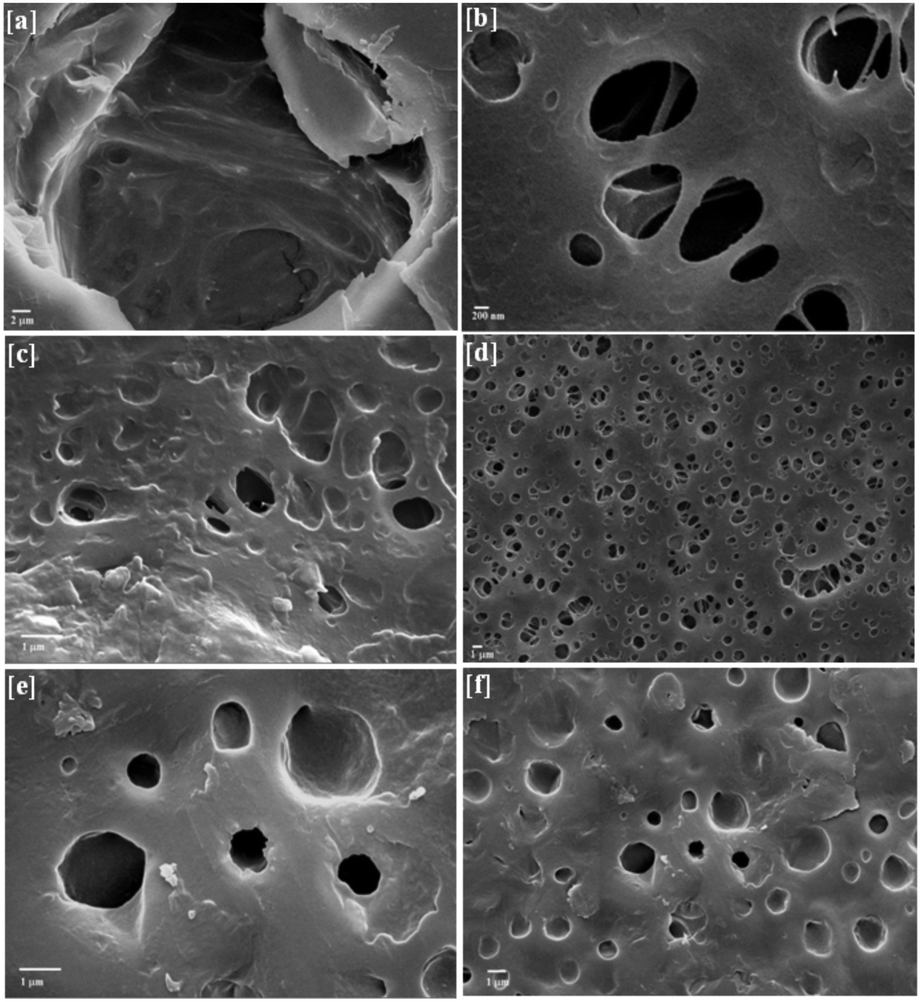 | ||
| Fig. 3 Scanning-electron micrographs of [a and b] BPM I, [c and d] BPM II and [e and f] BPM III. | ||
To confirm the size of nano-Fe3O4, TEM study was carried out (ESI 3†). It was observed that the size of nano-Fe3O4 was in the range of 20–60 nm, as observed in the SEM images.
3.3 Porosity and density measurement
The measured density and porosity of the BPMs is listed in Table 3. High porosities of 81.5%, 82.5% and 84.0% were observed for BPM I, BPM II and BPM III, respectively. The porosities observed for BPMs were in the range of cancellous bone (50–90% porosity).63 As porous structures of BPM I–III were obtained, the nanocomposites would have been able to favor cell adhesion and attachment with nutrient delivery to the site of tissue regeneration.64,65| Density (g cm−3) | Porosity (%) | |
|---|---|---|
| BPM I | 0.185 | 81.5 |
| BPM II | 0.175 | 82.5 |
| BPM III | 0.160 | 84.0 |
3.4 Mechanical properties
The mechanical properties were investigated for BPM I–III (Table 4). It was observed that an increase in the ratio of PEG led to an increase in the tensile strength of the nanocomposite. The highest tensile strength (18.31 MPa) was obtained for BPM I (40 wt% PEG) compared to BPM II (30 wt% PEG) and BPM III (20 wt% PEG) which had tensile strengths of 17.91 and 16.02 MPa, respectively, in the presence of a fixed amount of nano-Fe3O4. The total elongation at fracture of BPM I was 9.24%, higher than the total elongation at fracture of BPM II (8.28%) and BPM III (8.32%). BPM I also showed a much higher Young’s modulus compared to BPM II and BPM III. Recently, Shakir et al. prepared a CTS–PEG–HAP composite which has 6.7 MPa of compressive strength.66 A control sample (CBPM) was also prepared by us (ESI 4–6†) in the absence of nano-Fe3O4, by blending CTS (55 wt%), PEG (40 wt%) and nano-HAP (5 wt%) to observe any changes in mechanical behavior. It was observed that in the absence of nano-Fe3O4, the tensile strength of CBPM (8.15 MPa) was decreased significantly compared to BPM I–III (Table 4). Thus, the addition of nano-Fe3O4 into the CTS–PEG–HAP composite (BPM I–III) significantly enhanced the mechanical strength. It is well known that a highly porous scaffold is associated with low mechanical strength, which restricts its orthopedic application. However, for BPM I–III, we observed good mechanical strength along with high porosity. Notably, the tensile strength obtained for BPM I–III matches with the tensile strength of cancellous bone.63| Sample | Tensile strength (MPa) | Total elongation at fracture (%) | Young's modulus (MPa) |
|---|---|---|---|
| BPM I | 18.31 | 9.24 | 1462.43 |
| BPM II | 17.91 | 8.28 | 984.91 |
| BPM III | 16.02 | 8.32 | 617.19 |
| CBPM | 8.15 | 11.68 | 355.29 |
The mechanical properties of BPM I–III and CBPM were also measured under hydrated conditions after immersing the samples in phosphate buffer solution (PBS) at 37 °C for 24 hours (Table 5). In the hydrated state also, BPM I showed maximum tensile strength (16.16 MPa) compared to BPM II (14.5 MPa), BPM III (9.79 MPa) and CBPM (4.33 MPa). In the hydrated state, while BPM I showed 11.74% reduction in dry tensile strength, BPM II, BPM III and CBPM showed 23.5%, 38.8% and 46.87% reduction, respectively. The superior tensile strength obtained for BPM I could be due to the addition of a larger amount of PEG (40 wt%) into the nanocomposites, resulting in greater interaction between polar functional groups (–OH, –NH2 and –PO43−) present in CTS, PEG and HAP.
| Tensile strength (MPa) | Total elongation at fracture (%) | Young's modulus (MPa) | |
|---|---|---|---|
| BPM I | 16.16 | 7.58 | 1264 |
| BPM II | 14.5 | 6.50 | 550 |
| BPM III | 9.79 | 7.41 | 577 |
| CBPM | 4.33 | 14.3 | 239 |
3.5 Antimicrobial activities
The antimicrobial activities of BPM I–III were investigated and the results are shown in Fig. 4. We observed that the order of antimicrobial activity of the BPMs was BPM III > BPM II > BPM I.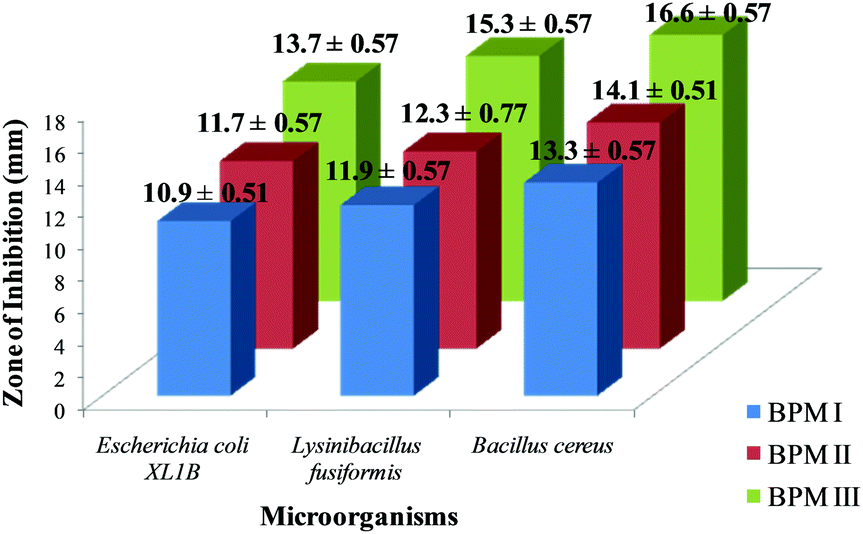 | ||
| Fig. 4 Zone of inhibition of BPM I–III against different bacterial strains. | ||
It was observed from the results that the cell-growth inhibition effect was more prominent in the case of Gram-positive bacteria strains than that of Gram-negative strains. Since CTS is a well known antimicrobial agent, cell inhibition was found to increase upon increasing the content of CTS in the nanocomposites. Also, Fe3O4 nanoparticles in the nanocomposites would provide additional support to reduce the risk of infection and prevent implant failure.67
As a fixed amount of nano-HAP–Fe3O4 was incorporated into BPM I–III, the maximum bactericidal activity (16.6 ± 0.57) was observed for the maximum CTS content (75%) film; BPM III against Bacillus cereus. For BPM I (55 wt% CTS) and BPM II (65 wt% CTs), the zones of inhibition for cell growth were 13.3 ± 0.57 and 14.1 ± 0.51, respectively, against Bacillus cereus. For Escherichia coli and Lysinibacillus fusiformis also, the maximum inhibition zones were observed for BPM III (13.7 ± 0.57 and 15.3 ± 0.77, respectively) compared to BPM I and BPM II. The images of the zones of inhibition of BPM I–III against Bacillus cereus, Escherichia coli and Lysinibacillus fusiformis are provided in ESI 7.†
3.6 Magnetization analysis
For magnetic characterization of BPM I, two types of measurement mode were used: temperature dependent measurements in a field (H = 200 Oe) and field dependent magnetization measurements at body temperature (T = 300 K). The temperature dependence of the magnetization of BPM I is shown in Fig. 5. The data was taken in a zero-field-cooling–field-heating sequence. The blocking temperature (TB) observed from these measurements was 212 K. Notably, addition transition at a lower temperature transition (6.05 K) was also observed. Initially, when the temperature rose from 2 K, the magnetization decreased, and then it reached a minimum point near the critical phase transition temperature (TC) of 6.05 K. After that, the magnetization started to increase up to the blocking temperature TB at 212 K.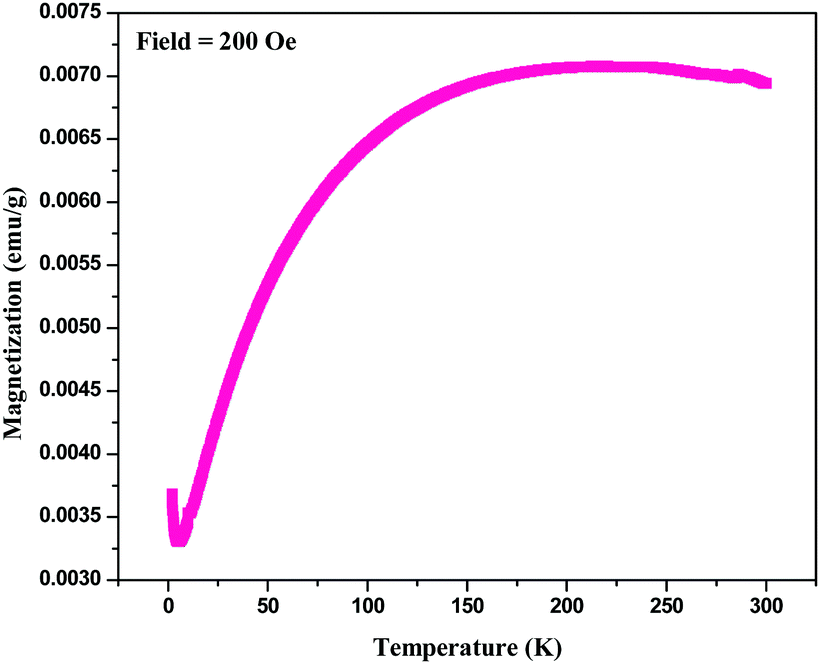 | ||
| Fig. 5 Temperature dependent magnetization of BPM I. | ||
Fig. 6 shows the field-dependent magnetization measurement of BPM I where magnetization (emu g−1) as a function of the applied field (Oe) is depicted with a confined field from −10000 to 10000 Oe.
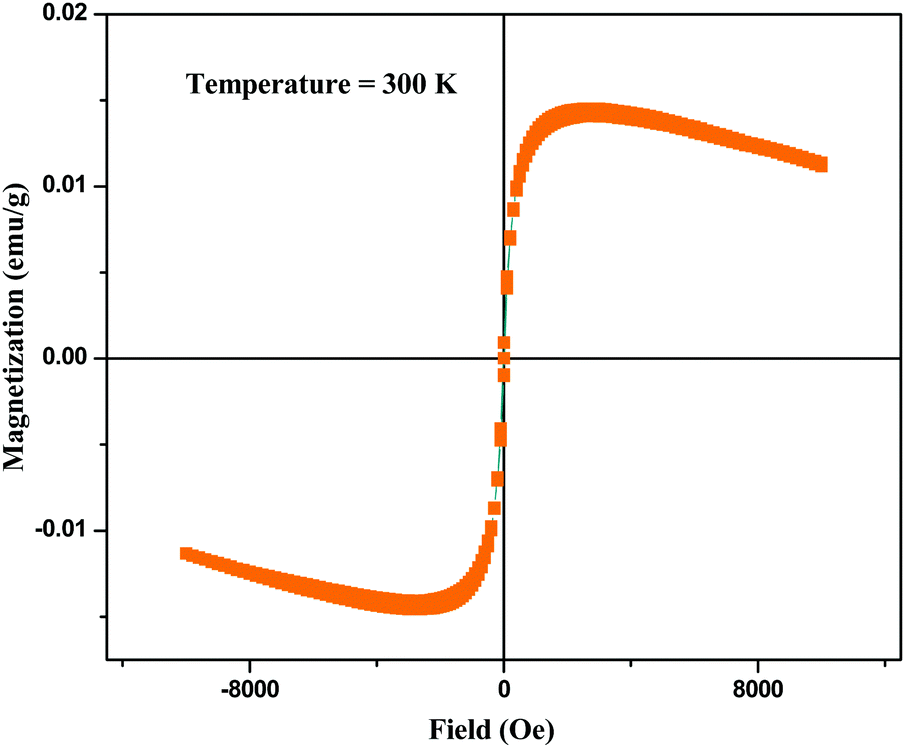 | ||
| Fig. 6 Field dependent magnetization of BPM I. | ||
The curves showed a low coercive field of 22 Oe at body temperature, typical of superparamagnetic behavior, which indicated that the presence of nano-Fe3O4 in HAP led to the superparamagnetism of the scaffolds.68 A diamagnetic background appeared, which was derived from the CTS–PEG and nano-HAP. Spontaneous remanent magnetization was obtained at 1 × 10−2 emu g−1.
These measurements confirmed the superparamagnetic character of the nanocomposite containing a very small weight percentage of nano-Fe3O4 (0.05 wt%), which was indicated by a very low coercive field and a distinct saturation magnetization. Therefore, these nanocomposites should be able to provide suitable magnetic therapy for bone tissue regeneration.
3.7 In vitro cell proliferation study (MTT assay)
In vitro cell proliferation and cytotoxicity studies of the prepared nanocomposites BPM I (with Fe3O4 NPs) and CBPM (without Fe3O4 NPs) were performed. MTT assay was done to check the toxicity of the prepared scaffolds (BPM I and CBPM). The cell viability of BPM I and CBPM was similar to that of the control (without the nanocomposite). As seen in Fig. 7, higher cell proliferation of human MG-63 cells was observed on BPM I, which contains CTS–PEG–HAP–Fe3O4, compared to CBPM (CTS–PEG–HAP) and the control (without nanocomposites). This result is in good accordance with previously reported literature indicating that the addition of HAP-coated Fe3O4 NPs increases osteoblast proliferation.69 The obtained results signified that the developed nanocomposites are beneficial for the osteoblast-like cells’ growth, attachment and proliferation. The biocompatibility assessment demonstrated that the modified nanocomposites had no negative effect on the osteoblast-like MG-63 cells and showed good cytocompatibility, having good prospects for bone regeneration.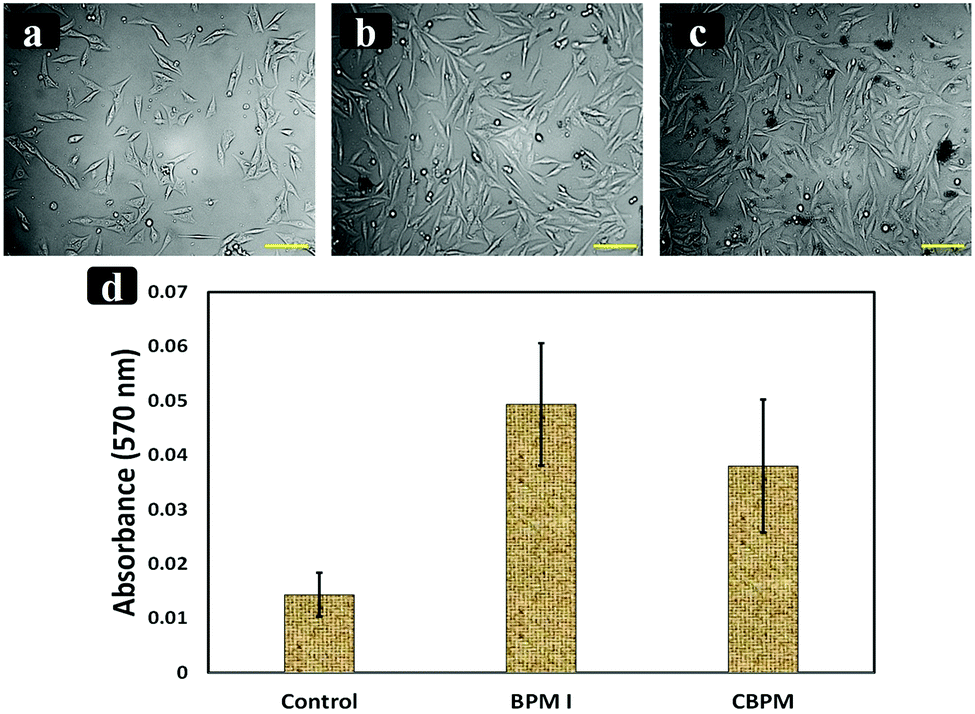 | ||
| Fig. 7 Inverted phase contrast micrographs of MG-63 cells grown over (a) non-coated (control), (b) BPM I coated and (c) CBPM coated tissue culture plate for 72 h. The scale bar measures 0.1 mm. All of the values are the mean ± SD of triplicates. (d) MTT assays for the proliferation of MG-63 cells cultured with scaffolds after 24 h, compared with the control (without scaffolds) under the same culture conditions. | ||
4 Conclusions
In conclusion, porous magnetic nanocomposites (BPM I–III) containing chitosan, polyethylene glycol and nano-hydroxyapatite–Fe3O4 were developed and characterized using Fourier transform infrared absorption spectra studies and X-ray diffraction studies. Scanning electron microscopy images revealed a highly interconnected macro- and micro-porous structure that could favor cell adhesion and attachment with nutrient delivery to the tissue regeneration site. The porous magnetic nanocomposites showed good mechanical and antimicrobial properties. It was observed from the water uptake study that with an increase in the hydrophilic polyethylene glycol content, the water uptake ability of the nanocomposites increased. Notably, the porosities and tensile strengths of the nanocomposite were in the range of cancellous bone. Additionally, these nanocomposites exhibited superparamagnetic properties, indicated by a very low coercive field even with a very small weight percentage of incorporated nano-Fe3O4. Finally, these nanocomposites supported human osteoblast-like MG-63 cells’ growth, attachment and proliferation with no negative effect on the MG-63 cells and they showed good cytocompatibility, suggesting positive prospects for bone regeneration. All of the above results suggest that these nanocomposites have great potential to be applied as bone tissue engineering materials.References
- M. Hirotaka, K. Toshihiro, N. Masayuki and U. Minoru, Sci. Technol. Adv. Mater., 2005, 6, 48 CrossRef.
- M. Freyman, I. V. Yannas and L. J. Gibson, Prog. Mater. Sci., 2001, 46, 273 CrossRef.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413 CrossRef CAS PubMed.
- J. L. Sang, J. L. Grace, J. W. Lee, A. Anthony and J. Y. James, Biomaterials, 2006, 27, 3466 CrossRef PubMed.
- Y. J. Wang, C. R. Yang, X. F. Chen and N. R. Zhao, Macromol. Mater. Eng., 2006, 291, 254 CrossRef CAS.
- C. Du, F. Z. Cui, W. Zhang, Q. L. Feng, X. D. Zhu and K. De Groot, J. Biomed. Mater. Res., Part A, 2000, 50, 518 CrossRef CAS.
- M. Kikuchi, S. Itoh, S. Ichinose, K. Shinomiya and J. Tanaka, Biomaterials, 2001, 22, 1705 CrossRef CAS PubMed.
- C. I. R. Boissard, P.-E. Bourban, A. E. Tami, M. Alini and D. Eglin, Acta Biomater., 2009, 5, 3316 CrossRef CAS PubMed.
- X. Cai, H. Tong, X. Shen, W. Chen, J. Yan and J. Hua, Acta Biomater., 2009, 5, 2693 CrossRef CAS PubMed.
- M. Farokhi, S. Sharifi, Y. Shafieyan, Z. Bagher, F. Mottaghitalab, A. Hatampoor, M. Imani and M. A. Shokrgozar, J. Biomed. Mater. Res., Part A, 2012, 100, 1051 CrossRef CAS PubMed.
- D. Huang, Y. Zuo, Q. Zou, Y. Wang, S. Gao, X. Wang, H. Liu and Y. Li, J. Biomed. Mater. Res., Part B, 2012, 100, 51 CrossRef PubMed.
- L. Jianguo, Z. Li, Z. Yi, W. Huanan, L. Jidong, Z. Qin and L. Yubao, J. Biomater. Appl., 2009, 24, 31 CrossRef PubMed.
- C. X. F. Lam, S. H. Teoh and D. W. Hutmacher, Polym. Int., 2007, 56, 718 CrossRef CAS.
- J. Russias, E. Saiz, R. K. Nalla and A. P. Tomsia, J. Mater. Sci., 2006, 41, 5127 CrossRef CAS.
- X. Xiao, R. Liu and Q. Huang, J. Mater. Sci.: Mater. Med., 2008, 19, 3429 CrossRef CAS PubMed.
- S. Xiongjun, R. Jianming, Z. Zhongchi, Z. Zhicheng and X. Lesi, Mater. Sci. Ed., 2009, 24, 566 Search PubMed.
- K. Ogawa, S. Hirano, T. Miyanishi, T. Yui and T. A. Watanabe, Macromolecules, 1984, 17, 973 CrossRef CAS.
- K. Okuyama, K. Noguchi, Y. Hanafusa, K. Osawa and K. Ogawa, Int. J. Biol. Macromol., 1999, 26, 285 CrossRef CAS PubMed.
- A. Sionkowska, Prog. Polym. Sci., 2011, 36, 1254 CrossRef CAS.
- N. Shanmugasundaram, P. Ravichandran, P. Reddy, N. N. Ramamurthy, S. Pal and K. P. Rao, Biomaterials, 2001, 22, 1943 CrossRef CAS PubMed.
- Y. Ding, J. Liu, H. Wang, G. Shen and R. Yu, Biomaterials, 2007, 28, 2147 CrossRef CAS PubMed.
- S. B. Bahrami, S. S. Kordestani, H. Mirzadeh and P. Mansoori, Iran. Polym. J., 2003, 12, 139 CAS.
- K. Desai and K. Kit, Polymer, 2008, 49, 4046 CrossRef CAS.
- L. V. Schueren, I. Steyaert, B. Schoenmaker and K. Clerck, Carbohydr. Polym., 2012, 88, 1221 CrossRef.
- M. Boissiere, P. Z. Meadows, R. Brayner, C. Helary, J. Livage and T. Coradin, J. Mater. Chem., 2006, 16, 1178 RSC.
- N. Pramanik, P. Bhargava, S. Alam and P. Pramanik, Polym. Compos., 2006, 27, 633 CrossRef CAS.
- M. Guiping, Y. Dongzhi, L. Qianzhu, W. Kemin, C. Binling, F. K. John and N. Jun, Carbohydr. Polym., 2010, 79, 620 CrossRef.
- X. Zhang, D. Yang and J. Nie, Int. J. Biol. Macromol., 2008, 43, 456 CrossRef CAS PubMed.
- M. Y. Santini, G. Rainaldi, A. Ferrante, P. L. Indovina, P. Vecchia and G. Donelli, Bioelectromagnetics, 2003, 24, 327 CrossRef CAS PubMed.
- K. J. McLeod and L. Collazo, Radiat. Res., 2000, 153, 706 CrossRef CAS PubMed.
- X. Y. Zhang, Y. Xue and Y. Zhang, Bioelectromagnetics, 2006, 27, 1 CrossRef CAS PubMed.
- K. F. Taylor, N. Inoue, B. Rafiee, J. E. Tis, K. A. McHale and E. Y. Chao, J. Orthop. Res., 2006, 24, 2 CrossRef PubMed.
- J. Meng, Y. Zhang and X. Qi, Nanoscale, 2010, 2, 2565 RSC.
- R. Houa, G. Zhanga, G. Dua, D. Zhanb and Y. Congb, Colloids Surf., B, 2013, 103, 318 CrossRef PubMed.
- J. M. Holzwarth and P. X. Ma, Biomaterials, 2011, 32, 9622 CrossRef CAS PubMed.
- D. Revi, V. P. Vineetha, J. Muhamed, A. Rajan and T. V. Anilkumar, J. Tissue Eng., 2013, 4, 2041731413518060 Search PubMed.
- H. J. Kim, I. H. Park, J. H. Kim, C. S. Cho and M. S. Kim, Tissue Eng. Regener. Med., 2013, 9, 63 CrossRef.
- C. G. Hadjipanayis, Small, 2008, 4, 1925 CrossRef CAS PubMed.
- R. B. Campbell, Nanomedicine, 2007, 2, 649 CrossRef CAS PubMed.
- S. Sun, H. Zeng and D. B. Robinson, J. Am. Chem. Soc., 2004, 126, 273 CrossRef CAS PubMed.
- Y. W. Jun, J. W. Seo and J. Cheon, Acc. Chem. Res., 2008, 41, 179 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS PubMed.
- M. Bañobre-López, Y. Piñeiro-Redondo and R. De Santis, J. Appl. Phys., 2011, 109, 07B313 Search PubMed.
- N. Bock, A. Riminucci and C. Dionigi, Acta Biomater., 2010, 6, 786 CrossRef CAS PubMed.
- R. De Santis, A. Gloria and T. Russo, J. Appl. Polym. Sci., 2011, 122, 3599 CrossRef CAS.
- J. Meng, B. Xiao, Y. Zhang, J. Liu and H. Xue, Sci. Rep., 2013, 3, 2655 Search PubMed.
- A. Tampieri, E. Landi and F. Valentini, Nanotechnology, 2011, 22, 015104 CrossRef CAS PubMed.
- A. Tampieri, T. D'Alessandro and M. Sandri, Acta Biomater., 2012, 8, 843 CrossRef CAS PubMed.
- A. Tampieri, M. Iafisco, M. Sandri, S. Panseri and C. Cunha, ACS Appl. Mater. Interfaces, 2014, 6, 15697 CAS.
- Y. Wei, X. Zhang and Y. Song, Biomed. Mater., 2011, 6, 055008 CrossRef PubMed.
- A. Bhowmick, R. Kumar, M. Das and P. P. Kundu, Adv. Polym. Technol., 2012, 33, 1391 Search PubMed.
- A. Bhowmick, A. Saha, N. Pramanik, S. Banerjee, M. Das and P. P. Kundu, RSC Adv., 2015, 5, 25437 RSC.
- H. Liu, J. Mao, K. Yao, G. Yang, L. Cui and Y. Cao, J. Biomater. Sci., Polym. Ed., 2004, 15, 25 CrossRef CAS PubMed.
- R. Hodgkinson and J. D. Curry, Proc. Inst. Mech. Eng., Part H, 2002, 204, 115 CrossRef.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337 CrossRef CAS PubMed.
- C. R. Nuttelman, M. A. Rice, A. E. Rydholm, C. N. Salinas, D. N. Shah and K. S. Anseth, Prog. Polym. Sci., 2008, 33, 167 CrossRef CAS PubMed.
- F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134 CrossRef CAS PubMed.
- T. M. Chang, Eur. J. Pharm. Biopharm., 1998, 45, 3 CrossRef CAS PubMed.
- M. Serra, C. Correia, R. Malpique, C. Brito, J. Jensen, P. Bjorquist, M. J. Carrondo and P. M. Alves, PLoS One, 2011, 6, e23212 CAS.
- Y. Man, P. Wang, Y. Guo, L. Xiang, Y. Yang, Y. Qu, P. Gong and L. Deng, Biomaterials, 2012, 33, 8802 CrossRef CAS PubMed.
- W. W. Thein-Han and R. D. K. Misra, Acta Biomater., 2009, 5, 1182 CrossRef CAS PubMed.
- A. Dagger, H. Lecomte and R. Moss, Scaffold with increased pore size, US Pat., US 2010/0143435 A1, Smith and Nephew Plc, London, 2010 Search PubMed.
- H. Liu and T. J. Webster, in Nanotechnology for the Regeneration of Hard and Soft Tissues, ed. T. J. Webster, World Scientific, 2007, pp. 1–52 Search PubMed.
- J. F. De Oliverira, P. F. De Aguiar, A. M. Rossi and G. A. Soares, Artif. Organs, 2003, 27, 406 CrossRef.
- E. Wintermantel, J. Mayer, J. Blum, K. L. Eckert, P. Lüscher and M. Mathey, Biomaterials, 1996, 17, 83 CrossRef CAS PubMed.
- M. Shakir, R. Jolly, M. S. Khan, N. Iram, T. K. Sharma and S. I. Al-Resayes, Polym. Adv. Technol., 2015, 26, 41 CrossRef CAS.
- S. Arokiyaraj, M. Saravanan, N. K. Udaya Prakash, M. V. Arasu, B. Vijayakumar and S. Vincent, Mater. Res. Bull., 2013, 48, 3323 CrossRef CAS.
- Y. Wu, W. Jiang and X. Wen, Biomed. Mater., 2010, 5, 15001 CrossRef PubMed.
- N. Tran and T. J. Webster, Acta Biomater., 2011, 7, 1298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Power XRD of BPM I–III; SEM of nano-Fe3O4; experimental procedure, FT-IR and XRD spectrum of the control sample (CBPM). See DOI: 10.1039/c6nj03358j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |