
A colloidal system of polythiophene-grafted edge-gold-coated silver nanoprisms with enhanced optical properties and stability†
Sugyeong
Jeong
,
Dongki
Lee
,
Joon Ki
Kim
and
Du-Jeon
Jang
*
Department of Chemistry, Seoul National University, NS60, Seoul 08826, Korea. E-mail: djjang@snu.ac.kr
First published on 16th November 2016
Abstract
Silver nanoprisms (Ag-PRs) have been epitaxially edge-coated with gold and subsequently covered fully with P3HT chains to fabricate poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms (P3HT@AuAg-PRs) which are miscible with organic solvents; each AuAg-PR with a typical edge length of 122 nm is surrounded by 4.4 × 103 P3HT chains. Both absorption and emission spectra have indicated that aggregated P3HT chains are bound on the surfaces of noble-metal nanoprisms. Picosecond emission kinetic profiles have suggested that the S1 relaxation time of P3HT in P3HT@AuAg-PRs is reduced due to energy transfer to AuAg-PRs. From nanosecond transient-absorption kinetic profiles, the reduced amplitude and the increased decay time of T1 excitons in P3HT@AuAg-PRs have been measured, indicating that neither the intersystem crossing of S1 into T1 nor the intersystem crossing of T1 into S0 occurs effectively in P3HT@AuAg-PRs owing to the stretched and rigid conformations of P3HT chains bound on the surfaces of AuAg-PRs. The surface-enhanced Raman scattering effect of P3HT@AuAg-PRs is as high as 4.6 because the sharp tips of AuAg-PRs which are maintained via gold coating serve as “hot spots”. The morphology deformation time of P3HT@AuAg-PRs in H2O2(aq) has been extended 48 times longer than that of Ag-PRs. Overall, our fabricated nanocomposites of P3HT@AuAg-PRs not only have feasible-process ability but also possess enhanced optical properties and high stability against oxidizing agents, extending the use of noble-metal nanoprisms for various optical applications.
1. Introduction
Colloidal noble-metal nanoparticles have been the focus of intense study for applications in catalysis, surface-enhanced Raman scattering (SERS), biosensors, and plasmonics because of their unique plasmonic properties, which originate from light absorption and scattering by localized surface-plasmon resonance (LSPR).1–5 LSPR, which is caused by the collective oscillation of electrons in the conduction band of a metal, can be excited by visible or near-infrared light and its resonance frequency is highly dependent on the shape and size of the metal.6 Among all the noble metals, silver is well known to exhibit superior optical properties such as high LSPR sensitivities and strong field enhancement.7,8 Thus, much effort has been devoted to synthesize silver nanoparticles with shapes varying from spheres to cubes, polyhedrons, plates, and wires, dramatically broadening the range of the LSPR wavelength.9 Particularly, the fabrication of silver nanoprisms (Ag-PRs) with controlled sizes has been explored extensively.10 Typically, Ag-PRs have three sharp vertices, which lead to strong near-field enhancement by the lightning-rod effect,11 making Ag-PRs useful for plasmonic applications such as SERS, metal-enhanced fluorescence (MEF), and plasmon-enhanced organic electronic devices.12 Despite such advantages, the utility of Ag-PRs is limited because the vertices of Ag-PRs are unstable and susceptible to truncation over time when they are exposed to water, halides, UV-irradiation, and heat, resulting in broadening and a blue shift of the LSPR band accompanied by the loss of surface-plasmon intensity.13,14A number of research studies have reported on the protection of Ag-PRs.15–17 For example, Du et al. utilized a protecting layer of TiO2 or SiO2 to coat the surface of Ag-PRs, protecting the edges of Ag-PRs from truncation over time.16 Xue et al. have incorporated 16-mercaptohexadecanoic acid for the stabilization of Ag-PRs against etching by silica coating.17 However, difficult control over the oxide-layer thickness and less sensitive LSPR of Ag-PRs limit the usefulness of the nanoprisms. Other groups have used gold to ensure the stability of Ag-PRs while maintaining shape asperities and optical properties such as high electric-field distribution around the nanoprisms.7,8 Coating Ag-PRs with less reactive noble metals such as gold has been investigated not only to protect against disturbance from the environment over a long period but also to retain the original plasmonic properties and surface characteristics of Ag-PRs. However, the dispersion of nanoprisms is still exclusively limited in aqueous media. Since many applications require nanoprisms to be blended or well dispersed into the host organic materials, it is crucial to make them miscible with organic solvents, thus, maximizing the utility of Ag-PRs.18,19
Recently, conjugated polymers have often been incorporated not only to protect metal nanoparticles from corrosion, chemical poisoning, and dissolution but also to make them miscible in organic media.20–27 Particularly, hybrid materials containing conjugated polymers and Ag-PRs are widely used for organic photovoltaic (OPV) devices having enhanced chemical and physical properties, which are not found in their single-component counterparts. For example, Kulkarni et al. have used Ag-PRs as optical antennas and scattering centers for OPV devices and investigated the optical properties of conjugated polymer-based cells having Ag-PRs under film conditions.28 Shahjamali et al. have also reported that the performance of an OPV cell was enhanced when the bulk heterojunction film of poly(3-hexylthiophene)/phenyl-C61-butyric acid methyl ester (P3HT/PCBM) was spin-coated on a glass substrate that had been immersed in a colloidal solution of edge-gold-coated silver nanoprisms.29 They have suggested that highly stable edge-gold-coated silver nanoprisms act as optical antennas to enhance incident-light absorption in the active layer of OPV devices.29 Likewise, interactions between the plasmon resonance of Ag-PRs and the exciton of conjugated polymers have been extensively explored by using various optical instruments; however, the study is often limited to the film state of these two materials so a profound study on the optical properties of such hybrid systems in the colloidal state is essential for the fundamental understanding of these nanocomposites.
In this paper, we have fabricated poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms (P3HT@AuAg-PRs) by blending thiol-functionalized P3HT (P3HT-SH) in tetrahydrofuran (THF) with edge-gold-coated silver nanoprisms (AuAg-PRs) dispersed in water (Fig. 1) and investigated their optical properties and stability in the colloidal state. As-prepared P3HT@AuAg-PRs have good miscibility with organic solvents and have enhanced optical properties such as a high SERS effect. The extremely extended deformation time of P3HT@AuAg-PRs in H2O2(aq) has demonstrated their highly improved stability, extending the use of noble-metal nanoprisms for various optical applications.
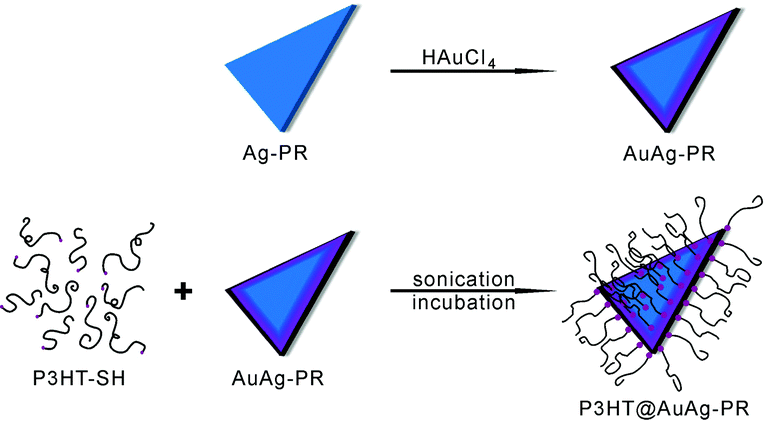 | ||
| Fig. 1 Scheme for the fabrication of a thiol-functionalized P3HT-grafted edge-gold-coated silver nanoprism (P3HT@AuAg-PR). | ||
2. Experimental
2.1 Chemicals and materials
Chemicals were used as received: gold chloride trihydrate (HAuCl4·3H2O), silver nitrate (AgNO3), L-ascorbic acid, sodium borohydride (NaBH4), and trisodium citrate dihydrate (TSC, Na3C6H5O7·2H2O) from Sigma-Aldrich; acetonitrile from Ducksan Pharmaceutical; 34% H2O2(aq) from Samchun Pure Chemicals; and THF from Daejung Chemicals & Metals. Ultrapure deionized water (>15 MΩ cm) from an Elga PURELAB option-S system was used throughout the experiments.2.2 Synthesis of Ag-PRs and AuAg-PRs
Ag-PRs were synthesized via a seed-mediated growth method30 with some modifications. Briefly, the silver seed solution was prepared by dissolving 12 mL of 75 mM TSC(aq), 0.20 mL of 0.10 M AgNO3(aq) and 0.42 mL of 34% H2O2(aq) in 200 mL of water. Then, 1.2 mL of freshly-made 0.10 M NaBH4(aq) was quickly injected to the above mixture and stirred vigorously for 2 h. Then, the prepared seed solution was aged for at least one week. The growth solution was prepared by adding 5.0 mL of acetonitrile(l), 75 μL of 0.10 M L-ascorbic acid(aq) and 0.20 mL of 75 mM TSC(aq) into 10 mL of water in an ice bath. In the growth solution, silver ions were reduced by L-ascorbic acid(aq), a weak reducing agent, that allows for silver seeds to grow slowly into well-defined AgPRs.31–33 Under vigorous stirring, 12 mL of the aged silver seed solution was added to the growth solution, followed by addition of 60 μL of 0.10 M AgNO3(aq) to initiate the growth of the silver seeds. After stirring for 5 min, the reaction mixture was aged without disturbance for an additional 55 min to produce Ag-PRs. AuAg-PRs were prepared by the drop-wise addition of 0.15 mL of 0.10 mM HAuCl4(aq) into 2.5 mL of the above Ag-PR colloid, which was then stirred for 15 min. The products, where the intended molar ratio of Au to Ag was 0.023, were centrifuged and washed with water at 9000 rpm for 10 min and dispersed in 0.16 mL of water.2.3 Synthesis of P3HT@AuAg-PRs
The detailed preparation and characterization of P3HT-SH having a Mn of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 565 g mol−1 have already been reported.20,34 P3HT@AuAg-PRs were synthesized under ambient conditions as described in detail elsewhere35 with little modification. First, 0.13 mL of concentrated 0.25 g L−1 P3HT-SH in THF was added to 40 μL of the AuAg-PR colloid. The intended molar ratio of P3HT-SH to Ag was calculated to be 55 in this case. Then, the mixed colloid was sonicated for 10 min and undisturbed for at least 24 h in the dark. The as-prepared P3HT@AuAg-PRs were centrifuged and washed twice with THF at 12000 rpm for 10 min and redispersed in 0.30 mL of THF. For synthesis of the P3HT-grafted silver nanoprisms (P3HT@Ag-PRs), the Ag-PR colloid was used instead of the AuAg-PR colloid with the same procedure.
565 g mol−1 have already been reported.20,34 P3HT@AuAg-PRs were synthesized under ambient conditions as described in detail elsewhere35 with little modification. First, 0.13 mL of concentrated 0.25 g L−1 P3HT-SH in THF was added to 40 μL of the AuAg-PR colloid. The intended molar ratio of P3HT-SH to Ag was calculated to be 55 in this case. Then, the mixed colloid was sonicated for 10 min and undisturbed for at least 24 h in the dark. The as-prepared P3HT@AuAg-PRs were centrifuged and washed twice with THF at 12000 rpm for 10 min and redispersed in 0.30 mL of THF. For synthesis of the P3HT-grafted silver nanoprisms (P3HT@Ag-PRs), the Ag-PR colloid was used instead of the AuAg-PR colloid with the same procedure.
2.4 Characterization
Transmission electron microscopy (TEM) images were obtained using a H-7600 Hitachi microscope while high-resolution TEM (HRTEM) and scanning TEM (STEM) images, as well as energy-dispersive X-ray (EDX) elemental maps and profiles, were obtained using a JEOL JEM-2100F microscope. X-ray photoelectron spectroscopy (XPS) spectra, as well as static and time-resolved spectra of absorption and emission, were obtained as reported.34 For the SERS measurement of P3HT@AuAg-PRs, a quartz plate (0.5 cm × 0.5 cm) was washed with acetone three times and dried under N2(g). Then, 10 μL of P3HT@AuAg-PRs dispersed in THF was dropped on the quartz plate and dried for at least 5 min for complete solvent evaporation. This process was repeated three times. SERS spectra were measured with excitation at 532 nm by using an OLYMBUS BX41 confocal microscope Raman system with a SLOC Laser GL532RA-100 DPSS laser. Each SERS spectrum was averaged over four measurements. The stability of each colloid in the presence of 0.82 mM H2O2(aq) was measured by monitoring absorption spectral changes at scheduled intervals.3. Results and discussion
The as-prepared P3HT@AuAg-PRs were facilely fabricated by blending P3HT-SH in THF with AuAg-PRs dispersed in water and these nanocomposites were further investigated for their optical properties and stability in the colloidal state. Fig. 2a, b and Fig. S1 (ESI†) show that the sharp tips of AuAg-PRs with an average edge length of 122 ± 10 nm having a typical thickness of ∼8 nm were maintained in water, whereas the tips of Ag-PRs became rounded with an average edge length of 79 ± 10 nm. According to a previous report,29 introducing HAuCl4 would induce the galvanic replacement with silver atoms preferentially around the edges of the Ag-PRs. The relative surface energies associated with the crystallographic planes of a face-centered cubic silver are in the order of γ110 > γ100 > γ111 and gold atoms initially replace the silver atoms at the edges which are mainly represented by (110) and (100) planes.7 Subsequently, the silver/gold alloyed structure at the edges of the Ag-PRs would start to form due to their close lattice-spacing values;7 the incorporated gold would make a great contribution to stabilize the sharp tips of the Ag-PRs. The TEM images in Fig. 2c and d show that the sizes of the Ag-PRs and AgAu-PRs have hardly changed after grafting P3HT chains on the surfaces of the nanoprisms. However, the TEM images in Fig. 2c and d also demonstrate that the tip rounding of P3HT@Ag-PRs has taken place to a certain degree in THF whereas the sharp tips of P3HT@AuAg-PRs have been still maintained. | ||
| Fig. 2 TEM images of (a) Ag-PRs, (b) AuAg-PRs, (c) P3HT@Ag-PRs, and (d) P3HT@AuAg-PRs. | ||
Furthermore, line-scanned EDX elemental profiles as well as EDX elemental maps have been measured to confirm the exact structure and compositions of P3HT@AgAu-PRs (Fig. 3). The STEM image and EDX elemental maps of Fig. 3a reveal that sulfur atoms, which represent the presence of P3HT chains, exist on the surface of AuAg-PRs with a full coverage, while gold atoms epitaxially coat the edges of Ag-PRs with a frame-like structure. The EDX elemental profiles in Fig. 3b indicate that silver atoms have gathered as a solid structure whereas gold or sulfur atoms, having two characteristic peaks, are present in a hollow or shell structure. Fig. 3b has also revealed that the line-scanned edge lengths of silver, gold, and sulfur are 38, 42, and 45 nm, respectively. Based on these observations, we suggest that Ag-PRs are epitaxially coated with gold atoms and that P3HT chains cover AuAg-PRs fully, making P3HT@AuAg-PRs miscible with organic solvents such as THF. The EDX analysis spectrum in Fig. S2 (ESI†) provides the specific molar percentages of silver, gold, and sulfur as 88.8%, 8.7%, and 2.5%, respectively. Considering this and the average size of the Ag-PRs, we have calculated that 4.4 × 103 P3HT chains surround one AuAg-PR, meaning that the molar mass of the P3HT@AuAg-PRs is 4.7 × 107 g mol−1. Then, the weight ratio of P3HT chains to AuAg-PRs can be calculated as 0.14 because the molar masses of AuAg-PRs and P3HT have been found to be 3.3 × 108 g mol−1 and 1.1 × 104, respectively.
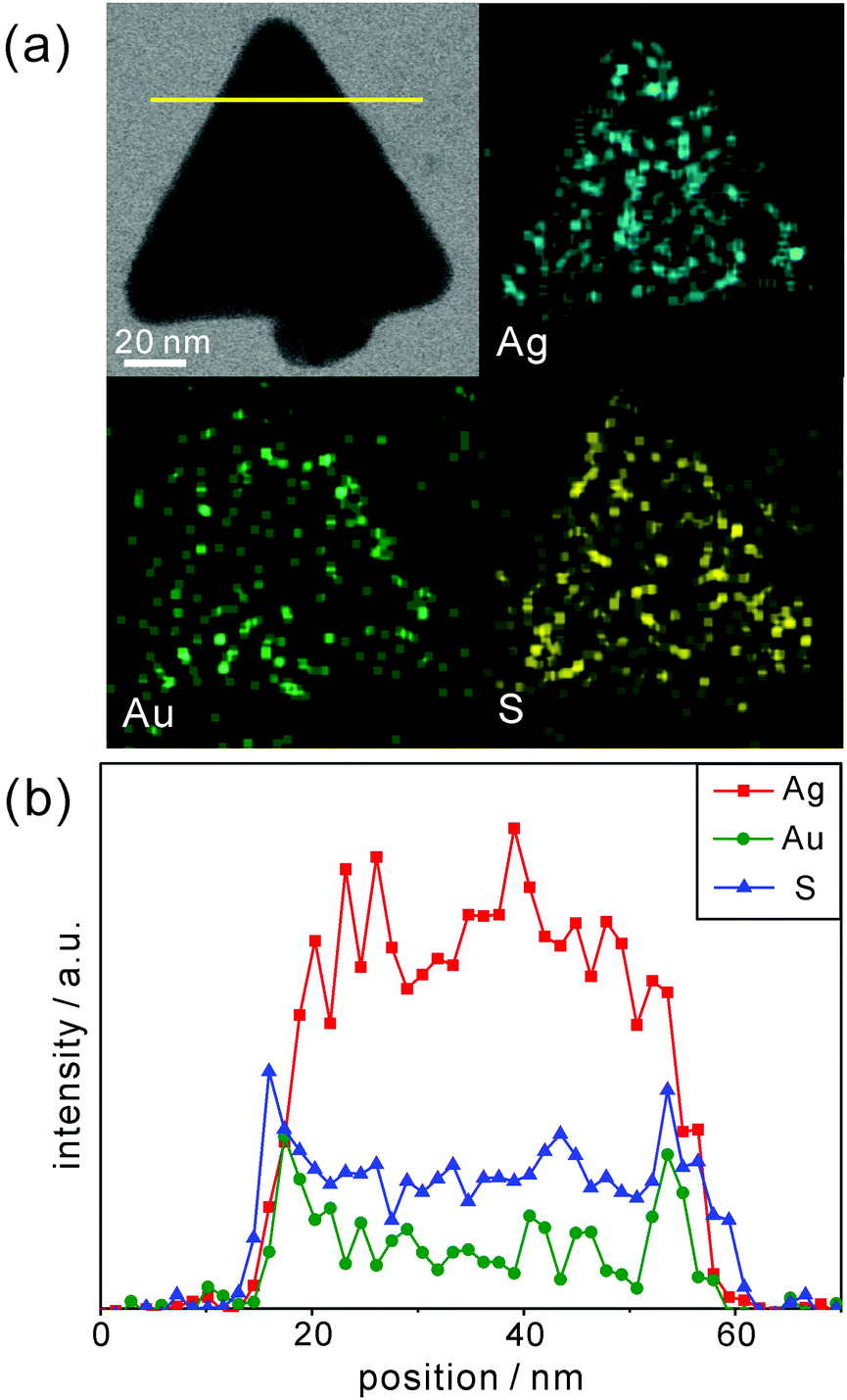 | ||
| Fig. 3 (a) STEM image and EDX maps of P3HT@AuAg-PRs. (b) EDX profiles of P3HT@AuAg-PRs. | ||
The XPS data in Fig. 4 can provide further significant information on the chemical bonds between the surfaces of the AuAg-PRs and the thiol groups of P3HT. The S 2p XPS spectrum of pristine P3HT in Fig. 4a has been resolved via curve deconvolution into two sets of doublet peaks having a splitting of 1.18 eV and an area ratio of 1:2 in agreement with the value from the theoretical spin–orbit effect (Table S1, ESI†). These two sets of peaks account for two types of sulfur species in pristine P3HT; a set of S 2p1/2 and S 2p3/2 corresponds to thiophene rings while the other set of S′ 2p1/2 and S′ 2p3/2 corresponds to free terminal thiol groups. As shown in Table S1 (ESI†), the peak positions of the S species of P3HT@AuAg-PRs are quite similar to those of pristine P3HT, supporting the fact that sulfur atoms in the thiophene rings of the P3HT chains have hardly been affected by forming nanocomposites with AuAg-PRs. However, the peak positions of the S′ species of P3HT@AuAg-PRs are shifted to higher binding energies by 0.12 eV than those of P3HT-SH, providing evidence that free terminal thiol groups are interacting with the surfaces of the nanoprisms. In particular, the positive shifts of the binding energies suggest that the sulfur atoms of the terminal thiol groups of the P3HT chains are mostly attached to gold because the electronegativity of gold (2.54) is higher than that of the hydrogen atom (2.20). Note that the electronegativity of silver (1.93) is even smaller than that of the hydrogen atom. This also supports the fact that the surfaces of Ag-PRs have been well-coated with gold and that most P3HT-SH chains bind chemically with gold. The abundance percentage of the S′ species of P3HT@AuAg-PRs related to free terminal thiol groups is 16.3% higher than that of pristine P3HT-SH, also demonstrating that the terminal thiol groups of P3HT molecules are chemically bound to the surfaces of AuAg-PRs.36
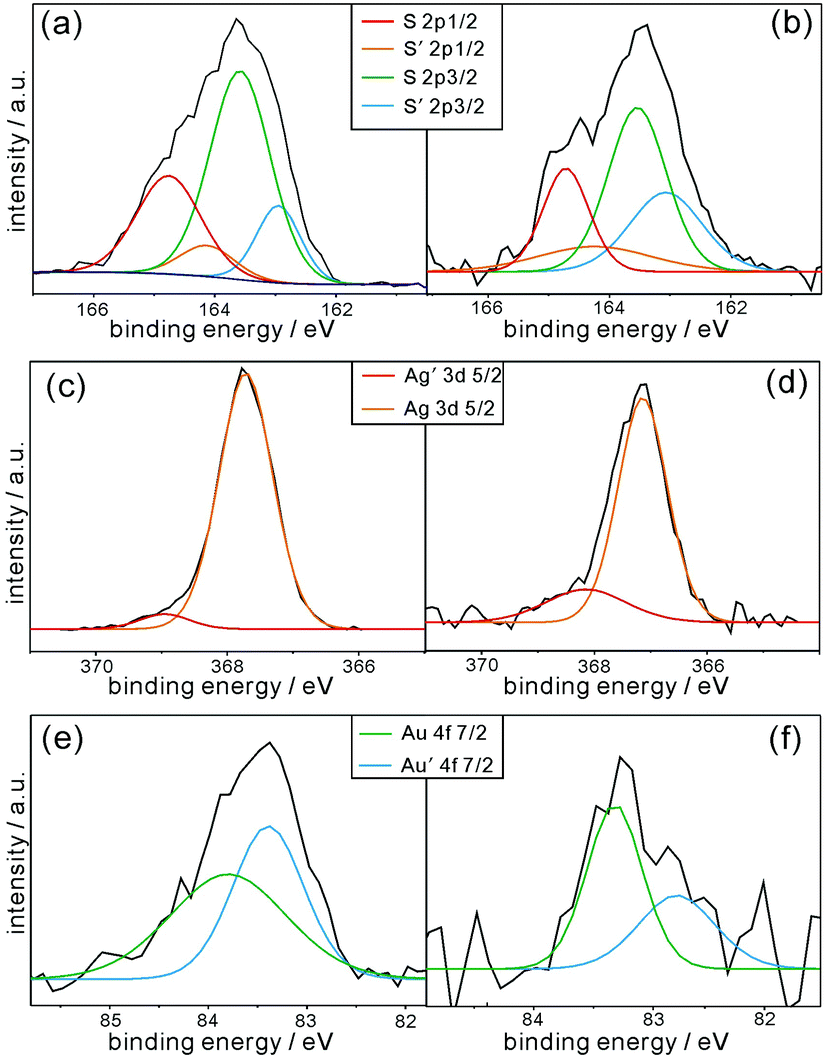 | ||
| Fig. 4 XPS spectra of (a) pristine P3HT-SH, (b, d and f) P3HT@AuAg-PRs, and (c and e) AuAg-PRs for sulfur (top), silver (middle), and gold (bottom). | ||
The individual Ag 3d and Au 4f XPS spectra of P3HT@AuAg-PRs and AuAg-PRs in Fig. 4 have also been deconvoluted into doublet peaks (Table S2, ESI†), suggesting that gold atoms as well as silver atoms have two different environments. In the case of silver, the binding energy of Ag′ 3d5/2 is higher than that of Ag 3d5/2 because Ag′ atoms are neighboring with gold atoms rather than other silver atoms. Note that the electronegativity of silver is lower than that of gold as we mentioned. This is also supported by the fact that the abundance percentage of Ag′ 3d5/2 (5.2%) is much smaller than that of Ag 3d5/2 (94.8%) because only a small amount of silver atoms are interacting with gold atoms on the edges of the nanoprisms. On the other hand, the gold atoms of Au′ 4f7/2 with a lower binding energy indicate that they are neighboring with silver atoms because of the aforementioned reason. Fig. 4 and Table S2 (ESI†) also indicate that the XPS spectra of both Ag 3d and Au 4f in P3HT@AuAg-PRs have been shifted from the respective ones in AuAg-PRs, also supporting the fact that the thiol terminal groups of P3HT molecules interact chemically with the surfaces of nanoprisms.
Fig. 5a shows that each absorption spectrum of pristine P3HT, P3HT@Ag-PRs, and P3HT@AuAg-PRs in THF contains a broad band at 450 nm due to the characteristic intrachain π–π transition of P3HT chains. In addition, each absorption spectrum of P3HT@Ag-PRs and P3HT@AuAg-PRs contains two characteristic bands at 556 and 606 nm. It has been reported that when a poly(3-alkylthiophene) is mixed with a poor solvent, the absorption bands in the range of 490–610 nm appear due to energy hopping between neighboring chains.37 Since the solvent of P3HT@Ag-PRs or P3HT@AuAg-PRs is the same as the solvent of pristine P3HT, the advent of these two bands can be assigned to P3HT interchain aggregates, resulting from the grafting of P3HT-SH chains on the surfaces of noble-metal nanoprisms. The absorption spectrum of Ag-PRs dispersed in water (Fig. S3, ESI†) exhibits peaks at 331, 544, and 804 nm corresponding to the out-of-plane quadrupole, in-plane quadrupole, and in-plane dipole modes, respectively, of surface-plasmon resonances (SPRs).19 A red shift by 18 nm of the dipole mode with 3 nm broadening has been observed after the incorporation of gold into Ag-PRs, indicating the galvanic replacement of silver with gold. The in-plane dipole modes of P3HT@Ag-PRs and P3HT@AuAg-PRs in THF have been observed as broad featureless bands. It is noteworthy that the absolute absorption strength of P3HT@AuAg-PRs shown in Fig. 5a is much less pronounced than that of AuAg-PRs shown in Fig. S3 (ESI†) due to the high density of grafted P3HT on the surfaces of the nanoprisms.
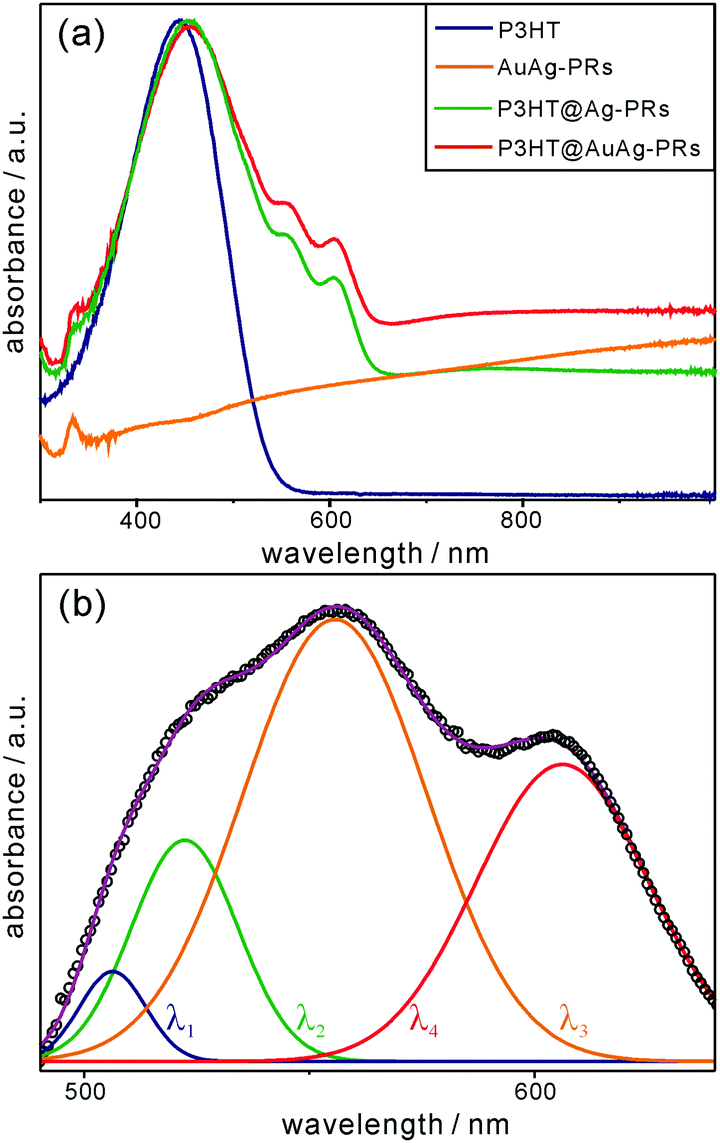 | ||
| Fig. 5 (a) Absorption spectra of P3HT, AuAg-PRs, P3HT@Ag-PRs, and P3HT@AgAu-PRs dispersed in THF. (b) Absorption difference spectrum from 490 nm to 640 nm (circles) obtained by subtracting the absorption spectra of pristine P3HT and AuAg-PRs from the absorption spectrum of P3HT@AuAg-PRs in (a), fitted with Gaussian curves of λ1, λ2, λ3, and λ4. | ||
For more profound observations of the chemical interactions between the thiol groups of P3HT-SH and the surfaces of the nanoprisms, we have attained the absorption difference spectrum by subtracting the absorption spectra of pristine P3HT and AuAg-PRs from the absorption spectrum of P3HT@AuAg-PRs (Fig. 5b). The appearance of absorption peaks from 470 nm to 650 nm has been assigned to P3HT aggregates having strong interchain and intrachain interactions.38Fig. 5b and Table S3 (ESI†) show that the subtracted absorption spectrum can be deconvoluted into four curves of λ1, λ2, λ3, and λ4 having maxima at 506, 522, 556, and 606 nm, respectively. According to a previous report,34 the λ1 curve originates from amorphous chains whereas the λ2, λ3, and λ4 curves correspond to the 0–2, 0–1, and 0–0 transitions of aggregated P3HT molecules, respectively. Thus, this also supports the fact that the thiol terminal groups of P3HT molecules bind chemically to the surfaces of nanoprisms, making P3HT@AuAg-PRs stable and miscible with THF.
As shown in Fig. 6, each emission spectrum of pristine P3HT, P3HT@Ag-PRs, and P3HT@AuAg-PRs contains three main parts: the peak around 550 nm ascribed to the 0–0 transition of free P3HT chains originating from the recombination of S1 excitons to the ground state, a shoulder around 620 nm, and a tail around 690 nm. The shoulder and the tail are attributed to transitions from the S1 state to the lowest vibrational level of the ground state (0–0) and the first-excited vibrational level of the ground state (0–1), respectively, of aggregated P3HT chains.39 Particularly, both the shoulder and the tail of P3HT@Ag-PRs and P3HT@AuAg-PRs are stronger than the respective ones of pristine P3HT, supporting the fact that the thiol groups of P3HT are bound on the surfaces of the nanoprisms, inducing P3HT chains aggregation as described in Fig. 1. Note that the shoulder as well as the tail in the emission spectra of P3HT@Ag-PRs and P3HT@AuAg-PRs is much more prominent with an excitation of 532 nm than with an excitation of 355 nm because the aggregated P3HT molecules are excited more efficiently by photons with a lower energy.39 This also supports the fact that P3HT chains are compactly surrounding the surfaces of noble-metal nanoprisms, as presented in Fig. 1. In addition, the shoulder and tail of P3HT@AuAg-PRs are more protrudent than the respective ones of P3HT@Ag-PRs. This implies that the thiol groups of P3HT molecules bind more strongly with gold surfaces than silver surfaces,40 making P3HT@AuAg-PRs more stable than P3HT@Ag-PRs (see below).
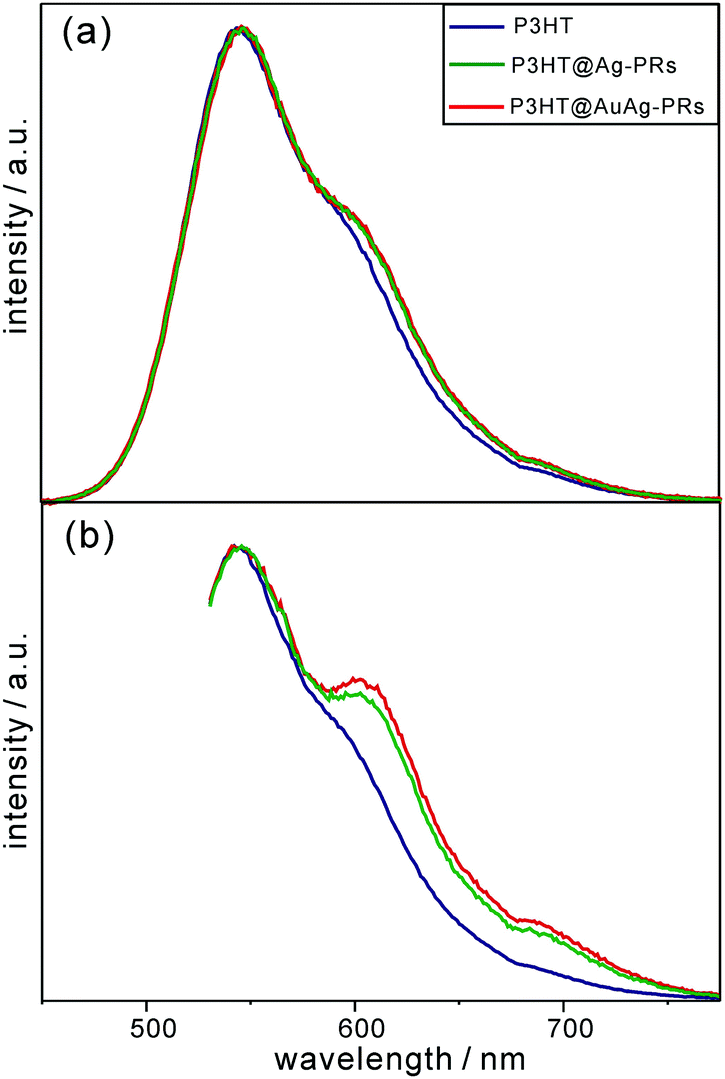 | ||
| Fig. 6 Fluorescence spectra of pristine P3HT, P3HT@Ag-PRs, and P3HT@AuAg-PRs in THF, after excitation at (a) 355 nm and (b) 532 nm. | ||
We have obtained the fluorescence and transient-absorption decay profiles of pristine P3HT and P3HT@AuAg-PRs in THF to understand the photophysical interactions between the excitons of P3HT and SPRs of AuAg-PRs. Fig. 7 shows that the picosecond emission decay kinetic profiles can be fitted into a single component, which arises from the relaxation of S1 excitons, of 450 ps for pristine P3HT and 437 ps for P3HT@AuAg-PRs (Table 1). In a previous paper,20 the relaxation time of S1 excitons in P3HT-coated gold nanoparticles has been found to be shorter than that in pristine P3HT owing to energy transfer from P3HT to gold nanoparticles. Accordingly, the shorter decay time of P3HT@AuAg-PRs demonstrates that energy transfer to the SPR states of AuAg-PRs reduces the relaxation time of S1 excitons in P3HT to some extent through chemical bonds between the thiol terminal groups of P3HT molecules and the noble-metal surfaces of AuAg-PRs.
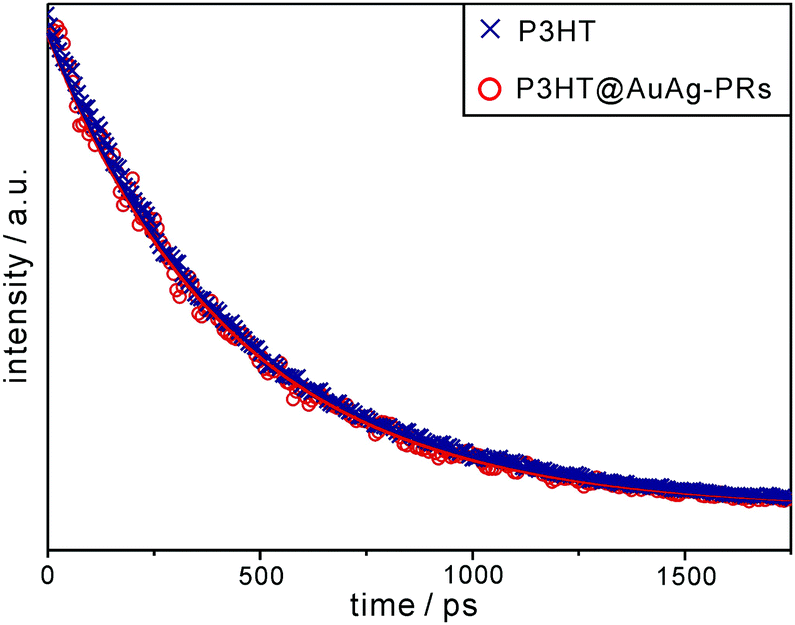 | ||
| Fig. 7 Picosecond emission decay profiles of P3HT and P3HT@AuAg-PRs in THF. Samples have been excited with 355 nm pulses and probed at 580 nm, and solid lines indicate best-fitted curves. | ||
Likewise, each nanosecond transient-absorption decay profile of pristine P3HT and P3HT@AuAg-PRs dispersed in THF (Fig. 8) has been fitted into a single component of 170 ns for P3HT and 180 ns for P3HT@AuAg-PRs (Table 1), showing the lifetime of T1 excitons. The amplitude of T1 absorption in P3HT@AuAg-PRs is less than that in pristine P3HT, revealing that T1 excitons in P3HT@AuAg-PRs are less populated due to the inefficient intersystem crossing of S1 excitons into the T1 state. In contrast to the obtained decay times of S1 excitons, T1 excitons in P3HT@AuAg-PRs decay slightly slower than those in pristine P3HT because the aggregated P3HT chains bound on the surfaces of AuAg-PRs have rigid and stretched conformations.31 Considering the reduced amplitude and the increased decay time of T1 excitons in P3HT@AuAg-PRs, we assert that neither the intersystem crossing of S1 into T1 nor the intersystem crossing of T1 into S0 takes place effectively in P3HT@AuAg-PRs owing to the high rigidity of the aggregated P3HT chains.
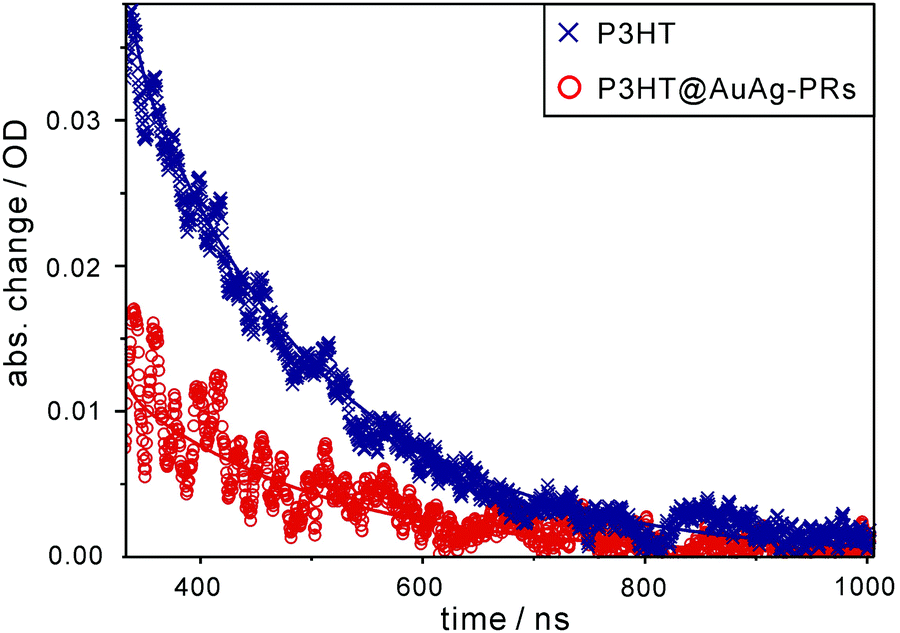 | ||
| Fig. 8 Nanosecond transient-absorption decay profiles of P3HT and P3HT@AuAg-PRs in THF. Samples have been excited at 355 nm and probed at 820 nm, and solid lines show best-fitted curves. | ||
As shown in Fig. 9, the Raman signals of pristine P3HT, P3HT@Ag-PRs, and P3HT@AuAg-PRs have been measured to examine the SERS effect of the nanoprisms. Previous approaches to the fabrication of a thin film with Ag-PRs have typically required the surface functionalization of a glass substrate with 3-aminopropyltrialkoxysilane and the subsequent adhesion of Ag-PRs; these processes are time-consuming and difficult to control.18,28 On the other hand, each of our Raman samples have been prepared by just dropping a colloid on a quartz slide since the surfaces of the nanoprisms have already been functionalized with P3HT. Under 532 nm excitation, which can resonant with the SPR absorption of the nanoprisms, the enhancement of Raman scattering was observed for both P3HT@Ag-PRs and P3HT@AuAg-PRs. The Raman intensity at 1464 cm−1, which arises from the in-phase stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C thiophene ring, of P3HT@Ag-PRs is higher by a factor of 1.5 than that of pristine P3HT due to the typical SERS effect of Ag-PRs.18 It has been reported that the sharp corners of silver nanoprisms have a relatively high field so that an enhanced plasmonic electric field would be generated within the gap between silver nanoprisms, enhancing the SERS effect.7 The Raman intensity of P3HT@AuAg-PRs is even 3.1 times higher than that of P3HT@Ag-PRs because the sharp tips of AuAg-PRs generate higher field enhancement than the edge-rounded Ag-PRs do, as expected from the lightning-rod effect. Thus, the protected sharp tips of AuAg-PRs act as “hot spots” to enhance the SERS effect as high as 4.6 (Fig. S4, ESI†).
C thiophene ring, of P3HT@Ag-PRs is higher by a factor of 1.5 than that of pristine P3HT due to the typical SERS effect of Ag-PRs.18 It has been reported that the sharp corners of silver nanoprisms have a relatively high field so that an enhanced plasmonic electric field would be generated within the gap between silver nanoprisms, enhancing the SERS effect.7 The Raman intensity of P3HT@AuAg-PRs is even 3.1 times higher than that of P3HT@Ag-PRs because the sharp tips of AuAg-PRs generate higher field enhancement than the edge-rounded Ag-PRs do, as expected from the lightning-rod effect. Thus, the protected sharp tips of AuAg-PRs act as “hot spots” to enhance the SERS effect as high as 4.6 (Fig. S4, ESI†).
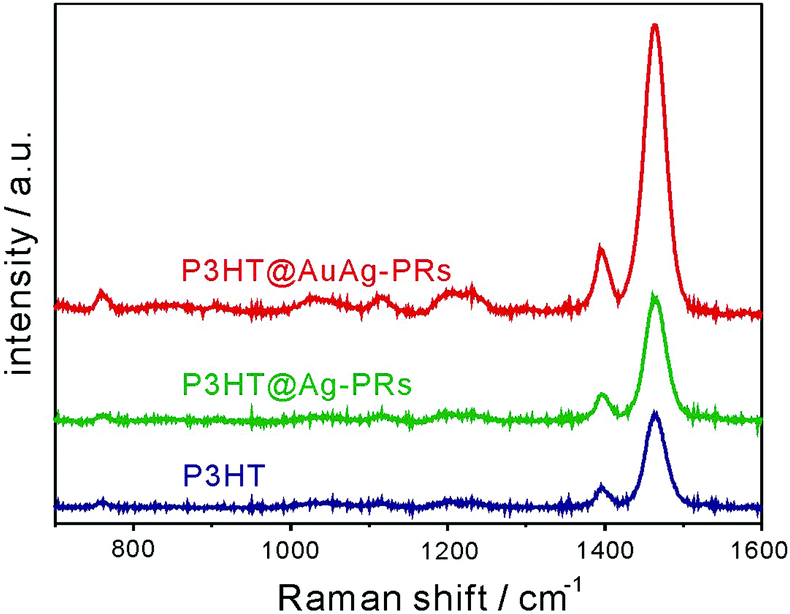 | ||
| Fig. 9 Raman spectra of pristine P3HT, P3HT@Ag-PRs, and P3HT@AuAg-PRs. The concentration of P3HT was fixed and all the samples were excited at 532 nm. | ||
Fig. 10a and b show that the SPR absorption of Ag-PRs has been blue-shifted enormously by 134 nm, whereas that of AuAg-PRs is blue-shifted by a small degree of 33 nm after treatment with H2O2. This demonstrates that gold coating on the edges of Ag-PRs has brought about structurally highly stable AuAg-PRs. Typically, gold coating on the surfaces of silver nanoparticles enhances structural stability because, compared with silver, gold is much more inert to oxidation.13,30Fig. 10c shows that the absorption of P3HT@AuAg-PRs in the range of 650–1000 nm, which arises from the SPR dipole mode of AuAg-PRs, has remained almost invariant after treatment with H2O2. It is noteworthy that P3HT@AuAg-PRs are even more stable than AuAg-PRs because P3HT chains act as a stabilizer, enhancing the structural stability extensively. To obtain a profound insight into the morphology deformation of nanoprisms over time, the absorption changes of nanoprisms after treatment with H2O2 have been fitted to the first-order kinetics; ln(A/A0) = −kt, where k is the deformation rate constant of nanoprisms, t is the monitored time, and A (A0) is the SPR absorbance of nanoprisms at time t(0). It is a pivotal point that the half lifetime (t1/2 = ln2k−1) for morphology deformation of P3HT@AuAg-PRs (290 min) has been extended 48 and 22 times longer than the half lifetimes of Ag-PRs (6 min) and AuAg-PRs (13 min), respectively (Table S4, ESI†). This suggests that the structural stability of Ag-PRs can be enhanced by gold coating on the edges of Ag-PRs, leading to AuAg-PRs whose stability can be further improved extensively by the protection of aggregated P3HT chains on the surfaces of AuAg-PRs. Thus, it has been shown that the edge gold coating and the subsequent thiol-functionalized P3HT grafting of Ag-PRs have enhanced not only the optical properties but also the feasible-process ability. Furthermore, the resultant P3HT@AuAg-PRs have enormously high stability against oxidizing agents, extending the use of noble-metal nanoprisms to various optical applications.
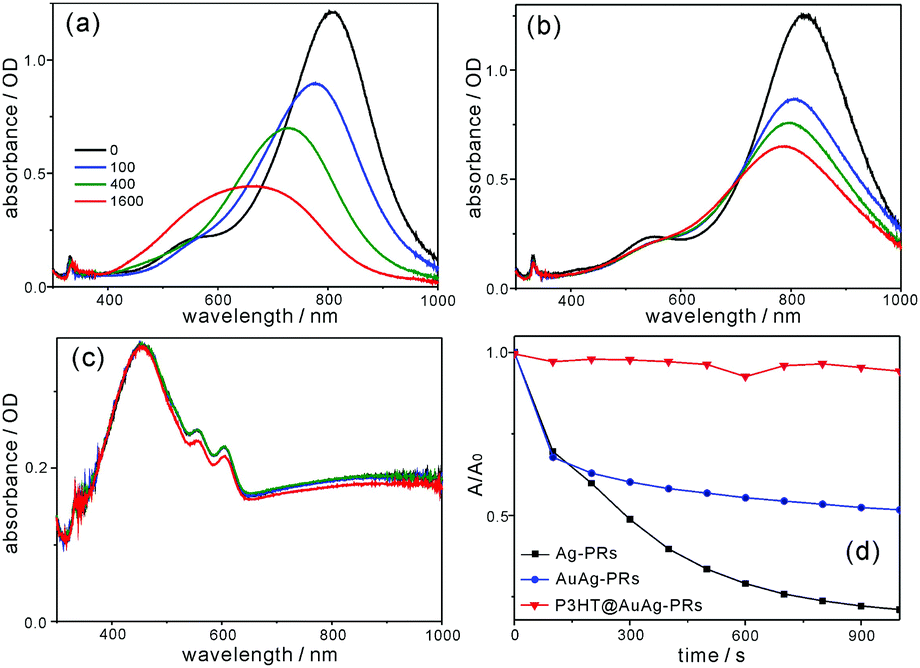 | ||
| Fig. 10 Absorption spectral evolutions of (a) Ag-PRs and (b) AuAg-PRs dispersed in water and (c) P3HT@AuAg-PRs dispersed in THF after treatment with H2O2. (d) A/A0vs. t at 804 nm for Ag-PRs, 823 nm for AuAg-PRs, and 998 nm for P3HT@AuAg-PRs. | ||
4. Conclusions
Silver nanoprisms have been epitaxially edge-coated with gold atoms and subsequently covered fully with poly(3-hexylthiophene) chains, leading to poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms which have good miscibility with organic solvents; individual AuAg-PRs with an average edge length of 122 nm and a typical thickness of 8 nm have been surrounded by 4.4 × 103 poly(3-hexylthiophene) chains. In the fluorescence spectra, the shoulder at 620 nm is much stronger in the poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms than in pristine poly(3-hexylthiophene), designating that the thiol groups of poly(3-hexylthiophene) molecules compactly surround the surfaces of the nanoprisms. In addition, picosecond emission kinetic profiles suggest that the S1 relaxation time of poly(3-hexylthiophene) in poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms is reduced due to energy transfer from poly(3-hexylthiophene) to edge-gold-coated silver nanoprisms. From the nanosecond transient-absorption kinetic profiles, the reduced amplitude and the increased decay time of T1 excitons in poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms have been measured, indicating that neither the intersystem crossing of S1 into T1 nor the intersystem crossing of T1 into S0 occurs effectively in poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms owing to the stretched and rigid conformations of P3HT chains bound on the surfaces of the edge-gold-coated silver nanoprisms. The SERS effect of the poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms is as high as 4.6 because the sharp tips of the edge-gold-coated silver nanoprisms which are maintained via gold coating serve as “hot spots”. The half lifetime for the morphology deformation of poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms in H2O2(aq) has been extended 48 times longer than that of the Ag-PRs. Overall, our fabricated poly(3-hexylthiophene)-grafted edge-gold-coated silver nanoprisms have not only feasible-process ability but also enhanced optical properties. Furthermore, the nanocomposites possess high stability against oxidizing agents, extending the use of noble-metal nanoprisms to various optical applications.Acknowledgements
This work was financially supported by research grants from the National Research Foundation of Korea (2015-051798 and 2014-057382).Notes and references
- B. Jana, S. Bhattacharyya and A. Patra, Phys. Chem. Chem. Phys., 2015, 17, 15392–15399 RSC.
- J. Lee and D.-J. Jang, J. Phys. Chem. C, 2016, 120, 4130–4138 CAS.
- J. Lee and D.-J. Jang, RSC Adv., 2015, 5, 64268–64273 RSC.
- M. A. Mahmoud, Langmuir, 2013, 29, 6253–6261 CrossRef CAS PubMed.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- E. Martinsson, M. M. Shahjamali, K. Enander, F. Boey, C. Xue, D. Aili and B. Liedberg, J. Phys. Chem. C, 2013, 117, 23148–23154 CAS.
- J. Sun, X. Wang, J. Liu, P. Wan, Q. Liao, F. Wang, L. Luo and X. Sun, RSC Adv., 2014, 4, 35263–35267 RSC.
- C. Gao, Z. Lu, Y. Liu, Q. Zhang, M. Chi, Q. Cheng and Y. Yin, Angew. Chem., Int. Ed., 2012, 51, 5629–5633 CrossRef CAS PubMed.
- Q. Zhang, J. Ge, T. Pham, J. Goebl, Y. Hu, Z. Lu and Y. Yin, Angew. Chem., Int. Ed., 2009, 121, 3568–3571 CrossRef.
- Q. Zhang, N. Li, J. Goebl, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 18931–18939 CrossRef CAS PubMed.
- Y. Gu, S. Kong, X. Diao, Y. Guo, K. Zhang and H. He, New J. Chem., 2016, 40, 7557–7563 RSC.
- M. Stavytska-Barba, M. Salvador, A. Kulkarni, D. S. Ginger and A. M. Kelley, J. Phys. Chem. C, 2011, 115, 20788–20794 CAS.
- D. Aherne, D. E. Charles, M. E. Brennan-Fournet, J. M. Kelly and Y. K. Gun’ko, Langmuir, 2009, 25, 10165–10173 CrossRef CAS PubMed.
- M.-S. Hsu, Y.-W. Cao, H.-W. Wang, Y.-S. Pan, B.-H. Lee and C.-L. Huang, ChemPhysChem, 2010, 11, 1742–1748 CrossRef CAS PubMed.
- B. Marta, E. Jakab, M. Potara, T. Simon, F. Imre-Lucaci, L. Barbu-Tudoran, O. Popescu and S. Astilean, Colloids Surf., A, 2014, 441, 77–83 CrossRef CAS.
- P. Du, P. Jing, D. Li, Y. Cao, Z. Liu and Z. Sun, Small, 2015, 11, 2454–2462 CrossRef CAS PubMed.
- C. Xue, X. Chen, S. J. Hurst and C. A. Mirkin, Adv. Mater., 2007, 19, 4071–4074 CrossRef CAS.
- L. Liu and T. L. Kelly, Langmuir, 2013, 29, 7052–7060 CrossRef CAS PubMed.
- B.-H. Lee, M.-S. Hsu, Y.-C. Hsu, C.-W. Lo and C.-L. Huang, J. Phys. Chem. C, 2010, 114, 6222–6227 CAS.
- D. Lee and D.-J. Jang, Polymer, 2014, 55, 5469–5476 CrossRef CAS.
- F. Monnaie, W. Brullot, T. Verbiest, J. De Winter, P. Gerbaux, A. Smeets and G. Koeckelberghs, Macromolecules, 2013, 46, 8500–8508 CrossRef CAS.
- R. C. Advincula, Dalton Trans., 2006, 2778–2784 RSC.
- E. Stratakis and E. Kymakis, Mater. Today, 2013, 16, 133–146 CrossRef CAS.
- L. Lu, Z. Luo, T. Xu and L. Yu, Nano Lett., 2012, 13, 59–64 CrossRef PubMed.
- D. H. Wang, D. Y. Kim, K. W. Choi, J. H. Seo, S. H. Im, J. H. Park, O. O. Park and A. J. Heeger, Angew. Chem., Int. Ed., 2011, 123, 5633–5637 CrossRef.
- S. Kazim, J. Pfleger, M. Procházka, D. Bondarev and J. Vohlídal, J. Colloid Interface Sci., 2011, 354, 611–619 CrossRef CAS PubMed.
- L. Scudiero, H. Wei and H. Eilers, ACS Appl. Mater. Interfaces, 2009, 1, 2721–2728 CAS.
- A. P. Kulkarni, K. M. Noone, K. Munechika, S. R. Guyer and D. S. Ginger, Nano Lett., 2010, 10, 1501–1505 CrossRef CAS PubMed.
- M. M. Shahjamali, M. Salvador, M. Bosman, D. S. Ginger and C. Xue, J. Phys. Chem. C, 2014, 118, 12459–12468 CAS.
- X. Liu, L. Li, Y. Yang, Y. Yin and C. Gao, Nanoscale, 2014, 6, 4513–4516 RSC.
- N. R. Jana, T. K. Sau and T. Pal, J. Phys. Chem. B, 1999, 103, 115–121 CrossRef CAS.
- K. Mallik, M. Mandal, N. Pradhan and T. Pal, Nano Lett., 2001, 1, 319–322 CrossRef CAS.
- N. R. Janafi, Z. L. Wangl and T. K. Sau, Curr. Sci., 2000, 79, 1367–1369 Search PubMed.
- D. Lee, S. Jeong, J.-H. Park, S. Y. Park and D.-J. Jang, J. Mater. Sci., 2016, 51, 9669–9678 CrossRef CAS.
- Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS PubMed.
- M. Turner, O. P. H. Vaughan, G. Kyriakou, D. J. Watson, L. J. Scherer, G. J. E. Davidson, J. K. M. Sanders and R. M. Lambert, J. Am. Chem. Soc., 2009, 131, 1910–1914 CrossRef CAS PubMed.
- M. He, L. Zhao, J. Wang, W. Han, Y. Yang, F. Qiu and Z. Lin, ACS Nano, 2010, 4, 3241–3247 CrossRef CAS PubMed.
- B. Ferreira, P. F. da Silva, J. S. S. de Melo, J. Pina and A. Macanita, J. Phys. Chem. B, 2012, 116, 2347–2355 CrossRef CAS PubMed.
- D. Lee, J. K. Kim and D.-J. Jang, Polymer, 2016, 99, 122–129 CrossRef CAS.
- H. Sellers, A. Ulman, Y. Shnidman and J. E. Eilers, J. Am. Chem. Soc., 1993, 115, 9389–9401 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures (Fig. S1–S3) and tables (Tables S1–S4). See DOI: 10.1039/c6nj02868c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |