
Fe/C and FeMo/C hybrid materials for the biphasic oxidation of fuel contaminants
Raquel V.
Mambrini
a,
Camila Z.
Maia
b,
José D.
Ardisson
c,
Patterson Patrício
de Souza
a and
Flávia C. C.
Moura
*b
aDepartamento de Química, Centro Federal de Educação Tecnológica de Minas Gerais, Av. Amazonas 5253 – Nova Suíça, Belo Horizonte, MG CEP 30421-169, Brazil
bDepartamento de Química, Universidade Federal de Minas Gerais, Belo Horizonte, MG 31270-901, Brazil. E-mail: flaviamoura@ufmg.br; Fax: +55-31-34095700; Tel: +55-31-34097556
cLaboratório de Física Aplicada, Centro de Desenvolvimento da Tecnologia Nuclear – CDTN, Belo Horizonte, MG 31270-901, Brazil
First published on 11th November 2016
Abstract
In the present work, hybrid magnetic nanoparticles based on Fe and FeMo coated with carbon nanostructures (Fe/C and FeMo/C) were produced by catalytic chemical vapor deposition at different temperatures using ethanol as the carbon source. Scanning electron microscopy, thermal analysis, Raman spectroscopy, X-ray diffraction, and Mössbauer spectroscopy analyses suggested that hematite was reduced by ethanol at 700 and 900 °C, producing carbon nanostructures such as nanofibers and nanotubes. The Fe/C and FeMo/C hybrid composites showed remarkable magnetic and amphiphilic properties. The composites were successfully used to oxidize sulfur and nitrogen contaminants present in fuels in a biphasic system.
1. Introduction
The importance of the removal of sulfur and nitrogen compounds from liquid fuels (desulfurization and denitrogenation, respectively) has gradually grown due to many governments putting specifications on the maximum sulfur and nitrogen contents permitted in the crude oil, the increased concentration of these compounds in the crude oil,1,2 and the increased demand for cleaner fuel. Organic sulfur molecules present in petroleum are mercaptans, represented by the general formula S–H, and heterocyclic compounds, such as sulfides, polysulfides, benzothiophenes and their derivatives.3 However, organic nitrogen molecules found in petroleum are mostly aromatic heterocyclic compounds in a neutral form, such as pyrroles, or in a basic form, such as pyridines and quinolines.4,5 The combustion of these compounds leads to the emission of SOx and NOx, which can cause critical health problems and are precursors of acid rain. Moreover, the presence of these contaminants in fuels provokes corrosion of the refining equipment and combustion engines.6,7 Hence, in recent decades, research into clean fuels, including desulfurization and denitrogenation, has become an important subject for environmental catalysis studies worldwide.8,9Hydrodesulfurization (HDS) is the main desulfurization technology10 and has been widely used to remove the majority of sulfur-containing compounds from fossil fuel. This technology is based on the removal of sulfur as hydrogen sulfide from the fuels using various types of catalysts at high temperatures (320–380 °C) and very high hydrogen pressure (30–70 atm).9 Among the currently used processes for removing nitrogen compounds, the featured ones are the catalytic process of hydrodenitrogenation (HDN) and adsorption by activated carbon with a high specific surface area and porosity, which is capable of adsorbing 15 to 19 mg of nitrogen per gram of the adsorbent.11,12
Although, the HDS process is highly efficient in removing aliphatic and acyclic thiophenic compounds, refractory benzothiophenic compounds, e.g. benzothiophene (BT), dibenzothiophene (DBT) and their alkyl derivatives, especially 4,6-dimethyldibenzothiophene (4,6-DMDBT), are very difficult to eliminate, probably due to their steric hindrance effect11,13 and high electron density around sulfur atom.14,15
In the HDN process, the use of a metal oxide/sulfide catalyst and zeolites has been promising.16,17 However, the unsaturated aromatic compounds (pyridine, quinoline, indole and carbazole), such as the ones present in the HDS process, are difficult to convert on the catalyst surface because of their steric hindrance.18 Moreover, this method is highly expensive because it requires high operating temperatures and pressures. To overcome these problems, many researchers are carrying out investigations on the environmentally benign, energy saving and more effective methods.
Currently, the legislation on the sulfur contaminant concentration limits in fuels is approximately 15 ppm in many developed countries. Moreover, the commonly used process are either expensive or less effective at achieving these levels. Therefore, to reach these specifications, a new desulfurization approach is needed. Among these new methods, oxidative desulfurization (ODS) is considered to be one of the most promising methods for the deep desulfurization of fuel oil.7
The ODS process consists of the oxidation of organosulfur compounds to obtain highly polar benzothiophenic sulfones and sulfoxides, which can be easily removed by extraction or adsorption. The ODS process has several advantages compared with the traditional HDS process, such as mild conditions (ambient pressure and relatively low temperatures) and high selectivity. Most of the oxidative desulfurization studies have focused on using oxidants such as peroxyacetic acid,19tert-butyl hydroperoxide,20,21 hydrogen peroxide22–26 and others.27–29
Oxidants such as hydrogen peroxide (H2O2) and organic carboxylic acids are commonly used in the ODS process. However, these reactants are soluble in polar solvents, whereas fuels with sulfur and nitrogen contaminants are in the non-polar oily phase, forming a biphasic liquid system; therefore, the oxidation of the contaminants is low because the interface between the two phases (oil/water) is limited, making the oxidation process very difficult.30
The presence of an efficient catalyst in the oxidation step of the ODS and ODN procedures is crucial for the success of the process. Therefore, our research group has developed hybrid catalysts for biphasic reactions in several applications.31–35 Hybrid materials are very interesting for biphasic reactions since they have an amphiphilic character, i.e. they are able to interact with aqueous and oily phases simultaneously, increasing the interface between the phases to form an emulsion.36
In this context, the objective in the present study was to explore a novel amphiphilic material and use it as a catalyst in the biphasic oxidation of S- and N-containing contaminants in the petroleum industry. The novel amphiphilic catalysts were based on iron or iron/molybdenum coated with carbon nanotubes and filaments produced by a CVD process using ethanol. Iron particles as catalysts play an important role in the reactions as they are very active in the CVD process to grow carbon filaments and are very active to promote H2O2 decomposition in heterogeneous Fenton's reaction,37–39 which makes them interesting to be used in the oxidation processes. Moreover, iron species can exhibit magnetic properties and can easily be removed from the reaction, promoting phase separation. Thus, after the oxidation process, the materials can be attracted by an external magnetic field and thereby promote the breaking of the emulsion, i.e., affect phase separation. In addition, the materials can easily be removed, allowing their reuse in the subsequent reactions.
2. Experimental
The hybrid composites were prepared by using hematite (Fe2O3) synthesized by heating [Fe(NO3)3]·9H2O at 400 °C for 3 hours. Molybdenum was impregnated on the surface of synthetic hematite in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Fe/Mo. Then, 0.713 g hematite was dispersed in a 100 mL solution containing 2.24 g (NH4)6Mo7O24·4H2O in water under stirring at 60 °C. After the water evaporation, the dried solid was calcined for 3 h at 400 °C, and the resulting solid was named as FeMo oxide.
:1 Fe/Mo. Then, 0.713 g hematite was dispersed in a 100 mL solution containing 2.24 g (NH4)6Mo7O24·4H2O in water under stirring at 60 °C. After the water evaporation, the dried solid was calcined for 3 h at 400 °C, and the resulting solid was named as FeMo oxide.
For the hydrophobization of hematite (in this case named Fe) or FeMo oxide surfaces, they were submitted to chemical vapor deposition (CVD) using ethanol as the carbon source. In the CVD process, a volatile carbon source is decomposed in the presence of these catalysts, depositing solid carbonaceous material on their surface.15–19 Here, about 200 mg of Fe oxide was placed in a 100 mm diameter quartz tube, heated at 10 °C min−1 with ethanol (80 mL min−1 N2 carrier gas flow) at up to 700 and 900 °C and kept at the final temperature for 1 h, followed by cooling down under a nitrogen flow. The materials were named according to the reaction temperature, i.e., Fe/C700 and Fe/C900 (CVD at 700 °C and 900 °C, respectively). The same process was repeated using FeMo oxides as the precursor, and the materials were again named according to the reaction temperature, i.e., FeMo/C700 and FeMo/900 (CVD at 700 °C and 900 °C, respectively).
The Fe/carbon (Fe/C) and FeMo/carbon (FeMo/C) composites were characterized by powder XRD in a Rigaku model Geigerflex using Cu Kα radiation scanning from 2 to 75° at a scan rate of 4° min−1. Scanning electron microscopy (SEM) analyses were carried out by a Jeol 840A and a Quanta 200 ESEM-FEG from FEI, and EDS microanalysis by a JEOL JXA-8900 RL at 15 kV. Raman spectra were obtained by using a SENTERRA Bruker spectrometer. Experimental data were obtained with the 633 nm line of a helium-neon laser (effective power of 6 mW at the sample surface) as the excitation source, diffraction gratings of 600 and 1800 grooves per mm, Peltier-cooled CCD detector, confocal Olympus microscope (100× objective), and an experimental resolution of typically 1 cm−1 for 10 accumulations of 30 s. Thermal analyses were carried out using a DTG60H Shimadzu, in which the samples were subjected to heating from 25 °C up to 900 °C at 10 °C min−1 rate in an air flow of 50 mL min−1.
Emulsion formation tests were performed using 4 mL of oily solution (80% mineral oil, 20% toluene and 200 ppm of Sudan IV dye), 4 mL of distilled water and 15 mg of the material. The purpose of using Sudan IV dye was simply to better identify the phases by optical microscopy. The system was vigorously stirred and optic microscopy images (Cole Parmer Instrument, 41500-50) were obtained. The oxidation was carried out for dibenzothiophene (DBT) and quinoline (QN) as model molecules for sulfur- and nitrogen-containing molecules, respectively. Initially, stable emulsions were formed and characterized by optical microscopy. These emulsions naturally broke (phases separated) within 15 min. However, in the presence of an external magnetic field, the emulsion can be separated in 2 min. Sulfur or nitrogen oxidation reactions were performed at 25 °C with stirring and in natural pH using 5 mL of organic phase (50 mg L−1 S of DBT or N of QN in cyclohexane), 1 mL of oxidant aqueous phase (H2O2 30%) and 15 mg of the catalyst.
The oxidation reactions of the sulfur compounds were followed over time using a gas chromatograph coupled to a mass spectrometer (GC-MS Shimadzu QP2010 – Plus with electron impact ionization at 70 eV and a non-polar capillary column RTX - 5MS). The oxidation of quinoline was analysed by a UV spectrometer (Shimadzu) and the intermediate species formed during the reaction were identified by using an ION-TRAP LCQ Fleet (Thermo Scientific, San Jose, CA) in the positive ion mode. The reaction samples were analyzed by introducing aliquots into the ESI-MS source using a syringe pump at 15 L min−1.
At the end of the reactions, metal leaching was measured by atomic absorption spectrometry using the AAS instrument (Hitachi-Z8200 equipment). Moreover, the catalysts were removed from the system with the assistance of a magnet and subsequently reused in three catalytic cycles.
3. Results and discussion
3.1. Preparation and characterization of the Fe/C and FeMo/C hybrid composites
The Fe/C and FeMo/C composites were characterized by XRD, TG, SEM and Raman spectroscopy to obtain the information about their metallic and carbon phases, as well as the morphology of the composites.The diffractograms of the Fe/C materials (Fig. 1a) showed that ethanol can reduce synthetic hematite (Fe2O3) to form magnetite (Fe3O4) at 700 °C, and after treatment at 900 °C, the hematite is reduced to metallic iron (Fe0) and iron carbide (Fe3C). After CVD, in the presence of Mo, the reduced phases of both iron and molybdenum were obtained (Fig. 1b). After treatment at 700 °C, the presence of magnetite (Fe3O4), iron carbide (Fe3C), and molybdenum(IV) oxide (MoO2) could be observed. Also, a mixed oxide of iron and molybdenum (Fe2Mo3O8), commonly formed above 700 °C in a reduced atmosphere, was observed in the FeMo/C700 material.40 After treatment at 900 °C (FeMo/C900), the presence of metallic iron (Fe0) and iron carbide (Fe3C) could be observed, as well as the presence of peaks related to Mo–Fe–C with low intensities. For this material obtained at 900 °C, the formation of molybdenum carbide (Mo2C) is also observed. The formation of Mo2C was not observed for the other materials.35 The initial precursor of FeMo was characterized by XRD (not shown), and it was possible to observe the presence of hematite (Fe2O3) and molybdenum oxide (MoO3). It is well known from the reports that molybdenum can act as an activator for nanotube growth in CVD processes, and it seems to have a very important effect on the characteristic and high yield production of the tubes.41–43
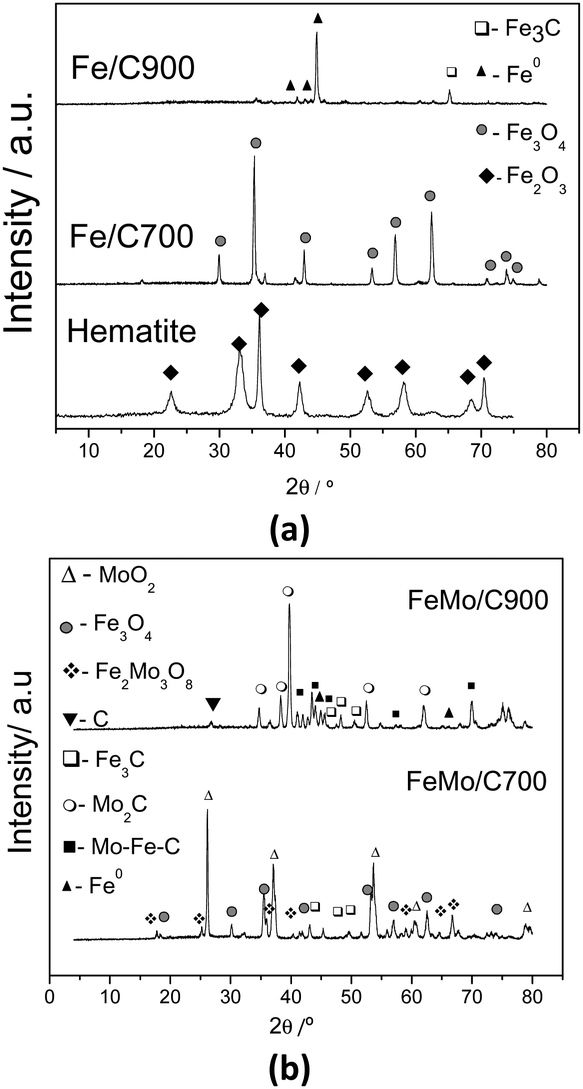 | ||
Fig. 1 XRD for Fe/C (a) and FeMo/C (b), obtained after the CVD of ethanol processed at different temperatures. Legend: ✦ Fe2O3, ▲ Fe0,  Fe3O4, □ Fe3C, ○ Mo2C, ■ Mo–Fe–C, △ MoO2, ▼ C, and Fe3O4, □ Fe3C, ○ Mo2C, ■ Mo–Fe–C, △ MoO2, ▼ C, and  Fe2Mo3O8. Fe2Mo3O8. | ||
The carbon amounts deposited on the Fe/C and FeMo/C materials were determined by thermal analyses of 0.5 and 3.0% for the samples of Fe/C obtained at 700 and 900 °C, respectively. Typically, carbon was oxidized at 350 °C in the presence of oxygen and showed a weight loss related to the process. For the samples with Mo (FeMo/C), the amount of C deposited increased considerably to 18 and 7% for the samples obtained at 700 and 900 °C, respectively.
Scanning electron micrographs of hematite before and after the CVD process (Fe/C and FeMo/C series) are shown in Fig. 2 and 3, respectively.
 | ||
| Fig. 2 Scanning electron micrographs for hematite before and after CVD (Fe/C) at different temperatures. | ||
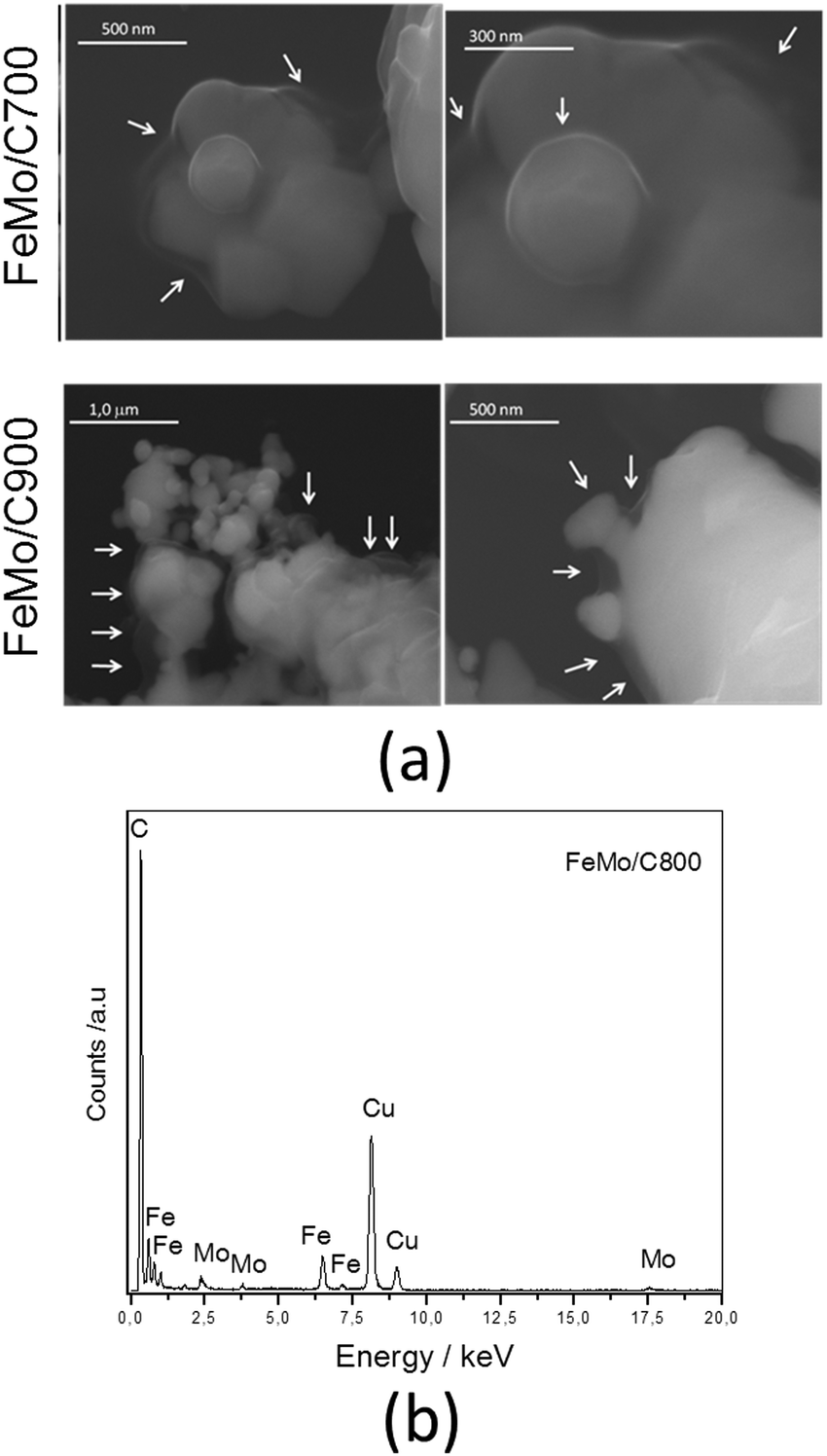 | ||
| Fig. 3 (a) Scanning electron micrographs for FeMo/C obtained after the CVD of ethanol at different temperatures, and (b) EDS spectrum of FeMo/C. | ||
The images for pure hematite in Fig. 2 are characteristic of iron oxides, showing very irregular surfaces with different grain sizes. The hematite after CVD, Fe/C, shows more rounded surfaces, probably due to sintering, particularly for the smaller particles. Fe/C900 showed a coated surface as well as the presence of carbon filaments (indicated by the arrows).
The impregnation of molybdenum significantly modified the surface of the material (Fig. 3). The presence of a thin layer covering all the materials can be noticed, and this is attributed to carbonaceous material (indicated by the arrows in Fig. 3), suggesting the formation of composites of FeMo/C.44,45 The presence of Mo favored the formation of this layer because it can act as a co-catalyst in the carbon decomposition process. EDS analysis associated with the SEM image (Fig. 3b) provides evidence of the presence of iron, molybdenum and carbon. The presence of copper is due to the sample port of the equipment.
Raman spectroscopy was performed to better characterize the metallic species, especially the carbon species present on the Fe/C and FeMo/C materials (Fig. 4).
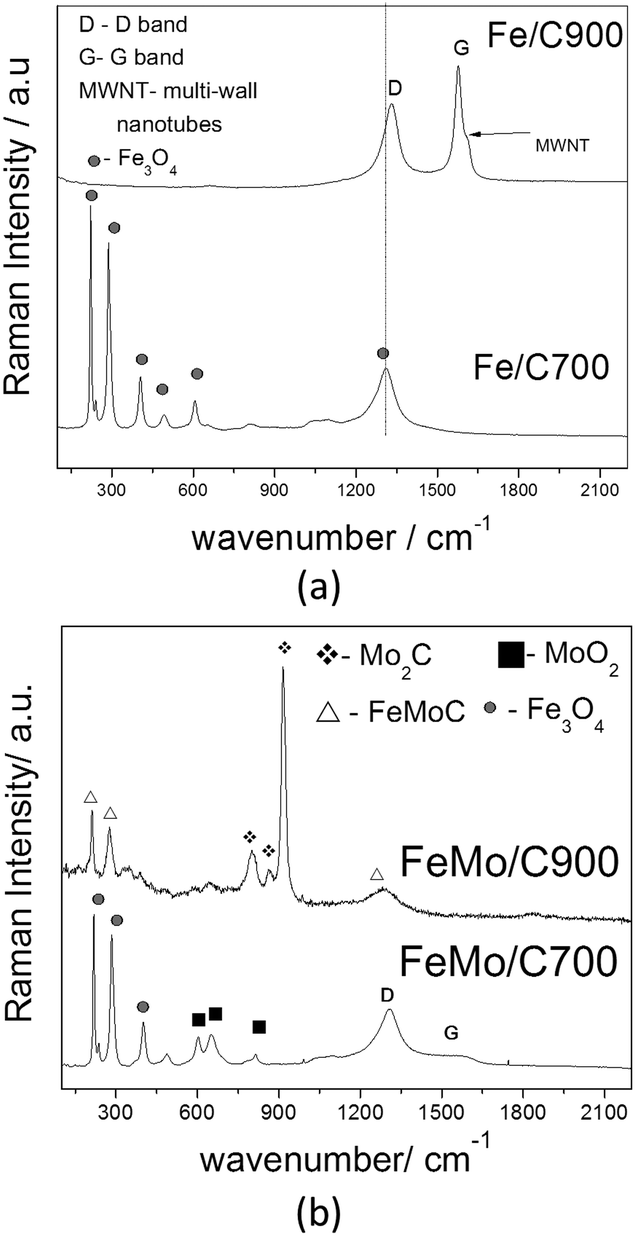 | ||
| Fig. 4 Raman spectra of the composites Fe/C (a) and FeMo/C (b), obtained after the ethanol/CVD process at 700 and 900 °C. | ||
The Fe/C900 material presented two important bands in the Raman spectra related to the carbon structures (Fig. 4a). The D band at 1340 cm−1 is characteristic of the defective or disorganized carbon structures, such as amorphous carbon, whereas the G band at 1580 cm−1 is related to the well-organized graphitic structures, i.e. carbon nanotubes and nanofibers.46 The shoulder of the G band is characteristic of the MWNTs (multi-walled carbon nanotubes).47,48 The higher the G band, the more organized the carbon structure formed on the surface of the materials. For the material obtained at the lowest temperature, no peaks related to carbon structures were observed. For the sample Fe/C700, only magnetite signs could be observed, and it was not possible to observe G or D bands due to the low carbon content in these samples.
The materials with Mo showed different characteristics. FeMo/C700 presented a G band with a very low intensity, suggesting the formation of more disorganized or amorphous carbon, as can be observed by the very intense D band at 1580 cm−1. Signals related to magnetite and Fe3O4, could also be observed besides MoO2, with both phases formed during the CVD process. The Raman spectrum of FeMo/C900 displayed only the signals related to iron and molybdenum carbide, suggesting that the reaction of Fe and Mo with the deposited carbon was favored at higher temperature.
3.2. Oxidation in a biphasic system
The synthesized hybrid materials, i.e., Fe/C and FeMo/C, were tested for the oxidation of sulfur and nitrogen compounds present in fossil fuels with hydrogen peroxide, which occurs in two-phase systems. In general, the oxidation of sulfur and nitrogen compounds in fuels, such as gasoline and diesel, shows a low conversion because the S- and N-containing contaminants and the oxidizing agent are in different phases. In this case, S- and N-containing compounds are found in the organic phase, whereas hydrogen peroxide is soluble in the aqueous phase. The limited interface between the two phases (oil/water) makes the oxidation process of the contaminants very difficult.The hybrid materials Fe/C and FeMo/C prepared in this study are very interesting for use in these biphasic reactions since they have an amphiphilic character, i.e. they are able to interact with both the aqueous and oily phases simultaneously, thereby increasing the interface between the phases. When the materials are introduced into a two-phase system, they will be located at the oil/water interface, and after stirring, the materials will preferably locate on the surface of the dispersed droplets of one phase into the other, thereby forming an emulsion. In an emulsion, the interface between the two phases is enhanced by promoting greater contact between them.
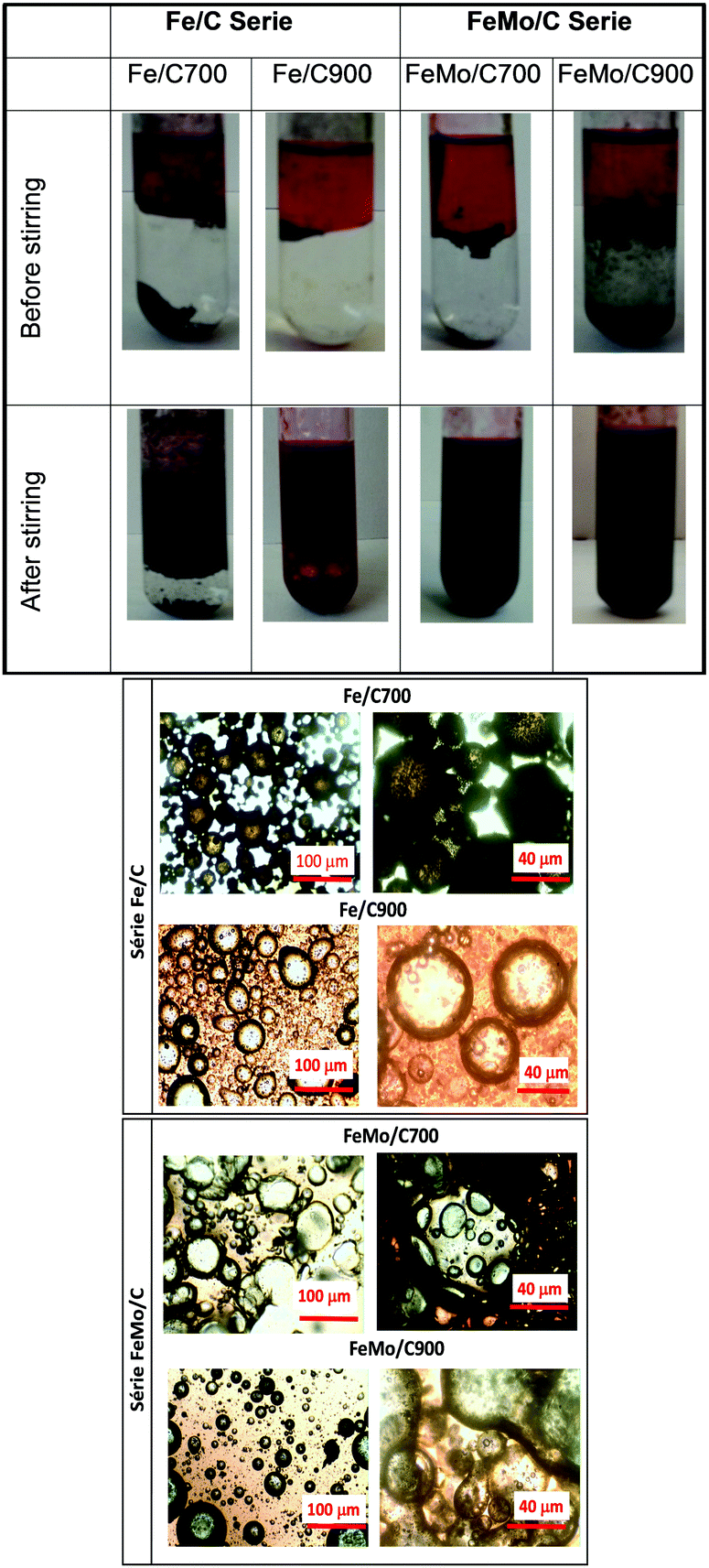 | ||
| Fig. 5 Initial system (before stirring) and emulsion formation (after stirring) using Fe/C and FeMo/C (above), and microscopic images of the formed emulsions for Fe/C and FeMo/C (below). | ||
As expected, it was observed that when Fe/C700 was introduced in the biphasic system before stirring, the material remained in the aqueous phase, whereas Fe/C900 was located at the interface of the two phases, showing an amphiphilic character. After vortex stirring, Fe/C700 showed a tendency to form an emulsion, where the nonpolar phase was dispersed into the polar phase, whereas for Fe/C900, a total dispersion of the two phases was observed, also indicating an emulsion formation.
The materials with molybdenum, such as FeMo/C, also seemed more amphiphilic as they were dispersed and located at the interface of the two phases before stirring. After stirring, a complete dispersion of the polar phase into the nonpolar phase occurred, indicating an emulsion formation, as can be observed by optical microscopy. In Fig. 5, emulsions of water-in-oil (W/O) and oil-in-water (O/W) can be seen. An emulsion of water-in-oil (W/O) is generally formed when the material interacts better with the non-polar phase.49–51
For Fe/C700, an emulsion of O/W formed, whereas for Fe/C900, an emulsion of W/O was observed. The amphiphilic behavior of the material at the interface of the two phases showing the formation of the emulsion is thus clear. When materials containing molybdenum was introduced, such as FeMo/C, emulsions like W/O were observed for both materials, i.e. the water droplets dispersed into the non-polar phase and the materials were located at the interface of the phases.
In addition to the intensification of the interface, the amphiphilic hybrid materials can also act as catalysts in the oxidation reactions promoted by the metallic phases, such as the reduced iron phases present in the materials, using hydrogen peroxide as the oxidizing agent. H2O2 can be activated by the Fe and Mo phases in the intensified interface of the phases to generate hydroxyl radicals (OH˙), which are extremely strong oxidants capable of oxidizing a wide range of organic contaminants in the oil phases. After the oxidation reaction, the oxidized products, typically more polar ones, migrated from the oily phase to the aqueous phase in an extraction process. Finally, the magnetic behavior of the materials played a decisive role at the end of the reaction as the materials could be easily removed from the reaction medium by an external magnetic field, thereby promoting breaking of the emulsion, i.e. phase separation. Moreover, after the reaction and magnetic separation, the materials could be easily reused in the subsequent reactions.
The hybrid materials were studied in the biphasic oxidation of fuel contaminants, using dibenzothiophene (DBT) and quinoline (QN) as model contaminants of sulfur and nitrogen containing compounds, respectively, and hydrogen peroxide as the oxidant.
The reactions were followed by analyzing the non-polar phase using a gas chromatograph coupled to a mass spectrometer (GC-MS). Fig. 6 shows a typical analysis using Fe/C900. Only one peak related to DBT was noted at the beginning of the reaction. After 60 minutes of reaction, it was possible to observe new peaks related to the oxidized products, identified by mass spectroscopy as dibenzosulfoxide and dibenzosulfone (Fig. 6a and b, respectively). After 90 minutes of reaction, it was observed that the intensity of the peaks related to the products increased, however, after 120 min, the intensity started to decrease. The final removal of DBT from the organic phase for Fe/C and FeMo/C can be seen in Fig. 7. Initially, the Fe2O3 and FeMo precursors were tested as catalysts in the S oxidation biphasic reaction, however, they did not show any catalytic activity.
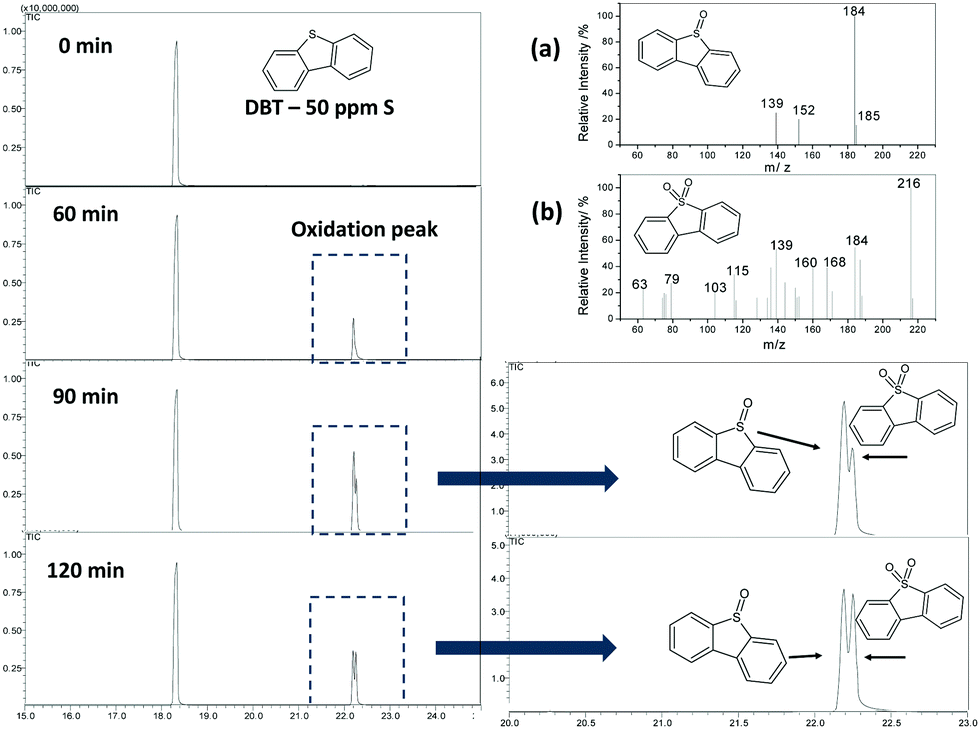 | ||
| Fig. 6 Chromatograms obtained by GC-MS for the evaluation of the oxidation reaction of DBT, catalyzed by Fe/C900. Detail: mass spectrum for the formed products: (a) dibenzosulfoxide and (b) dibenzosulfone. | ||
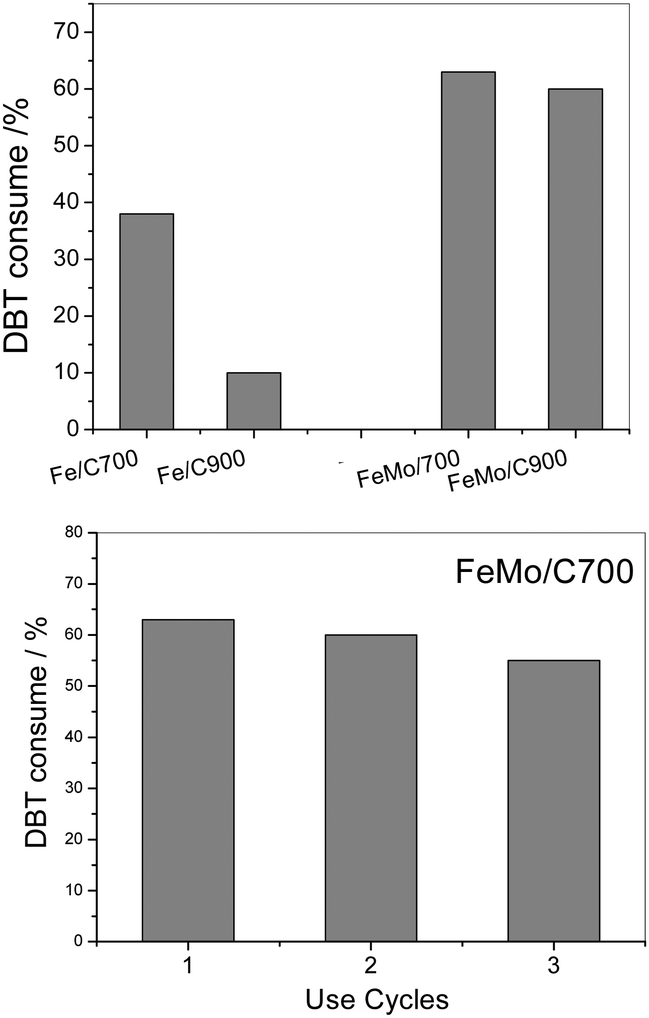 | ||
| Fig. 7 DBT removal using Fe/C and FeMo/C, measured after 120 minutes of reaction (above). Reuse tests of DBT removal using FeMo/C700 (below). | ||
A DBT removal of 38 and 10% from the organic phase was observed for Fe/C700 and Fe/C900, respectively. However, the materials with molybdenum showed higher activity to oxidize the DBT, reaching 63% of DBT removal from the organic phase with FeMo/C700 (Fig. 6). Here, the highest carbon coating present in the molybdenum-containing material enhanced the interaction of the catalyst with the organic phase, where the substrate was contained, and resulted in more stable emulsions being formed by these materials. During a typical oxidation process of DBT, the H2O2 molecule was catalytically decomposed to form hydroxyl radicals (OH˙), which then operated in the oxidation process. The DBT molecule in the presence of hydroxyl radicals initially underwent successive oxidations to form the sulfoxide and further a sulfonated derivative.
After the reaction and magnetic separation, the FeMo/C700 material was washed with distilled water and reused in three subsequent reactions, showing almost the same activity (Fig. 6). A small decrease in the activity was seen in the third cycle, which could be related to the loss of material following subsequent uses.
The emulsion formation between the two phases of the reaction and the presence of active metallic phases in Fe/C and FeMo/C increased the catalytic activity of the system and the oxidation of sulfur compounds, thus showing promising results, possibly due to the synergy of these factors.
It is very well known in the literature that the reduced iron phases, such as Fe3O4 and Fe3C, especially in the presence of Fe0, as well as molybdenum species are highly active in the oxidation of organic compounds with hydrogen peroxide.37,52 However, despite these phases being present in large amounts in Fe/C, this series shows a lower activity when compared with materials containing molybdenum, such as FeMo/C. This is probably due to the low content of carbon in the Fe/C materials (0.5–3% C) which does not favor a good dispersion between the oil/water phases, i.e., the systems are less emulsified with reduced interface. On the other hand, the materials with molybdenum, i.e., FeMo/C, with higher carbon contents (26–34% C) are capable of completely emulsifying the system, thus favoring a more efficient oil/water interface. Then, the active phases present in these materials, e.g. Fe3O4, Fe0, Fe3C and Fe–Mo–C, can take part in the oxidation of sulfur compounds by the activation of H2O2.
Based on the results, we propose that the hybrid materials Fe/C and FeMo/C acted in two steps. In the first step of the mechanism, the composites increased the interface between the organic phase (containing the contaminant) and the aqueous phase (containing the oxidant), forming an emulsion with a high interface. Then, hydrogen peroxide was activated by the reduced Fe and Mo species on the surface of the composites in a heterogeneous Fenton-like mechanism, forming hydroxyl radicals (˙OH). These radicals were then capable of rapidly oxidizing the DTB on the interface of the immiscible phases.
By intensifying the interface, the composites facilitated the contact between the radicals and S molecules. Consequently, the radicals could initiate the selective oxidation of the S contaminants present in the organic phase, and finally, the oxidized S contaminant was extracted in the aqueous phase due to its increased polarity. At the end of reaction, the composites located on the interface were attracted by an external magnet, which destabilized the emulsion formed and the phases were then separated. Moreover, the mixture was analyzed by atomic absorption spectrometry after the reaction and showed less than 1% Fe or Mo leaching.
The materials were also used in the biphasic oxidation of quinoline (QN), which was used as a model for nitrogen-containing contaminants present in different fuels, such as in gasoline and diesel.
The decreased concentration of quinoline in the organic phase was analysed using UV-Vis spectroscopy (Fig. 8). It could be observed in both the cases, Fe/C and FeMo/C, that the removal of quinoline reached 100% after 120 minutes of reaction. All the materials showed similar behavior. However, the material prepared at 900 °C, with and without molybdenum, showed a total removal in a few minutes.
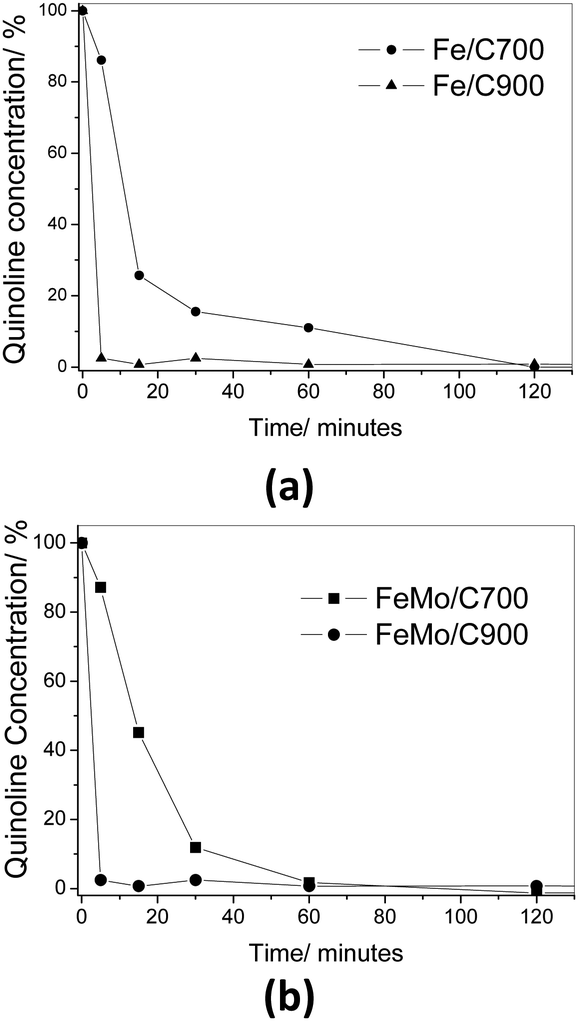 | ||
| Fig. 8 Consumption of quinoline (%) catalyzed by Fe/C (a) and FeMo/C (b) with H2O2 in a biphasic system. | ||
The catalytic oxidation of quinoline was also monitored by analyzing the aqueous phase by ESI-MS in the positive mode after 3 h of reaction (Fig. 9). Although the concentration of quinoline in the oily phase was very low, it was predominant in the aqueous phase (as indicated by the peak m/z = 130), and there was no formation of degradation peaks resulting from the oxidation of the quinoline (Fig. 9a). In Fig. 9b, it can be observed for the Fe/C materials that there was a great decrease in the intensity of the peak for quinoline (see the scale) and peaks related to oxidized products did not form. These results suggest a total mineralization of the quinoline present in the solution, which was expected since the active iron phases present in these materials were widely available in the materials once the samples showed low carbon coating. This made them catalytically very active to promote the mineralization of the contaminant molecule.
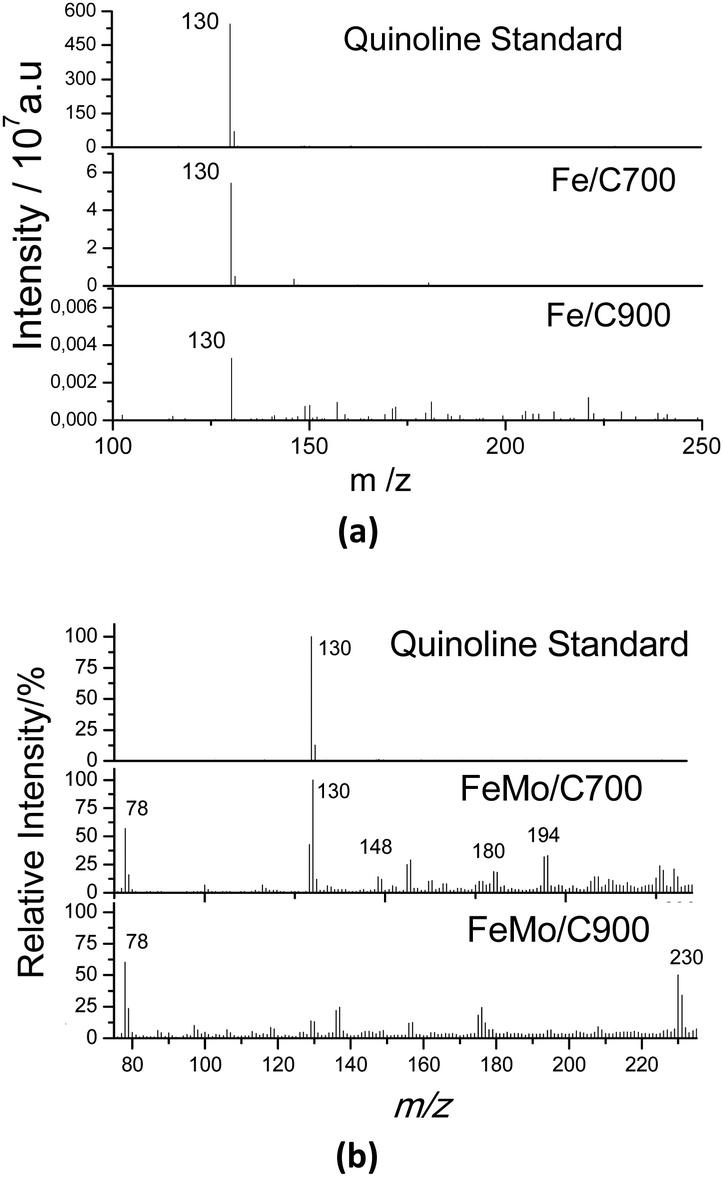 | ||
| Fig. 9 ESI(+)-MS of standard quinoline (50 ppm) and after 3 h of reaction in the presence of (a) Fe/C and (b) FeMo/C, with H2O2 in a biphasic system. | ||
For the reaction with the materials containing Mo, several oxidized intermediates could be observed. After the reaction with FeMo/C700, the peak associated with quinoline (m/z = 130) could be observed, which is possibly related to the migration to the aqueous phase since it is partly soluble in water. In addition, new peaks with m/z = 148, 180 and 194 were also be observed, corresponding to the sequential hydroxylations of the quinoline molecule. For FeMo/C900, it was also possible to observe oxidized products with peaks at m/z = 230 and 118. The material with FeMo/C900 looked more active since no signal related to quinoline (m/z = 130) was observed. The mechanism of the initial oxidation together with the possible structures for the reaction products identified by ESI-MS is proposed in Fig. 10.
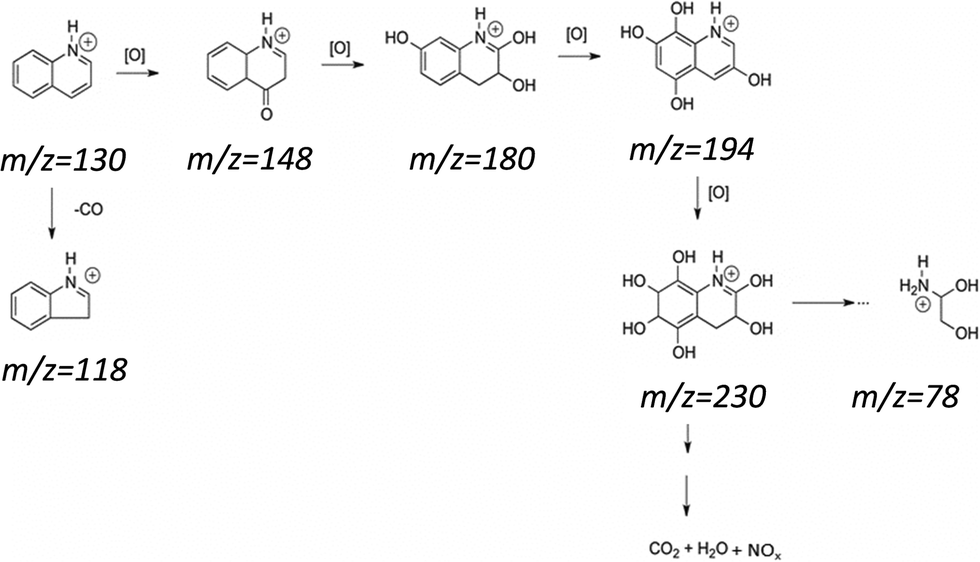 | ||
| Fig. 10 Schematic representation of quinoline oxidation with H2O2. | ||
The reactions involved in the biphasic oxidation of nitrogen in the presence of these materials indicated that the materials obtained at 900 °C (Fe/C900 and FeMo/C900) were quite promising for the degradation of quinoline and the formation of more oxidized products.
4. Conclusions
Two series of hybrid materials, Fe/C and FeMo/C, were obtained via iron and molybdenum oxides followed by a CVD process using ethanol as the carbon source. The results of XRD, SEM, and Raman spectroscopy showed that the iron and molybdenum oxides species were reduced during the CVD process with ethanol, leading to a deposition of carbon nanotubes and nanofibers and iron phases with magnetic behavior. The hybrid materials were amphiphilic and strongly interacted with different phases, i.e. polar and nonpolar, simultaneously. Studies on the use of these materials as catalysts in the biphasic oxidation of nitrogen and sulfur compounds showed very promising results, with consumptions of up to 63% of DBT and about 100% of QN. In addition to the high consumption, more oxidized products could be observed. These results can be explained by the versatility of the materials that can form emulsions in addition to acting as catalysts in the oxidation reactions. Finally, another advantage of these materials is that after their use, the composite can be easily removed by the approach of an external magnetic field.Acknowledgements
The authors are grateful to Petrobras, CNPq, FAPEMIG, CAPES for its support and to Centro de Microscopia da UFMG for providing the microscopy facilities.Notes and references
- X. H. Han, A. J. Wang, X. S. Wang, X. Li, Y. Wang and Y. K. Hu, Catal. Commun., 2013, 42, 6–9 CrossRef CAS.
- K. Nakano, S. A. Ali, H.-J. Kim, T. Kim, K. Alhooshani, J.-I. Park and I. Mochida, Fuel Process. Technol., 2013, 116, 44–51 CrossRef CAS.
- F. M. Collins, A. R. Lucy and C. Sharp, J. Mol. Catal. A: Chem., 1997, 117, 397–403 CrossRef CAS.
- I. Suelves, C. A. Islas, M. Millan, C. Galmes, J. F. Carter, A. A. Herod and R. Kandiyoti, Fuel, 2003, 82, 1–14 CrossRef.
- L. R. Snyder and B. E. Buell, Anal. Chem., 1968, 40, 1295–1302 CrossRef CAS.
- X. Tao, Y. Zhou, Q. Wei, G. Yu, Q. Cui, J. Liu and T. Liu, Fuel Process. Technol., 2014, 118, 200–207 CrossRef CAS.
- A. Akbari, M. Omidkhah and J. T. Darian, Ultrason. Sonochem., 2014, 21, 692–705 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- P. Tétényi, T. Ollár and T. Szarvas, Catal. Today, 2012, 181, 148–155 CrossRef.
- F.-t. Li, R.-h. Liu, W. Jin-hua, D.-s. Zhao, Z.-m. Sun and Y. Liu, Green Chem., 2009, 11, 883–888 RSC.
- P. Zeuthen, K. G. Knudsen and D. D. Whitehurst, Catal. Today, 2001, 65, 307–314 CrossRef CAS.
- A. L. Nuzhdin, K. A. Kovalenko, D. N. Dybtsev and G. A. Bukhtiyarova, Mendeleev Commun., 2010, 20, 57–58 CrossRef CAS.
- W. Zhu, C. Wang, H. Li, P. Wu, S. Xun, W. Jiang, Z. Chen, Z. Zhao and H. Li, Green Chem., 2015, 17, 2464–2472 RSC.
- W. Zhang, H. Zhang, J. Xiao, Z. X. Zhao, M. X. Yu and Z. Li, Green Chem., 2014, 16, 211–220 RSC.
- X. L. Ma, K. Y. Sakanishi and I. Mochida, Ind. Eng. Chem. Res., 1994, 33, 218–222 CrossRef CAS.
- H. Wang, C. Xie, S. Yu and F. Liu, Chem. Eng. J., 2014, 237, 286–290 CrossRef CAS.
- Y. Li, D. Liu and C. Liu, Energy Fuels, 2010, 24, 789–795 CrossRef CAS.
- X. Zhang, A. Guo, F. Wang and X. Duan, Energy Fuels, 2010, 24, 3772–3777 CrossRef CAS.
- S. Chehreh Chelgani and E. Jorjani, Fuel, 2011, 90, 3156–3163 CrossRef CAS.
- D. Wang, E. W. Qian, H. Amano, K. Okata, A. Ishihara and T. Kabe, Appl. Catal., A, 2003, 253, 91–99 CrossRef CAS.
- A. Ishihara, D. Wang, F. Dumeignil, H. Amano, E. W. Qian and T. Kabe, Appl. Catal., A, 2005, 279, 279–287 CrossRef CAS.
- L. C. Caero, J. F. Navarro and A. Gutiérrez-Alejandre, Catal. Today, 2006, 116, 562–568 CrossRef.
- L. Cedeño-Caero, H. Gomez-Bernal, A. Fraustro-Cuevas, H. D. Guerra-Gomez and R. Cuevas-Garcia, Catal. Today, 2008, 133–135, 244–254 CrossRef.
- J. L. García-Gutiérrez, G. A. Fuentes, M. E. Hernández-Terán, P. García, F. Murrieta-Guevara and F. Jiménez-Cruz, Appl. Catal., A, 2008, 334, 366–373 CrossRef.
- J. L. García-Gutiérrez, G. A. Fuentes, M. E. Hernández-Terán, F. Murrieta, J. Navarrete and F. Jiménez-Cruz, Appl. Catal., A, 2006, 305, 15–20 CrossRef.
- L. Yang, J. Li, X. Yuan, J. Shen and Y. Qi, J. Mol. Catal. A: Chem., 2007, 262, 114–118 CrossRef CAS.
- T. Shimada, T. Sugai, C. Fantini, M. Souza, L. G. Cancado, A. Jorio, M. A. Pimenta, R. Salto, A. Gruneis, G. Dresselhaus, M. S. Dresselhaus, Y. Ohno, T. Mizutani and H. Shinohara, Carbon, 2005, 43, 1049–1054 CrossRef CAS.
- T. Aida, D. Yamamoto, M. Iwata and K. Sakata, Heteroat. Chem., 2000, 22, 241–256 CAS.
- F. M. Collins, A. R. Lucy and C. Sharp, J. Mol. Catal. A: Chem., 1997, 117, 397–403 CrossRef CAS.
- A. A. S. Oliveira, I. F. Teixeira, L. P. Ribeiro, E. Lorençon, J. D. Ardisson, L. Fernandez-Outon, W. A. A. Macedo and F. C. C. Moura, Appl. Catal., A, 2013, 456, 126–134 CrossRef CAS.
- A. A. S. Oliveira, I. F. Teixeira, T. Christofani, J. C. Tristão, I. R. Guimarães and F. C. C. Moura, Appl. Catal., B, 2014, 144, 144–151 CrossRef CAS.
- D. A. S. Costa, R. V. Mambrini, L. E. Fernandez-Outon, W. A. A. Macedo and F. C. C. Moura, Chem. Eng. J., 2013, 229, 35–41 CrossRef CAS.
- A. A. S. Oliveira, D. A. S. Costa, I. F. Teixeira and F. C. C. Moura, Appl. Catal., B, 2015, 162, 475–482 CrossRef CAS.
- A. P. C. Teixeira, A. D. Purceno, A. S. Barros, B. R. S. Lemos, J. D. Ardisson, W. A. A. Macedo, E. C. O. Nassor, C. C. Amorim, F. C. C. Moura, M. G. Hernández-Terrones, F. M. Portela and R. M. Lago, Catal. Today, 2012, 190, 133–143 CrossRef CAS.
- R. V. Mambrini, A. L. M. Saldanha, J. D. Ardisson, M. H. Araujo and F. C. C. Moura, Appl. Clay Sci., 2013, 83–84, 286–293 CrossRef CAS.
- I. F. Teixeira, A. A. D. Oliveira, T. Christofani and F. C. C. Moura, J. Mater. Chem. A, 2013, 1, 10203–10208 RSC.
- R. C. C. Costa, F. C. C. Moura, J. D. Ardisson, J. D. Fabris and R. M. Lago, Appl. Catal., B, 2008, 83, 131–139 CrossRef CAS.
- A. P. C. Teixeira, J. C. Tristao, M. H. Araujo, L. C. A. Oliveira, F. C. C. Moura, J. D. Ardisson, C. C. Amorim and R. M. Lago, J. Braz. Chem. Soc., 2012, 23, 1579–1593 CrossRef CAS.
- P. Tetenyi, T. Ollar and T. Szarvas, Catal. Today, 2012, 181, 148–155 CrossRef CAS.
- H. Zhang, J. Shen and X. Ge, J. Solid State Chem., 1995, 117, 127–135 CrossRef CAS.
- W. E. Alvarez, B. Kitiyanan, A. Borgna and D. E. Resasco, Carbon, 2001, 39, 547–558 CrossRef CAS.
- M. Perez-Mendoza, C. Valles, W. K. Maser, M. T. Martinez and A. M. Benito, Nanotechnology, 2005, 16, S224–S229 CrossRef CAS.
- A. D. Purceno, B. R. Barrioni, A. Dias, G. M. da Costa, R. M. Lago and F. C. C. Moura, Appl. Clay Sci., 2011, 54, 15–19 CrossRef CAS.
- F. Wang, G. Yao, M. Xu, M. Zhao, Z. Sun and X. Song, J. Alloys Compd., 2011, 509, 5969–5973 CrossRef CAS.
- Y. Z. Chen, N. Shah, A. Braun, F. E. Huggins and G. P. Huffman, Energy Fuels, 2005, 19, 1644–1651 CrossRef CAS.
- K.-E. Jeong, T.-W. Kim, J.-W. Kim, H.-J. Chae, C.-U. Kim, Y.-K. Park and S.-Y. Jeong, Korean J. Chem. Eng., 2013, 30, 509–517 CrossRef CAS.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- T. Selvam, A. Machoke and W. Schwieger, Appl. Catal., A, 2012, 445, 92–101 CrossRef.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100–102, 503–546 CrossRef CAS.
- T. Tadros, P. Izquierdo, J. Esquena and C. Solans, Adv. Colloid Interface Sci., 2004, 108–109, 303–318 CrossRef CAS PubMed.
- M. Shen and D. E. Resasco, Langmuir, 2009, 25, 10843–10851 CrossRef CAS PubMed.
- F. C. C. Moura, M. H. Araujo, R. C. C. Costa, J. D. Fabris, J. D. Ardisson, W. A. A. Macedo and R. M. Lago, Chemosphere, 2005, 60, 1118–1123 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |