
Selenium in the prevention of atherosclerosis and its underlying mechanisms
Hongmei
Liu
ab,
Huibi
Xu
ab and
Kaixun
Huang
*ab
aSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, People's Republic of China. E-mail: hxxzrf@hust.edu.cn; Fax: +86 27 87543632; Tel: +86 27 87543032
bHubei Key Laboratory of Bioinorganic Chemistry & Materia Medica, Wuhan 430074, People's Republic of China
First published on 7th December 2016
Abstract
Atherosclerosis and related cardiovascular diseases (CVDs) represent the greatest threats to human health worldwide. Selenium, an essential trace element, is incorporated into selenoproteins that play a crucial role in human health and disease. Although findings from a limited number of randomized trials have been inconsistent and cannot support a protective role of Se supplementation in CVDs, prospective observational studies have generally shown a significant inverse association between selenium or selenoprotein status and CVD risk. Furthermore, a benefit of selenium supplementation in the prevention of CVDs has been seen in population with low baseline selenium status. Evidence from animal studies shows consistent results that selenium and selenoproteins might prevent experimental atherosclerosis, which can be explained by the molecular and cellular effects of selenium observed both in animal models and cell cultures. Selenoproteins of particular relevance to atherosclerosis are glutathione peroxidases, thioredoxin reductase 1, selenoprotein P, selenoprotein S. The present review is focusing on the existing evidence that supports the concept that optimal selenium intake can prevent atherosclerosis. Its underlying mechanisms include inhibiting oxidative stress, modulating inflammation, suppressing endothelial dysfunction, and protecting vascular cells against apoptosis and calcification. However, the benefit of selenium supplementation in the prevention of atherosclerosis remains insufficiently documented so far. Future studies with regard to the effects of selenium supplementation on atherosclerosis should consider many factors, especially the baseline selenium status, the dose and forms of selenium supplementation, and the selenoprotein genotype. Additionally, much more studies are needed to confirm the roles of selenoproteins in atherosclerosis prevention and clarify the underlying mechanisms.
1 Introduction
Selenium (Se) is an essential trace element that plays a crucial role in human health and disease.1,2 The biological functions of Se are mainly carried out by selenoproteins, in which Se is incorporated as the amino acid selenocysteine (Sec), the 21st “naturally occurring” amino acid. The insertion of Sec to form a selenoprotein is encoded by the UGA codon in mRNA under specific conditions.3 At present, 25 selenoproteins have been identified in humans (Table 1).4 Among the selenoproteins, 13 have known functions: GPX1, GPX2, GPX3, GPX4 and GPX6 (glutathione peroxidases), TrxR1, TrxR2, and TrxR3 (thioredoxin reductases), DIO1, DIO2, and DIO3 (iodothyronine deiodinases), SelR (selenoprotein R) and SPS2 (selenophosphate synthetase 2). At least 12 of them serve as oxidoreductases, wherein Sec is the catalytic redox-active residue.3,5–7 The best-known function is the reduction of H2O2 and damaging lipid and phospholipid hydroperoxides to harmless products (water and alcohols) by the family of Se-dependent GPXs. This function helps to maintain membrane integrity, and reduces oxidative damage to biomolecules such as lipids, lipoproteins, and DNA with the associated increased risk of conditions such as atherosclerotic cardiovascular diseases (CVDs) and cancer.1,2,8 Expression of selenoproteins is differentially regulated by the Se status.3,9 While expression of some selenoproteins is significantly decreased under Se-deficient conditions, e.g., GPX1 and SelR (referred to as stress-related selenoproteins), expression of the other group of selenoproteins including TrxR1 and GPX4 (referred to as housekeeping selenoproteins) is less regulated by dietary Se. Likewise, available Se is not equally supplied to all tissues.9 Under conditions of Se-deficiency, brain, testes and thyroid maintain near normal Se levels, whereas other tissues, such as liver, lung and those of the immune system, exhibit a significant decline in Se and selenoprotein synthesis. For recent reviews focused in detail on selenoproteins, please refer to the reviews.3,5–7| Selenoprotein (abbreviation) | Localization | Function |
|---|---|---|
| a Adapted from ref. 3 and 5–7. | ||
| Cytosolic glutathione peroxidase (GPX1) | Cytosol | Reduces cellular H2O2 |
| Gastrointestinal glutathione peroxidase (GPX2) | Cytosol | Reduces peroxide in gut |
| Plasma glutathione peroxidase (GPX3) | Plasma | Reduces peroxide in blood |
| Phosholipid hydroperoxide glutathione peroxidase (GPX4, PHGPX) | Cytosol; mitochondria; nucleus (testis-specific) | Reduces phospholipid peroxide |
| Olfactory glutathione peroxidase (GPX6) | Cytosol | Reduces cellular H2O2 in the olfactory epithelium |
| Thioredoxin reductase 1 (TrxR1) | Cytosol | Reduces the oxidized form of cytosolic thioredoxin |
| Thioredoxin reductase 2 (TrxR3) | Cytosol | Has a glutaredoxin domain; catalyzes a variety of reactions, specific for thioredoxin and glutaredoxin systems; expressed in spermatids |
| Thioredoxin reductase 3 (TrxR2) | Mitochondria | Reduces the oxidized form of mitochondrial thioredoxin and glutaredoxin 2 |
| Thyroid hormone deiodinase 1 (DIO1) | Plasma membrane | Removes iodine from the outer ring of T4 to produce plasma T3; catalyzes deiodination and thus inactivation of T3 |
| Thyroid hormone deiodinase 2 (DIO2) | Endothelial reticulum | Converts T4 to T3 locally in tissues |
| Thyroid hormone deiodinase 3 (DIO3) | Plasma membrane | Catalyzes deiodination of T4 to T3 in peripheral tissues |
| Selenoprotein R (SelR, MsrB1) | Cytosol | Reduces methionine-R-sulfoxide residues in proteins to methionine |
| Selenophosphate synthetase (SPS2) | Cytosol | Synthesis of selenophosphate |
| Selenoprotein P (SelP) | Plasma | Se transport to peripheral tissues and antioxidant function |
| 15 kDa selenoprotein (Sep15) | Endoplasmic reticulum (ER) | Has thioredoxin (Trx)-like fold; regulated by ER stress; interacts with UDP-glucose:glycoprotein glucosyl-transferase; potentially involved in glycoprotein folding |
| Selenoprotein M (SelM) | ER | Has Trx-like fold; protects neurons from oxidative stress |
| Selenoprotein T (SelT) | ER and Golgi | Has Trx-like fold; redox regulation; plays a role in cell adhesion |
| Selenoprotein S (SelS) | ER membrane | Upregulated upon treatment with pro-inflammatory cytokines; component of ER associated degradation; regulate ER stress; function as an efficient reductase and a moderate peroxidase in vitro |
| Selenoprotein K (SelK) | ER membrane | Modulates Ca2+ influx that affects immune cell function; component of ER associated degradation |
| Selenoprotein N (SelN) | ER membrane | Expressed in skeletal muscle, heart, lung, and placenta; controls redox state of the intracellular calcium-release channel (ryanodine receptor (RyR)), and therefore affects Ca2+ homeostasis; mutations in SelN gene cause congenital myopathy |
| Selenoprotein H (SelH) | Nucleus | Has Trx-like fold; protects cells from H2O2, increases mitochondrial biogenesis and cytochrome c production; AT-hook family protein and facilitates synthesis of genes responsible for de novo GSH synthesis and phase II detoxification in response to redox changes |
| Selenoprotein I (SelI) | Membrane | Unknown function |
| Selenoprotein O (SelO) | Mitochondria | Unknown function |
| Selenoprotein V (SelV) | Cytosol | Has Trx-like fold; unknown function; expressed in spermatids |
| Selenoprotein W (SelW) | Cytosol | Has Trx-like fold; unknown function; expressed in skeletal muscle and other tissues |
Atherosclerotic CVDs are the biggest cause of mortality and a major cause of morbidity and disability worldwide. Atherosclerosis is a progressive disease in the arterial wall that develops over many years, characterized by the deposition of fatty material and cholesterol in large- and medium-sized arteries.10–12 The initial event in atherogenesis is the increased transcytosis of low density lipoprotein (LDL), and its subsequent deposition, retention and modification (in particular oxidative modification) in the arterial intima. Additionally, endothelial cell dysfunction enhances the expression of chemoattractants or cell adhesion molecules, which induces adhesion of circulating monocytes to endothelial cells. Adhered monocytes migrate into the arterial intima and differentiate into macrophages. The intimal macrophages engulf excess lipid derived from oxidized LDL (oxLDL) by scavenger receptors, and transform into foam cells. Excessive foam cell formation results in focal accumulation of cholesterol esters, which is called a fatty streak. In a fatty streak, macrophages and T lymphocytes secrete inflammatory cytokines to enhance vascular smooth muscle cell (VSMC) migration from media to intima. Migrated VSMCs thicken the arterial wall, and the fatty streak evolves into a stable atherosclerotic plaque. In the advanced stage of atherosclerosis, VSMCs in plaques proliferate and secrete extracellular matrix proteins, which generate a fibrous cap, and apoptotic or necrotic cell death of lipid-laden foam cells (originated from VSMCs and macrophages) produce a necrotic core. The necrotic core contains cholesterol ester deposits and dead cells or their debris. Continuous accumulation of foam cells and T cells in the intima induces chronic inflammation, which recruits more VSMCs and inflammatory cells into the intima. During this process, fibrous cap thinning, calcification, and arterial wall injury by cholesterol crystals in the necrotic core transform stable plaques into unstable fibrous plaques. Finally, the unstable plaque with a thin fibrous cap and a large necrotic core begins to rupture and induce the formation of a thrombus or blood clot, which results in heart attack or stroke. These two clinical manifestations of atherosclerosis are often referred to as coronary artery disease and cerebrovascular disease. For simplicity, they are commonly referred to by the collective term “CVD” in this review.
It is widely known that oxidative stress is a major factor leading to atherosclerosis and related CVDs.11,12 Therefore, in the past over 30 years, particular interest has been given to the preventive role of Se in atherosclerosis and CVDs owing to its antioxidant properties. More recently, since a better understanding of the relationship between Se and CVDs requires research focusing on the role of selenoproteins, a lot of studies were conducted to investigate whether selenoproteins, especially GPX1, have protective roles in the cardiovascular system. The present review is focusing on the existing evidence about the effects of Se and selenoproteins on atherosclerosis and its related mechanisms. Since the clinical manifestations of atherosclerosis are CVDs, the current knowledge from human studies regarding the relationship between Se and atherosclerotic CVDs is also summarized.
2 Human studies of Se and CVDs
2.1 Se and CVDs
The first hint about the role of Se in the cardiovascular system should trace back to Keshan disease, a congestive cardiomyopathy occurred in Se deficient areas of China before 1980, which is the first reported human disease associated with Se deficiency,13 and can be completely prevented by supplementation of sodium selenite.14 In the past over 30 years, a considerable number of prospective observational studies investigated the association of Se status with CVDs, and a limited number of randomized trials investigated whether Se supplementation (most as Se-enriched yeast) prevents CVDs. In addition, it is worth mentioning that the topic of Se and CVDs has been extensively reviewed by others.15–19 Therefore, this review will just do some summary regarding this topic and introduce new studies published recently.Current knowledge derived from prospective observational studies showed a significant inverse association between blood Se status and CVD risk within a narrow Se range of 55–145 μg L−1.1,2,19–21 This result suggests that Se deficiency or low Se status (because of inadequate Se intake) can be considered a risk factor for CVDs. However, findings from a limited number of randomized trials are inconsistent. In 2006, a meta-analysis of 6 trials (published between 1989 and 2004, a total of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 766 participants) showed that participants taking supplements containing Se had a nonsignificant 11% reduction in CVD risk. But the trials were small and Se was given in combination with other vitamins or minerals in all but 2 trials.21 In 2013, another meta-analysis of 12 trials (published up to October 2012, a total of 19715 participants) reported that there were no statistically significant effects of Se supplementation as a single ingredient on CVD mortality, and all fatal and non-fatal CVD events.22 Recently, Zhang et al. conducted a meta-analysis of 15 trials (published up to December 2013) and showed that oral Se supplementation (median dose: 200 μg per day) for 2 weeks to 144 months significantly raised the blood Se concentrations by 56.4 μg L−1, whereas oral Se supplementation (median: 100 μg per day) for 6 to 114 months caused no effect on CVDs.20 However, 2 intervention trials published recently showed a protective effect of Se supplementation on CVDs to some extent. A trial from 230 UK pregnant women showed that UK pregnant women had relatively low Se concentration at the baseline and Se treatment (60 μg Se per day, as Se-enriched yeast) from mean gestational age 12.3 weeks until the delivery of the baby significantly reduced the odds ratio for pre-eclampsia and pregnancy-induced hypertension.23 Another trial among elderly Swedish citizens (443 participants, 70–88 years old) showed that supplementation with Se (200 μg Se per day, as Se-enriched yeast) and coenzyme Q10 (200 mg per day) for 4 years significantly reduced cardiovascular mortality and enhanced the cardiac function during a follow up time of 5.2 years.24 Significantly reduced cardiovascular mortality was still observed during a follow up time of 10 years after intervention, demonstrating that the protective action of Se and coenzyme Q10 was not confined to the intervention period, but persisted during the follow-up period.25 In addition, this research group determined whether the effects of supplementation with Se and coenzyme Q10 on cardiovascular mortality during a four-year intervention were dependent on the basal level of Se. They found that supplementation was cardio-protective in participants with a low Se basal concentration (≤85 μg L−1). In those with serum Se basal concentration >85 μg L−1 and no apparent deficiency, there was no effect of supplementation.26 These results suggest that Se plays a pivotal role in the protective action of Se and coenzyme Q10. However, the 2 recent trials are small, restricted in specific people (pregnant women and elderly people), and Se was given in combination with coenzyme Q10 in one trial.
766 participants) showed that participants taking supplements containing Se had a nonsignificant 11% reduction in CVD risk. But the trials were small and Se was given in combination with other vitamins or minerals in all but 2 trials.21 In 2013, another meta-analysis of 12 trials (published up to October 2012, a total of 19715 participants) reported that there were no statistically significant effects of Se supplementation as a single ingredient on CVD mortality, and all fatal and non-fatal CVD events.22 Recently, Zhang et al. conducted a meta-analysis of 15 trials (published up to December 2013) and showed that oral Se supplementation (median dose: 200 μg per day) for 2 weeks to 144 months significantly raised the blood Se concentrations by 56.4 μg L−1, whereas oral Se supplementation (median: 100 μg per day) for 6 to 114 months caused no effect on CVDs.20 However, 2 intervention trials published recently showed a protective effect of Se supplementation on CVDs to some extent. A trial from 230 UK pregnant women showed that UK pregnant women had relatively low Se concentration at the baseline and Se treatment (60 μg Se per day, as Se-enriched yeast) from mean gestational age 12.3 weeks until the delivery of the baby significantly reduced the odds ratio for pre-eclampsia and pregnancy-induced hypertension.23 Another trial among elderly Swedish citizens (443 participants, 70–88 years old) showed that supplementation with Se (200 μg Se per day, as Se-enriched yeast) and coenzyme Q10 (200 mg per day) for 4 years significantly reduced cardiovascular mortality and enhanced the cardiac function during a follow up time of 5.2 years.24 Significantly reduced cardiovascular mortality was still observed during a follow up time of 10 years after intervention, demonstrating that the protective action of Se and coenzyme Q10 was not confined to the intervention period, but persisted during the follow-up period.25 In addition, this research group determined whether the effects of supplementation with Se and coenzyme Q10 on cardiovascular mortality during a four-year intervention were dependent on the basal level of Se. They found that supplementation was cardio-protective in participants with a low Se basal concentration (≤85 μg L−1). In those with serum Se basal concentration >85 μg L−1 and no apparent deficiency, there was no effect of supplementation.26 These results suggest that Se plays a pivotal role in the protective action of Se and coenzyme Q10. However, the 2 recent trials are small, restricted in specific people (pregnant women and elderly people), and Se was given in combination with coenzyme Q10 in one trial.
In summary, the results from randomized trials to date have been inconsistent and the role of Se supplementation in CVD prevention is inconclusive. The difference in the baseline Se status of the populations studied and the dose of Se supplementation might partially account for the lack of consistency in trial studies. Se supplementation may benefit people with low baseline Se status, but have no effect or even an adverse effect on the cardiovascular system in people with adequate-to-high status. For example, supplementation of additional Se in people who already have adequate Se intake might increase their risk of type-2 diabetes.27 Thus, a U-shaped association between Se status and CVD risk may be reasonable28 and is accepted by more and more investigators.1,16,19 The baseline Se status of the target population and the dose of Se supplementation should be importantly considered in further research in order to obtain optimal CVD prevention without side-effects (e.g. type-2 diabetes).
2.2 Selenoproteins and CVDs
A better understanding of the relationship between Se and CVDs requires research to focus on the role of selenoproteins in the cardiovascular system. To date, the protecting role of GPXs in the cardiovascular system has been studied in most detail.GPX1, the most abundant selenoprotein in mammals, is a ubiquitous enzyme expressed in all cell types. It consumes reduced glutathione to convert H2O2 to water and lipid peroxides to their respective alcohols. Many studies implicate a potential protective effect of GPX1 against CVDs, particularly in individuals who had CVDs at the baseline.29 In a prospective study among 636 patients with a history of CVDs, those with low erythrocyte GPX1 activities had an increased incident of recurrent cardiovascular events, suggesting that baseline erythrocyte GPX1 activity may be a strong predictor of the risk of a subsequent cardiovascular event. Additionally, increasing GPX1 activity might lower the risk of cardiovascular events.30 In another prospective study among 643 patients with CVDs conducted by Schnabel et al., individuals with high GPX1 activity were more protected from the deleterious effects of elevated homocysteine on the cardiovascular system.31 Further, Espinola-Klein et al. found that in 508 patients with atherosclerosis, individuals with the lowest levels of GPX1 and the most extensive atherosclerosis were at the greatest risk for cardiovascular events.32 In addition, a meta-analysis of 22 case-control and 2 prospective studies identified a strong and statistically significant inverse associations of GPX1 activity with CVD outcomes. The pooled odds ratio for CVDs associated with a modest increase in GPX1 activity was 0.51.33 However, Yang et al. recently reported that GPX1 activity was not associated with CVDs in generally healthy women.34 This discrepancy is likely due to the different role of GPX1 in individuals with CVDs versus those without CVDs at the baseline. In an overall healthy population, the level of oxidative stress is low and GPX1 may not be the major antioxidant enzyme to neutralize free radicals. However, in individuals with existing CVDs, the level of oxidative stress can be exceptionally high, and GPX1 may play an important role in protecting cardiovascular tissue from oxidative damage after a cardiovascular event. Thus, an exhaustion of GPX1 activity can result in recurrent CVD events.
Besides GPX1, GPX3 also appears to prevent CVDs. GPX3 is a major antioxidant enzyme in plasma and the extracellular space that scavenges reactive oxygen species (ROS) produced during normal metabolism or after oxidative insult. Buijsse et al. have performed a nested, case-control analysis of stored serum samples from the Minnesota Heart Survey including 240 controls and 130 participants who had died of CVDs after 5 to 12 years of follow-up.35 They found that the serum GPX3 activity was inversely and linearly correlated with CVD mortality, and low serum GPX3 activity was associated with increased CVD mortality in individuals with low high density lipoprotein cholesterol.
2.3 Polymorphisms of selenoprotein genes and CVDs
In the past over 10 years, the discovery of disease-associated polymorphisms in selenoprotein genes has drawn attention to the relevance of selenoproteins to health.36 Genetic data showed that risk for CVDs was linked to a number of single nucleotide polymorphisms (SNPs) of selenoproteins. These SNPs could alter Se metabolism (including Se bioavailability), selenoprotein synthesis or activity, as well as the response to environmental stresses. Additionally, SNPs in selenoprotein genes were found to interact with biological markers of the Se status (including plasma Se and SelP levels) and genetic variants in other selenoprotein genes or other protein genes (such as superoxide dismutase).36 These observations reveal an extra level of complexity when considering the relationship between Se and health, and thus offer another explanation for the lack of consistency in clinical trials regarding Se supplementation and CVD prevention. To date, genetic polymorphisms in GPX1, GPX3 and SelS have been reported to be associated with CVDs.Overall, 38 SNPs have been reported for the GPX1 gene. One common polymorphism (at SNP rs1050450, commonly referred to as Pro198Leu) involves a Leu substitution in place of the more common Pro at amino acid 198 due to a substitution of T (codon, CTC) for a C (codon, CCC). Many studies have performed association analyses for Pro198Leu polymorphisms in GPX1 gene and CVDs. In one of these studies in Japan, the GPX1 Pro197Leu variant was found to associate with CVDs, including the increased intima-media thickness of the carotid arteries, coronary heart disease, and peripheral vascular disease in a small cohort of type-2 diabetics.37 Similarly, in another small study of 91 type 2 diabetic subjects in Japan, coronary artery calcification in individuals with the Pro197Leu genotype of the GPX1 gene was significantly higher than in those with Pro197Pro.38 Furthermore, GPX1 Pro197Leu variant genotypes are significantly associated with an increased risk of CVDs in a case-control study from China.39 In 2014, based on two cohort studies and 8 case-control studies (involving 1430 cases and 3767 controls), a meta-analysis showed that GPX1 gene Pro198Leu polymorphisms are related to significantly increased risks of CVDs, especially in East Asian populations.40 Recently, 2 case control studies showed different results with respect to association between Pro198Leu polymorphisms in GPX1 gene and CVDs. In a case control study from Sri Lanka, individuals possessing GPX Pro198Leu polymorphism were found to be associated with low erythrocyte GPX1 activity and increased susceptibility for CVDs.41 However, a case-control study from Tunisia showed a lack of association of GPX Pro198Leu polymorphism with CVD risk.42 The association of the GPX1 Pro197Leu variant with CVD risk may be due to the decrease in GPX1 expression. In many different cell types including aortic endothelial cells and cardiomyocytes, this Pro198Leu variant was found to be less responsive to the stimulation of Se supplementation and has reduced GPX1 enzymatic activity.29
Voetsch et al. identified a novel polymorphism in the promoter region of the GPX3 gene.43 They found that this genetic variant had decreased expression of GPX3 and was associated with an increased incidence of arterial ischemic stroke in children and young adults. Further, this genetic variant was also a strong, independent risk factor for cerebral venous thrombosis.44
SelS is localized to the ER membrane and has been implicated in the ER-associated degradation of misfolded proteins. Several studies have shown that SelS functions in cell survival by regulating ER stress.3 In addition, a large body of evidence implicated SelS in inflammation and the immune response. Moreover, SelS was proposed to function as an efficient reductase as well as a moderate peroxidase in vitro.3 The effect of genetic polymorphisms in SelS on CVD risk was first investigated in two independent prospective Finnish cohorts by Alanne et al.45 They found a significant association of SelS gene polymorphism with CVD risk, especially in females. Further, several SelS SNPs were observed to be associated with risk for subclinical CVDs in European Americans enriched for type-2 diabetes.46 These results suggest a potential role for SelS in the prevention of CVDs, since polymorphisms have been described to significantly decrease the expression of SelS.47
3 Animal studies for Se in the prevention of atherosclerosis
3.1 Se supplementation and atherosclerosis in animal studies
Evidence from animal studies showed consistent results that Se and selenoproteins might prevent experimental atherosclerosis. Atherosclerotic lesions as they occur in humans or as induced by high-fat diet feeding in experimental animals share some common properties. Three studies have considered the potential for Se supplementation or Se combined with vitamin E to inhibit experimental atherosclerosis induced by a high-fat diet in rabbits. In 1991, the first animal study reported that areas of atherosclerotic lesions in the total aorta of rabbits were reduced 49% by Se (22 μg Se per day, for 12 weeks), 63% by Se combined with vitamin E, and slightly (25%) but not significantly reduced by vitamin E.48 Later, Schwenke et al. showed the consistent results that areas of atherosclerotic lesions in the total aorta of rabbits were reduced 68% by Se (22 μg Se per day, for about 152 day) combined with vitamin E and 35% by vitamin E.49 Also, the inhibitory action of Se supplementation (the diet containing 1 μg sodium selenite per g, for 3 months) on the formation of atherosclerotic plaque in rabbits was further confirmed by Mehta et al.50 In addition, in a combined atherosclerosis and diabetes hamster model, the yeast with the highest Se (the diet containing 5.65 μg Se g−1, for 3 months) and glutathione levels was reported to inhibit the formation of atherosclerosis better than low Se/glutathione yeast supplementation.51 Very recently, a high-Se lentil diet (containing 0.33 μg Se per g, for 13 weeks) has been shown to protect against arsenic-induced atherosclerosis in apolipoprotein E knockout (ApoE−/−) mice.52 ApoE−/− mice have spontaneously increased plasma cholesterol levels in response to even a normal chow diet and develop atherosclerotic lesions with morphologic features and an arterial distribution resembling those in humans.53Table 2 summarizes animal studies with the dose, forms and time of Se supplementation and their effects on atherosclerosis. Thus, it is concluded from the above-mentioned animal studies that Se supplementation can prevent atherosclerosis.| Animals | Dose and forms of Se supplementation | Time of Se supplementation | Effects on atherosclerosis |
|---|---|---|---|
| a The forms of Se were unknown. | |||
| Rabbits | 22 μg Se per daya | 12 weeks | Areas of atherosclerotic lesions in the total aorta were reduced 49%48 |
| Rabbits | 22 μg Se per day plus 146 IU per day vitamin Ea | 152 ± 2 days | Areas of atherosclerotic lesions in the total aorta were reduced 68%49 |
| Rabbits | The diet containing 1 μg sodium selenite per g | 3 months | Development of atherosclerotic plaques was not visible; vascular cell apoptosis and oxidative stress were inhibited50,55 |
| Hamsters | The diet containing 5.65 μg Se per g plus 45.5 μg GSH per g, given as enriched yeast | 3 months | Areas of atherosclerotic lesions in the total aorta were reduced 87%; protected against oxidative stress and oxidation of LDL51 |
| ApoE−/− mice | High-Se lentil diet containing 0.33 μg Se per g | 13 weeks | Protected against arsenic-induced atherosclerosis by preventing oxidative stress52 |
However, it should be noted that since the safety window for Se intake is very narrow, the content of Se supplementation should be moderate, considering its toxicity.1,8,54 For example, an adequate Se level in the diet is 0.2 μg g−1, and the subtoxic level of Se is 2 μg g−1 for rabbits.50 Furthermore, choosing the most effective Se species is also essential because the biological activity of Se is dependent upon its speciation.54 Therefore, the different choices of Se supplementation forms may partially explain the inconsistent results observed in animal studies and clinical trials regarding Se supplementation and CVD prevention. Se-enriched yeast (predominantly as selenomethionine) has been the preferred form of Se for use in human studies. Yet animal studies use different Se forms such as selenite, Se-enriched yeast, and a high-Se diet, as shown in Table 2.
3.2 Selenoproteins and atherosclerosis in animal studies
Several selenoproteins have been shown to exist in the arterial wall. Among them, the most abundant selenoproteins are GPX1, GPX4 and TrxR1 (most prominent). Beyond, the plasma selenoproteins, GPX3 and SelP, may bind to endothelial cells in the arterial wall.56,57 Recently, our laboratory found that SelS was highly expressed in the arterial wall in rats.58 Meiler et al. found that SelK was expressed in large arteries of LDL receptor knockout (LDL R−/−) mice59 (LDL R−/− mice are mildly hypercholesterolemic due to the absence of hepatic LDL receptors which prolongs the plasma half lives of very low-density lipoprotein and LDL, and when fed a potent cholesterol-rich diet, they become severely hypercholesterolemic and develop aortic atherosclerosis60). These selenoproteins existing in the arterial wall may be involved in atherosclerotic lesion formation by different mechanisms.Because of its major role in the prevention of oxidative stress, GPX1 may be an important antiatherosclerotic enzyme. The most direct evidence that established a protective role for GPX1 in atherosclerosis comes from GPX1 and ApoE double-knockout mice (ApoE−/−/GPX1−/−). Two independent studies showed that GPX1 deficiency accelerated atherosclerotic lesion progression in ApoE−/− mice.61,62 One study found that after fed for 24 weeks with a Western-type diet, ApoE−/−/GPX1−/− mice developed significantly more atherosclerosis than control ApoE−/− mice.61 In the other study, ApoE−/−/GPX1−/− mice and ApoE−/− mice were made diabetic with streptozotocin. The results showed that atherosclerotic lesions within the aortic sinus region, as well as arch, thoracic, and abdominal lesions, were significantly increased in diabetic ApoE−/−/GPX1−/− aorta compared with diabetic ApoE−/− aorta.62 In support of a role for GPX1 in protecting against atherosclerosis, subsequent studies showed that ebselen, a seleno-organic GPX1 mimetic, substantially reduced atherosclerotic lesions in most regions of diabetic ApoE−/− aorta.63 However, in C57BL/J6 mice, GPX1 deficiency only did not accelerate atherosclerotic lesion development after 12 and 20 weeks on a high-fat diet,64 but resulted in structural alterations in the arterial wall, such as neointima formation and periadventitial inflammation.65 Moreover, it should be noted that overexpression of GPX1 has no benefit for the prevention of atherosclerosis. Instead, GPX1-overexpressing mice were shown to develop hyperglycemia and hyperinsulinemia, and they also developed high levels of blood insulin and increased islet β-cell mass, similar to phenotypes observed in Type-2 diabetes models.66,67
Among GPXs, GPX4 stands out because of its broad substrate specificity and ability to reduce complex lipid hydroperoxides.3 GPX4 is widely expressed in all normal tissues, including arteries, and is distributed among multiple subcellular sites such as the cytoplasm, ER, plasma membrane, mitochondria, and nucleus. As GPX4 is involved in the reduction of membrane-bound phospholipid and cholesterol hydroperoxides, inhibition of lipid peroxidation is considered to be its primary function in most tissues. It is well established that accumulation of oxidized lipids in the arterial wall contributes to atherosclerosis, but limited data are available regarding a protective role for GPX4 in the progression of atherogenesis. One study has demonstrated that overexpression of GPX4 in ApoE−/− mice inhibited atherosclerotic lesions in the aortic tree and aortic sinus, and also diminished signs of advanced lesions (fibrous caps and acellular areas) in the aortic sinus.68 Given the high levels of expression of this enzyme, the protective role of GPX4 in atherosclerosis and related CVDs warrants more attention.
However, it should be mentioned that SelK seems to promote atherosclerosis. SelK is also an ER membrane protein, and its expression is sensitive to dietary Se levels. Meiler et al. investigated the role of Selk in atherosclerotic lesion formation with the use of lethally irradiated LDL R−/− mice (fed with a high-fat diet) reconstituted with bone marrow from SelK knockout (Selk−/−) mice.59 They found that atherosclerotic lesion formation in the aorta of LDL R−/− mice reconstituted with Selk−/− bone marrow was decreased significantly compared with LDL R−/− mice transplanted with wild type bone marrow, which suggested that SelK deficiency reduced the formation of atherosclerotic lesions under the experimental conditions.
4 Mechanisms for Se in the prevention of atherosclerosis
Many studies have been performed to explore the molecular and cellular mechanisms by which Se and selenoproteins prevent atherosclerosis. The results of these works have showed that Se and selenoproteins may block many key processes in the development of atherosclerosis, including ROS-induced oxidative stress, inflammation (in particular adhesion and migration of monocytes, foam cell formation, abnormal eicosanoid metabolism), endothelial dysfunction (in particular the decrease of bioavailable NO), vascular cell apoptosis and vascular calcification (Fig. 1). In particular, Table 3 summarizes cellular studies with the dose, forms and pretreatment time of Se supplementation and their effects on atherosclerosis.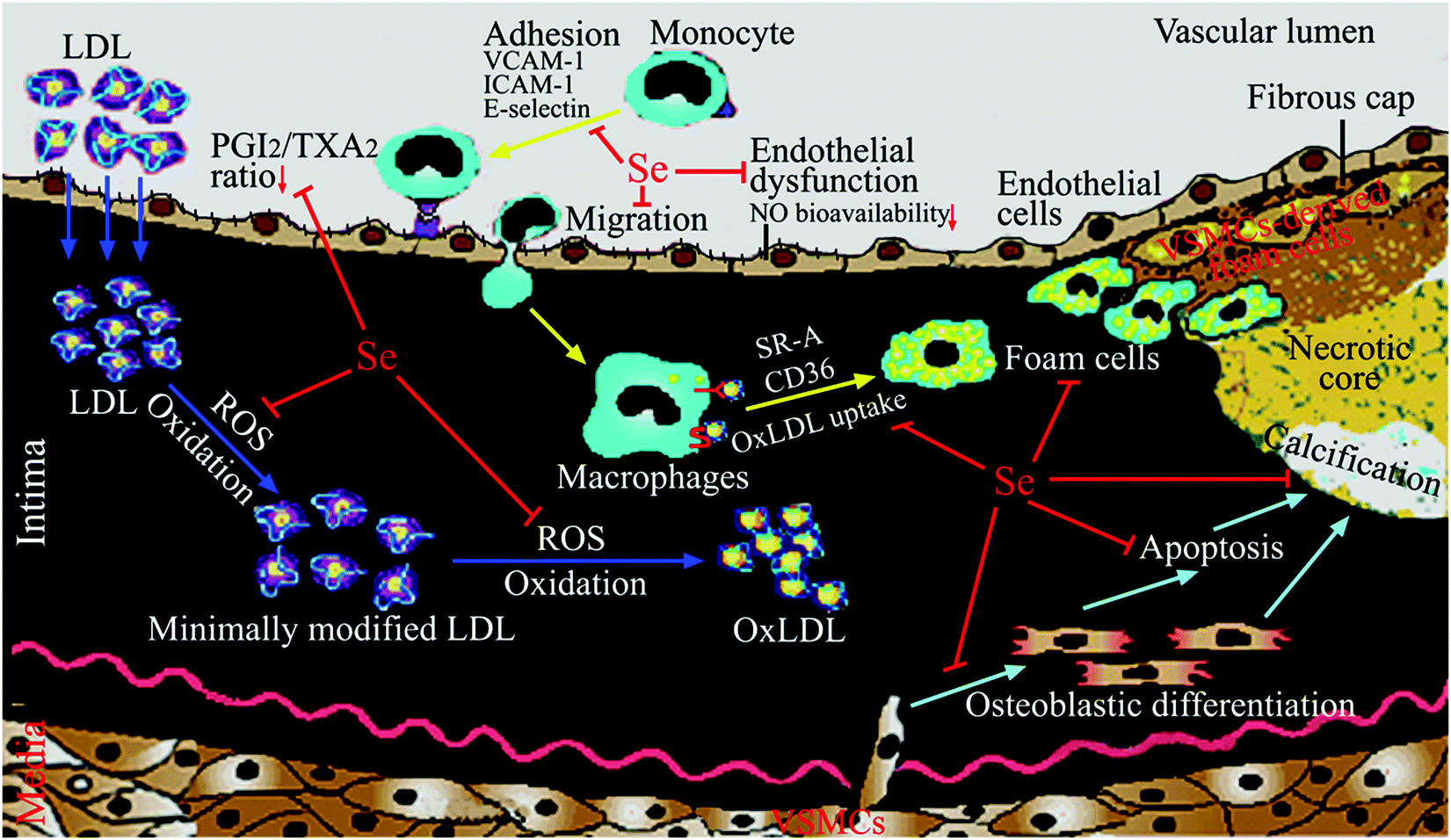 | ||
| Fig. 1 Mechanisms for Se in the prevention of atherosclerosis. Se and selenoproteins may inhibit ROS-induced LDL oxidation, adhesion and migration of monocytes, foam cell formation, abnormal eicosanoid metabolism, endothelial dysfunction (in particular the decrease of bioavailable NO), vascular cell apoptosis and vascular calcification. | ||
| Cells | Dose, forms and pretreatment time of Se supplementation | Key processes (crucial to atherosclerosis) modulated by Se | Molecular and cellular action of Se |
|---|---|---|---|
| a The forms of Se were unknown. | |||
| Human umbilical vein endothelial cells | 0.5, 1.5, and 2 μM sodium selenite for 24 h | Reduction of recruitment of inflammatory cells | Inhibited TNF-α-induced adhesion molecule expression69 |
| Human umbilical vein endothelial cells | 100 nM Se for 48 ha | Reduction of recruitment of inflammatory cells | Inhibited high glucose- and high insulin-induced adhesion molecule expression by inhibiting activation of p38 MAPK70 |
| Bovine aorta endothelial cells | Cultured in media supplemented with 10 ng ml−1 sodium selenite (57.8 nM) | Reduction of recruitment of inflammatory cells | Reduced fatty acid hydroperoxide-induced adhesion molecule expression accompanying by increased both GPX1 and TrxR1 enzymatic activities while decreased ROS accumulation71 |
| Human umbilical vein endothelial cells | 50, 100, or 200 nM Se for 12 ha | Inhibition of endothelial dysfunction and endothelial cell apoptosis | Reversed homocysteine-induced reduction of NO release by increasing the expression and phosphoylation of eNOS via AKT pathway; inhibited homocysteine-induced apoptosis of endothelial cells by downregulating caspase-3 activity, expression of caspase-3 and Bax, and by stimulating Bcl-2 expression72 |
| Rat VSMCs | 50 nM sodium selenite for 12 or 24 h | Reduction of VSMCs apoptosis | Inhibited oxysterol-induced VSMC apoptosis by reducing oxidative stress and intracellular Ca2+ level, whereas increasing activities of GPX1 and superoxide dismutase, and expression of c-myc, Bcl-2, GPX1, and TrxR173,74 |
| Rat VSMCs | 100 nM sodium selenite for 24 h | Suppression of vascular calcification | Suppressed oxidative stress-enhanced VSMC calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress75,76 |
4.1 Inhibition of oxidative stress by Se
An imbalance between oxidants (including ROS, reactive nitrogen species (RNS)) and antioxidants in favor of the oxidants, potentially leading to damage, is termed ‘oxidative stress’.77 Plenty of evidence from data of laboratory and clinical studies suggests that oxidative stress plays a pivotal role in the pathogenesis of atherosclerosis.11,12 Indeed, atherogenic risk factors such as hypercholesterolemia, hypertension, and diabetes mellitus enhance ROS generation in the arterial wall, resulting in oxidative stress. This leads to oxidative modification of lipids, particularly the formation of oxLDL, that contribute to atherogenesis.11,12Several pieces of evidence support that the anti-atherosclerosis effects of Se are mainly due to its antioxidant properties. In vitro studies from other laboratories and ours showed that Se supplementation (most as sodium selenite) increased expression and activity of GPX1, GPX4, TrxR1, and SelP in vascular endothelial cells or VSMCs and thus inhibited oxidative stress, LDL oxidation, cell damage and apoptosis from oxLDL or oxysterol.57,74,78–82 Oxysterol, oxygenated molecules derived from steroid compounds and a component of oxLDL, has been found in high concentrations in human atherosclerotic lesions and may have potent effects in atherosclerotic plaque formation.11 Similarly, animal studies from other groups exhibited that Se supplementation elevated GPX1 activity in the arterial wall, protected against oxidative stress and oxidation of LDL, and thus inhibited the atherosclerotic process.50,51,55,83 Data from our laboratory revealed that long-term Se deficiency in rats severely decreased the activity and expression of GPX1 and the total antioxidant capacity, and increased both physiological and oxysterol-induced oxidative stress and vascular damage.84–87 These effects could be reversed by dietary supplementation of Se (drinking water containing 1 μg Se per ml, given as sodium selenite).85–87 In Se-deficient humans, Se supplementation has also been shown to increase enzymatic antioxidant activity and decrease lipid peroxidation.21 Finally, Se supplementation may suppress atherosclerosis induced by toxic metals, such as mercury and arsenic, by preventing metal-induced oxidative damage or by forming nontoxic complexes with metals.52,88
A further indication linking the antioxidant capacity of Se and atherosclerosis is demonstrated in knockout and transgenic studies. In ApoE−/− mice, GPX1 knockout augmented markers associated with oxidative stress including ROS production and nitrotyrosine levels in the arterial wall.61,62 These effects could be reversed by Ebselen treatment.63 Conversely, overexpression of GPX4 significantly reduced lipid peroxidation and inhibited the sensitivity of vascular cells to oxidized lipids in the atherosclerosis mouse model.68
4.2 Modulation of inflammation by Se
Atherosclerosis has been considered to be a chronic inflammatory disease resulting from the interaction between modified lipoproteins (in particular oxidative modification), monocyte-derived macrophages, T cells, and the normal cellular elements of the arterial wall.10 Dietary Se, mainly through its incorporation into selenoproteins, plays an important role in inflammation and immunity.89 Many studies supported that Se could also block inflammatory events crucial to atherosclerosis, including recruitment and migration of monocytes, foam cell formation, and eicosanoid metabolism, thus inhibiting the progress of atherosclerosis.The inhibition of Se on adhesion molecule expression may be mainly due to the antioxidant and redox-regulating properties of GPX1. In human microvascular endothelial cells, Lubos et al. found that GPX1 deficiency and its accompanying oxidative stress augmented TNF-α-induced ICAM-1 and VCAM-1 expression primarily via activation of nuclear transcription factor kappa B (NF-κB) and c-Jun N-terminal kinase (JNK), whereas overexpression of GPX1 attenuated these TNF-α-mediated responses.92 Similarly, they also found that GPX1 deficiency enhanced lipopolysaccharide (LPS)-induced intracellular ROS accumulation, and expression of VCAM-1, ICAM-1, in part, by altering CD14-mediated effects. Conversely, overexpression of GPX1 significantly diminished LPS-induced adhesion molecule expression. Consistent with these results, LPS responses were also greater in endothelial cells derived from GPX1-knockout mice, whereas decreased in cells from GPX1-overexpressing transgenic mice.93 In addition, Wagner et al. showed that knockdown of GPX1 expression in endothelial cells reinforced H2O2 generation, expression of monocyte chemoattractant protein-1 and VCAM-1, and subsequent monocyte adhesion to endothelial cells after cyclic stretch.94 Also GPX mimics, such as ebselen, BXT-51072 and BXT-51077, were reported to prevent TNF-α-induced expression of adhesion molecules in endothelial cells.95,96 Recently, Sharma et al. demonstrated that GPX1 deficiency resulted in sustained activation of MAPK (extracellular-regulated kinase (ERK), p38 and JNK) and NF-κB pathways, and increased leukocyte adhesion to the vascular endothelium, all of which are rescued with ebselen treatment.97 Collectively, all these findings suggest a crucial role for GPX1 in inhibiting the expression of adhesion molecules in relation to atherosclerosis.
Besides, GPX4 may also be involved in modulating the expression of adhesion molecules. The SNP (GPX4c718t) in the GPX4 gene, which decreased the expression of this enzyme, was found to enhance monocyte adhesion to endothelial cells.98 In contrast, overexpression of GPX4 inhibited IL-1-induced NF-κB activation99 and VCAM-1 expression.100
Based on the above discussion, it could be concluded that the anti-inflammatory properties of Se play a vital role in its prevention of atherosclerosis. However, it should be noted the anti-inflammatory properties of Se still rely on its antioxidant properties. In fact, the vicious circle between oxidative stress and inflammation exists in atherosclerosis.110 For example, ox-LDL activates circulating monocytes, thereby increasing their ability to migrate into the vascular wall and differentiate into macrophages; meanwhile activated macrophages further increase the production of ROS and ox-LDL by secreting oxidant enzymes. Thus, the antioxidant properties and anti-inflammatory properties of Se are promotive interdependent.
4.3 Inhibition of endothelial dysfunction by Se
Vascular endothelial cells play a crucial role in regulating vascular homeostasis, in part, through their production of nitric oxide (NO). Under normal physiological conditions, endothelial cells generate NO from arginine catalyzed by endothelial nitric oxide synthase (eNOS). NO is the most potent endogenous vasodilator. It also inhibits cytokine-mediated adhesion molecule expression, and attenuates VSMC growth and platelet aggregation. Thus, normal production of NO is antithrombotic and antiatherogenic. Oxidative stress can counteract these protective actions by promoting endothelial dysfunction. Endothelial dysfunction implies a loss of normal endothelial function characterized by a decrease in bioavailable NO and a loss in normal endothelium-dependent vasorelaxation responses111 (broadly speaking, in addition to decreased NO bioavailability, endothelial dysfunction is also characterized by the impairment of endothelial barrier function and promotion of monocyte adhesion and migration,112,113 which has been discussed above). NO can interact with superoxide resulting in the production of the more reactive peroxynitrite that further enhances oxidant production; modifies DNA, proteins, and lipids; and has other cytotoxic effects. An overproduction of superoxide, as present in inflammatory processes, lowers and inactivates locally produced NO, leading to endothelial dysfunction. Hydrogen peroxide can also contribute to endothelial dysfunction by a number of mechanisms that may ultimately influence NO bioavailability.114 Se supplementation for 3 days (intraperitoneal injection of 4.33 μmol sodium selenite per kg body weight per day) was shown to enhance acetylcholine-induced aortic ring relaxation by increasing NO release in rats.115 This effect has been achieved probably by increasing the expression of TrxR1 since TrxR1 can regenerate reduced Trx and overexpression of Trx could prevent inactivation of eNOS in porcine pulmonary artery endothelial cells.116 Recently, Ren et al. reported that Se supplementation reversed homocysteine-induced impairment of endothelium-dependent thoracic aortic ring relaxation and reduction of NO release.72 This effect has been achieved by increasing the expression and phosphorylation of eNOS via an AKT pathway. Furthermore, Se also inhibited homocysteine-induced apoptosis of endothelial cells by downregulating caspase-3 activity and expression of caspase-3 and Bax, and by stimulating Bcl-2 expression.72More attention has been paid to the role of GPX1 in the regulation of endothelial function. Homocysteine inhibits the expression of GPX1 in vitro and in vivo, which can lead to an increase in ROS that inactivate NO and promote endothelial dysfunction. GPX1 deficiency exacerbated endothelial dysfunction in hyperhomocysteinemic mice,117 whereas overexpression of GPX1 in hyperhomocysteinemic mice restored the normal endothelial function by increasing bioavailable NO.118 Additional studies suggest that GPX1 protects against angiotensin II-induced endothelial dysfunction. In vascular cells, angiotensin II can stimulate ROS through the activation of NADPH oxidase, leading to a reduction in bioavailable NO and subsequent endothelial dysfunction.119 GPX1 deficiency was found to augment endothelial dysfunction in response to angiotensin II in carotid arteries of heterozygous GPX1+/− mice, whereas GPX1 overexpression decreased the harmful effects of angiotensin II on endothelium-dependent relaxation. An elegant study reported by Oelze et al. highlighted the important role of GPX1 in aging-associated endothelial dysfunction. In their study, GPX1 deficiency was shown to exacerbate the decrease in NO bioavailability because of eNOS uncoupling and the reduction in acetylcholine-induced endothelium-dependent vasodilation during the aging process.120 Further study using primary aortic endothelial cells isolated from GPX1 knockout mice showed that GPX1 deficiency decreases NO bioavailability by reducing AKT signaling and subsequently inactivating eNOS.97 Taken together, these studies suggest that GPX1 plays an essential role in preserving normal endothelial function and NO bioavailability.
4.4 Reduction of vascular cell apoptosis by Se
Vascular cell apoptosis, especially apoptotic VSMCs seen in the advanced atherosclerotic, results in plaque instability and rupture, leading to thrombosis with clinical manifestations of heart attack or stroke.121–123 Oxidative stress and ER stress, two intracellular stresses that have been implicated in the pathogenesis of atherosclerosis,11,12,124 can induce vascular cell apoptosis.125–128 An earlier study showed that Se supplementation inhibited apoptosis of vascular cells, particularly VSMCs, in experimental atherosclerosis in rabbits.55 Our previous studies showed that sodium selenite inhibited oxysterol-induced apoptosis of rat VSMCs by reducing oxidative stress and the intracellular Ca2+ level, whereas increasing the activities of GPX1 and superoxide dismutase, and the mRNA expression of c-myc, Bcl-2, GPX1, and TrxR1.73,74 Recently, we found that SelS gene silence by small interference RNA (siRNA) rendered VSMCs more sensitive to oxidative stress-induced apoptosis by activating p38 MAPK and JNK pathways. SelS silence also aggravated ER stress-induced apoptosis of VSMCs.58 These results suggested that SelS highly expressed in the blood vessel might protect VSMCs from apoptosis by inhibiting oxidative stress and ER stress. Moreover, overexpression of GPX4 inhibits H2O2-induced oxidation, NF-κB activation and apoptosis of rabbit VSMCs.129 Collectively, these findings suggest that Se may inhibit VSMC apoptosis thereby maintaining plaque stability, which provides another mechanistic explanation for the potential preventive role of Se in atherosclerosis.4.5 Suppression of vascular calcification by Se
Vascular calcification, the deposition of hydroxyapatite in the arterial wall, is a highly prevalent pathological phenotype associated with atherosclerosis.130–132 This pathology underlies substantial cardiovascular morbidity and mortality, through adverse mechanical effects on vascular compliance, vasomotion, and, most likely, plaque stability. Accumulating evidence supports the concept that vascular calcification is an active, highly regulated process similar to osteogenesis, instead of a passive deposition of hydroxyapatite in the arterial wall.130–132 VSMCs play an integral role in mediating vascular calcification.123,131,132 Apoptotic bodies derived from VSMCs can act as nucleating structures for calcium phosphate deposition to initiate vascular calcification.115,124,125 Additionally, VSMCs undergo osteoblastic differentiation and express Runx2, a key osteogenic transcription factor, and many other bone formation-associated proteins, such as type I collagen, alkaline phosphatase, and osteocalcin, which results in the deposition of calcium phosphate mineral in the extracellular matrix.131 Increasing evidence suggests that oxidative stress has been implicated in the pathogenesis of vascular calcification and promotes osteoblastic differentiation of VSMCs.131 For example, Byon et al. demonstrated that oxidative stress induced vascular calcification through modulation of Runx2 by phosphatidylinositol 3-kinase (PI3K)/AKT signaling.133 Minimally modified oxidized LDL and different lipid peroxidation products induced VSMCs to undergo osteoblastic differentiation.134 Our previous experiments demonstrated that oxysterol increased intracellular ROS and enhanced osteoblastic differentiation of VSMCs, and that these effects were attenuated by antioxidant vitamin C plus vitamin E.135 Moreover, oxidative stress can activate ER stress, which is another mechanism by which VSMCs undergo osteoblastic differentiation.131 Liberman et al. found that ER stress stimulated by bone morphogenetic protein-2-induced oxidative stress promoted VSMC calcification.136 Other studies found that ER stress-mediated apoptosis was activated in vascular calcification137 and activating transcription factor 4 is involved in ER stress-mediated apoptosis contributing to vascular calcification.138Considering the antioxidant properties of Se, we examined the effect of sodium selenite on oxidative stress-induced vascular calcification, by using H2O2 or xanthine/xanthine oxidase (XXO) as oxidative stress inducers.75,76 The results showed that Se suppressed osteoblastic differentiation and calcification of VSMCs induced by H2O2 or XXO, as demonstrated by the decrease of alkaline phosphatase activity, the expression of Runx2, type I collagen and osteocalcin, and calcium deposition in the extracellular matrix. This finding indicates a potential preventive role for Se in vascular calcification, suggesting that vascular calcification may be another target of the Se action in anti-atherosclerosis. Based on the results of mechanistic studies, we proposed a potential mechanism by which Se suppressed oxidative stress-enhanced vascular calcification (Fig. 2). Se first suppresses oxidative stress by increasing the activity of antioxidant selenoenzymes such as GPX1. Subsequently, Se inhibits the activation of redox-sensitive PI3K/AKT and ERK signaling pathways, resulting in decreased osteoblastic differentiation of VSMCs. Additionally, Se alleviated oxidative stress-activated ER stress, most likely via ER-resident selenoproteins, thereby leading to decreased apoptosis. Taken together, the decreased osteoblastic differentiation and apoptosis of VSMCs result in the reduction of calcium deposition in the extracellular matrix.
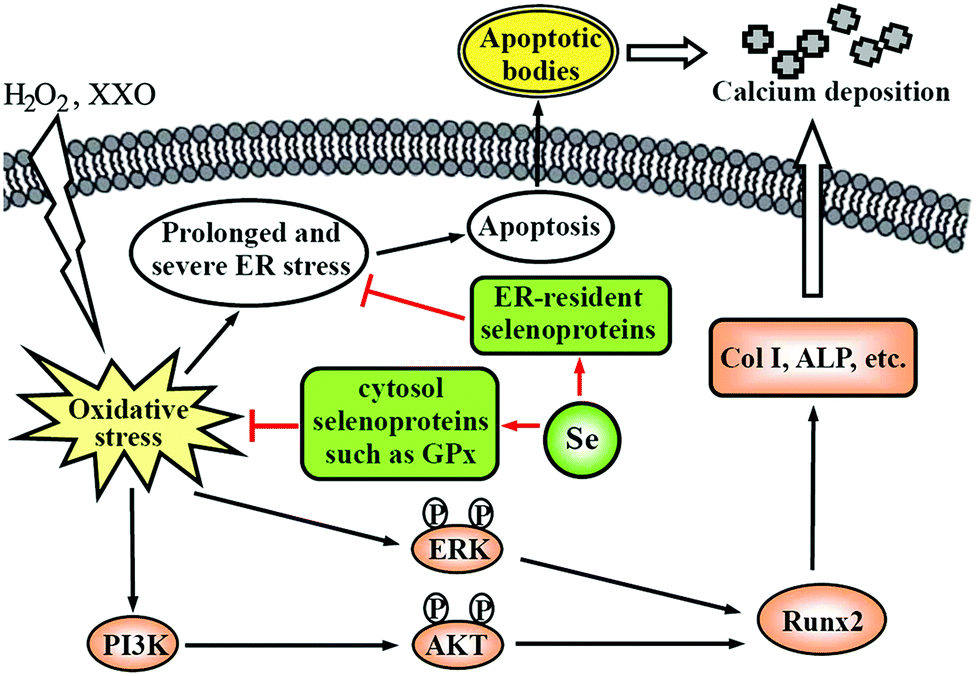 | ||
| Fig. 2 A potential mechanism by which Se suppresses oxidative stress-enhanced VSMC calcification. Col I: type I collagen; ALP: alkaline phosphatase. This scheme is adapted from ref. 76. | ||
5 Conclusion and future research
Although findings from a limited number of randomized trials are inconsistent and the protective effect of Se supplementation on CVDs is inconclusive, Se supplementation can confer benefit in population with low baseline Se status. Furthermore, most of the available data from observational studies have shown a significant inverse association between Se status and CVDs. Besides, the low activities and polymorphisms of some selenoproteins (GPX1, GPX3 and SelS) have been shown to be associated with CVDs. Additionally, a great deal of laboratory studies support the concept that optimal Se intake can prevent atherosclerosis by inhibiting oxidative stress, inflammation, endothelial dysfunction, vascular cell apoptosis, and vascular calcification. These actions of Se are mainly executed by selenoproteins existing in the arterial wall. Selenoproteins that may be relevant to atherosclerosis prevention are described in Table 4 and include GPX1, GPX3, GPX4, TrxR, SelP and SelS. Therefore, it can be concluded that the optimal Se status may benefit cardiovascular health and prevent atherosclerotic CVDs.| Selenoprotein | Functions by which prevent atherosclerosis | Important references |
|---|---|---|
| GPX1 | Antioxidant enzyme in intracellular space: remove H2O2 and lipid peroxides thereby inhibiting oxidative stress, expression of cell adhesion molecules, macrophage foam cell, and endothelial dysfunction; regulate eicosanoid metabolism | 29–33, 37–41, 61, 65, 92–97, 102 and 117–120 |
| GPX3 | Antioxidant enzyme in plasma and the extracellular space: bind to endothelial cells in the arterial wall and remove H2O2 and lipid peroxides thereby inhibiting oxidative stress | 35, 43 and 44 |
| GPX4 | Antioxidant enzyme in cytoplasm, ER, plasma membrane, mitochondria, and nucleus: remove membrane-bound phospholipid and cholesterol hydroperoxides thereby inhibiting oxidative stress, expression of cell adhesion molecules, and vascular cell apoptosis; regulate eicosanoid metabolism | 68, 98–100 and 129 |
| SelS | ER membrane protein and component of ER associated degradation: inhibit oxidative stress, ER stress, and vascular cell apoptosis | 45, 46 and 58 |
| SelP | Bind to endothelial cells in the arterial wall and inhibit oxidative stress | 57, 80 and 82 |
| TrxR1 | The most prominent selenoprotein in vascular endothelial cell: regeneration of Trx and maintenance of intracellular redox state thereby inhibiting oxidative stress and endothelial dysfunction | 78, 81 and 116 |
However, there are many challenges in research into Se as a supplement for CVD prevention. First, the relationship between the Se status and CVD risk can be represented by a U-shaped curve, but the markers of the optimal Se status still remain to be determined. Although the Se level in blood is an easily accessible marker of the human Se nutritional status, the Se level itself does not reflect its functional significance. The plasma SelP level and plasma GPX3 activity have been widely used in discriminating of the Se status, but it has been suggested recently that the expression of selenoprotein mRNA in circulating blood leukocytes could differently reflect the Se status, due to prioritization of specific selenoprotein synthesis in response to dietary Se supplementation. The use of selenoprotein transcripts as a molecular biomarker of the Se status requires further studies on a large group of healthy individuals with different baseline Se, including data regarding genetic polymorphism of selenoproteins and data regarding the various metabolic pathways of Se compounds.139,140 Secondly, Se speciation is vitally important to the health outcomes of Se supplementation studies. Based on the current understanding of the metabolism of common dietary Se compounds, Weekley and Harris suggest that SeMet is a good choice for Se supplementation to correct a deficiency; the methylated forms of Se, with lower toxicities compared to selenite and in the absence of a route for adventitious incorporation into proteins, may be a good choice for future clinical trials of Se supplementation in disease prevention.54 However, further investigation of the metabolism and distribution of the different dietary forms of Se in vivo is needed in order to determine which form is the most effective. Thirdly, while the protective role of GPX1 in atherosclerosis has been established, other selenoproteins (including GPX3, GPX4, TrxR, SelP and SelS) have been studied in relation to atherosclerosis with findings worthy of mention. Much more studies are needed to confirm their roles in atherosclerosis and clarify their underlying mechanisms. In particular, screening for relevant polymorphisms of other selenoproteins with atherosclerotic CVDs is essential, which may also lead to a greater understanding of individual variation in Se supplementation.
In summary, there is a long way to enable the clinical use of Se in CVD prevention. In the future, more and large randomized trials of Se supplementation are needed to confirm the beneficial effect of Se on CVDs. Considering the complexity of Se biology, the dose and forms of Se supplementation, the baseline Se status and the selenoprotein genotype of the target population should be specifically emphasized in the design of a prevention strategy. In continuing laboratory studies, much attention should be paid to the roles of some selenoproteins (including GPX3, GPX4, TrxR, SelP and SelS) in atherosclerosis prevention and their underlying mechanisms.
Acknowledgements
The research studies conducted by the authors were supported by the National Natural Science Foundation of China (grant no. 29741003, 29971012, 30700136, and 31170775), Excellent Youth Foundation of Hubei Scientific Committee (grant no. 2014CFA022), and the “Youth Chen-Guang Project” of Wuhan Bureau of Science and Technology (grant no. 2015070404010184).References
- M. P. Rayman, Selenium and human health, Lancet, 2012, 379, 1256–1268 CrossRef CAS.
- S. J. Fairweather-Tait, Y. Bao, M. R. Broadley, R. Collings, D. Ford, J. E. Hesketh and R. Hurst, Selenium in human health and disease, Antioxid. Redox Signaling, 2011, 14, 1337–1383 CrossRef CAS PubMed.
- V. M. Labunskyy, D. L. Hatfield and V. N. Gladyshev, Selenoproteins: molecular pathways and physiological roles, Physiol. Rev., 2014, 94, 739–777 CrossRef CAS PubMed.
- G. V. Kryukov, S. Castellano, S. V. Novoselov, A. V. Lobanov, O. Zehtab, R. Guigo and V. N. Gladyshev, Characterization of mammalian selenoproteomes, Science, 2003, 300, 1439–1443 CrossRef CAS PubMed.
- D. L. Hatfield, P. A. Tsuji, B. A. Carlson and V. N. Gladyshev, Selenium and selenocysteine: roles in cancer, health, and development, Trends Biochem. Sci., 2014, 39, 112–120 CrossRef CAS PubMed.
- M. V. Kasaikina, D. L. Hatfield and V. N. Gladyshev, Understanding selenoprotein function and regulation through the use of rodent models, Biochim. Biophys. Acta, 2012, 1823, 1633–1642 CrossRef CAS PubMed.
- A. H. Rose and P. R. Hoffmann, Selenoproteins and cardiovascular stress, Thromb. Haemostasis, 2015, 113, 494–504 CrossRef PubMed.
- M. P. Rayman, The importance of selenium to human health, Lancet, 2000, 356, 233–241 CrossRef CAS.
- L. Schomburg and U. Schweizer, Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium, Biochim. Biophys. Acta, 2009, 1790, 1453–1462 CrossRef CAS PubMed.
- C. K. Glass and J. L. Witztum, Atherosclerosis. the road ahead, Cell, 2001, 104, 503–516 CrossRef CAS PubMed.
- R. Stocker and J. F. Keaney, Jr., Role of oxidative modifications in atherosclerosis, Physiol. Rev., 2004, 84, 1381–1478 CrossRef CAS PubMed.
- F. Bonomini, S. Tengattini, A. Fabiano, R. Bianchi and R. Rezzani, Atherosclerosis and oxidative stress, Histol. Histopathol., 2008, 23, 381–390 CAS.
- B. Keshan and Disease Research Group of the Chinese Academy of Medical Sciences, Epidemiologic studies on the etiologic relationship of selenium and Keshan disease, China Med. J., 1979, 92, 477–482 Search PubMed.
- B. Keshan and Disease Research Group of the Chinese Academy of Medical Sciences, Observations on effect of sodium selenite in prevention of Keshan disease, China Med. J., 1979, 92, 471–476 Search PubMed.
- A. Navas-Acien, J. Bleys and E. Guallar, Selenium intake and cardiovascular risk: what is new, Curr. Opin. Lipidol., 2008, 19, 43–49 CrossRef CAS PubMed.
- S. Stranges, A. Navas-Acien, M. P. Rayman and E. Guallar, Selenium status and cardiometabolic health: state of the evidence, Nutr., Metab. Cardiovasc. Dis., 2010, 20, 754–760 CrossRef CAS PubMed.
- S. Tanguy, S. Grauzam, J. de Leiris and F. Boucher, Impact of dietary selenium intake on cardiac health: experimental approaches and human studies, Mol. Nutr. Food Res., 2012, 56, 1106–1121 CAS.
- J. Joseph and J. Loscalzo, Selenistasis: epistatic effects of selenium on cardiovascular phenotype, Nutrients, 2013, 5, 340–358 CrossRef CAS PubMed.
- C. Benstoem, A. Goetzenich, S. Kraemer, S. Borosch, W. Manzanares, G. Hardy and C. Stoppe, Selenium and its supplementation in cardiovascular disease – what do we know, Nutrients, 2015, 7, 3094–3118 CrossRef CAS PubMed.
- X. Zhang, C. Liu, J. Guo and Y. Song, Selenium status and cardiovascular diseases: meta-analysis of prospective observational studies and randomized controlled trials, Eur. J. Clin. Nutr., 2016, 70, 162–169 CrossRef CAS PubMed.
- G. Flores-Mateo, A. Navas-Acien, R. Pastor-Barriuso and E. Guallar, Selenium and coronary heart disease: a meta-analysis, Am. J. Clin. Nutr., 2006, 84, 762–773 CAS.
- K. Rees, L. Hartley, C. Day, N. Flowers, A. Clarke and S. Stranges, Selenium supplementation for the primary prevention of cardiovascular disease, Cochrane Database Syst. Rev., 2013, CD009671, DOI:10.1002/14651858.CD009671.pub2.
- M. P. Rayman, S. C. Bath, J. Westaway, P. Williams, J. Mao, J. J. Vanderlelie, A. V. Perkins and C. W. Redman, Selenium status in U.K. pregnant women and its relationship with hypertensive conditions of pregnancy, Br. J. Nutr., 2015, 113, 249–258 CrossRef CAS PubMed.
- U. Alehagen, P. Johansson, M. Bjornstedt, A. Rosen and U. Dahlstrom, Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: a 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens, Int. J. Cardiol., 2013, 167, 1860–1866 CrossRef PubMed.
- U. Alehagen, J. Aaseth and P. Johansson, Reduced cardiovascular mortality 10 years after supplementation with selenium and coenzyme Q10 for four years: follow-up results of a prospective randomized double-blind placebo-controlled trial in elderly citizens, PLoS One, 2015, 10, e0141641 Search PubMed.
- U. Alehagen, J. Alexander and J. Aaseth, Supplementation with selenium and coenzyme Q10 reduces cardiovascular mortality in elderly with low selenium status. A secondary analysis of a randomised clinical trial, PLoS One, 2016, 11, e0157541 Search PubMed.
- M. P. Rayman and S. Stranges, Epidemiology of selenium and type 2 diabetes: can we make sense of it, Free Radical Biol. Med., 2013, 65, 1557–1564 CrossRef CAS PubMed.
- J. Bleys, A. Navas-Acien and E. Guallar, Serum selenium levels and all-cause, cancer, and cardiovascular mortality among US adults, Arch. Intern. Med., 2008, 168, 404–410 CrossRef CAS PubMed.
- E. Lubos, J. Loscalzo and D. E. Handy, Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities, Antioxid. Redox Signaling, 2011, 15, 1957–1997 CrossRef CAS PubMed.
- S. Blankenberg, H. J. Rupprecht, C. Bickel, M. Torzewski, G. Hafner, L. Tiret, M. Smieja, F. Cambien, J. Meyer and K. J. Lackner, Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease, N. Engl. J. Med., 2003, 349, 1605–1613 CrossRef CAS PubMed.
- R. Schnabel, K. J. Lackner, H. J. Rupprecht, C. Espinola-Klein, M. Torzewski, E. Lubos, C. Bickel, F. Cambien, L. Tiret, T. Munzel and S. Blankenberg, Glutathione peroxidase-1 and homocysteine for cardiovascular risk prediction: results from the AtheroGene study, J. Am. Coll. Cardiol., 2005, 45, 1631–1637 CrossRef CAS PubMed.
- C. Espinola-Klein, H. J. Rupprecht, C. Bickel, R. Schnabel, S. Genth-Zotz, M. Torzewski, K. Lackner, T. Munzel and S. Blankenberg, Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis, Am. J. Cardiol., 2007, 99, 808–812 CrossRef CAS PubMed.
- G. Flores-Mateo, P. Carrillo-Santisteve, R. Elosua, E. Guallar, J. Marrugat, J. Bleys and M. I. Covas, Antioxidant enzyme activity and coronary heart disease: meta-analyses of observational studies, Am. J. Epidemiol., 2009, 170, 135–147 CrossRef PubMed.
- S. Yang, M. K. Jensen, E. B. Rimm, W. Willett and T. Wu, Erythrocyte superoxide dismutase, glutathione peroxidase, and catalase activities and risk of coronary heart disease in generally healthy women: a prospective study, Am. J. Epidemiol., 2014, 180, 901–908 CrossRef PubMed.
- B. Buijsse, D. H. Lee, L. Steffen, R. R. Erickson, R. V. Luepker, D. R. Jacobs, Jr. and J. L. Holtzman, Low serum glutathione peroxidase activity is associated with increased cardiovascular mortality in individuals with low HDLc's, PLoS One, 2012, 7, e38901 CAS.
- C. Meplan, Selenium and chronic diseases: a nutritional genomics perspective, Nutrients, 2015, 7, 3621–3651 CrossRef CAS PubMed.
- T. Hamanishi, H. Furuta, H. Kato, A. Doi, M. Tamai, H. Shimomura, S. Sakagashira, M. Nishi, H. Sasaki, T. Sanke and K. Nanjo, Functional variants in the glutathione peroxidase-1 (GPx-1) gene are associated with increased intima-media thickness of carotid arteries and risk of macrovascular diseases in japanese type 2 diabetic patients, Diabetes, 2004, 53, 2455–2460 CrossRef CAS PubMed.
- M. Nemoto, R. Nishimura, T. Sasaki, Y. Hiki, Y. Miyashita, M. Nishioka, K. Fujimoto, T. Sakuma, T. Ohashi, K. Fukuda, Y. Eto and N. Tajima, Genetic association of glutathione peroxidase-1 with coronary artery calcification in type 2 diabetes: a case control study with multi-slice computed tomography, Cardiovasc. Diabetol., 2007, 6, 23 CrossRef PubMed.
- N. P. Tang, L. S. Wang, L. Yang, H. J. Gu, Q. M. Sun, R. H. Cong, B. Zhou, H. J. Zhu and B. Wang, Genetic variant in glutathione peroxidase 1 gene is associated with an increased risk of coronary artery disease in a Chinese population, Clin. Chim. Acta, 2008, 395, 89–93 CrossRef CAS PubMed.
- J. X. Zhang, Z. M. Wang, J. J. Zhang, L. L. Zhu, X. F. Gao and S. L. Chen, Association of glutathione peroxidase-1 (GPx-1) rs1050450 Pro198Leu and Pro197Leu polymorphisms with cardiovascular risk: a meta-analysis of observational studies, J. Geriatr. Cardiol., 2014, 11, 141–150 Search PubMed.
- D. Wickremasinghe, H. Peiris, L. G. Chandrasena, V. Senaratne and R. Perera, Case control feasibility study assessing the association between severity of coronary artery disease with Glutathione Peroxidase-1 (GPX-1) and GPX-1 polymorphism (Pro198Leu), BMC Cardiovasc. Disord., 2016, 16, 111 CrossRef PubMed.
- Y. Souiden, H. Mallouli, S. Meskhi, Y. Chaabouni, A. Rebai, F. Cheour and K. Mahdouani, MnSOD and GPx1 polymorphism relationship with coronary heart disease risk and severity, Biol. Res., 2016, 49, 22 CrossRef PubMed.
- B. Voetsch, R. C. Jin, C. Bierl, K. S. Benke, G. Kenet, P. Simioni, F. Ottaviano, B. P. Damasceno, J. M. Annichino-Bizacchi, D. E. Handy and J. Loscalzo, Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: a novel risk factor for arterial ischemic stroke among young adults and children, Stroke, 2007, 38, 41–49 CrossRef CAS PubMed.
- B. Voetsch, R. C. Jin, C. Bierl, L. Deus-Silva, E. C. Camargo, J. M. Annichino-Bizacchi, D. E. Handy and J. Loscalzo, Role of promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene as a risk factor for cerebral venous thrombosis, Stroke, 2008, 39, 303–307 CrossRef CAS PubMed.
- M. Alanne, K. Kristiansson, K. Auro, K. Silander, K. Kuulasmaa, L. Peltonen, V. Salomaa and M. Perola, Variation in the selenoprotein S gene locus is associated with coronary heart disease and ischemic stroke in two independent Finnish cohorts, Hum. Genet., 2007, 122, 355–365 CrossRef CAS PubMed.
- A. J. Cox, A. B. Lehtinen, J. Xu, C. D. Langefeld, B. I. Freedman, J. J. Carr and D. W. Bowden, Polymorphisms in the Selenoprotein S gene and subclinical cardiovascular disease in the Diabetes Heart Study, Acta Diabetol., 2013, 50, 391–399 CrossRef CAS PubMed.
- J. E. Curran, J. B. Jowett, K. S. Elliott, Y. Gao, K. Gluschenko, J. Wang, D. M. Abel Azim, G. Cai, M. C. Mahaney, A. G. Comuzzie, T. D. Dyer, K. R. Walder, P. Zimmet, J. W. MacCluer, G. R. Collier, A. H. Kissebah and J. Blangero, Genetic variation in selenoprotein S influences inflammatory response, Nat. Genet., 2005, 37, 1234–1241 CrossRef CAS PubMed.
- J. Wojcicki, L. Rozewicka, B. Barcew-Wiszniewska, L. Samochowiec, S. Juzwiak, D. Kadlubowska, S. Tustanowski and Z. Juzyszyn, Effect of selenium and vitamin E on the development of experimental atherosclerosis in rabbits, Atherosclerosis, 1991, 87, 9–16 CrossRef CAS PubMed.
- D. C. Schwenke and S. R. Behr, Vitamin E combined with selenium inhibits atherosclerosis in hypercholesterolemic rabbits independently of effects on plasma cholesterol concentrations, Circ. Res., 1998, 83, 366–377 CrossRef CAS PubMed.
- U. Mehta, B. P. Kang, R. S. Kukreja and M. P. Bansal, Ultrastructural examination of rabbit aortic wall following high-fat diet feeding and selenium supplementation: a transmission electron microscopy study, J. Appl. Toxicol., 2002, 22, 405–413 CrossRef CAS PubMed.
- G. A. Agbor, J. A. Vinson, S. Patel, K. Patel, J. Scarpati, D. Shiner, F. Wardrop and T. A. Tompkins, Effect of selenium- and glutathione-enriched yeast supplementation on a combined atherosclerosis and diabetes hamster model, J. Agric. Food Chem., 2007, 55, 8731–8736 CrossRef CAS PubMed.
- R. M. Krohn, M. Lemaire, L. F. Negro Silva, C. Lemarie, A. Bolt, K. K. Mann and J. E. Smits, High-selenium lentil diet protects against arsenic-induced atherosclerosis in a mouse model, J. Nutr. Biochem., 2016, 27, 9–15 CrossRef CAS PubMed.
- Y. Nakashima, A. S. Plump, E. W. Raines, J. L. Breslow and R. Ross, ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree, Arterioscler. Thromb., 1994, 14, 133–140 CrossRef CAS PubMed.
- C. M. Weekley and H. H. Harris, Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease, Chem. Soc. Rev., 2013, 42, 8870–8894 RSC.
- U. Mehta, B. P. Kang, G. Bansal and M. P. Bansal, Studies of apoptosis and bcl-2 in experimental atherosclerosis in rabbit and influence of selenium supplementation, Gen. Physiol. Biophys., 2002, 21, 15–29 CAS.
- R. Brigelius-Flohe, A. Banning and K. Schnurr, Selenium-dependent enzymes in endothelial cell function, Antioxid. Redox Signaling, 2003, 5, 205–215 CrossRef CAS PubMed.
- R. F. Burk and K. E. Hill, Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis, Annu. Rev. Nutr., 2005, 25, 215–235 CrossRef CAS PubMed.
- Y. Ye, F. Fu, X. Li, J. Yang and H. Liu, Selenoprotein S is highly expressed in the blood vessels and prevents vascular smooth muscle cells from apoptosis, J. Cell. Biochem., 2016, 117, 106–117 CrossRef CAS PubMed.
- S. Meiler, Y. Baumer, Z. Huang, F. W. Hoffmann, G. J. Fredericks, A. H. Rose, R. L. Norton, P. R. Hoffmann and W. A. Boisvert, Selenoprotein K is required for palmitoylation of CD36 in macrophages: implications in foam cell formation and atherogenesis, J. Leukocyte Biol., 2013, 93, 771–780 CrossRef CAS PubMed.
- S. Ishibashi, J. L. Goldstein, M. S. Brown, J. Herz and D. K. Burns, Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice, J. Clin. Invest., 1994, 93, 1885–1893 CrossRef CAS PubMed.
- M. Torzewski, V. Ochsenhirt, A. L. Kleschyov, M. Oelze, A. Daiber, H. Li, H. Rossmann, S. Tsimikas, K. Reifenberg, F. Cheng, H. A. Lehr, S. Blankenberg, U. Forstermann, T. Munzel and K. J. Lackner, Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice, Arterioscler., Thromb., Vasc. Biol., 2007, 27, 850–857 CrossRef CAS PubMed.
- P. Lewis, N. Stefanovic, J. Pete, A. C. Calkin, S. Giunti, V. Thallas-Bonke, K. A. Jandeleit-Dahm, T. J. Allen, I. Kola, M. E. Cooper and J. B. de Haan, Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice, Circulation, 2007, 115, 2178–2187 CrossRef CAS PubMed.
- P. Chew, D. Y. Yuen, P. Koh, N. Stefanovic, M. A. Febbraio, I. Kola, M. E. Cooper and J. B. de Haan, Site-specific antiatherogenic effect of the antioxidant ebselen in the diabetic apolipoprotein E-deficient mouse, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 823–830 CrossRef CAS PubMed.
- J. B. de Haan, P. K. Witting, N. Stefanovic, J. Pete, M. Daskalakis, I. Kola, R. Stocker and J. J. Smolich, Lack of the antioxidant glutathione peroxidase-1 does not increase atherosclerosis in C57BL/J6 mice fed a high-fat diet, J. Lipid Res., 2006, 47, 1157–1167 CrossRef CAS PubMed.
- M. A. Forgione, A. Cap, R. Liao, N. I. Moldovan, R. T. Eberhardt, C. C. Lim, J. Jones, P. J. Goldschmidt-Clermont and J. Loscalzo, Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure, Circulation, 2002, 106, 1154–1158 CrossRef CAS PubMed.
- J. Zhou, K. Huang and X. G. Lei, Selenium and diabetes – evidence from animal studies, Free Radical Biol. Med., 2013, 65, 1548–1556 CrossRef CAS PubMed.
- J. P. McClung, C. A. Roneker, W. Mu, D. J. Lisk, P. Langlais, F. Liu and X. G. Lei, Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8852–8857 CrossRef CAS PubMed.
- Z. Guo, Q. Ran, L. J. Roberts, 2nd, L. Zhou, A. Richardson, C. Sharan, D. Wu and H. Yang, Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice, Free Radical Biol. Med., 2008, 44, 343–352 CrossRef CAS PubMed.
- F. Zhang, W. Yu, J. L. Hargrove, P. Greenspan, R. G. Dean, E. W. Taylor and D. K. Hartle, Inhibition of TNF-alpha induced ICAM-1, VCAM-1 and E-selectin expression by selenium, Atherosclerosis, 2002, 161, 381–386 CrossRef CAS PubMed.
- H. T. Zheng, L. N. Zhou, C. J. Huang, X. Hua, R. Jian, B. H. Su and F. Fang, Selenium inhibits high glucose- and high insulin-induced adhesion molecule expression in vascular endothelial cells, Arch. Med. Res., 2008, 39, 373–379 CrossRef CAS PubMed.
- L. M. Sordillo, K. L. Streicher, I. K. Mullarky, J. C. Gandy, W. Trigona and C. M. Corl, Selenium inhibits 15-hydroperoxyoctadecadienoic acid-induced intracellular adhesion molecule expression in aortic endothelial cells, Free Radical Biol. Med., 2008, 44, 34–43 CrossRef CAS PubMed.
- H. Ren, J. Mu, J. Ma, J. Gong, J. Li, J. Wang, T. Gao, P. Zhu, S. Zheng, J. Xie and B. Yuan, Selenium inhibits homocysteine-induced endothelial dysfunction and apoptosis via activation of AKT, Cell. Physiol. Biochem., 2016, 38, 871–882 CrossRef CAS PubMed.
- R. Tang and K. Huang, Inhibiting effect of selenium on oxysterols-induced apoptosis of rat vascular smooth muscle cells, J. Inorg. Biochem., 2004, 98, 1678–1685 CrossRef CAS PubMed.
- R. Tang, H. Liu, T. Wang and K. Huang, Mechanisms of selenium inhibition of cell apoptosis induced by oxysterols in rat vascular smooth muscle cells, Arch. Biochem. Biophys., 2005, 441, 16–24 CrossRef CAS PubMed.
- H. Liu, Q. Lu and K. Huang, Selenium suppressed hydrogen peroxide-induced vascular smooth muscle cells calcification through inhibiting oxidative stress and ERK activation, J. Cell. Biochem., 2010, 111, 1556–1564 CrossRef CAS PubMed.
- H. Liu, X. Li, F. Qin and K. Huang, Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress, JBIC, J. Biol. Inorg. Chem., 2014, 19, 375–388 CrossRef CAS PubMed.
- H. Sies, Oxidative stress: oxidants and antioxidants, Exp. Physiol., 1997, 82, 291–295 CrossRef CAS PubMed.
- S. Miller, S. W. Walker, J. R. Arthur, F. Nicol, K. Pickard, M. H. Lewin, A. F. Howie and G. J. Beckett, Selenite protects human endothelial cells from oxidative damage and induces thioredoxin reductase, Clin. Sci., 2001, 100, 543–550 CrossRef CAS PubMed.
- J. P. Thomas, P. G. Geiger and A. W. Girotti, Lethal damage to endothelial cells by oxidized low density lipoprotein: role of selenoperoxidases in cytoprotection against lipid hydroperoxide- and iron-mediated reactions, J. Lipid Res., 1993, 34, 479–490 CAS.
- H. Traulsen, H. Steinbrenner, D. P. Buchczyk, L. O. Klotz and H. Sies, Selenoprotein P protects low-density lipoprotein against oxidation, Free Radic. Res., 2004, 38, 123–128 CrossRef CAS PubMed.
- L. Campbell, F. Howie, J. R. Arthur, F. Nicol and G. Beckett, Selenium and sulforaphane modify the expression of selenoenzymes in the human endothelial cell line EAhy926 and protect cells from oxidative damage, Nutrition, 2007, 23, 138–144 CrossRef CAS PubMed.
- H. Steinbrenner, E. Bilgic, L. Alili, H. Sies and P. Brenneisen, Selenoprotein P protects endothelial cells from oxidative damage by stimulation of glutathione peroxidase expression and activity, Free Radic. Res., 2006, 40, 936–943 CrossRef CAS PubMed.
- B. Ruseva, M. Atanasova, R. Tsvetkova, T. Betova, M. Mollova, M. Alexandrova, P. Laleva and A. Dimitrova, Effect of selenium supplementation on redox status of the aortic wall in young spontaneously hypertensive rats, Oxid. Med. Cell. Longevity, 2015, 2015, 609053 Search PubMed.
- K. Huang, H. Liu, Z. Chen and H. Xu, Role of selenium in cytoprotection against cholesterol oxide-induced vascular damage in rats, Atherosclerosis, 2002, 162, 137–144 CrossRef CAS PubMed.
- Q. Wu, K. Huang and H. Xu, Effects of long-term selenium deficiency on glutathione peroxidase and thioredoxin reductase activities and expressions in rat aorta, J. Inorg. Biochem., 2003, 94, 301–306 CrossRef CAS PubMed.
- Q. Wu and K. Huang, Effect of long-term Se deficiency on the antioxidant capacities of rat vascular tissue, Biol. Trace Elem. Res., 2004, 98, 73–84 CrossRef CAS PubMed.
- Q. Wu and K. Huang, Effect of selenium compounds on the damage induced by oxysterol on rat arterial walls, Biol. Trace Elem. Res., 2006, 112, 273–282 CrossRef CAS PubMed.
- M. C. Houston, Role of mercury toxicity in hypertension, cardiovascular disease, and stroke, J. Clin. Hypertens., 2011, 13, 621–627 CrossRef CAS PubMed.
- Z. Huang, A. H. Rose and P. R. Hoffmann, The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities, Antioxid. Redox Signaling, 2012, 16, 705–743 CrossRef CAS PubMed.
- T. Collins, M. A. Read, A. S. Neish, M. Z. Whitley, D. Thanos and T. Maniatis, Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers, FASEB J., 1995, 9, 899–909 CAS.
- J. F. Maddox, K. M. Aherne, C. C. Reddy and L. M. Sordillo, Increased neutrophil adherence and adhesion molecule mRNA expression in endothelial cells during selenium deficiency, J. Leukocyte Biol., 1999, 65, 658–664 CAS.
- E. Lubos, N. J. Kelly, S. R. Oldebeken, J. A. Leopold, Y. Y. Zhang, J. Loscalzo and D. E. Handy, Glutathione peroxidase-1 deficiency augments proinflammatory cytokine-induced redox signaling and human endothelial cell activation, J. Biol. Chem., 2011, 286, 35407–35417 CrossRef CAS PubMed.
- E. Lubos, C. E. Mahoney, J. A. Leopold, Y. Y. Zhang, J. Loscalzo and D. E. Handy, Glutathione peroxidase-1 modulates lipopolysaccharide-induced adhesion molecule expression in endothelial cells by altering CD14 expression, FASEB J., 2010, 24, 2525–2532 CrossRef CAS PubMed.
- A. H. Wagner, O. Kautz, K. Fricke, M. Zerr-Fouineau, E. Demicheva, B. Guldenzoph, J. L. Bermejo, T. Korff and M. Hecker, Upregulation of glutathione peroxidase offsets stretch-induced proatherogenic gene expression in human endothelial cells, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 1894–1901 CrossRef CAS PubMed.
- M. Moutet, P. d'Alessio, P. Malette, V. Devaux and J. Chaudiere, Glutathione peroxidase mimics prevent TNFalpha- and neutrophil-induced endothelial alterations, Free Radical Biol. Med., 1998, 25, 270–281 CrossRef CAS PubMed.
- P. d'Alessio, M. Moutet, E. Coudrier, S. Darquenne and J. Chaudiere, ICAM-1 and VCAM-1 expression induced by TNF-alpha are inhibited by a glutathione peroxidase mimic, Free Radical Biol. Med., 1998, 24, 979–987 CrossRef.
- A. Sharma, D. Yuen, O. Huet, R. Pickering, N. Stefanovic, P. Bernatchez and J. B. de Haan, Lack of glutathione peroxidase-1 facilitates a pro-inflammatory and activated vascular endothelium, Vascs. Pharmacol., 2016, 79, 32–42 CrossRef CAS PubMed.
- L. K. Crosley, S. Bashir, F. Nicol, J. R. Arthur, J. E. Hesketh and A. A. Sneddon, The single-nucleotide polymorphism (GPX4c718t) in the glutathione peroxidase 4 gene influences endothelial cell function: interaction with selenium and fatty acids, Mol. Nutr. Food Res., 2013, 57, 2185–2194 CAS.
- R. Brigelius-Flohe, B. Friedrichs, S. Maurer, M. Schultz and R. Streicher, Interleukin-1-induced nuclear factor kappa B activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line, Biochem. J., 1997, 328(Pt 1), 199–203 CrossRef CAS PubMed.
- A. Banning, K. Schnurr, G. F. Bol, D. Kupper, K. Muller-Schmehl, H. Viita, S. Yla-Herttuala and R. Brigelius-Flohe, Inhibition of basal and interleukin-1-induced VCAM-1 expression by phospholipid hydroperoxide glutathione peroxidase and 15-lipoxygenase in rabbit aortic smooth muscle cells, Free Radical Biol. Med., 2004, 36, 135–144 CrossRef CAS PubMed.
- H. Kaur and M. P. Bansal, Studies on scavenger receptors under experimental hypercholesterolemia: modulation on selenium supplementation, Biol. Trace Elem. Res., 2011, 143, 310–319 CrossRef CAS PubMed.
- F. Cheng, M. Torzewski, A. Degreif, H. Rossmann, A. Canisius and K. J. Lackner, Impact of glutathione peroxidase-1 deficiency on macrophage foam cell formation and proliferation: implications for atherogenesis, PLoS One, 2013, 8, e72063 CAS.
- S. A. Mattmiller, B. A. Carlson and L. M. Sordillo, Regulation of inflammation by selenium and selenoproteins: impact on eicosanoid biosynthesis, J. Nutr. Sci., 2013, 2, e28 CAS.
- V. Capra, M. Back, S. S. Barbieri, M. Camera, E. Tremoli and G. E. Rovati, Eicosanoids and their drugs in cardiovascular diseases: focus on atherosclerosis and stroke, Med. Res. Rev., 2013, 33, 364–438 CrossRef CAS PubMed.
- G. Hampel, K. Watanabe, B. B. Weksler and E. A. Jaffe, Selenium deficiency inhibits prostacyclin release and enhances production of platelet activating factor by human endothelial cells, Biochim. Biophys. Acta, 1989, 1006, 151–158 CrossRef CAS.
- Y. Z. Cao, C. C. Reddy and L. M. Sordillo, Altered eicosanoid biosynthesis in selenium-deficient endothelial cells, Free Radical Biol. Med., 2000, 28, 381–389 CrossRef CAS PubMed.
- M. Meydani, Modulation of the platelet thromboxane A2 and aortic prostacyclin synthesis by dietary selenium and vitamin E, Biol. Trace Elem. Res., 1992, 33, 79–86 CrossRef CAS PubMed.
- A. Haberland, K. Neubert, I. Kruse, D. Behne and I. Schimk, Consequences of long-term selenium-deficient diet on the prostacyclin and thromboxane release from rat aorta, Biol. Trace Elem. Res., 2001, 81, 71–78 CrossRef CAS PubMed.
- J. Arnaud, M. Bost, D. Vitoux, J. Labarere, P. Galan, H. Faure, S. Hercberg, J. C. Bordet, A. M. Roussel and P. Chappuis, Effect of low dose antioxidant vitamin and trace element supplementation on the urinary concentrations of thromboxane and prostacyclin metabolites, J. Am. Coll. Nutr., 2007, 26, 405–411 CrossRef CAS PubMed.
- M. Hulsmans and P. Holvoet, The vicious circle between oxidative stress and inflammation in atherosclerosis, J. Cell. Mol. Med., 2010, 14, 70–78 CrossRef CAS PubMed.
- D. G. Harrison, Cellular and molecular mechanisms of endothelial cell dysfunction, J. Clin. Invest., 1997, 100, 2153–2157 CrossRef CAS PubMed.
- H. Lum and K. A. Roebuck, Oxidant stress and endothelial cell dysfunction, Am. J. Physiol.: Cell Physiol., 2001, 280, C719–741 CAS.
- M. Yoshizumi, Y. Fujita, Y. Izawa, Y. Suzaki, M. Kyaw, N. Ali, K. Tsuchiya, S. Kagami, S. Yano, S. Sone and T. Tamaki, Ebselen inhibits tumor necrosis factor-alpha-induced c-Jun N-terminal kinase activation and adhesion molecule expression in endothelial cells, Exp. Cell Res., 2004, 292, 1–10 CrossRef CAS PubMed.
- H. Cai, Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences, Cardiovasc. Res., 2005, 68, 26–36 CrossRef CAS PubMed.
- X. Lu, S. Y. Liu and R. Y. Man, Enhancement of endothelium dependent relaxation in the rat aortic ring by selenium supplement, Cardiovasc. Res., 1994, 28, 345–348 CrossRef CAS PubMed.
- J. Zhang, Y. D. Li, J. M. Patel and E. R. Block, Thioredoxin overexpression prevents NO-induced reduction of NO synthase activity in lung endothelial cells, Am. J. Physiol., 1998, 275, L288–293 CAS.
- S. Dayal, K. L. Brown, C. J. Weydert, L. W. Oberley, E. Arning, T. Bottiglieri, F. M. Faraci and S. R. Lentz, Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 1996–2002 CrossRef CAS.
- N. Weiss, Y. Y. Zhang, S. Heydrick, C. Bierl and J. Loscalzo, Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12503–12508 CrossRef CAS PubMed.
- W. J. Welch, Angiotensin II-dependent superoxide: effects on hypertension and vascular dysfunction, Hypertension, 2008, 52, 51–56 CrossRef CAS PubMed.
- M. Oelze, S. Kroller-Schon, S. Steven, E. Lubos, C. Doppler, M. Hausding, S. Tobias, C. Brochhausen, H. Li, M. Torzewski, P. Wenzel, M. Bachschmid, K. J. Lackner, E. Schulz, T. Munzel and A. Daiber, Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging, Hypertension, 2014, 63, 390–396 CrossRef CAS PubMed.
- M. M. Kockx and A. G. Herman, Apoptosis in atherosclerosis: beneficial or detrimental, Cardiovasc. Res., 2000, 45, 736–746 CrossRef CAS PubMed.
- M. C. Clarke, N. Figg, J. J. Maguire, A. P. Davenport, M. Goddard, T. D. Littlewood and M. R. Bennett, Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis, Nat. Med., 2006, 12, 1075–1080 CrossRef CAS PubMed.
- M. C. Clarke, T. D. Littlewood, N. Figg, J. J. Maguire, A. P. Davenport, M. Goddard and M. R. Bennett, Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration, Circ. Res., 2008, 102, 1529–1538 CrossRef CAS PubMed.
- I. Tabas, The role of endoplasmic reticulum stress in the progression of atherosclerosis, Circ. Res., 2010, 107, 839–850 CrossRef CAS PubMed.
- K. R. Brunt, K. K. Fenrich, G. Kiani, M. Y. Tse, S. C. Pang, C. A. Ward and L. G. Melo, Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT, Arterioscler., Thromb., Vasc. Biol., 2006, 26, 2027–2034 CrossRef CAS PubMed.
- Y. Okura, M. Brink, H. Itabe, K. J. Scheidegger, A. Kalangos and P. Delafontaine, Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques, Circulation, 2000, 102, 2680–2686 CrossRef CAS PubMed.
- K. M. Spitler and R. C. Webb, Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms, Hypertension, 2014, 63, e40–45 CrossRef CAS PubMed.
- A. Appukuttan, S. A. Kasseckert, S. Kumar, H. P. Reusch and Y. Ladilov, Oxysterol-induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase, Cardiovasc. Res., 2013, 99, 734–742 CrossRef CAS PubMed.
- R. Brigelius-Flohe, S. Maurer, K. Lotzer, G. Bol, H. Kallionpaa, P. Lehtolainen, H. Viita and S. Yla-Herttuala, Overexpression of PHGPx inhibits hydroperoxide-induced oxidation, NFkappaB activation and apoptosis and affects oxLDL-mediated proliferation of rabbit aortic smooth muscle cells, Atherosclerosis, 2000, 152, 307–316 CrossRef CAS PubMed.
- A. P. Sage, Y. Tintut and L. L. Demer, Regulatory mechanisms in vascular calcification, Nat. Rev. Cardiol., 2010, 7, 528–536 CrossRef CAS PubMed.
- J. A. Leopold, Vascular calcification: Mechanisms of vascular smooth muscle cell calcification, Trends Cardiovasc. Med., 2015, 25, 267–274 CrossRef CAS PubMed.
- F. Otsuka, K. Sakakura, K. Yahagi, M. Joner and R. Virmani, Has our understanding of calcification in human coronary atherosclerosis progressed?, Arterioscler., Thromb., Vasc. Biol., 2014, 34, 724–736 CrossRef CAS PubMed.
- C. H. Byon, A. Javed, Q. Dai, J. C. Kappes, T. L. Clemens, V. M. Darley-Usmar, J. M. McDonald and Y. Chen, Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling, J. Biol. Chem., 2008, 283, 15319–15327 CrossRef CAS PubMed.
- F. Parhami, A. D. Morrow, J. Balucan, N. Leitinger, A. D. Watson, Y. Tintut, J. A. Berliner and L. L. Demer, Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients, Arterioscler., Thromb., Vasc. Biol., 1997, 17, 680–687 CrossRef CAS.
- H. Liu, L. Yuan, S. Xu, T. Zhang and K. Wang, Cholestane-3beta, 5alpha, 6beta-triol promotes vascular smooth muscle cells calcification, Life Sci., 2004, 76, 533–543 CrossRef CAS PubMed.
- M. Liberman, R. C. Johnson, D. E. Handy, J. Loscalzo and J. A. Leopold, Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification, Biochem. Biophys. Res. Commun., 2011, 413, 436–441 CrossRef CAS PubMed.
- X. Duan, Y. Zhou, X. Teng, C. Tang and Y. Qi, Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification, Biochem. Biophys. Res. Commun., 2009, 387, 694–699 CrossRef CAS PubMed.
- X. H. Duan, J. R. Chang, J. Zhang, B. H. Zhang, Y. L. Li, X. Teng, Y. Zhu, J. Du, C. S. Tang and Y. F. Qi, Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification, Apoptosis, 2013, 18, 1132–1144 CrossRef CAS PubMed.
- R. Elsom, P. Sanderson, J. E. Hesketh, M. J. Jackson, S. J. Fairweather-Tait, B. Akesson, J. Handy and J. R. Arthur, Functional markers of selenium status: UK Food Standards Agency workshop report, Br. J. Nutr., 2006, 96, 980–984 CrossRef CAS PubMed.
- E. Reszka, E. Jablonska, J. Gromadzinska and W. Wasowicz, Relevance of selenoprotein transcripts for selenium status in humans, Genes Nutr., 2012, 7, 127–137 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |