
Modulating the catalytic activity of enzyme-like nanoparticles through their surface functionalization†
Roberto
Cao-Milán
 a,
Luke D.
He
a,
Spencer
Shorkey
a,
Gulen Y.
Tonga
a,
Li-Sheng
Wang
a,
Xianzhi
Zhang
a,
Imad
Uddin
b,
Riddha
Das
a,
Mine
Sulak
c and
Vincent M.
Rotello
*a
a,
Luke D.
He
a,
Spencer
Shorkey
a,
Gulen Y.
Tonga
a,
Li-Sheng
Wang
a,
Xianzhi
Zhang
a,
Imad
Uddin
b,
Riddha
Das
a,
Mine
Sulak
c and
Vincent M.
Rotello
*a
aDepartment of Chemistry, University of Massachusetts Amherst, 710 North Pleasant Street, Amherst, Massachusetts 01003, USA. E-mail: rotello@chem.umass.edu
bDepartment of Chemistry, Hazara University, Mansehra 21120, Pakistan
cSchool of Applied Science, Pamukkale University, 20600, Çivril, Denizli, Turkey
First published on 24th October 2017
Abstract
The inclusion of transition metal catalysts into nanoparticle scaffolds permits the creation of catalytic nanosystems (nanozymes) able to imitate the behaviour of natural enzymes. Here we report the fabrication of a family of nanozymes comprised of bioorthogonal ruthenium catalysts inserted in the protective monolayer of gold nanoparticles. By introducing simple modifications to the functional groups at the surface of the nanozymes, we have demonstrated control over the kinetic mechanism of our system. Cationic nanozymes with hydrophobic surface functionalities tend to replicate the classical Michaelis Menten model, while those with polar groups display substrate inhibition behaviour, a key mechanism present in 20% of natural enzymes. The structural parameters described herein can be used for creating artificial nanosystems that mimic the complexity observed in cell machinery.
Design, System, ApplicationNanocatalysis is an important tool for sustainable chemistry and biology. Maximizing the efficiency of nanocatalysts is crucial, however little work has been done towards developing nanosystems able to mimic the sophisticated kinetic behaviors of natural enzymes. Here we report the construction of nanocatalysts comprised of bioorthogonal ruthenium catalysts inserted into the protecting monolayer of gold nanoparticles. By introducing simple modifications in their surface functional groups, these “nanozymes” could be engineered to display kinetic behaviors observed in natural enzymes, including both “simple” classical Michaelis–Menten as well as substrate inhibition mechanisms. These studies provide initial steps toward understanding the structural parameters that modulate the catalytic behavior of nanozymes required for developing new nanodevices with controlled kinetic characteristics. |
Introduction
Enzymes have evolved to perform a wide variety of transformations,1 coupling efficiency with a complex suite of regulatory mechanisms.2 These properties make enzymes important catalysts for synthetic processes, particularly in the area of sustainable chemistry.3 There are, however, many limitations for enzymatic catalysis ranging from instability to the fact that despite their enormous breadth of mechanisms, there are many reactions that enzymes cannot catalyze.4Bioorthogonal catalysis has opened a promising direction, with transition metal catalysts (TMCs)5–8 providing access to transformations using chemical processes that are orthogonal to biocatalysis.9–14 Loading of TMCs into nanomaterial scaffolds provides water solubility and a protective environment for TMCs,12,14,15 playing a role similar to that of the protein scaffold in enzymatic catalysis. These scaffolds also have the potential to access more complex attributes of enzymatic behaviour, an area that to date has not been well-explored.
In our recent studies we have developed a family of gold nanoparticles (2 nm core, ∼7 nm overall diameter) loaded with ruthenium or palladium catalysts, and shown that these “nanozymes” (NZs) have catalytic behaviour that can be described as consistent with enzymes, and can be characterized using classical Michaelis–Menten kinetics.15 We report here the extension of this biomimetic capability to more complex kinetic behaviours. In this study, structural motifs present on the monolayer were used to regulate catalysis, providing systems that feature substrate inhibition, an important mechanism observed in processes that regulate neutrotransmission16 and DNA methylation,17 among others.18 We believe this study constitutes a starting point for the creation of nanozyme-based systems that could eventually replicate the complexity of cellular pathways.
Experimental section
Materials
All chemicals used were purchased from Fischer Scientific and were used without any previous treatment. Synthesis of nanoparticle scaffolds (NPs) can be found in ESI-2.† The substrate (Pro-Rho) was prepared as early reported.9,15Instrumentation
All fluorescence determinations were carried out in a SpectraMax M5 microplate spectrophotometer. The hydrodynamic diameter of NZs was measured in a Malvern Zetasizer Nano ZS instrument. The ICP-MS analyses were performed on a Perkin-Elmer NexION 300X ICP mass spectrometer.Nanozyme preparation
Nanozymes were synthesized following a previous method.15 A 3 mg mL−1 solution of the ruthenium catalyst ([Cp*Ru(cod)Cl]) in acetone was mixed (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) with an aqueous solution of NPs (20 μM). The acetone was slowly removed by evaporation to favour the encapsulation of the catalyst in the particle monolayer. The mixture was then submitted to filtration (Millex-GP filter; 25 mm PES, pore Size: 0.22 μm) to remove the precipitated catalyst. The nanozyme solutions were subsequently submitted to 5 filtration sessions (Amicon® ultra 4, 10 K) and dialysed (Snake Skin® dialysis tubing, 10 K) against water (5 L, 24 h) to remove unbound catalyst. The amount of encapsulated catalyst was measured by ICP-MS by tracking 101Ru relative to 197Au.15 (ESI-3†).
:1 v/v) with an aqueous solution of NPs (20 μM). The acetone was slowly removed by evaporation to favour the encapsulation of the catalyst in the particle monolayer. The mixture was then submitted to filtration (Millex-GP filter; 25 mm PES, pore Size: 0.22 μm) to remove the precipitated catalyst. The nanozyme solutions were subsequently submitted to 5 filtration sessions (Amicon® ultra 4, 10 K) and dialysed (Snake Skin® dialysis tubing, 10 K) against water (5 L, 24 h) to remove unbound catalyst. The amount of encapsulated catalyst was measured by ICP-MS by tracking 101Ru relative to 197Au.15 (ESI-3†).
Results and discussion
Design and synthesis of the nanozymes
Gold nanoparticles with core diameters of 2 nm capped with a monolayer (∼2.5 nm thick) were used as the scaffold for the catalysts, with the goal of creating protein-sized systems (Fig. 1a).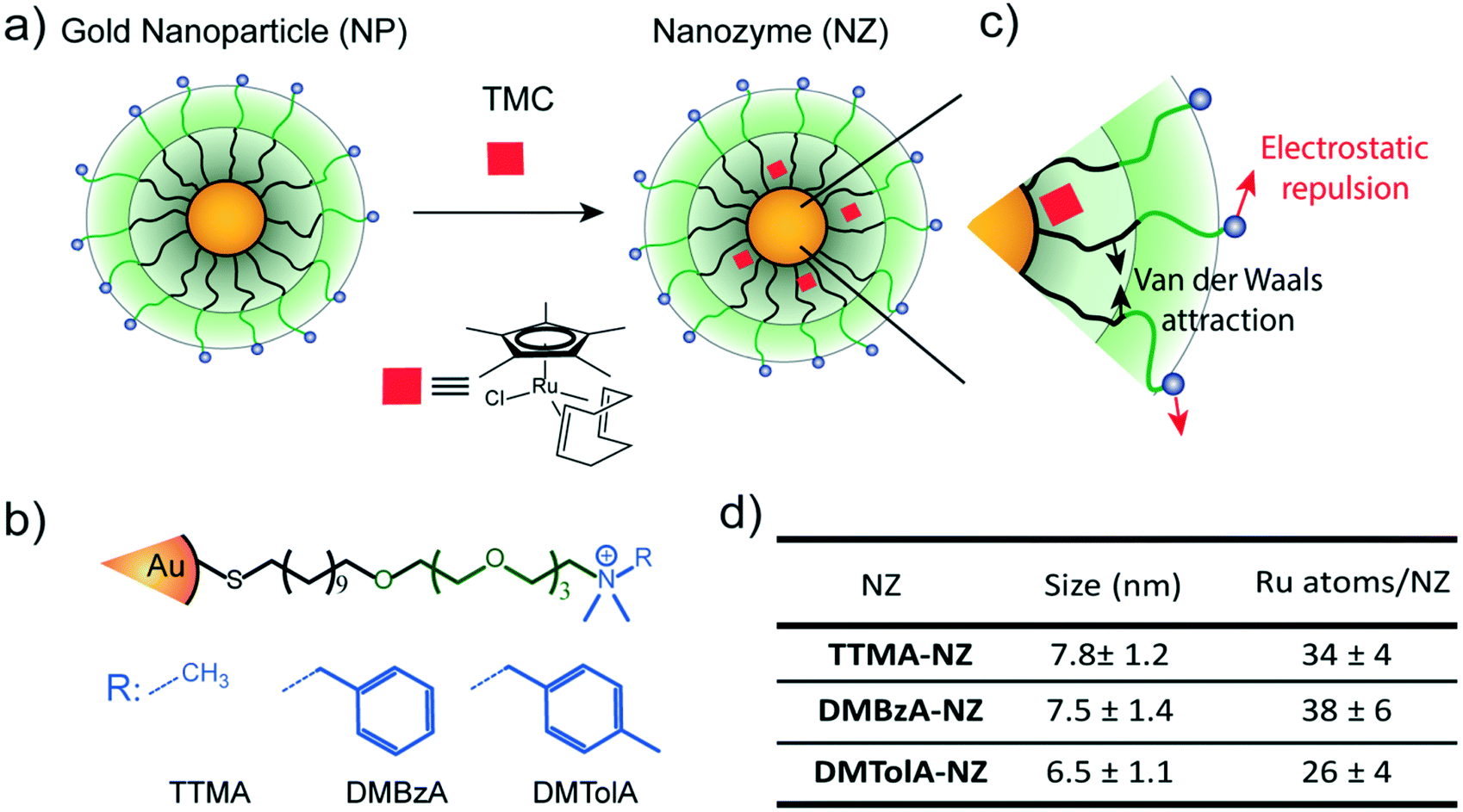 | ||
| Fig. 1 Structure of nanozymes. a) Encapsulation of TMCs in the hydrophobic pocket of gold nanoparticles. b) Structure of the nanoparticle scaffolds for generating cationic nanozymes with different hydrophobic surface functional groups. c) Schematic representation of the interacting forces among the ligands in the monolayer. Black arrows represent Van der Waals attraction. Red arrows represent repulsive electrostatic interaction. d) Table containing the size (determined via dynamic light scattering) and the amount of Ru catalyst per nanozyme. Mean values ± standard deviation, N = 3. | ||
The activity of most of enzymes is modulated by the amino acids residues located on their surfaces.19 Mutations on these amino acids can produce drastic changes in their tertiary structure, thus influencing their kinetic behaviour.19 With these concepts in mind, the effect of various modifications in the hydrophobicity of nanozyme ligands was explored (Fig. 1b). The structure of the protecting monolayer of the nanozyme platform is ruled by two major interactions: the long hydrophobic segments will interact through attractive Van der Waals forces, while the alkyl ammonium groups generate electrostatic repulsions between these ligands of the monolayer. The balance between these opposing forces should define the degree of compaction of the nanozyme monolayer, and thus, their kinetic behaviour (Fig. 1c). We reasoned that increasing the hydrophobicity of the positively charged head groups would produce a favourable effect towards the compaction of the monolayer, thus impacting the kinetic behaviour of the nanozymes. In this regard, ligands bearing methyl (TTMA), benzyl (DMBzA) and tolyl (DMTolA) substituents were used for constructing the NP scaffolds (Fig. 1b). More hydrophobic ligands were also included in the designs; however, the resulting nanozymes proved insoluble in PBS (see ESI-4†).
Fig. 1d shows some of the characteristics of our family of nanozymes. It can be observed that the three types of nanozymes used were dispersed in PBS and display sizes comparable with proteins (see also ESI-4†).
Kinetic behaviour of nanozymes
The kinetic behaviour of our nanozymes was studied using an allylcarbamate-protected derivative of rhodamine 110 (Pro-Rho), as substrate (Fig. 2a). Pro-Rho is a hydrophobic molecule with low fluorescence. After the catalytic deallylation, Pro-Rho is transformed into the zwitterionic, strongly fluorescent rhodamine 110 (Fig. 2a).9 The fluorogenesis of this dye provides a direct way for monitoring reaction kinetics.9 In addition, the zwitterionic nature of catalysed Pro-Rho molecules drastically increases its hydrophilic behaviour, presumably reducing affinity for the hydrophobic pocket of our nanozymes.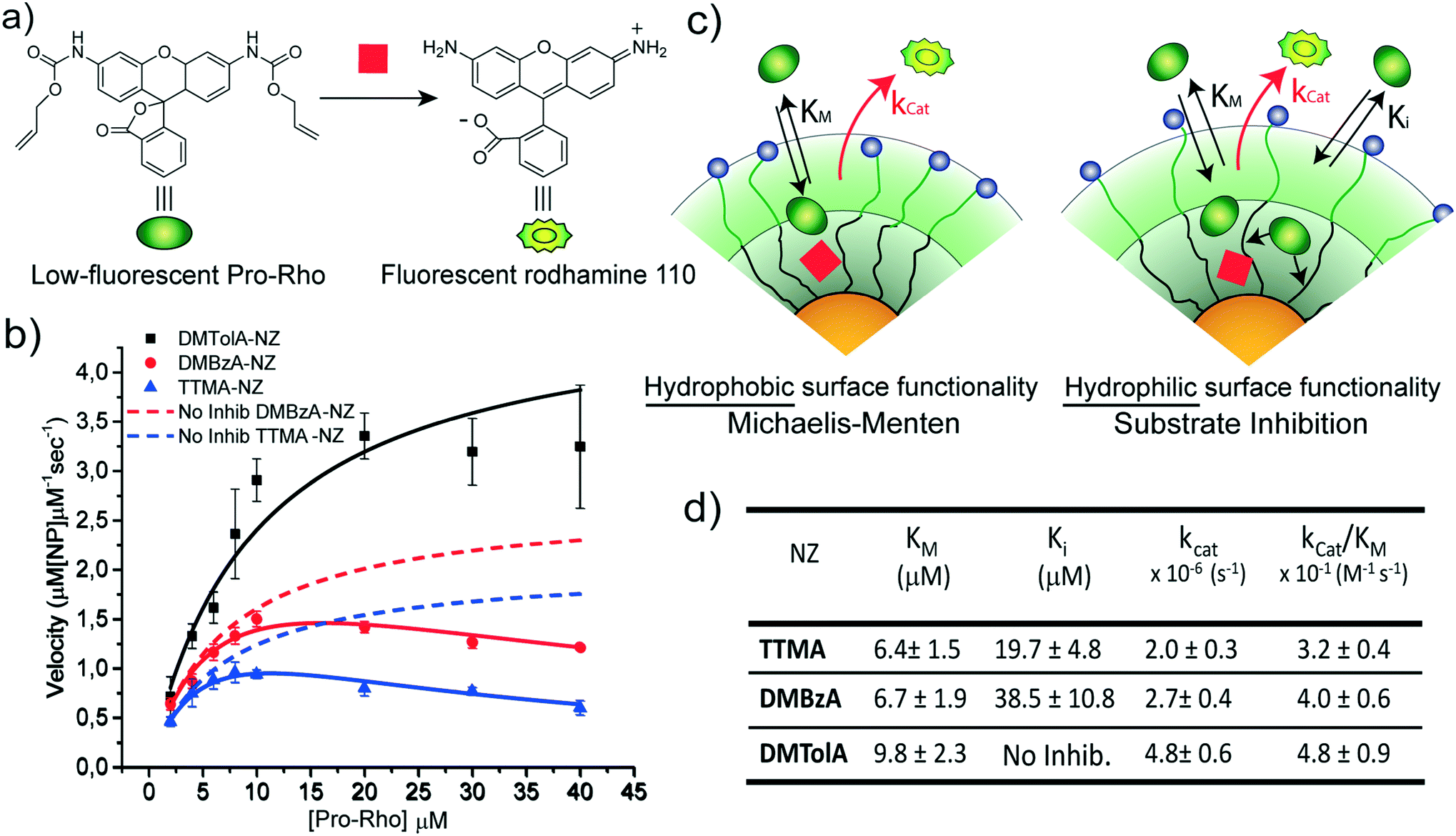 | ||
| Fig. 2 Kinetic study of nanozymes. a) Schematic representation of the activation of allyl carbamate protected rhodamine 110 (Pro-Rho). b) Nanozyme kinetics are shown as a function of substrate concentration. Solid lines are the regression curves corresponding to each mechanism. Dashed lines correspond to simulated kinetic curves for TTMA-NZ and DMBzA-NZ without substrate inhibition effect. c) Schematic representations of two possible kinetic behaviours of nanozymes. Left, nanozymes with monolayers bearing hydrophobic quaternary alkyl ammonium groups display the Michaelis–Menten mechanism. Right, nanozymes bearing hydrophilic quaternary alkyl ammonium groups display a substrate inhibition mechanism. d) Kinetic parameters for each nanozyme. Mean values ± standard deviation, N = 3. | ||
Fig. 2b shows the kinetic behaviour of the nanozymes in PBS. DMTolA-NZ displayed a classical Michaelis–Menten kinetic model, where the rate of conversion tends to asymptote to a steady-state level at high concentration of substrates. Such relationship demonstrates saturation of all nanozyme active sites. The kinetic behaviour of DMTolA-NZ can be represented by the classical Michaelis–Menten expression:
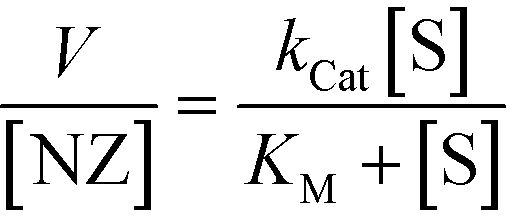 | (1) |
Interestingly, at high concentrations of substrate, DMBzA-NZ and TTMA-NZ tend to reduce the conversion rate of substrate. This type of catalytic behaviour is known as substrate inhibition and is also present in a large group of natural enzymes.18 In such cases, at high concentrations, substrate can bind inactive pockets of the enzyme, producing antagonistic effects.16–18 In the case of the NZs, any hydrophobic pocket without the ruthenium catalyst can be, in principle, able to bind substrate, producing the abovementioned allosteric attenuation of catalysis (Fig. 2c right).
The behaviour of DMBzA-NZ and TTMA-NZ was modelled by the equation used for describing substrate inhibition mechanism:16
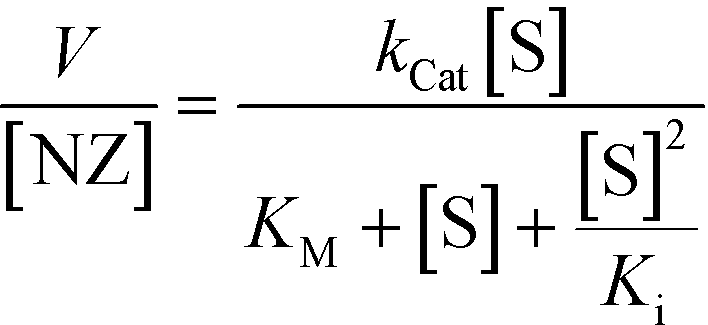 | (2) |
In eqn (2), Ki represents the dissociation constant of substrate binding the allosteric sites (Fig. 2c right) and producing a 50% reduction of catalytic activity (for comparison, see in Fig. 2a simulated curves for TTMA-NZ and DMBzA-NZ without substrate inhibition effect).
With the increase in the hydrophobicity of the substituent of the alkyl ammonium head groups, the value of Ki increases until no additional substrate can influence the activity of nanozymes, as in the case of DMTolA-NZ (Fig. 2d. Note also that for large Ki values eqn (2) tends to eqn (1)). An increase in the hydrophobicity of the positively charged head group also tended to increase the value of KM. However, variations in KM are not as pronounced as in Ki.
The observed kinetic results suggest that increased compaction of the monolayer, driven by an increasingly hydrophobic ligand, induces nanozyme behaviour to follow the classical Michaelis–Menten model. To test this hypothesis a nanozyme bearing an uncharged head group (TEGOH) was synthesized and studied (see ESI 7†). In this design, ligands in the protecting monolayer of the nanozyme do not experience electrostatic repulsion; thus, attractive Van der Waals forces should dominate. ESI 7† shows that TEGOH-NZs displayed classical Michaelis–Menten behaviour. These results are consistent with our understanding of the effect of monolayer compaction on the kinetic mechanism of the nanozyme.
Substrate adsorption experiments
The observed trend for magnitudes of both KM and Ki suggests that the affinity of substrate for nanozymes tends to decrease with increasing hydrophobicity of the alkyl ammonium head group. Therefore, an experiment to explore the capacity of our NP scaffolds to adsorb substrate was carried out. Adsorption capacity of NPs was determined in PBS by monitoring the quenching effect of gold particles on the residual fluorescence of adsorbed Pro-Rho. It should be noted that adsorption experiments are carried out in equilibrium conditions; thus, only the NP scaffolds (without the ruthenium catalyst) can be used.Fig. 3a shows the number of molecules of Pro-Rho adsorbed in the monolayer of each of our NPs at different concentrations of this substrate. Increasing the hydrophobicity of the alkyl ammonium group was observed to reduce the adsorption capacity of the corresponding NP. In addition, Pro-Rho adsorption curves shift to sigmoidal shapes for the NPs bearing the most hydrophobic surface functionalities (Fig. 3a). This sigmoidal behaviour (as DMTolA-NP) indicates that the inclusion of Pro-Rho into the monolayer of NPs is not favoured at low concentrations of the substrate. However, insertion of the first few molecules into this monolayer contributes to subsequent adsorption of the substrate via a cooperative effect (Fig. 3b). In the case of TTMA-NPs, the linear behaviour suggests that the amount of adsorbed Pro-Rho is only dependent on the concentration of substrate in solution (Fig. 3c).
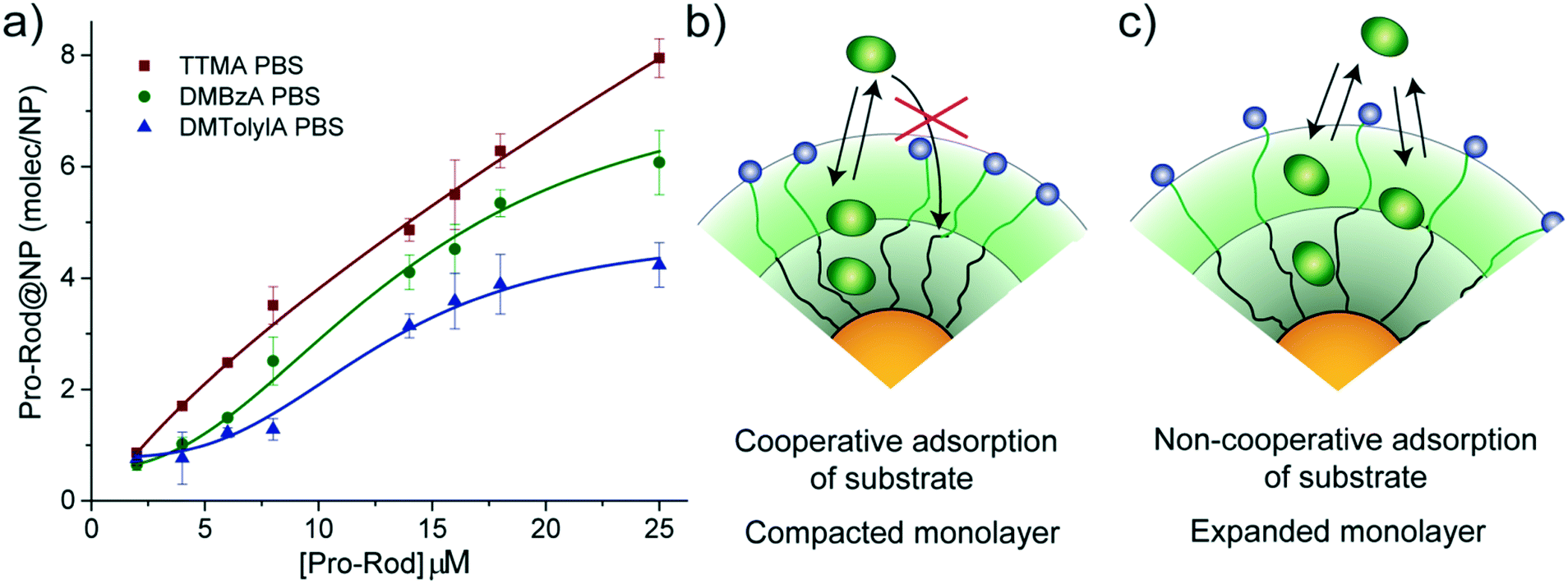 | ||
| Fig. 3 Adsorption of Pro-Rho on the nanoparticle scaffolds used. a) Adsorption curves of Pro-Rho on each NP in PBS. Adsorbed Pro-Rho molecules on the NPs were plotted as function of Pro-Rho concentration. b) Schematic representations of the cooperative adsorption mechanism observed in DMTolA-NP featuring compact monolayer: the first adsorbed molecules favour the further adsorption of substrate c) non-cooperative adsorption of substrate on TTMA-NP. The adsorption of Pro-Rho molecules is only dependent on its concentration in solution. Adsorbed molecules do not influence the further adsorption of substrate. Mean values ± standard deviation, N = 3. | ||
The trend observed for Pro-Rho adsorption could be caused by different levels of compaction of the monolayer of our NP scaffolds, with DMTolA-NP displaying the most compact one and TTMA-NP the least. To confirm this assumption, the same adsorption experiment was carried out in deionized water. Under low ionic strength conditions, the monolayer of our NP scaffolds should acquire a lower compaction degree.20,21 All NPs scaffolds displayed increased substrate adsorption capacity in deionized water than PBS (see ESI-4†). These results suggest that the more expanded the monolayer of our NPs the greater their capacity to associate substrates (see ESI-5†).
The results obtained by these adsorption experiments with our NPs scaffolds shed some light on how DMTolA-NZ displays Michaelis–Menten kinetic behaviour while DMBzA-NZ and TTMA-NZ exhibit substrate inhibition. We hypothesized that the higher compaction of the monolayer of DMTolA-NZ reduces the affinity for Pro-Rho; thus, decreases the possibility of insertion of substrate in allosteric sites. However, in the case of DMBzA-NZ and TTMA-NZ, with a more opened monolayer, at high concentrations Pro-Rho can bind both active and allosteric sites. Our next steps will focus on answering questions regarding the key structural parameters that modulate the efficiency and selectivity of these bioorthogonal nanozymes.
Conclusion
In conclusion, we have demonstrated the construction of bioorthogonal nanozymes featuring different kinetic behaviours present in natural enzymes. The kinetic mechanism of nanozymes was observed to be strongly influenced by their surface functionalities. The ability to fabricate bioorthogonal nanozymes featuring sophisticated enzymatic behaviours is essential for developing a platform of nanodevices able to modulate complex cell bioprocesses through abiotic catalytic reactions, with envisaged applications in both fundamental science and biomedical procedures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the NIH (EB022641) and the NSF (CHE-1506725). M. S. acknowledges TÜBİTAK for the fellowship 1059B191501588.Notes and references
- H. Bisswanger, in Enzyme Kinetics: Principles and Methods, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2008, ch. 2.6–2.12, pp. 124–193 Search PubMed.
- M. J. Evans and B. F. Cravatt, Chem. Rev., 2006, 106, 3279–3301 CrossRef CAS PubMed.
- Q. Husain, Crit. Rev. Biotechnol., 2006, 26, 201–221 CrossRef CAS PubMed.
- (a) T. Carell and M. Vrabel, Top. Curr. Chem., 2016, 374, 9 CrossRef PubMed; (b) Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097–1105 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- A. Unciti-Broceta, E. M. V. Johansson, R. M. Yusop, R. M. Sánchez-Martín and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- J. H. Do, H. N. Kim, J. Yoon, J. S. Kim and H.-J. Kim, Org. Lett., 2010, 12, 932–934 CrossRef CAS PubMed.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Köster, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- T. Völker and E. Meggers, Curr. Opin. Chem. Biol., 2015, 25, 48–54 CrossRef PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 Search PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- I. Winge, J. A. Mckinney, M. Ying, C. S. D'Santos, R. Kleppe, P. M. Knappskog and J. Haavik, Biochem. J., 2008, 410, 195–204 CrossRef CAS PubMed.
- P. A. Jones and D. Takai, Science, 2001, 293, 1068–1070 CrossRef CAS PubMed.
- M. C. Reed, A. Lieb and H. F. Nijhout, BioEssays, 2010, 32, 422–429 CrossRef CAS PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- A. A. Volkert, V. Subramaniam, M. R. Ivanov, A. M. Goodman and A. J. Haes, ACS Nano, 2011, 5, 4570–4580 CrossRef CAS PubMed.
- R. Mout, G. Y. Tonga, L. S. Wang, M. Ray, T. Roy and V. M. Rotello, ACS Nano, 2017, 11, 3456–3462 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00055c |
| This journal is © The Royal Society of Chemistry 2017 |