
Modern fluorescence-based concepts and methods to study biomolecular interactions
Maria
Strianese
a,
Maria
Staiano
b,
Alessandro
Capo
b,
Gabriella
Pinto
c,
Claudio
Pellecchia
a and
Sabato
D'Auria
 *b
*b
aDepartment of Chemistry & Biology, University of Salerno, Italy
bInstitute of Food Science, CNR, Via Roma, 6483100 Avellino, Italy. E-mail: sabato.dauria@cnr.it; Fax: +39 0825299100; Tel: +39 0825299101
cUniversity of Naples Federico II, Italy
First published on 10th February 2017
Abstract
Molecular interactions are of high interest to molecular biologists because they help to understand the function and behavior of proteins and they can also contribute to the prediction of biological processes that a protein of unknown function is involved in. In fact, protein interactions are critical for many biological processes since they mediate most of the cellular functions. Molecular interactions include both stable associations of proteins within multi-subunit protein complexes and transient associations of biomolecules with a regulatory function. Currently several methods are used to investigate protein–protein interactions. Affinity purification methods combined with mass spectrometry experiments are the most used methods even if they allow the study of molecular interactions only in cellular lysates and not in intact cells. Super-resolution microscopy approaches are applied to study protein interactions in vivo, but these approaches still suffer from a lack of availability of highly bright and stable fluorescent probes as well as the diffusion effects of labeled molecules inside the cell matrix. It appears clear that each of the actual proposed methods has its own strengths and weaknesses, especially with regard to the possibility of revealing molecular interactions in large protein complexes or among different subunits of a large protein molecule. This review article reports on the development of strategies which appear as potential tools to monitor molecular interactions in complex real matrices for advanced diagnostic purposes. More specifically, the goal of this article is not to cover all proposed methods present in the literature to study molecular interactions in large protein complexes, but to highlight the advances in fluorescence spectroscopy, nanotechnology and probe chemistry to investigate molecular interactions.
 Maria Strianese | Maria Strianese (B.S./M.S. & Ph.D, Salerno University; scientist at Leiden University). She was born in Salerno in 1979. She is an assistant professor of Inorganic Chemistry at the University of Salerno. Her current research is focused on optical sensors for bio-medical and environmentally relevant molecules. |
 Maria Staiano | Maria Staiano has a doctorate degree in Biology (University of Naples Federico II) and PhD in Cellular Biology (University of Siena, Italy). She is a scientist with a tenured position at the Italian National Research Council. Her research interests are focused on the design of biomolecule-based sensors. |
 Alessandro Capo | Alessandro Capo (B.S/M.S. in Medical Biotechnology at the University of Naples Federico II) holds a PhD in System Biology from Salerno University and is a fellow at the Italian National Research Council. He works as a post-doctoral scientist at the Institute of Food Science, National Research Council. He was born in 1984 in Salerno. |
 Gabriella Pinto | Gabriella Pinto holds a first PhD in Biotechnology Sciences from the Department of Organic Chemistry and Biochemistry and a second PhD from the Department of Agriculture (University of Naples “Federico II”). She worked as a post-doctoral scientist at University College London, UK, within the “Proteomics & Molecular Cell Dynamics” research group. She worked as a researcher at the Institute of Food Science, National Research Council (ISA-CNR) in the molecular bio-sensing field. Over the years, her research interests have focused mainly on food proteomics, large-scale quantitative sub-cellular proteomics, and bio-sensing. |
 Claudio Pellecchia | Claudio Pellecchia (B.S./M.S. and Ph.D., University of Naples). He was born in Avellino in 1959. Since 2001 he has been a full Professor of Inorganic Chemistry at the University of Salerno. His research interests deal with the design of novel catalysts and development of optical sensors. |
 Sabato D'Auria | Sabato D'Auria has a doctorate degree in biology (University of Naples Federico II). He holds a tenured position of Science Director at the Italian National Research Council and he is also the Director of the Institute of Food Science, CNR, Avellino, Italy. His scientific interests are focused on the study of molecular interactions and design of advanced sensing methodologies. |
Design, System, ApplicationThe present mini-review focuses on new theoretical and experimental approaches developed in our labs that enable monitoring of molecular interactions both in vitro and in vivo. Among these approaches we present the use of near-infrared fluorescent probes to covalently label antibodies, the potential application of novel infrared proteins as cellular biomarkers for total body imaging and new photonic concepts that allow long-distance RET analyses both in vitro and in vivo. These frontier procedures are also based on the use of nanostructured metal particles, ad hoc engineered biomolecules and graphene structures. More specifically, the aim of this paper is not to cover all proposed methods present in the literature to study molecular interactions in large protein complexes, but to highlight the advances in fluorescence spectroscopy, nanotechnology and probe chemistry to investigate molecular interactions in biology. In the introduction section we also discussed various different literature methodologies currently used for studying protein–protein interactions and molecular interactions to give a more complete picture of all the approaches used. |
Introduction
The function of a protein is largely mediated by its interactions with other molecules. Consequently, molecular interactions are responsible for the regulatory processes of cellular functions. Hence the mapping of protein–protein interactions (PPIs) is of critical importance for understanding cellular mechanisms of action.The analysis of protein complexes and protein–protein interaction networks and the dynamic behavior of these networks as a function of time and cell state are therefore of central importance in biological research. Furthermore, many human diseases can be traced to aberrant protein–protein interactions, either through the loss of an essential interaction or through the formation of a protein complex at an inappropriate time or in an incorrect cell location.1 Therefore, mapping PPIs and their interaction interfaces in living cells is critical not only for comprehensive understanding of protein function and regulation, but also for describing the molecular mechanisms underlying human pathologies and identifying potential targets for better therapeutics.
Current interactomes have been mainly obtained by making use of data generated by yeast cells (Y2H) and (tandem) affinity purification methods combined with mass spectrometry experiments.2
Although very robust and highly efficient, these approaches do not allow studying PPI modulation because they do not offer the proper context for mammalian cellular PPI analysis, e.g. they operate in Y2H or make use of cell lysates and not in intact cells. Therefore, our current understanding of these multi-component systems is limited because of a lack of tools for characterizing their function in cellular contexts. These remarks support the need for approaches that allow PPIs to be assayed in their native cellular environment. NMR spectroscopy and X-ray crystallography are powerful techniques for studies on PPIs, providing structural data at the atomic level.3 However, both techniques are restricted by various factors such as protein solubility, protein size and protein location. Apart from the high-throughput methods mentioned above, a number of approaches devised to study PPIs actually operate in mammalian cells.1,4,5 A potentially powerful technique is represented by FTIR (Fourier transform IR) spectroscopy.3 Historically, IR spectroscopy is one of the earliest techniques used for protein structure analysis. For protein analysis, the vibration of the peptide bond has been most productively used for characterization of protein secondary structures. However, like other spectroscopic methods, IR spectroscopy suffers from some disadvantages such as the overlap of absorption peaks that hinder accurate assignments of structural elements.3
Super-resolution microscopy techniques may possibly overcome some of the limitations of the currently used techniques for studying PPIs and molecular interactions.6 In addition, FRET (Förster resonant energy transfer) and BRET, which rely on fluorescence or bioluminescence energy transfer between interacting fusion proteins, have also been exploited to develop assays with high spatial–temporal resolution.7,8 A variety of protein fragment complementation assays (PCAs) have been reported, including split fluorescent protein or reporter enzyme technologies, that are able to capture aspects of PPI dynamics in a mammalian background.9,10 A recent addition is represented by an infrared fluorescent PCA that, unlike previous fluorescent PCAs, exhibits a reversible complementation, thus enabling spatial–temporal analysis of dynamic PPIs.11
In this article, we aim at highlighting the progress that has been achieved in our labs on the implementation of potential approaches for the monitoring of dynamic PPIs.
FRET and molecular interactions
(a) Basics of FRET
Fluorescence resonance energy transfer (FRET) is a technique half a century old, yet, due to recent advances, it is undergoing a renaissance. FRET, which relies on the distance-dependent transfer of energy from a donor fluorophore to an acceptor fluorophore, is widely used in biology as a sensitive method for measuring inter- and intramolecular distances (“spectroscopic ruler”).12,13The FRET phenomenon occurs when the emission of a so-called donor exhibits spectral overlap with the absorption of a so-called acceptor molecule. When the absorption spectrum of the acceptor changes, the amount of FRET related transfer of energy changes as well, leading to a concomitant change of the fluorescence of the donor. The direct transfer of energy by a radiation-less process from the excited state of the donor to the acceptor is responsible for the decrease of the fluorescence of the donor.
Efficiency of the FRET process (E) drops off as the inverse sixth power of the distance between the donor and acceptor, R,
| E = R06/(R06 + R6) |
(b) Use of FRET to study molecular interactions
By making use of the latter approach in the past few years we and others devised a number of efficient sensing devices.14–21 In particular, we focused on naturally occurring gases, most significant examples of which are nitric oxide (NO) and hydrogen sulfide (H2S). Both NO and H2S have a number of physiological and/or pathophysiological functions which constitutes a challenging motivation for devising new ways to monitor their production, trafficking, and consumption in living systems. More specifically, the sensing schemes exploit the use of metallo-proteins (i.e. cytochrome c peroxidase, myoglobin, hemocyanin, tyrosinase, cobalt peptide deformylase) able to bind the target analytes and whose absorption features are different in the presence and absence of the target analytes. By attaching a fluorescent label to the protein, whose emission spectrum overlaps with one of the absorption bands of the protein (which changes upon analyte binding at the enzyme cofactor), this change in absorption can be translated into a change of the fluorescence intensity of the label through a FRET mechanism. The fluorescent label acts as a passive “beacon” reflecting the presence (or absence) of the analyte. By applying this strategy, it was showed that cytochrome c peroxidase from baker's yeast can be used as a “turn on” NO FRET-based biosensor,17 whereas myoglobin15 and cobalt containing peptide deformylase14 can be implemented as H2S sensing elements; on the other hand tyrosinase and hemocyanin function as oxygen sensing proteins (Fig. 1). | ||
| Fig. 1 Schematic representation of the sensing methodology. Left: Dye labeled cuvette filled with CcP–NO free; right: dye-labeled cuvette filled with CcP–NO bound. Adapted with permission from ref. 16. | ||
Despite all the advantages of the above mentioned probes, the main weakness of the scheme deals with the distance of the donor–acceptor molecules involved in the FRET process which cannot exceed 100 Å. This feature makes the application of this approach not always possible when aiming at the study of protein–protein interactions that occur at larger distances. To date, using FRET-based fluorescence microscopy, great achievements have been made in the study of protein interactions in cells which occur on the scale of 80 Å to 90 Å. However, there are still many important spatial/proximal interactions that take place at distances larger than 100 Å. Indeed, in this case the FRET efficiency strongly decreases with the increase of the donor–acceptor distance.
One of the approaches that has been proposed to extend FRET to greater distances is the use of high quantum-yield and very long-lived lanthanide donors.22–25Fig. 2 displays typical examples of lanthanide probes. Their organic moiety (chromophore) acts as an antenna or sensitizer absorbing the excitation light and donating the energy to the lanthanide ion.
 | ||
| Fig. 2 Typical examples of lanthanide probes. Adapted with permission from ref. 22. | ||
Their millisecond luminescence lifetime allows for the suppression of nanosecond fluorescence from both the acceptor and the background, allowing the observation of acceptor-sensitized emission with time-gated detection. The long lifetime of this emission enables the observation of significantly smaller changes in the lifetime induced by FRET, allowing monitoring of interactions that occur at distances much greater than R0. Lanthanides are also quite useful because of their remarkably sharp line emission that is suitable for multiplexing. Using lanthanide donors matched to the proper organic fluorophore acceptors, it is possible to measure distances over 110 Å. Using acceptors such as quantum dots (QDs) and phycobiliproteins (PEs), which possess significantly higher absorption and molar extinction coefficients, R0 values of 90 to 100 Å have been measured when using lanthanide donors. When looking at these results it would seem that, in principle, it should be possible to monitor FRET at distances close to 150 Å under practical experimental conditions if using proper donor–acceptor pairs. However, it should be noted that both chelated lanthanide donors and acceptors, such as QD and PE molecules, are significantly larger than conventional organic acceptor molecules. Because the measured donor–acceptor separation refers to the distance between the centers of the interacting entities, a practical consequence of their relatively larger sizes is that the donor and acceptor molecules will occupy up to 50 to 60 Å of the measured distance. This will result in limiting the effective observation distance to around 100 Å under most experimental conditions. Yet among the drawbacks of lanthanide-based probes are their quite low molar extinction coefficients and the need to excite in the UV/near-visible spectrum (typically between 325 and 360 nm).
Another innovative approach designed to extend the FRET distance to beyond 100 Å exploits the use of nanostructured silver (or gold) islands in the proximity of the fluorophore.26 Metal island films consist of subwavelength size patches of metal on inert substrates where “metal” refers to a highly conducting particle or surface, and not metal ions or oxides (see Fig. 3).
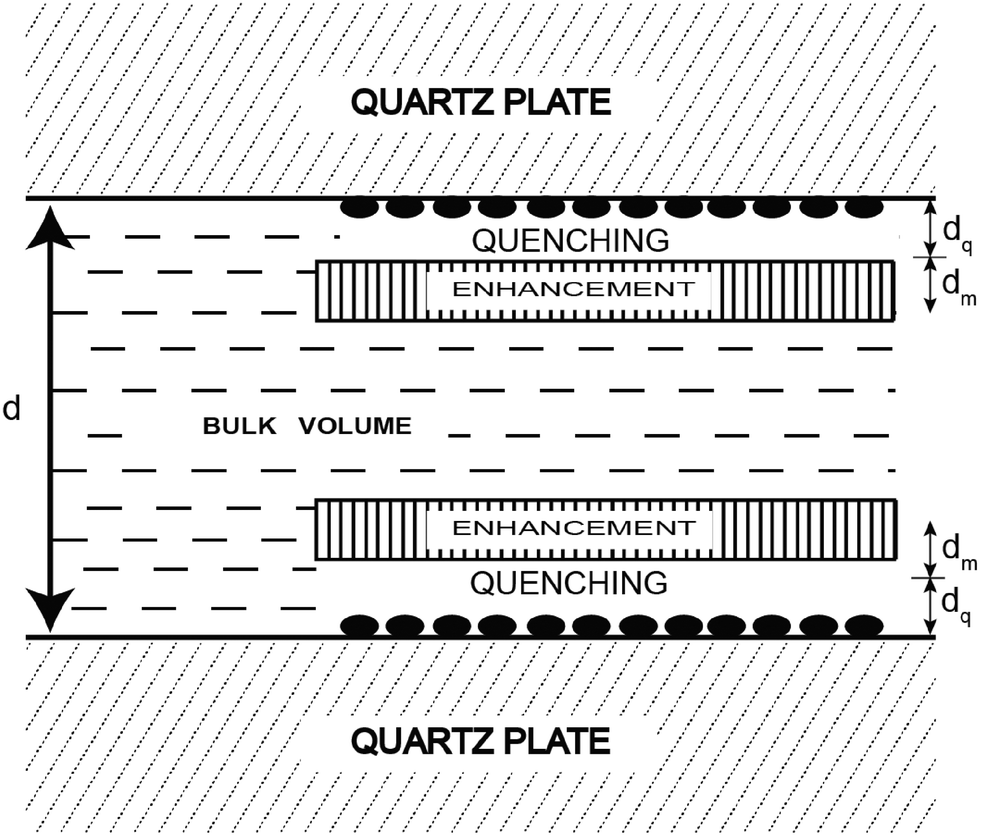 | ||
| Fig. 3 Representation of a fluorophore solution between two silver island films. The solid ellipsoids represent the silver island. Adapted with permission from ref. 26. | ||
In this approach metal nanoparticles modulate the fluorescence of fluorophores. The presence of metals substantially modifies the electromagnetic field in the neighborhood of excited molecules which leads, under specific conditions, to significant FRET interactions. The process has been described as nanomaterial surface energy transfer (NSET). By applying this method it was shown that the effects of metallic silver island films on resonance energy transfer (RET) between the donor 4,6-diamidino-2-phenylindole (DAPI) and the acceptor propidium iodide (PI), both of them bound to a double-helical DNA molecule, resulted in a dramatic increase of the Förster distance from 3.5 nm to an apparent value of 16.6 nm (Fig. 4).27 However there are several reasons why the use of silver nanoparticles has severe restrictions for in vivo studies. Just to name a few, the difficulty of introducing silver-labeled fluorescent proteins into the cell and the poisoning action of silver nanoparticles on the cell cycle. Developing a metal-free method that would allow the use of FRET over distances longer than those presently accessible to investigate the assembly/disassembly of large protein complexes directly in cells is currently a challenging task.
 | ||
| Fig. 4 Emission spectra of DAPI-labeled DNA and DAPI-PI labeled DNA donor–acceptor on quartz (top) and silver island films (bottom). Adapted with permission from ref. 27. | ||
A different method where the single acceptor molecule of the FRET pair is replaced by many linked and closely located molecules to which resonance energy transfer is much more effective has been also proposed for extending the utilizable range of FRET.28–31 When labeling a suitably sized single protein/antibody/small particle with multiple acceptors in place of a single acceptor, the observed energy transfer efficiency enhances. The observed FRET is the sum of the individual transfer rates to each acceptor and can be obtained as the same value of FRET efficiency as in the donor–single acceptor case at longer distances. The working hypothesis is based on the use of several rather closely placed acceptors that are bound to a single bio/macromolecule (e.g., an antibody 1) and can amplify energy transfer as an antenna system. Acceptors will collect excitation energy that is transferred to them from a single donor molecule localized on a different bio/macromolecule (e.g., an antibody 2). Further extension of the range of energy transfer can be obtained by preferential nonrandom distribution of acceptor transition moments toward the donor transition moment in emission. By means of Monte Carlo (MC) simulations we showed that it is possible to mimic the biological antenna systems and, as a result, we can simply obtain not only the average value of FRET efficiency for a given system, but also the decay curve of donor and acceptor emission, which can be helpful in the interpretation and design of FRET experiments. The simple antenna model is built by placing the donor molecule, D, at the center of the Cartesian coordinate system and the group of acceptors, A, randomly positioned on a surface of a globular protein, approximated by a sphere with radius r. The donor and the protein are connected via a link of a certain length, R. The vectors of donor and acceptor transition moments, as well as the explicit distances between the donor and each acceptor, are determined during each step of the MC simulation. The parameters strongly influencing the FRET efficiency are both the orientation and the number of fluorescent probes. To examine the effect of the number of acceptors on the FRET efficiency, we carried out a number of suitable simulations. We analyzed the dependence of the FRET efficiency in the presence of 1, 5 and 10 acceptors attached to the spherical protein with a 40 Å diameter, as a function of the distance R. R0 was assumed to be 80 Å and the distance is approximated by a rigid linker. Two orientation cases, random and parallel, were considered. We found that increasing the number of acceptors to only five, for the distance R = 150 Å, results in a FRET efficiency of 6%, and for ten acceptors it is 11% (almost 40% in the parallel case), whereas for a single acceptor the value of FRET efficiency is close to zero. This means that using multiple acceptors, it is possible to obtain measurable values of FRET efficiency even at the distance of 150 Å: the useful range of energy transfer can be extended as a spectroscopic ruler. Other aspects that we investigated as influencing the FRET efficiency are the size and the shape of proteins. To simplify the calculations, a spherical shape of the protein and a fixed donor–protein distance were assumed. In order to experimentally test the model, a 30-mer oligonucleotide with biotin on the 3′-end and Alexa568 (A568) on the 5′-end was used (Fig. 5).29
 | ||
| Fig. 5 Structure of the labeled 30-mer oligonucleotide used. Adapted with permission from ref. 29. | ||
Alexa568 (A568) acted as the donor and DyLight-649 (DL649) or DyLight750 (DL750) acted as the acceptor. The calculated R0 are 71.5 Å and 61 Å for the A568-DL649 and A568-DL750 pairs, respectively. In the presence of labeled avidin, both a decrease in the longest lifetime values and an increase in the amplitudes of the shorter lifetime components were observed. The extent of FRET was about 9% in the case of the A568-DL750 pair (A = 8) and 12% and 14.2% in the cases of A568-DL649 with 4.5 and 6 acceptors, respectively. It is clear that the presence of multiple acceptors increases the transfer efficiency regardless of R under the presented experimental conditions. In fact, a higher transfer as the R0 value increased with increasing number of acceptors was observed. The FRET efficiency depends on the number of acceptors, but our experimental results show that the lower FRET efficiency of a fraction of molecules with a smaller number of acceptors is compensated by an equivalent fraction with a higher than average number of acceptors. It should be beneficial to use a higher labeling efficiency in order to avoid contributions from pairs without acceptor-labeled proteins.
GFP targeting and fluorescent proteins
A milestone in the application of FRET-based fluorescence microscopy to the understanding of cellular functioning was the advent of fluorescent proteins (FPs) which act as ideal FRET pairs. In the first instance, green fluorescent protein (GFP) targeting (Fig. 6) gained popularity both for the implementation of biosensors and for the study of PPIs.32–43 | ||
| Fig. 6 Structure of GFP. At the N-terminus of the protein, the fluorophore rhodamine 6G has been covalently added. | ||
Tsien and coworkers have developed several GFP-FRET constructs that are used to monitor the biochemical environment inside living cells.44 GFP-based constructs do, however, suffer from limited sensitivity, as they are relatively large, thereby limiting spatial resolution. Then labeling with several fluorescent proteins became possible and some of the GFP-based constructs' limitations could be overcome. From here live-cell imaging experiments were totally revolutionized and lots of intracellular biochemical mechanisms were elucidated. Mutations of GFPs45,46 and the discovery of coral-derived proteins47 have led to a broad selection of different colored proteins48 and marked another important point in the history of live-cell imaging experiments (Table 1). It should be mentioned however that many of the problems generally associated with FRET (e.g. mutual resonance energy transfer or fluorescence quenching by photoinduced electron transfer) are also met by FP-FRET. First because the excitation and emission spectra of many FPs are broad thus inducing significant cross-talk; second the large size of FPs occupies much of the FRET distance resulting in lower FRET efficiencies.
| Class | Protein | Source laboratory | Excitation (nm) | Emission (nm) | Brightness | Photostability | pKa | Oligomerization |
|---|---|---|---|---|---|---|---|---|
| Far-red | mPlum | Tsien | 590 | 649 | 4.1 | 53 | <4.5 | Monomer |
| Red | mCherry | Tsien | 587 | 610 | 16 | 96 | <4.5 | Monomer |
| tdTomato | Tsien | 554 | 581 | 95 | 98 | 4.7 | Tandem dimer | |
| mStramberry | Tsien | 574 | 596 | 26 | 15 | <4.5 | Monomer | |
| J-Red | Evrogen | 584 | 610 | 8.8 | 13 | 5.0 | Dimer | |
| DsRed-monomer | Clonetech | 556 | 586 | 3.5 | 16 | 4.5 | Monomer | |
| Orange | mOrange | Tsien | 548 | 562 | 49 | 9.0 | 6.5 | Monomer |
| mKo | MBL Intl. | 548 | 559 | 31 | 122 | 5.0 | Monomer | |
| Yellow-green | mCitrine | Tsien | 516 | 529 | 59 | 49 | 5.7 | Monomer |
| Venus | Miyawaki | 515 | 528 | 53 | 15 | 6.0 | Weak dimer | |
| Ypet | Daugherty | 517 | 530 | 80 | 49 | 5.6 | Weak dimer | |
| EYFP | Invitrogen | 514 | 527 | 51 | 60 | 6.9 | Weak dimer | |
| Green | Emerald | Invitrogen | 487 | 509 | 39 | 0.69 | 6.0 | Weak dimer |
| EGFP | Clonetech | 488 | 507 | 34 | 174 | 6.0 | Weak dimer | |
| Cyan | CyPet | Daugherty | 435 | 477 | 18 | 59 | 5.0 | Weak dimer |
| mCFPm | Tsien | 433 | 475 | 13 | 64 | 4.7 | Monomer | |
| Cerulean | Piston | 433 | 475 | 27 | 36 | 4.7 | Weak dimer | |
| UV-excitable green | T-Sapphire | Griesbeck | 399 | 511 | 26 | 25 | 4.9 | Weak dimer |
The first FRET pair developed was a blue FP (BFP) coupled with a GFP.49 Other pairs include GFPs or YFPs as donors coupled with orange or red derivatives such as mKO or mCherry.48,50 Non-genetically encoded, but clever, FRET pairs can also be used to monitor cellular events in live cells. To overcome some of the limitations of GFP-based constructs, Tsien and coworkers developed several membrane permeable dyes useful for FRET. One construct contains a blue-emitting coumarin donor coupled via a cleavable linker to a green-emitting fluorescein derivative.51
Fluorescence lifetime imaging
A conventional method for determining FRET is to compare the donor intensity of the donor–acceptor sample to that of the donor sample, and possibly to compare the acceptor intensity of the donor–acceptor sample to that of the acceptor sample. The problem is that this requires matching concentrations of different samples, which is difficult to do accurately. Lifetime measurements have the advantage of being concentration independent and, if multiple lifetimes can be resolved, of being able to differentiate subpopulations with different amounts of energy transfer. Clegg, Jovin and others have pioneered a method to measure donor lifetimes on a point by point basis in the image of a cell (or a microscopic sample); this is called fluorescence lifetime imaging microscopy (FLIM).52 The idea is that the excitation light is modulated rapidly on the order of the lifetime of the donor's excited state, which is in the nanosecond timescale for organic dyes. For dyes with lifetimes significantly faster than the modulation time, the emission intensity will simply follow the excitation intensity, with little or no phase lag, and a large modulation amplitude. Dyes that have a much longer lifetime cannot follow the rapidly changing excitation intensity and will have very little modulation. For dyes on the order of the modulation time, their emission will have an intermediate modulation amplitude and a significant phase lag. A CCD with a tuning image intensifier is used to detect this phase and modulation at every pixel—that is, in a spatially resolved manner.12Near-infrared (NIR) fluorescent proteins
There are also several commercially available reactive near-infrared probes that can be used to enhance the transfer efficiency and allow larger distances to be measured. Indeed long-wavelength probes with emission in the NIR region are optimum for biological imaging applications in living systems because of minimum photo damage to biological samples and minimum interference from background auto-fluorescence.Snakes possess a unique sensory system for infrared radiation detection.53 Current research efforts are devoted to the understanding of how snakes detect and transduce infrared signals into nerve impulses, most likely they express particular proteins able to do this job.53 Hopefully in the next years these near-infrared sensing proteins will be isolated and used as cellular biomarkers for implementing total body imaging techniques. The design and synthesis of new near-infrared probes with high quantum yield is also very challenging. Indeed the near-infrared band represents a spectral window where the human body is transparent. It is well known that hemoglobin in the blood absorbs visible wavelengths shorter than 650 nm, while water molecules absorb wavelengths above 900 nm. It appears evident that the possibility to have a protein with an emission window in the region between 650 nm and 900 nm means that it could be possible to develop an advanced human body marker, since near-infrared radiation penetrates easily into and out of tissues.
Anti-Stokes fluorescence emission
Up-conversion (UC) is an anti-Stokes process in which longer wavelength radiation, usually NIR or IR light, is converted to shorter wavelength radiation such as UV or visible (vis) radiation via a two-photon or multi-photon mechanism. In other words the output photons have shorter wavelength than the input ones. There are several benefits of using UC for biological studies (and in particular biological imaging applications in living systems): less photo-toxicity and deeper penetration depth due to a longer wavelength pump, increased signal-to-noise ratio due to low auto fluorescence, reduction of light scattering, low photo-bleaching, and multiple labeling capabilities under the same excitation.54 For a fluorophore to be useful for in vivo imaging, it is critical that the material does not impart excessive toxicity to the organism. Especially for up-converting nano-materials to be used as in vivo imaging agents, several criteria such as near infrared excitation and emission, water solubility, biocompatibility, and means of attachment of additional optical reporters or targeting molecules should be met.55 In the last decade there has been a tremendous amount of effort to synthesize or prepare up-conversion nanoparticles (UCNPs) for UCL.56–59 Most common types of nanoparticles used for up-conversion luminescence are those doped with rare-earth lanthanide. There have been a few reports using quantum dots (QDs) for UC bio-imaging applications.60,61 The major difference of QDs from UCNPs is the mechanism of excitation and relaxation of electrons that eventually leads to the emission of photons. QDs have been estimated to exhibit 20 times brighter luminescence and found to be 100 times more stable than traditional fluorescent reporters. Tunability and narrow emission profile are also two major advantages of QDs. Recently graphene has received much attention because of its many unique and novel properties such as superior mechanical flexibility, excellent thermal/chemical stability and environmentally friendly nature. Thus graphene quantum dots (GQDs) found applications also in the field of UC bio-imaging applications.62,63 Some other materials such as gold nano-rods and quantum rods have also been investigated as alternatives to traditional fluorophores for their two-photon luminescence for UC bio-imaging applications.64,65Perspectives and final remarks
By highlighting specific examples, this article illustrates a variety of possible optical approaches that can be used for the study of protein interactions and molecular interactions. In particular we described a number of methods proposed during the last decade to enhance FRET measurements to beyond 100 Å which could be very useful in the field of live-cell imaging experiments. We are certain that with our approach exploiting the replacement of the single acceptor molecule of the FRET pair by many linked and closely located molecules FRET measurements can be easily extended to about 140–150 Å with over 10% FRET efficiency. However, from an experimental point of view it may be useful to limit the number of acceptors to not risk the occurrence of phenomena such as aggregation, self-quenching, energy homotransfer. Of course the highest advisable number of acceptors depends on several factors such as the size of macromolecules and fluorophores, the eventual tendency of the macromolecule to dimerization and/or aggregation, and the number of active sites on the macromolecules. Any future development that improves R0 will help in the investigation of even larger distances. We expect our approach to be easily adapted within the framework of FRET-based cellular florescence microscopy and help cell biologists to monitor different components within organisms and manipulate these in vivo and in the whole animal. There are also some other promising potential FRET acceptors that can serve the same purpose of enhancing the FRET distance. For example, very small silica nanoparticles and dendrimers (which are only a few nm in size) can bring many probes and a very large molar extinction coefficient to a very small area and can be functionalized for use as acceptor labels. In practical terms, these particles are similar to QDs, though smaller. One of the parameters governing transfer efficiency is the size of the acceptor-labeled protein/macromolecule/particle. The smaller the particle, the greater the transfer efficiency for any given R0 and number of acceptors. The RET of lanthanides has many useful attributes and we foresee that this approach should be also applicable for FRET-based cellular florescence microscopy (with lanthanides as potential donors). One of the less realized advantages of lanthanides is their single lifetime, which makes the interpretation of data simpler.Finally, the use of new near-infrared fluorescent probes or NIR proteins could have a great impact in biology and medicine.
Acknowledgements
This Project was partially funded by the project CTN01_00230_240864 “SAFE &SMART-Nuove tecnologie abilitanti per la food safety e l'integrità delle filiere agro-alimentari in uno scenario globale” (SD, MS, AC).References
- D. P. Ryan and J. M. Matthews, Curr. Opin. Struct. Biol., 2005, 15, 441–446 CrossRef CAS PubMed.
- A. C. Gingras, M. Gstaiger, B. Raught and R. Aebersold, Nat. Rev. Mol. Cell Biol., 2007, 8, 645–654 CrossRef CAS PubMed.
- P. Haris, Biochem. Soc. Trans., 2010, 38, 940 CrossRef CAS PubMed.
- R. M. Kaake, X. Wang, A. Burke, C. Yu, W. Kandur, Y. Yang, E. J. Novtisky, T. Second, J. Duan, A. Kao, S. Guan, D. Vellucci, S. D. Rychnovsky and L. Huang, Mol. Cell. Proteomics, 2014, 13, 3533–3543 CAS.
- S. Lievens, S. Gerlo, I. Lemmens, D. J. De Clercq, M. D. Risseeuw, N. Vanderroost, A. S. De Smet, E. Ruyssinck, E. Chevet, C. S. Van and J. Tavernier, Mol. Cell. Proteomics, 2014, 13, 3332–3342 CAS.
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS PubMed.
- D. W. Piston and G. J. Kremers, Trends Biochem. Sci., 2007, 32, 407–414 CrossRef CAS PubMed.
- K. D. Pfleger and K. A. Eidne, Nat. Methods, 2006, 3, 165–174 CrossRef CAS PubMed.
- B. Stynen, H. Tournu, J. Tavernier and D. P. Van, Microbiol. Mol. Biol. Rev., 2012, 76, 331–382 CrossRef CAS PubMed.
- S. W. Michnick, P. H. Ear, E. N. Manderson, I. Remy and E. Stefan, Nat. Rev. Drug Discovery, 2007, 6, 569–582 CrossRef CAS PubMed.
- E. Tchekanda, D. Sivanesan and S. W. Michnick, Nat. Methods, 2014, 11, 641–644 CrossRef CAS PubMed.
- P. R. Selvin, Nat. Struct. Biol., 2000, 7, 730–734 CrossRef CAS PubMed.
- L. Stryer and R. P. Haugland, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719–726 CrossRef CAS.
- M. Strianese, G. J. Palm, S. Milione, O. Kuhl, W. Hinrichs and C. Pellecchia, Inorg. Chem., 2012, 51, 11220–11222 CrossRef CAS PubMed.
- M. Strianese, F. De Martino, C. Pellecchia, G. Ruggiero and S. D'Auria, Protein Pept. Lett., 2011, 18, 282–286 CrossRef CAS PubMed.
- M. Strianese, A. Varriale, M. Staiano, C. Pellecchia and S. D'Auria, Nanoscale, 2011, 3, 298–302 RSC.
- M. Strianese, F. De Martino, V. Pavone, A. Lombardi, G. W. Canters and C. Pellecchia, J. Inorg. Biochem., 2010, 104, 619–624 CrossRef CAS PubMed.
- M. Strianese, S. Milione, V. Bertolasi, C. Pellecchia and A. Grassi, Inorg. Chem., 2011, 50, 900–910 CrossRef CAS PubMed.
- M. Strianese, G. Zauner, L. C. Tabares, A. W. Tepper, M. F. De, C. Pellecchia, T. J. Aartsma and G. W. Canters, Chem. – Eur. J., 2013, 19, 14977–14982 CrossRef CAS PubMed.
- G. Zauner, M. Strianese, L. Bubacco, T. J. Aartsma, A. W. Tepper and G. W. Canters, Inorg. Chim. Acta, 2008, 361, 1116–1121 CrossRef CAS.
- S. Milione, C. Capacchione, C. Cuomo, M. Strianese, V. Bertolasi and A. Grassi, Inorg. Chem., 2009, 48, 9510–9518 CrossRef CAS PubMed.
- P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275–302 CrossRef CAS PubMed.
- A. N. Kapanidis, Y. W. Ebright, R. D. Ludescher, S. Chan and R. H. Ebright, J. Mol. Biol., 2001, 312, 453–468 CrossRef CAS PubMed.
- E. Heyduk, T. Heyduk, P. Claus and J. R. Wisniewski, J. Biol. Chem., 1997, 272, 19763–19770 CrossRef CAS PubMed.
- H. E. Rajapakse, N. Gahlaut, S. Mohandessi, D. Yu, J. R. Turner and L. W. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13582–13587 CrossRef CAS PubMed.
- J. R. Lakowicz, Y. Shen, S. D'Auria, J. Malicka, J. Fang, Z. Gryczynski and I. Gryczynski, Anal. Biochem., 2002, 301, 261–277 CrossRef CAS PubMed.
- J. Lakowicz, J. Kuśba, Y. Shen, J. Malicka, S. D'Auria, Z. Gryczynski and I. Gryczynski, J. Fluoresc., 2003, 13, 69–77 CrossRef CAS.
- P. Bojarski, L. Kulak, K. Walczewska-Szewc, A. Synak, V. M. Marzullo, A. Luini and S. D'Auria, J. Phys. Chem. B, 2011, 115, 10120–10125 CrossRef CAS PubMed.
- B. P. Maliwal, S. Raut, R. Fudala, S. D'Auria, V. M. Marzullo, A. Luini, I. Gryczynski and Z. Gryczynski, J. Biomed. Opt., 2012, 17, 011006 CrossRef PubMed.
- K. Walczewska-Szewc, P. Bojarski and S. D'Auria, J. Mol. Model., 2013, 19, 4195–4201 CrossRef CAS PubMed.
- S. D'Auria, E. Apicella, M. Staiano, G. S. Di, G. Ruggiero, M. Rossi, P. Sarkar, R. Luchowski, I. Gryczynski and Z. Gryczynski, Anal. Biochem., 2012, 425, 13–17 CrossRef PubMed.
- A. Miyawaki, J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura and R. Y. Tsien, Nature, 1997, 388, 882–887 CrossRef CAS PubMed.
- G. Patterson, R. N. Day and D. Piston, J. Cell Sci., 2001, 114, 837–838 CAS.
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802–805 CAS.
- T. Nagai, S. Yamada, T. Tominaga, M. Ichikawa and A. Miyawaki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10554–10559 CrossRef CAS PubMed.
- B. Ponsioen, J. Zhao, J. Riedl, F. Zwartkruis, K. G. van der, M. Zaccolo, W. H. Moolenaar, J. L. Bos and K. Jalink, EMBO Rep., 2004, 5, 1176–1180 CrossRef CAS PubMed.
- P. W. Vanderklish, L. A. Krushel, B. H. Holst, J. A. Gally, K. L. Crossin and G. M. Edelman, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2253–2258 CrossRef CAS PubMed.
- R. D. Mitra, C. M. Silva and D. C. Youvan, Gene, 1996, 173, 13–17 CrossRef CAS PubMed.
- L. G. Tertoolen, C. Blanchetot, G. Jiang, J. Overvoorde, T. W. Gadella Jr., T. Hunter and H. J. Den, BMC Cell Biol., 2001, 2, 8 CrossRef CAS PubMed.
- M. C. Overton and K. J. Blumer, Curr. Biol., 2000, 10, 341–344 CrossRef CAS PubMed.
- I. A. Tonaco, J. W. Borst, S. C. de Vries, G. C. Angenent and R. G. Immink, J. Exp. Bot., 2006, 57, 33–42 CrossRef CAS PubMed.
- R. G. Immink, T. W. Gadella Jr., S. Ferrario, M. Busscher and G. C. Angenent, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2416–2421 CrossRef CAS PubMed.
- R. N. Day, Mol. Endocrinol., 1998, 12, 1410–1419 CrossRef CAS PubMed.
- R. Y. Tsien and A. Miyawaki, Science, 1998, 280, 1954–1955 CrossRef CAS PubMed.
- R. Heim, D. C. Prasher and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12501–12504 CrossRef CAS.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- M. V. Matz, A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov and S. A. Lukyanov, Nat. Biotechnol., 1999, 17, 969–973 CrossRef CAS PubMed.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- R. Heim and R. Y. Tsien, Curr. Biol., 1996, 6, 178–182 CrossRef CAS PubMed.
- S. Karasawa, T. Araki, T. Nagai, H. Mizuno and A. Miyawaki, Biochem. J., 2004, 381, 307–312 CrossRef CAS PubMed.
- G. Zlokarnik, P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. Feng, M. Whitney, K. Roemer and R. Y. Tsien, Science, 1998, 279, 84–88 CrossRef CAS PubMed.
- J. Gadella, T. M. Jovin and R. M. Clegg, Biophys. Chem., 1993, 48, 221–239 CrossRef.
- E. O. Gracheva, N. T. Ingolia, Y. M. Kelly, J. F. Cordero-Morales, G. Hollopeter, A. T. Chesler, E. E. Sanchez, J. C. Perez, J. S. Weissman and D. Julius, Nature, 2010, 464, 1006–1011 CrossRef CAS PubMed.
- J. Zhou, Y. Sun, X. Du, L. Xiong, H. Hu and F. Li, Biomaterials, 2010, 31, 3287–3295 CrossRef CAS PubMed.
- S. A. Hilderbrand, F. Shao, C. Salthouse, U. Mahmood and R. Weissleder, Chem. Commun., 2009, 4188–4190 RSC.
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Nature, 2005, 437, 121–124 CrossRef CAS PubMed.
- H. X. Mai, Y. W. Zhang, R. Si, Z. G. Yan, L. D. Sun, L. P. You and C. H. Yan, J. Am. Chem. Soc., 2006, 128, 6426–6436 CrossRef CAS PubMed.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642–5643 CrossRef CAS PubMed.
- S. Wu, G. Han, D. J. Milliron, S. Aloni, V. Altoe, D. V. Talapin, B. E. Cohen and P. J. Schuck, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10917–10921 CrossRef CAS PubMed.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- Y. Chen and H. Liang, J. Photochem. Photobiol., B, 2014, 135, 23–32 CrossRef CAS PubMed.
- S. Zhuo, M. Shao and S. T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- J. Shen, Y. Zhu, C. Chen, X. Yang and C. Li, Chem. Commun., 2011, 47, 2580–2582 RSC.
- N. J. Durr, T. Larson, D. K. Smith, B. A. Korgel, K. Sokolov and A. Ben-Yakar, Nano Lett., 2007, 7, 941–945 CrossRef CAS PubMed.
- K. T. Yong, J. Qian, I. Roy, H. H. Lee, E. J. Bergey, K. M. Tramposch, S. He, M. T. Swihart, A. Maitra and P. N. Prasad, Nano Lett., 2007, 7, 761–765 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |