
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deracemisations under kinetic and thermodynamic control
A. R. A.
Palmans

Laboratory of Macromolecular and Organic Chemistry, Institute for Complex Molecular Systems, TU Eindhoven, PO Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl
First published on 26th January 2017
Abstract
Deracemisation reactions occur when a racemic mixture is converted into a nonracemic mixture by increasing the quantity of one enantiomer at the expense of the other. This process can take place under thermodynamic control, but when combined with crystallisation processes kinetic factors also play a role. This review summarises the different approaches that have been taken to achieve efficient deracemisations. Starting from examples in which spontaneous symmetry breaking was found to occur, attrition enhanced deracemisation will be discussed in which solid-solution equilibria drive the deracemisation process. The combination of detailed experimental studies and mathematical models resulted in a profound understanding of this complex process, which is applicable to all congomerate forming compounds with a racemisable stereocenter. Then, we focus on deracemisations that occur under full thermodynamic control. Especially the combination of supramolecular interactions with a racemisation process gives interesting results, albeit that they are less predictable. The review will end with the possibilities supramolecular helical structures that show dynamic helicity can offer in conjunction with asymmetric catalysis. Herein, the helical preference induced by a minute amount of chiral compound is relayed to high enantiomeric excesses in a variety of reactions.
 Anja Palmans | Anja Palmans obtained a degree in chemical engineering (1992) at the Eindhoven University of Technology (The Netherlands) and continued with a PhD on the topic of supramolecular chemistry (1997). After a postdoc at the ETH Zürich (Switzerland) and working as a research scientist at DSM Research (The Netherlands), she became assistant professor at the Laboratory of Macromolecular and Organic Chemistry in Eindhoven in 2002. Since 2010 she is an associate professor, and focusses on the controlled folding of macromolecules, supramolecular chemistry, and the design and synthesis of synthetic enzymes for cascade catalysis. |
Design, System, ApplicationAccess to enantiomerically pure intermediates is essential for obtaining pharmaceutically active products. Both asymmetric catalysis as well as resolutions of racemates have been applied in the past decades to develop efficient routes to achieve this goal. Here, we focus on deracemising racemic mixtures into one, preferred enantiomer with complete consumption of the undesired enantiomer. In deracemisations that are coupled to crystallisations, this feat is accomplished by combining the formation of conglomerate crystals and a chemical racemisation of the stereocenter, with complex solid-solution equilibria that drive the system to a homochiral end-state. However, not all compounds crystallise as conglomerates. An alternative is found by eliminating the energetic degeneracy between two enantiomers by diastereomer formation. Especially the thermodynamically controlled formation of transient diastereomers by non-covalent, supramolecular interactions has emerged as an interesting alternative to achieve deracemisations, but the design parameters of these systems are only beginning to be explored. Finally, supramolecular helical conformations can be deracemised into one preferential helicity in thermodynamically controlled conditions. Coupling these biased helical structures to transition-metal-based catalysts can afford either enantiomer by simply changing the conformation of the catalyst carrier in response to environmental changes. Such fusion between fields may open up new avenues for asymmetric catalysis. |
1. Introduction
Deracemisation was defined by Pirkle as “the conversion of a racemic mixture into a nonracemic mixture by increasing the quantity of one enantiomer at the expense of the other”1 and is an important tool in the production of optically active compounds. The process requires a change of configuration of the stereocenter of the undesired enantiomer. Deracemisation reactions can occur under thermodynamic as well as under kinetic control.1 Although asymmetric catalysis is tremendously important to prepare enantiomerically pure compounds, deracemisation reactions have their own merits and advantages.2In recent years, Faber proposed to broaden the definition of deracemisation to “constitute any process during which a racemate is converted into a non-racemic product in 100% theoretical yield without intermediate separation of material”.3 As a result, many more processes have been developed which induce a deracemisation, and these have been covered in excellent recent reviews.4–9 In many examples, highly enantioselective enzymes play a crucial role in the success of the deracemisation reactions. In combination with synthetic racemisation catalysts beautiful examples of complete deracemisations of enantiomer mixtures into homochiral compounds have been achieved.
This review will predominantly focus on examples that follow the original definition of deracemisation given by Pirkle. Special emphasis will be placed on deracemisations that occur during crystallisation, a process in which kinetic factors may play an important role. Furthermore, deracemisations in thermodynamically controlled conditions governed by supramolecular interactions will be discussed. The latter is less established as a method to obtain scalemic from racemic mixtures, but could prove versatile in a variety of applications. Some recent examples in which a bias in supramolecular helicity induces an increase in the enantiomeric excess of the building blocks of the supramolecular structure will be discussed as well. The improved understanding of the physical mechanisms underlying deracemisation reactions has been crucial to further develop the field.
We end this review with examples in which the possibilities are explored of (supramolecular) helical structures that show dynamic helicity. These systems open up exciting new avenues in asymmetric catalysis. In a next step, ligands that adopt a preferential conformation in response to changes in the environment could produce switchable ligands for asymmetric catalysis and afford both enantiomers by simply changing operation conditions.
2. Deracemisation by crystallisation
2.1 Crystallisation of racemic mixtures
Crystallisation is a complex process, governed by both thermodynamic and kinetic factors which makes it a difficult process to control. Crystallising a racemic mixture of enantiomers can result in the formation of racemate crystals, conglomerate crystals or, in rare cases, solid solutions. In the vast majority of cases (90%), a racemic mixture crystallises as a racemate—crystals that comprise equal amounts of both the enantiomers—whereas a minority (5–10%) crystallises as a conglomerate—each crystal is homochiral but equal amounts of both types of crystals are formed (Fig. 1). In solid solutions, both enantiomers are randomly distributed in the crystal. In case a conglomerate is formed, the first crystal formed is by definition of one handedness, thus if this handedness could bias the whole mixture, symmetry breaking—i.e. the formation of 1 enantiomer out of a racemic starting condition—can be achieved. Preferential crystallisation takes advantage of the fact that both of the enantiomers can be separately isolated from a conglomerate forming compound.10–12 However, this results in a yield that is usually around 15–20% of either enantiomer in a single cycle.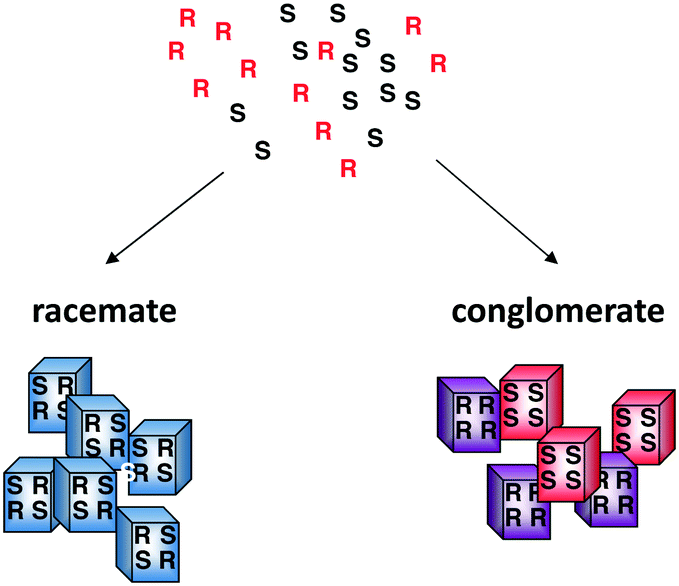 | ||
| Fig. 1 Formation of a racemate or a conglomerate upon crystallisation of a racemic mixture. | ||
Since Pasteur's first experiments on tartaric acids, the formation of conglomerate crystals received a lot of attention. Nowadays the difference between conglomerates and racemates can be readily made by a number of techniques (infrared spectroscopy, X-ray powder diffraction analysis, differential scanning calorimetry, second harmonic generation).13,14 In contrast, it still remains impossible to predict in which type of crystal a racemic mixture of enantiomers will crystallise.
2.2 Spontaneous deracemisation during crystallisation
In 1941, a summary of the lecture given by Prof. Havinga from Leiden University (The Netherlands) on the topic “spontaneous asymmetric synthesis” appeared in “Chemisch Weekblad”.15 Herein, Havinga postulated that it should be possible to convert a racemic mixture in solution to crystals of predominantly one enantiomorphic form if the following prerequisites were fulfilled: (1) the compound crystallises as a conglomerate, (2) there is a high rate of racemisation when the compound is in solution, which can be enhanced by a catalyst, (3) the rate at which the crystals are nucleated should be low whereas the rate of crystal growth should be high, (4) the rate of racemisation should be higher than the rate of crystal growth. Then, as soon as the first crystal is nucleated, this will grow fast and the chance of new nucleation events will be reduced.The crystallisation of a chiral ammonium salt, Me(Et)N+(All)Ph−, was investigated since it possesses an asymmetric nitrogen centre and was known to rapidly racemise in chloroform (Fig. 2A).16 In addition, Me(Et)N+(All)Ph− forms a conglomerate from chloroform.17,18 Gratifyingly, racemisation was significantly slowed down when the compound was dissolved in water or ethanol, permitting the determination of the optical rotation of the crystals after isolation. Remarkably, it appeared that in 14 crystallisations, 12 resulted in the formation of dextrorotatory and none in levorotatory crystals. This implied not only that deracemisation occurred in these systems but also that there was a preference for the dextrorotatory isomer. The fact that the outcome of the crystallisations was not random, i.e. equal amounts of crystallisations resulting in either dextrorotatory or levorotatory isomers, was surprising and attributed to “latent” nucleating agents or inhibitors of natural origins that either promoted crystallisation of the dextrorotatory crystals or prevented crystallisation of the levorotatory crystals. In fact, upon careful filtration of the solutions, also levorotatory crystals were obtained although the nucleation rate of the crystals was significantly reduced. Almost 60 years later, the results of Havinga were successfully repeated by Lyssenko and coworkers.19 Additional stirring of the crystallising solutions or the melt increased the ee significantly, up to 98.5%.
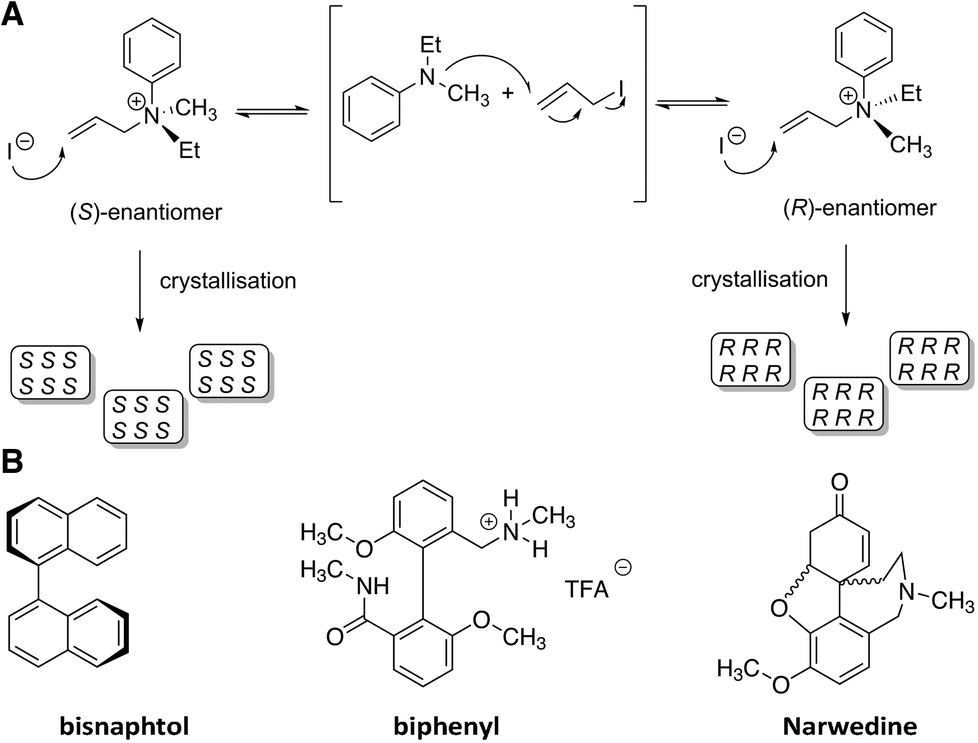 | ||
| Fig. 2 A) Chemical structure of Me(Et)N+(All)PhI−, its racemisation mechanism in chloroform and its crystallisation as a conglomerate;15 B) chemical structures of atropisomers that show deracemisation by a primary nucleation process: bisnaphtol,20 biphenyl derivative,21 and narwedine.23 | ||
After this landmark example, other systems were found to deracemise upon crystallisation either from the melt or from solution via a primary nucleation process (Fig. 2B).20–23 Interestingly, both enantiomers of the pharmaceutical intermediate narwedine, a precursor for (−)-galanthamine, could be readily obtained by initiating the crystallisation with a nucleating crystal.23
The importance of stirring to enhance the formation of one type of crystals was highlighted in the seminal work of Kondepudi and co-workers in the 1990s.24,25 Although achiral, NaClO3 crystallises in a chiral space group as a conglomerate, and it was long realized that a crystallisation in “normal” conditions, i.e. heating up a mixture of solid in solvent and after complete dissolution slow cooling, resulted in the formation of close to equal numbers of dextrorotatory and levorotatory crystals in each crystallisation.26 The probability distribution for the crystal enantiomeric excess (cee) in unstirred crystallisations was monomodal, with the maximum centred at zero (Fig. 3, top graphs). In contrast, stirring during crystallisation in supersaturated conditions resulted in an outcome in which either almost all crystals were levorotatory or almost all dextrorotatory for each crystallisation (Fig. 3, bottom graphs). The resulting probability distribution for the crystal enantiomeric excess was bimodal, with close to equal amounts of both forms. The remarkable symmetry breaking in these stirred crystallisations was attributed to a secondary nucleation process. Upon the formation of the first crystals, the first one that is struck by the stirrer clones many new nuclei that are enantiomerically identical to the “mother” crystal. The growth of these nuclei lowers the concentration of the NaClO3 in solution, preventing the formation of new, primary nuclei that have an equal probability of being levorotatory or dextrorotatory. This secondary nucleation process was beautifully illustrated by McBride using video recordings.27
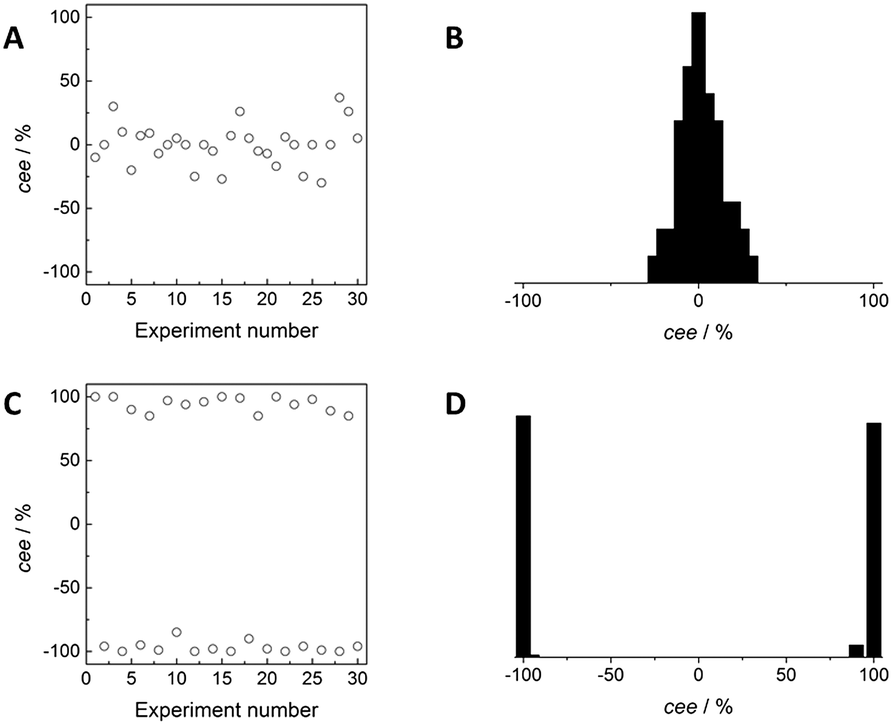 | ||
| Fig. 3 Hypothetical results to illustrate the outcome of a crystallisation of NaClO3 in the absence (A and B) and presence (C and D) of stirring. The crystal enantiomer excess, cee, of the crystals in the different hypothetical experiments is represented in A and C which corresponds to either a monomodal (B) or bimodal (D) distribution. The hypothetical results are inspired by the figures given in ref. 25. | ||
Since Havinga's first observations, spontaneous deracemisation of conglomerate forming compounds has been discovered for several compounds comprising asymmetric centra28–30 but also for atropisomers,21,31 metal complexes,32,33 helical coordination polymers,34 and even helical oligomers.35 Deracemisations often proceeded faster and more reliably when stirring was conducted during the crystallisation process.21,29,31
2.3 Deracemisation in conditions of abrasive grinding
Following the work of Kondepudi, Viedma stirred a slurry of NaClO3 crystals comprising equal amounts of levorotatory and dextrorotatory crystals in a saturated solution in the presence of glass beads. The glass beads induced abrasive grinding or attrition, resulting in a constant crushing of the crystals.36 All slurries showed total symmetry breaking and chiral purity after 24 h, a remarkable and unexpected outcome. The handedness of the crystals was randomly either levorotatory or dextrorotatory. In the presence of a small excess (5%) of one of the enantiomorphs, the outcome was always towards the excess enantiomer.The fascinating outcome of these experiments was rationalised by considering that the combination of abrasive grinding and stirring induces a constant dissolution of crystals, which is thermodynamically driven. At the same time, growth and secondary nucleation/crystallisation takes place. The solubility of a crystal in a solution depends on its size (Gibbs–Thompson rule): smaller crystals show a higher solubility than larger crystals. As a result, the smallest crystals easily dissolve, feeding the larger ones. In addition, the larger crystals grow faster than smaller crystals (Ostwald ripening). Consequently, the smallest crystals more easily dissolve, and feed the larger ones. This, in combination with secondary nucleation processes, results in a complete symmetry breaking process that does not require any external chiral bias.37 The driving force of this process was attributed to “chiral amnesia”38 of the solution phase, which remains achiral, and a consequence of the interplay between a kinetic departure from equilibrium in dynamic crystallisation/dissolution and the system's attempt to re-establish equilibrium.38,39 Such abrasive grinding processes are currently referred to as “Viedma ripening” or “attrition-enhanced deracemisation” and constitute a prime example of symmetry breaking from an optically inactive starting condition. This finding was not limited to only NaClO3, other achiral compounds forming conglomerates such as NaBrO3,37 and ethylene diammonium sulfate40 were completely deracemised in abrasive grinding conditions as well.
The scientific impact of Viedma's work is tremendous, but the crystallisation of an intrinsically achiral compound into an optically pure crystal is of limited practical use. This changed with ground-breaking experiments performed by Noorduin et al. in 2008. By derivatising phenylglycine to a conglomerate forming imine (PhgI) (Fig. 4A) and subjecting a near racemic mixture (ee = 3%) to the abrasive grinding conditions developed by Viedma, complete deracemisation of the amino acid derivative was achieved in a few weeks.41 In this case, the stereocenter had to be racemised to keep the solution racemic. This was accomplished by adding a base, 1,8-diazabicycloundec-7-ene (DBU), after establishing the solution-solid equilibrium in MeOH or acetonitril. Importantly, deracemisation was only observed in the presence of glass beads (i.e. under conditions of abrasive grinding or attrition) and not upon only stirring. Also, seeding a racemic mixture with 0.1% of an enantiopure additive, phenylglycine, sufficed to direct the outcome of the reaction to a homochiral end state. Interestingly, the chiral additive obeyed Lahav's rule of inversion,42i.e. the R-impurity favoured deracemisation into the S-enantiomer. Although the deracemisation process was rather slow, the ramifications of this experiment were immense because every racemic compound (or close derivative thereof) capable of forming a conglomerate could, in principle, be deracemised into one enantiomer by a simple abrasive grinding procedure of conglomerate crystals in contact with a solution in which racemisation occurs.
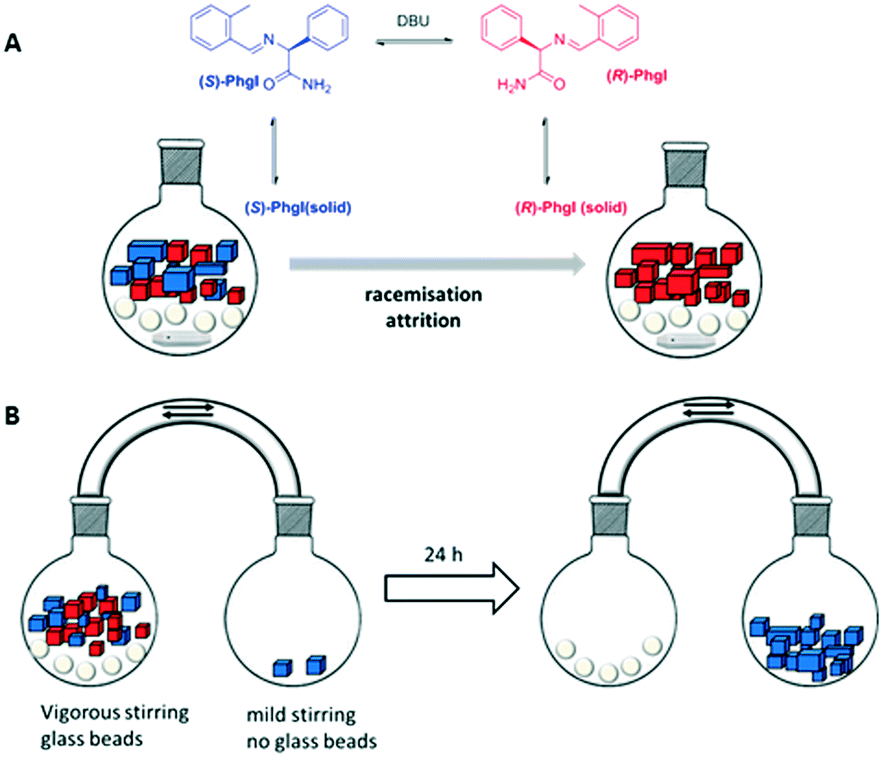 | ||
| Fig. 4 A) Chemical structure of the phenylglycine derivate that deracemised in abrasive grinding conditions. Depending on the exact conditions, either the R or the S enantiomer is obtained;41 B) deracemisation using crystal-size-induced solubility as a driving force.52 | ||
In the ensuing years, the mechanism of this process was studied in detail both experimentally as well as theoretically and the factors enhancing the rate of the deracemisation process were identified.43–45 In the beginning, deracemisation starting from a racemic mixture tended to produce the R-enantiomer, indicating that the behaviour of the system was not random.46 However, it was found that the outcome of deracemisations was sensitive to the order of the addition of the different reagents.47 Moreover, the outcome could be biased by impurities or additives which inhibit the crystal growth of one of the enantiomorphs,41,48,49 by mixing enantiomorphs of different crystal sizes50 and even by subjecting the stirring slurries to circularly polarized light.51
Detailed mechanistic studies by the group of Blackmond using 15N labelled derivatives of PhGI showed that complete exchange of mass between the solid and solution phase under non-racemising conditions occurred over a relatively short period of time (hours) in a highly reproducible manner.52 This means that under conditions of attrition, the crystals are ablated to such an extent that even the molecules at the interior of crystals are exposed to the solution phase. In contrast, the net movement of converting one enantiomorph into the other remained a random process: the evolution of the net ee of the solid phase was erratic and in some cases no deracemisation occurred. From this, it was concluded that the solid-solution physical movement of molecules as well as the interconversion between enantiomers are facile but the attrition-induced solubility difference is a stochastic process which is difficult to control.
To enhance reproducibility of the attrition-enhanced deracemisation, an alternative approach was suggested which takes advantage of crystal-size-induced solubility as the driving force (Fig. 4B).52 Herein, the solution phase was circulated between two vessels. One vessel contained a racemic mixture of crystals of PhGI, glass beads and was stirred vigorously. The other contained a few large, and therefore less soluble, crystals of (S)-PhGI and was gently stirred. In the first vessel constant attrition occurred, keeping the crystals small. The only driving forces in the system were the solution-phase racemisation and the crystal-size-induced solubility differences. Over time, this resulted in a complete net movement of the crystals in the vessel comprising the racemic mixture to the other vessel only comprising solid (S)-PhGI. Thus, the driving force of deracemisation is solely based on the differential enantiomer solubility induced by crystal size differences as dictated by the Gibbs–Thompson rule.
Apart from gaining a better understanding on the mechanism of attrition-enhanced deracemisation, it also became clear that some compounds could also be deracemised even if they crystallised as epitaxial racemates53 or without a base present.54 Particularly interesting is a study by Viedma, Blackmond and co-workers in which the natural amino acid, aspartic acid, was deracemized by combining abrasive grinding and in situ imine formation with salicylaldehyde in acidic media (Fig. 5A).55 Although elevated temperatures were required, complete deracemisation occurred. Also glutamic acid was readily deracemised using a similar process (Fig. 5B).56 Moreover, achiral staring materials prone to Mannich condensations57,58 or Michael additions59,60 were combined with abrasive grinding, resulting in enantiomerically enriched products (Fig. 5C and D). Viedma ripening is not limited to compounds with a racemisable sp3 stereocenter: metal-based complexes that racemise spontaneously in solution were readily deracemised in abrasive grinding conditions as well.33,61 Finally, the groups of Vlieg and Kellogg showed that precursors to important pharmaceuticals such as naproxen,62 clopidogrel,63 and prasugrel64 can be readily deracemised using the Viedma ripening process highlighting the industrial relevance of attrition-enhanced deracemisations (Fig. 6).
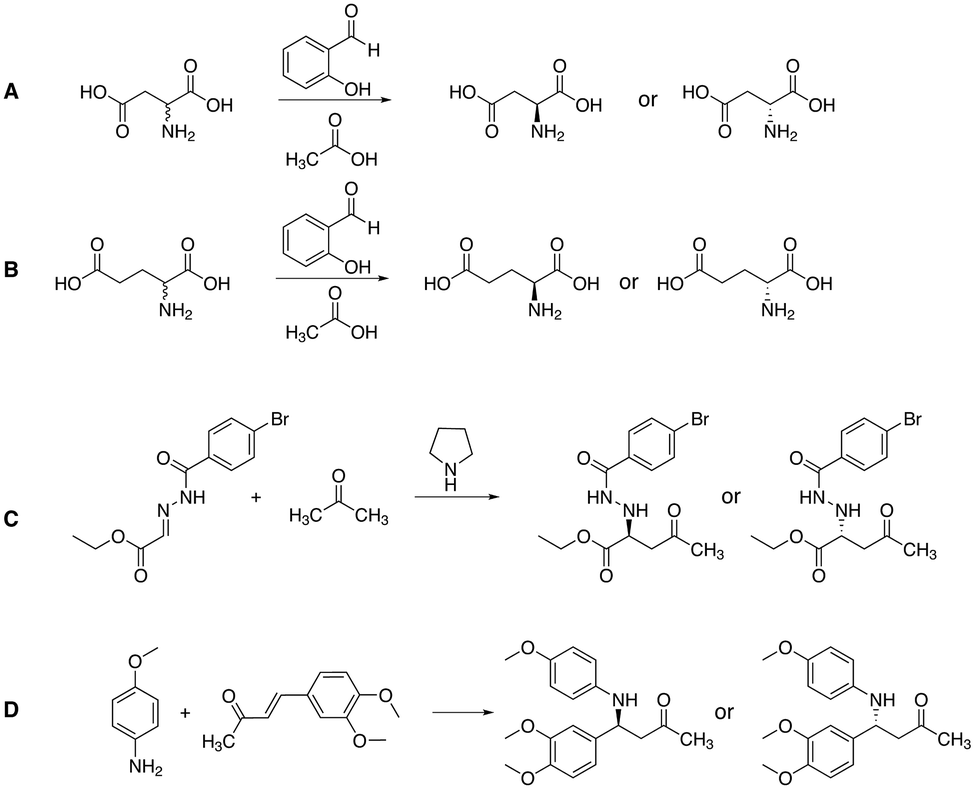 | ||
| Fig. 5 Chemical structures of compounds that have been deracemised by combining in situ imine formation (A and B),55,56 a Mannich reaction (C)57 or a Michael addition (D)59 with Viedma ripening of the solid state. | ||
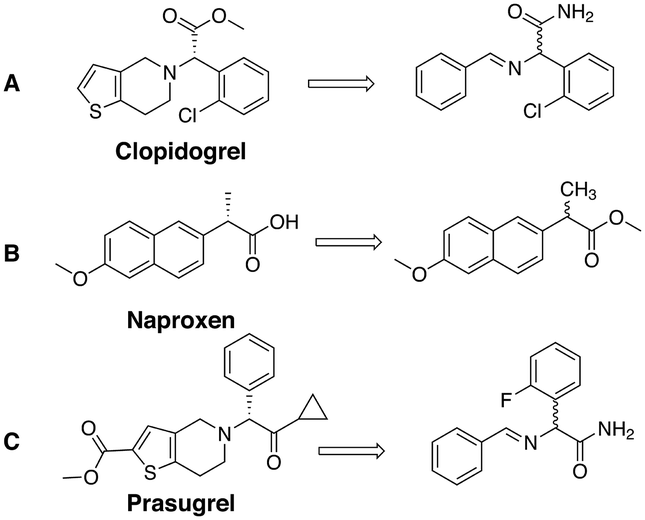 | ||
| Fig. 6 Chemical structure of pharmaceutically active naproxen (A),62 clodiprogel (B)63 and prasugrel (C)64 and their precursors that deracemise using Viedma ripening. | ||
To date, attrition-enhanced deracemisation is not yet applied in industrial processes, but the application of more efficient grinding mechanisms significantly increased the deracemisation rates making the whole process highly applicable for large scale production.65 Therefore, it is only a matter of time before this elegant and simple process will find industrial application.
3. Deracemisations by eliminating the energetic degeneracy between enantiomers
The Viedma ripening process requires constant energy to be put into the system by means of mechanical stirring and grinding, driving the system to a homochiral end state. An intriguing question is in how far deracemisations can be achieved in conditions in which no crystallisation occurs. In principle, eliminating the energetic degeneracy of the two enantiomers suffices to alter the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equilibrium ratio of interconverting enantiomers. Thus, in situ formation of diastereomers favours an equilibrium in which one of the diastereomers is formed in preference. The formation of such diastereomers can be accomplished by using either supramolecular, non-covalent interactions (such as hydrogen bonding and π-stacking interactions) or covalent bond formation.
:1 equilibrium ratio of interconverting enantiomers. Thus, in situ formation of diastereomers favours an equilibrium in which one of the diastereomers is formed in preference. The formation of such diastereomers can be accomplished by using either supramolecular, non-covalent interactions (such as hydrogen bonding and π-stacking interactions) or covalent bond formation.
3.1 The use of supramolecular interactions to induce deracemisations
In 1987, Pirkle and Reno reported on the deracemisation of racemic thioester 2 in the presence of (R)-1 and a chiral stationary phase based on (R)-3 in basic solution (Fig. 7).1 This stationary phase exhibited an effective separation of the enantiomers of 2 and (R)-1. At the same time, the more acidic stereocenter in 2 racemised in the presence of triethylamine, conditions in which the stereocentres of 1 and 3 were configurationally stable. A solution of (R)-1, 2 and TEA was periodically examined by HPLC using a column packed with chiral stationary phase (R)-3. After a period of 28 days, equilibrium was reached with an ee of 78% for (R)-2.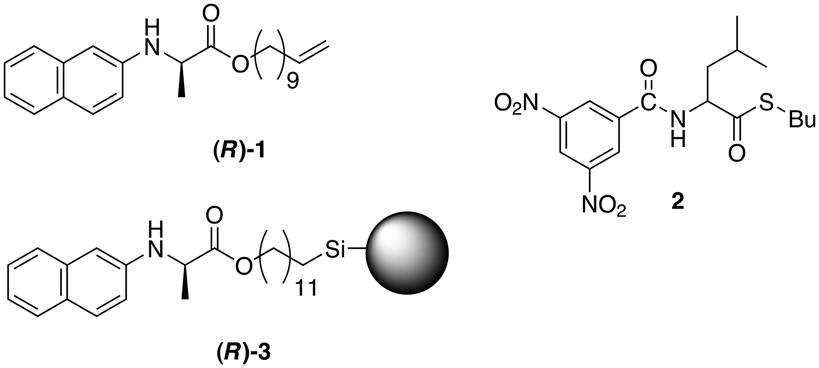 | ||
| Fig. 7 Chemical structures of non-racemisable glycine derivative (R)-1, racemisable leucine derivative (2) and the structure of the chiral stationary phase (R)-3 used in the column chromatography for separating 1 and 2.1 | ||
The equilibrium position of the deracemisation depended on the conditions employed; more polar solvents reduced the ee whereas lower temperatures increased the ee. Also, an increase in (R)-1 increased the ee of (R)-2 although there was a saturation level after which no further increase was observed. The triethylamine concentration enhanced the deracemisation rate of 2 but had little effect on the final ee.
The success of the deracemisation was attributed to the differential complexation of (R)-1 with the enantiomers of 2 which supplied the energetic driving force for the deracemisation. The molecular recognition resulted from hydrogen bonding between the amides in combination with donor–acceptor interaction between the aromatic systems. Since (R)-1 and (S)-2 could not achieve all these bonding interactions simultaneously, this complex was of lesser stability.66 As a result, a shift takes place towards the formation of more (R)-2 which is stabilised by complex formation with the excess of (R)-1. The fact that more apolar solvents and lower temperatures resulted in an increase of the ee and that a saturation effect in the attainable ee was present for the concentration of (R)-1 was consistent with strong complex formation.
Similarly, Roussel and coworkers67 and Lindner and coworkers68 deracemised axially chiral biphenyl derivatives using HPLC with a chiral stationary phase. In these cases, there was no need for a racemisation catalyst. 2,2′-Diiodobiphenyl, for example, racemises at slightly elevated temperatures (50 °C). Cycling the temperature of the column between 8 and 50 °C and performing a repeated elution protocol in combination with a very large separation and capacity factor resulted in an optically pure compound with a yield of 90%. Later, the principle of deracemising tropos ligands on a chiral column and freezing in one stereoisomer by decreasing the temperature was elegantly coupled to asymmetric catalysis by Trapp and coworkers (see section 4).69,70
Deracemisation was also achieved by using a chiral host. Tsunoda and co-workers investigated the base-catalysed deracemisation of configurationally labile α-substituted cyclohexanones 4 and 5 in the presence of porous crystals made by dioxolane derivative (R,R)-6 (Fig. 8), which functioned as a chiral host.71 Based on preferred host–guest inclusion complexation of the (R)-enantiomers in the cavity of the crystals of (R,R)-6, the (S)-enantiomers were more prone to racemisation, feeding the formation of more of the (R)-enantiomer. In fact, the (R,R)-6 crystals functioned as a “thermodynamic sink” which complexed more and more of the (R)-enantiomer, which was constantly supplied by the concomitant racemisation of the (S)-enantiomer in solution. The results were quite promising; ees up to 94% and near quantitative yields were achieved. Clearly, the efficacy of the deracemisation crucially depends on the molecular recognition properties of the host for the guest. Crystal elucidation studies showed that in case of the inclusion complex of (R,R)-6 with (R)-4, hydrogen bonding and van der Waals contacts cooperate to stabilise the inclusion complex. Later, the concept of thermodynamically controlled deracemisation applying an optically active host was broadened to acyclic ketones72 and configurationally labile N-nitrosamines.73
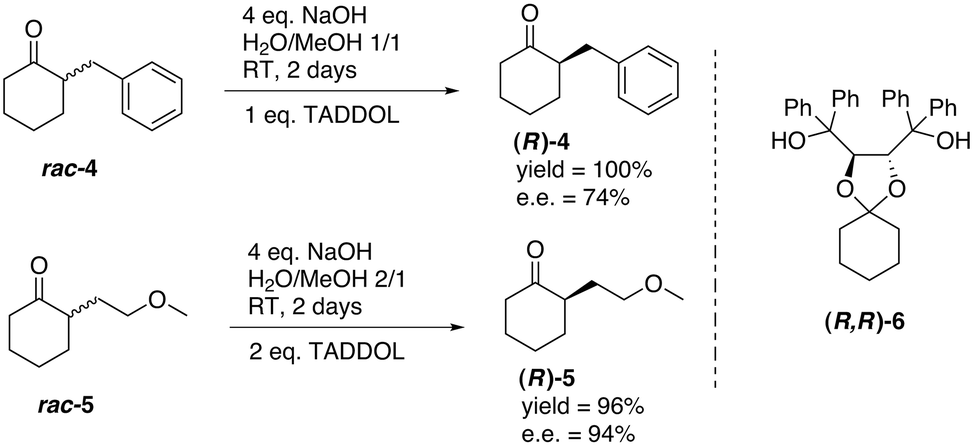 | ||
| Fig. 8 Deracemisation of cyclic α-substituted cyclohexanones 4 and 5 using molecular recognition with chiral host (R,R)-6.71 | ||
Finally, Vidal-Ferran and coworkers reported the supramolecular-directed chiral induction of biaryl derivatives.74 Herein enantiopure 1,2-diaminocyclohexane derivatives were applied to complex to biphenyl-2,2′diol derivatives which freely rotate. Because of hydrogen-bond formation, one diastereomer of the supramolecular complex was preferentially formed by chirality transfer from the enantiopure building block to a dynamically racemic biaryl derivative. Such an approach could prove very useful to create enantiomerically pure ligands for asymmetric catalysis, similar to the results presented by Trapp and coworkers.69,70
3.2 Deracemisation of amino acids by imine formation
Deracemisation of natural and non-natural amino acids is of high synthetic value and in many cases accomplished using enzymatic methods. However, enzymes typically do not easily accept non-natural amino acids. Although not strictly a deracemisation in the Pirkle definition, because covalent diastereomer intermediates are formed, the complementarity of this technique to Viedma ripening for accessing optically pure amino acids is very interesting. As already stated in section 2.3, Viedma ripening is an elegant approach to obtain homochiral amino acids but the procedure is limited to conglomerate forming compounds.An elegant alternative for deracemisation of amino acids was provided by the work of Chin et al., Cavallo et al. and Soloshonok et al. These groups focused on the deracemisation of amino acids utilising the formation of an imine with a non-racemisable, chiral ligand in combination with epimerization of the labile stereocentre of the amino acid part (Fig. 9). This procedure affords the thermodynamically more stable diastereomer. After deracemisation is complete, the enantio-enriched amino acid can be set free by hydrolysis of the imine.
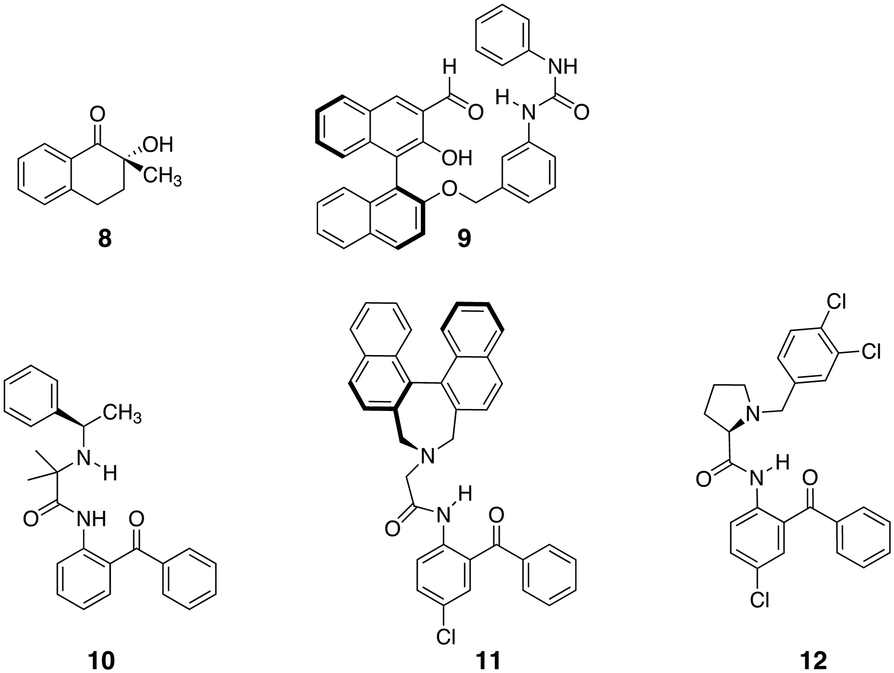 | ||
| Fig. 9 Chemical structures of optically pure ligand applied in the deracemisation of amino acids.75,76,78 | ||
Several approaches were evaluated and over the years the scope of amino acids and the achievable ees significantly increased. Solladié-Cavilla and coworkers reported the use of tetralone 8 to deracemize racemic amino acids (Fig. 9).75 Heating of enantiopure (S)-8 under reflux with an excess of the methyl ester of alanine in the presence of a catalytic amount of BF3.OEt2 provided around 95% of the trans-(S,S) isomer, which, after purification and hydrolysis, gave the enantiopure (S)-alanine with recovery of the chiral auxiliary 8 and at least 90% yield of amino acid. Later, Chin et al. introduced a biomimetic approach to amino acid deracemisation inspired by pyridoxal phosphate dependent enzymes that racemize amino acids.76 In the enzyme, the first step of the racemization reaction involves the formation of a special type of imines with internal resonance-assisted hydrogen bonds. The strong hydrogen bonds activate the bound amino acids and convert L-amino acids to D-amino acids by epimerization of the imines with no need for a base (Fig. 10A). Following this principle, ligand 9 was developed (Fig. 9). The stereoselectivity of 9 for binding a wide range of R-amino acids was remarkable. Amino acids with basic (His) and acidic (Glu) side chains were tolerated as well as those with hydrophobic (Tyr, Trp, Phe, Leu, Ala) and hydrophilic (Ser, Thr, Gln, Arg, Asn, Met) side chains. The stereoselectivity was the highest for threonine because of the steric bulk next to the R-carbon (Fig. 10B). Later the system was optimised by applying highly stereoselective supramolecular self-assembly of diphenylethylenediamine-based guanidine with imines based on Phe, Ala, Trp, Val and PhG (Fig. 10C). Here, epimerisation of the α-carbon of the amino acid residue occurred at room temperature within 2 hours and excellent stereoselectivities were obtained.77 Simple addition of HCl hydrolysed the imine and extraction with water permitted to isolate the pure amino acid in high ees. This approach reduced the number of reaction steps and, in principle, the amino acid could be isolated through continuous extraction, leaving the aldehyde and guanidine derivative in the organic layer for the next round of deracemisation.
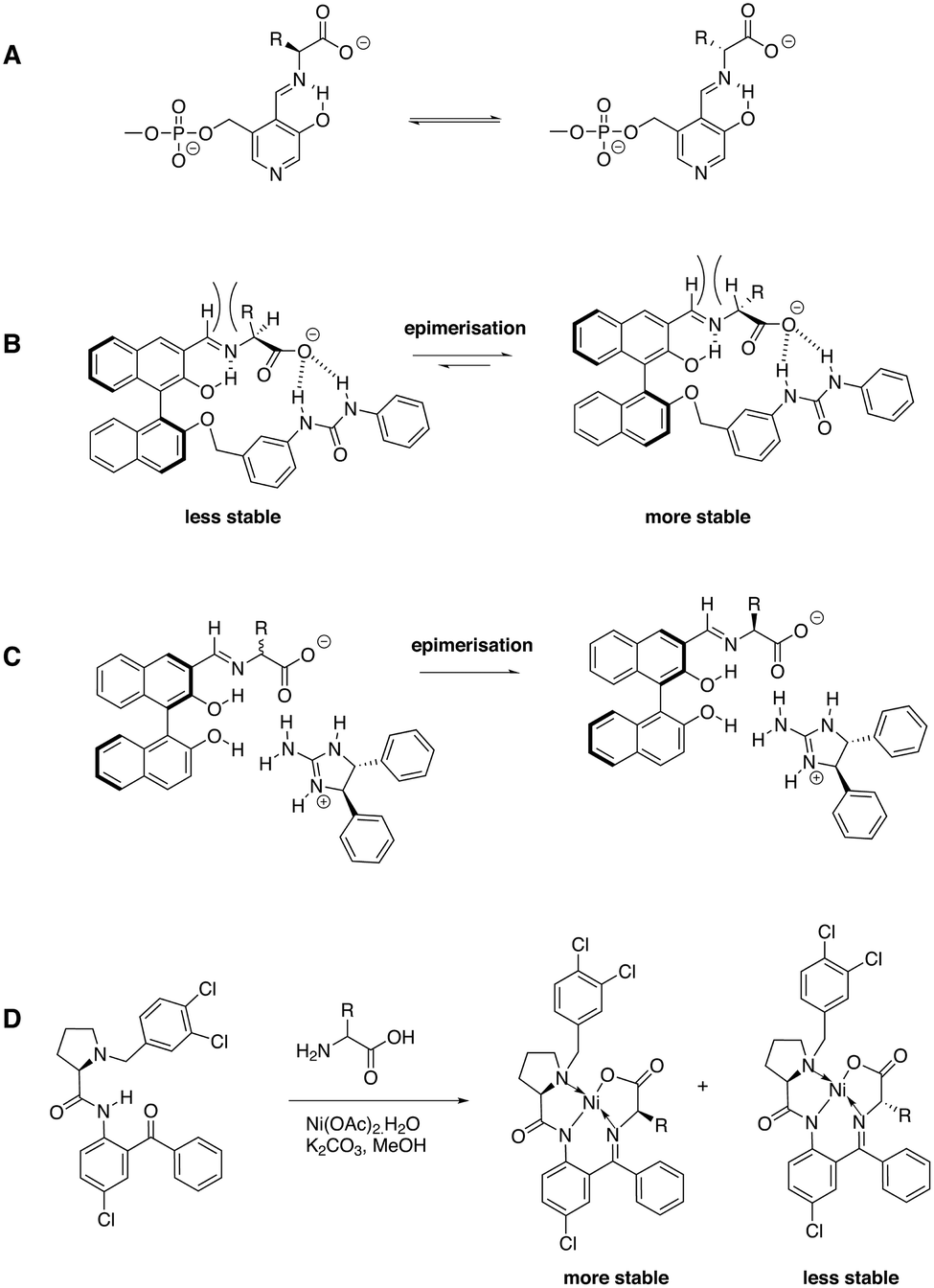 | ||
| Fig. 10 A) Mechanism of pyridoxal phosphate dependent enzymes that deracemise amino acids; B) imine formation of ligand 9 with amino acids, the hydrogen-bond formation enhances the acidity of the amino acid stereocenter and epimerisation occurs in the applied reaction conditions; because of steric interactions the equilibrium is shifted to the right;76 C) stereoselective self-assembly of dipenylethylenediamine-based guanidine with an amino acid-based imine that spontaneously deracemises by imine exchange;77 D) imine formation with ligand 12 and subsequent formation of Ni(II)-based complexes induces deracemisation.81 | ||
Soloshonok and coworkers applied a slightly different concept by using Ni(II) complexation instead of resonance-assisted hydrogen bonding in addition to imine formation. Ligands 10–12 were developed, which deracemised a wide variety of natural and non-natural amino acids.78–81 In the presence of Ni(OAc)2 and K2CO3 the labile stereocenter of the amino acid was racemised and within a few hours, near quantitative conversion to near enantiopure amino acid derivatives was achieved. Treating the isolated diastereomer with HCl in MeOH afforded the enantiopure amino acid and the ligand in high yields. In case of ligand 12, an impressive number of natural and non-natural amino acids was fully deracemised into optically pure enantiomers (Fig. 10D).81
3.3 Using amplification of chirality effects to induce deracemisation in thermodynamically controlled conditions
In dynamic helical polymers, it is well-established that a small amount of a chiral, nonracemic monomer having a helical preference, can dominate the helical sense preference of achiral monomers. This effect was coined “the sergeants-and-soldiers” (SaS) effect by Green in 1989.82 Later, Green found that mixing enantiomers in different ees resulted in a helical sense preference of the majority enantiomer. This was referred to as the “Majority-rules” (MR) effect.83 These effects are commonly known as “amplification of (supramolecular) chirality” because the optical activity of the helical polymer non-proportionally increases with the amount of chiral compound added or with the ee.In contrast to amplification of chirality observed in synthetic systems,84,85 or chiral symmetry breaking during Viedma ripening, the net ee of the monomer in helical polymers does not increase, only the excess helicity does. As a result, “amplification of chirality” in the field of helical polymers implies that only at the supramolecular level a full expression of a chiral superstructure exists. In contrast, in organic chemistry the term “amplification of chirality” is used when the ee of the reaction product increases relative to the substrate or catalyst used.
Mark Green postulated that “in a helical polymer the minority units residing in the helix sense of the majority, and vice versa, would be of higher energy. Even a small excess of majority units could therefore lead to a preference for epimerization of the minority units to the configuration of the majority units, which would lead to a fuller excess of the majority helical sense and thus to a cycle of increasing enantiomeric excess of the units making up the chain”.86 Unfortunately, this hypothesis was never experimentally verified in helical polyisocyanates.
Following up on the seminal work of Green and coworkers, the SaS and MR effects were also found to occur in helically aggregating supramolecular polymers, first in C3-symmetrical bipyridine-based systems developed in the group of Meijer87,88 and later in many more self-assembling systems.89 However, although optical activity non-proportionally increased, also in supramolecular polymers the ee at the molecular level was not altered.
To investigate if these two approaches could be united, a benzene-1,3,5-tricarboxamide (BTA) derivative based on phenylglycine (PhG-BTA) was developed by Cantekin et al. which comprised one configurationally labile stereocenter (Fig. 11A).90 This compound forms helical supramolecular aggregates stabilised by threefold intermolecular hydrogen bonding. Scalemic mixtures of both enantiomers show a pronounced MR effect (Fig. 11B) with a critical point, eecr, at |ee| = 35%. Between |ee| = 100% and |ee| = 35% all supramolecular polymers in solution have the same handedness, either P or M. At |eecr| = 35%, the M helices contain both (R)- and (S)-enantiomers (72.5 and 37.5%, respectively). In contrast, both helical senses occur between eecr and ee = 0%.
 | ||
| Fig. 11 A) Chemical structure of PhG-BTA and B) majority-rules effect in scalemic mixtures of (R)- and (S)-PhG-BTA; C) racemisation of (S)-PhG-BTA at 80 °C; D) racemisation of (S)-PhG-BTA at 20 °C. Racemisation was conducted at c = 1.2 mM in MCH in the presence of 1 eq. of DBU. The red line is the model fit to the experimental data. | ||
Enantiomerically enriched (R)-PhG-BTA (ee = 80%) was first racemised in de presence of the base DBU. Two conditions were investigated, (1) when all molecules were molecularly dissolved in methylcyclohexane (MCH) at a temperature of 80 °C (Fig. 11C) and (2) when the molecules were predominantly aggregated at 20 °C (Fig. 11D). At 20 °C, the molecularly dissolved PhG-BTA concentration is around 1000× lower than the total PhG-BTA concentration. The racemisation followed first-order reaction kinetics, with complete racemisation in 3 h at 80 °C (Fig. 11C). In contrast, the kinetic profile of the racemisation was completely different at 20 °C and two kinetic regimes were identified. In the first part, the rate was relatively fast but after reaching an ee of around 35%, the rate significantly slowed down (Fig. 11D). A mathematical model was developed by ten Eikelder and Markvoort which assumed that racemisation only occurs for molecularly dissolved PhG-BTA but does not happen when PhG-BTA is present in a helical aggregate. This assumption, together with the MR effect, resulted in a perfect fit of the experimental results to the model. This finding highlighted the dramatic reduction in the rate of base-catalysed racemisation of the labile stereocenter if the molecule is fixed in an aggregate.
The addition of a non-racemisable additive, (S)-BTA, which has a preference for forming M helical stacks, to the supramolecular polymers formed by racemic PhG-BTA (Fig. 12A), resulted in a coassembled structure and the stabilisation of M helical polymers (Fig. 12B). This resulted in a depletion of the (R)-PhG-BTA in solution. Upon the addition of DBU (Fig. 12C), an increase in the |ee| was observed, indicative of deracemisation. Although the process was very slow, this was the first time a deracemisation was observed in a supramolecular polymer in which the helical bias of an added “sergeant” induced a configurational change of the product's stereocenter. The system was under full thermodynamic control. After equilibrium was reached, the |ee| levelled off at around 35% (Fig. 12D). Even when the starting ee was 21%, (S)-BTA was capable of driving the deracemisation to an equilibrium ee of −35% (Fig. 12E).
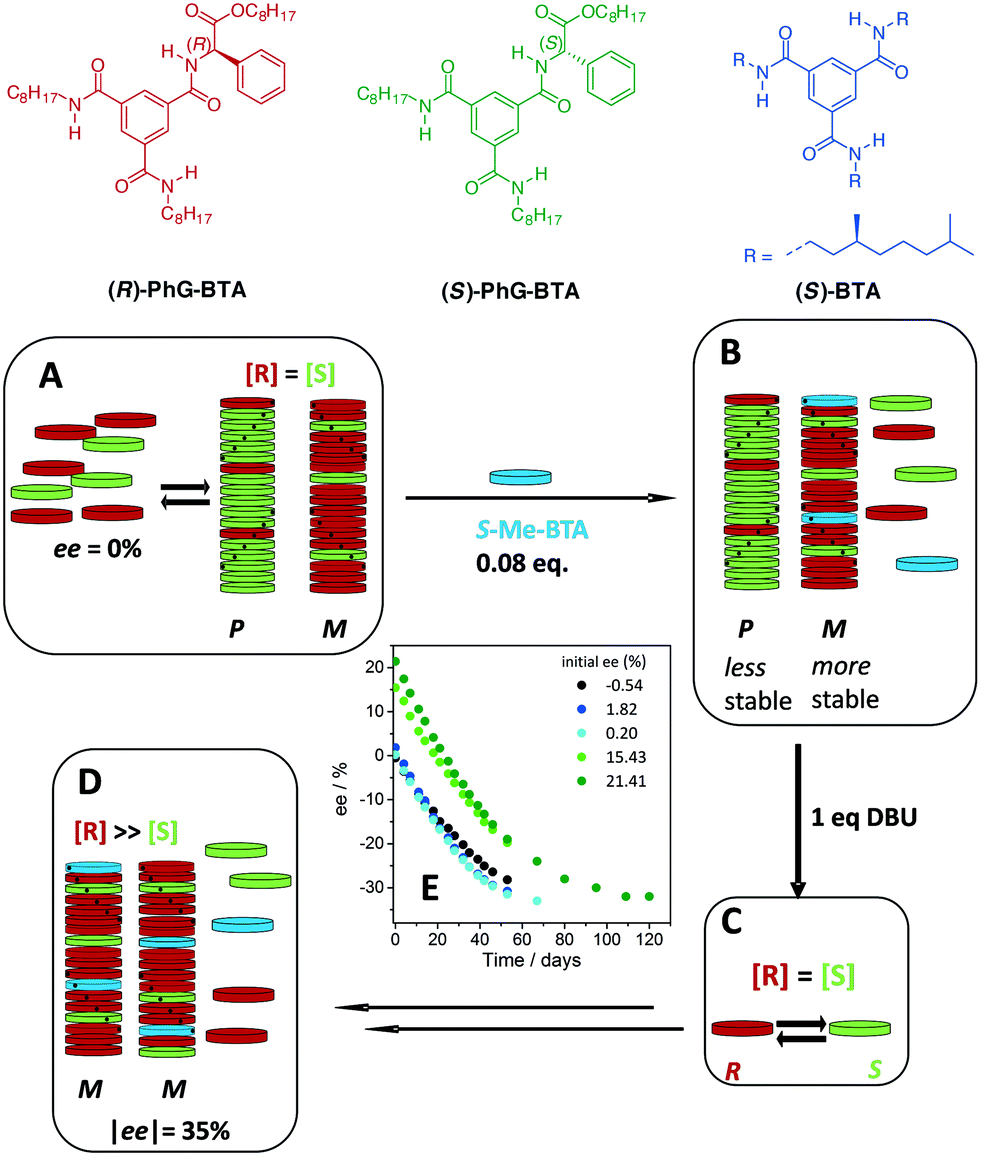 | ||
| Fig. 12 Colour coding of both PhG-BTA enantiomers and chiral additive (S)-BTA. A) Mixing of the two enantiomers in equimolar amounts provides equal amount of P and M supramolecular aggregates. Both types of aggregates accept around 32.5% of the wrong monomer because of the MR effect. B) Addition of a chiral non-racemisable (S)-BTA with a preference for M helices, resulting in a stabilisation of M helices in solution. C) Addition of DBU equilibrates the two enantiomers present in the monomerically dissolved state. D) In time, the M helical polymers function as a thermodynamic sink, attracting more (R)-PhG-BTA and as a result increasing the ee towards an excess of (R)-PhG-BTA. Because of the MR effect equilibrium is reached at an ee of 35%. E) The change in ee in time for mixtures of Phg-BTA with DBU added (1 eq.) and (S)-BTA (8 mol%) at 20 °C, ctot (Phg-BTA) = 1.2 mM in MCH, with different initial ee values. | ||
In the investigated system equilibrium was reached at a modest |ee| of approximately 35%. The mathematical models revealed why the |ee| remained so low and how it could be further enhanced. The crucial factor to achieve high ees was to increase the mismatch penalty between the two enantiomers of the system, i.e. reduce the tolerance of helical polymers to accept the other enantiomer. In BTAs, the majority rules principle is operative which means that the majority enantiomer biases the helical preference to which the minority adopts. It also means that a certain amount of the other enantiomer is accepted in helical aggregates which limits the attainable ee. To date, unfortunately, systems of racemising, cooperatively self-assembling monomers which show very high mismatch penalties have not been evaluated in deracemisations. In fact, conglomerate systems are characterised by an infinitely high mismatch penalty. As a result, the deracemisation proceeds to enantiopure products. Unfortunately, the translation of the factors responsible for conglomerate formation (or high mismatch penalties) into the molecular design of self-assembling units is not trivial.
Recently, an interesting alternative was proposed by Feringa and coworkers in which amplification of molecular chirality through the induction of supramolecular chirality was investigated using light.91 A solution of prochiral, ring-open diarylethenes (DAopen) was doped with a small amount of a chiral, ring-closed product (DAclosed). The ring-opened and ring-closed molecules co-assembled into a gel, formed by helical fibres that are stabilised by hydrogen bonding and π-stacking interactions. The handedness of the helical fibres was biased by the chiral, ring-closed diarylethene, which thus acted as a sergeant. The photochemical ring closure of the open diarylethene yielded the ring-closed product, which adopted the same stereochemical signature as the sergeant (Fig. 13A, templated pathway). Thus, the sergeant induced its own formation. The induction of ee in this process was remarkably high: the ee increased from 10% (amount of sergeant added) to 50% during the photochemical ring closure. This represents an ee induction of 40%. A higher amount of sergeant resulted in a slightly higher induction (46%) whereas lower concentrations reduced the ee induction. When the sergeants had the opposite configuration, the other enantiomer of the product was obtained. Importantly, in the absence of the sergeant, the photochemical ring closure afforded a racemic mixture of helical aggregates and the ee remained zero (Fig. 13B, untemplated pathway). In this example, no base is required for racemisation because the soldier racemises spontaneously.
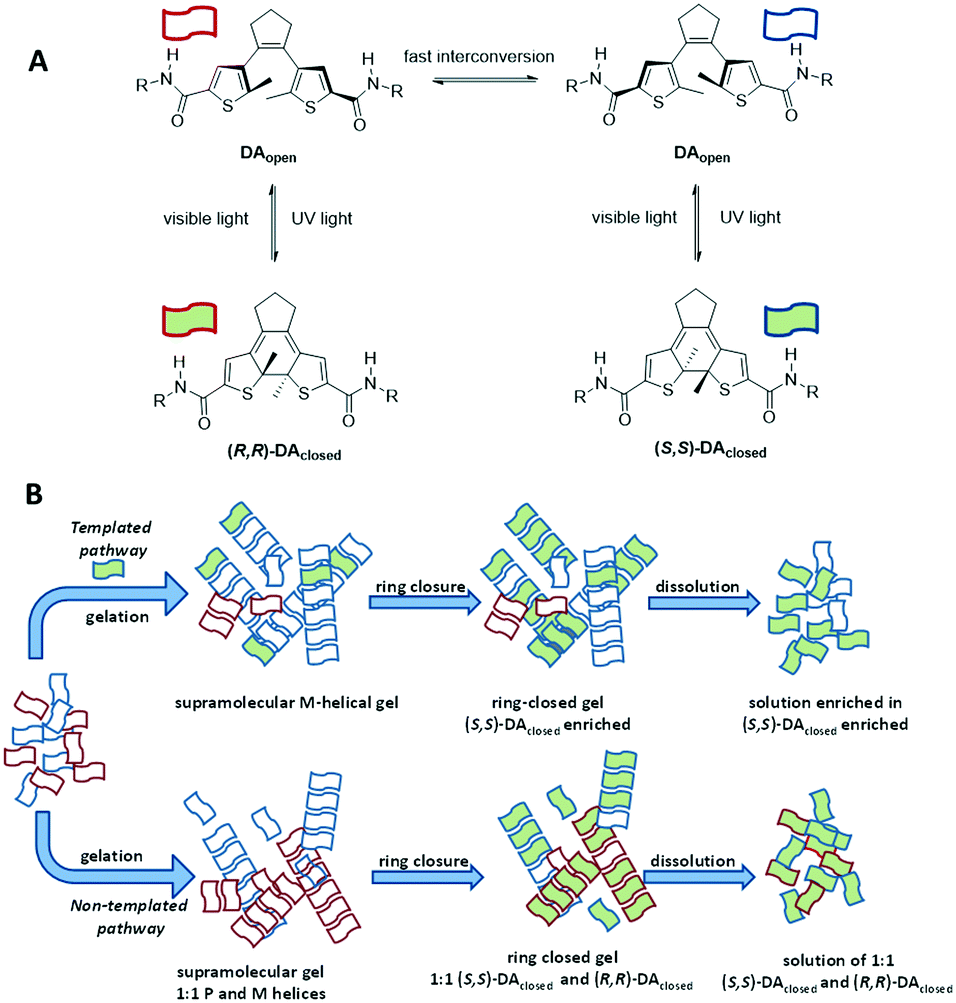 | ||
| Fig. 13 A) Chemical structures of the diarylethenes (DAs) applied in the work of Feringa;91 B) schematic representation of the gelation of ring opened DAs. | ||
Attempts to further enhance the ee induction of the system by changing the solvent, concentration of sergeant or total concentration did not result in improved values for the induction. In fact, the data indicated that the closed form is better soluble than the open form, which limits the amount of sergeant present in the fibres and rationalises the origin of the modest asymmetric induction.
Despite the promising results shown in the above examples and the availability of mathematical models to extract thermodynamic parameters that define a thermodynamically controlled deracemisation, there is still a remarkable paucity in deracemisations that relay supramolecular chirality back into an increase of the net ee of the monomers forming the supramolecular aggregate. It is likely that this is caused by our inability to predict self-assembly behaviour in one and more component systems from the molecular structure of a compound, similar to our inability to predict when a compound will form a conglomerate. Recently, the importance of developing predictive tools for supramolecular chemistry has been realised,92,93 and this together with the advance of adequate and accessible mathematical models94,95 will likely steer this field in a more design-driven direction.
4. From thermodynamically controlled conformations to asymmetric catalysis
Resolutions of racemates and asymmetric catalysis are often studied as separate disciplines in academic research, although both have the same aim, namely to procure optically pure compounds. For asymmetric catalysis, the availability of enantiopure ligands is crucial, and often these are accessed via lengthy synthetic procedures that require resolution steps to achieve optical purity. Tropos ligands, axially chiral systems that racemise at ambient conditions, can easily be biased to adopt a preferential conformation by using supramolecular interactions with chiral “selectors”, which can be a chiral stationary LC phase or the interaction with a small amount of chiral compound. Trapp and co-workers elegantly combined a thermodynamically controlled resolution of ligands with an asymmetric reaction.69 Combining such conformational preferences of biphenyl-based ligands may give new vistas for asymmetric synthesis.Not only tropos ligands can be deracemised to adopt one preferential conformation. In the field of helical polymers, biasing helical conformation by external triggers (solvent, guests, temperature,...) has been studied in great detail.96 Especially dynamic helical polymers which are formed under thermodynamic control and in which in the absence of chiral information equal amounts of P and M helical polymers are present, are an interesting class of compounds. Suginome and coworkers impressively showed that the helical sense bias of dynamic poly(quinoxalines) comprising chiral side chains can be altered by the solvent's polarity to either P or M (Fig. 14A).97–99 In addition, when phosphine-based ligands were embedded into the polymer, asymmetric hydrosilylation reactions showed high enantiomeric excesses (ee up to 97%). Switching the helical sense preference between P and M solely by changing the solvent polarity, permitted to access both enantiomers in high yield and ee (Fig. 14A). Moreover, the SaS effect was operative in the polymers; only small amounts of chiral monomer were required to bias the polymer's helical sense preference.100–103 Several asymmetric reaction (hydrosilylations, Suzuki–Miyaura cross-couplings) were compatible with these helical polymer systems, producing a variety of products in very high ee.104 Intriguingly, not only solvent polarity but also solvent shape could alter the helical sense preference. For example, whereas in heptane a P helix was preferred, the M was preferred in methylcyclohexane.105 In all of the examples presented to date, thermodynamic control of the helical preference of the dynamic helical polymer, requiring only a small amount of chiral input, resulted in excellent values for the enantiomeric excess. The ultrasensitivity of the helical preference of these poly(quinoxalines) to solvent polarity and shape offers many opportunities in biasing the outcome of an asymmetric reaction.
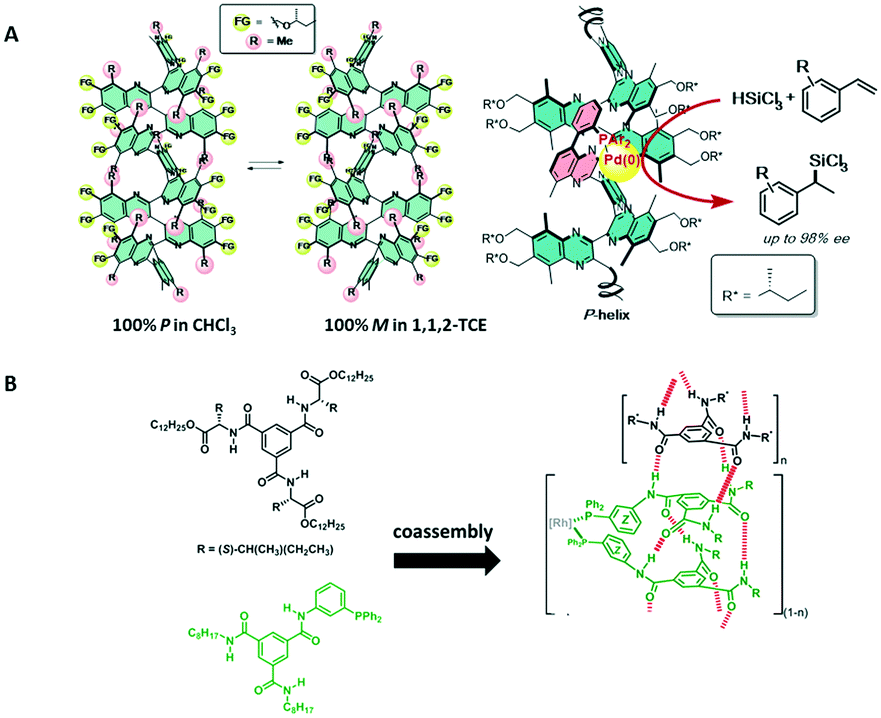 | ||
| Fig. 14 A) Left: the helical preference of poly(quinoxaline)s depends on the configuration of the chiral side chains in combination with the solvent's polarity; right: when Pd(0) is complexed to P-helical poly(quinoxaline), the hydrosilylation reaction becomes highly asymmetric and affords one enantiomer in high ee; B) chemical structures of BTA derivatives and their Rh(II) complexes after forming helical co-assemblies (reprinted with permission from ref. 107. Copyright (2016) American Chemical Society). | ||
Finally, Raynal and Bouteiller and coworkers presented a system based on achiral 1,3,5-benzenetricarboxamide-based monomers which contained one triphenylphosphine-based ligand (Fig. 14B).106,107 Mixing these achiral ligands with a few% of a chiral BTA derivative biased the helical sense preference of the formed supramolecular polymer. These supramolecular polymers are stabilised by helical, threefold hydrogen bonding between consecutive monomers and the chiral packing of the BTAs and as a result the phosphine ligands reside in a chiral environment. After complexation with Rh(II), good selectivity was observed in the asymmetric hydrogenation of dimethyl itaconate (ee = 85%). Also in this case, the helical preference is thermodynamically controlled and relays stereoselectivity into the asymmetric reduction.
5. Conclusions
Deracemisation reactions can occur under kinetic and thermodynamic control and permit to procure relevant, optically pure products in high yields and ees without the need of complex catalytic systems. Whereas deracemisations in attrition-enhanced crystallisations yield high ees, are scalable and are easy to operate, the underlying fundamental physical processes are complex and depend on a multitude of factors. Some of these factors occur randomly and are difficult to control. The detailed experimental and theoretical studies that were conducted in the past decade contributed greatly in achieving a fundamental understanding of these complex crystallisation processes, facilitating future large-scale application in industrial processes. However, despite all the progress made in this area, the attrition-enhanced deracemisation process crucially depends on the ability of the racemic mixture to form conglomerate crystals, which is still impossible to predict. Apart from the importance of this process for obtaining optically pure pharmaceutical intermediates, it is also believed that symmetry breaking processes occurring in solid-solution conglomerate mixtures may have contributed to the origin of homochirality.Deracemisations that occur under thermodynamic control, in contrast, always require the formation of (transient) diastereomers from the to-be separated racemic mixture, either by supramolecular interactions or by the formation of (weak) covalent bonds. Detailed understanding on a molecular level in this area is still limited and the challenges lie in the ability to predict which supramolecular recognition process may shift the equilibrium between interconverting enantiomers to such an extent that high ees can be obtained for structurally similar compounds. Nevertheless, with the use of molecular recognition processes, chiral hosts and imine formation, high ees have been achieved in families of compounds. Combining this field with the advent of predictive supramolecular chemistry undoubtedly will result in exciting developments in the coming years.
Finally, the interest for switchable asymmetric catalysts to procure both enantiomers by an asymmetric catalytic reactions is growing. The understanding of dynamic helical (supramolecular) polymers has increased in recent years. The insights obtained in their ability to switch helical conformation in response to medium and temperature changes or as a result of minute chiral information, could become a versatile platform for developing novel asymmetric catalytic methods.108 In a next step, ligands that adopt a preferential conformation in response to changes in the environment could produce switchable ligands for asymmetric catalysis and afford both enantiomers by simply changing operation conditions.
Acknowledgements
The author greatly acknowledges the support and inspiration from Prof. “Bert” Meijer. Prof. Suginome from Kyoto University is gratefully acknowledged for providing the artwork for Fig. 14A. The Dutch Science foundation (NWO) is acknowledged for financial support.Notes and references
- W. H. Pirkle and D. S. Reno, J. Am. Chem. Soc., 1987, 109, 7189–7190 CrossRef CAS.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed.
- K. Faber, Chem. – Eur. J., 2001, 7, 5005–5010 CrossRef.
- M. Rachwalski, N. Vermue and F. P. J. T. Rutjes, Chem. Soc. Rev., 2013, 42, 9268–9282 RSC.
- C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 CrossRef CAS.
- J. Steinreiber, K. Faber and H. Griengl, Chem. – Eur. J., 2008, 14, 8060–8072 CrossRef CAS PubMed.
- W. K. Lee, Y. S. Park and P. Beak, Acc. Chem. Res., 2009, 42, 224–234 CrossRef CAS PubMed.
- Dynamic stereochemistry of chiral compounds: Principles and Applications, by C. Wolf, 2007, RSC, Chapter 7: Asymmetric resolution and transformation of chiral compounds under thermodynamic and kinetic control..
- D. B. Amabilino and R. M. Kellogg, Isr. J. Chem., 2011, 51, 1034–1040 CrossRef CAS.
- G. Coquerel, Top. Curr. Chem., 2007, 269, 1–51 CrossRef CAS PubMed.
- R. Yoshioka, Top. Curr. Chem., 2007, 269, 83–132 CrossRef CAS PubMed.
- K. M. J. Brands and A. J. Davies, Chem. Rev., 2006, 106, 2711–2733 CrossRef CAS PubMed.
- G. Coquerel, in Novel optical resolution technologies, Springer, Heidelberg, 2007 Search PubMed.
- A. Galland, V. Dupray, B. Berton, S. Morin-Grognet, M. Sanselme, H. Atmani and G. Coquerel, Cryst. Growth Des., 2009, 9, 2713–2718 CAS.
- E. Havinga, Chemisch Weekblad, November 1941, 3846, pp. 642–644 Search PubMed.
- E. Havinga, Biochim. Biophys. Acta, 1954, 13, 171–174 CrossRef CAS.
- E. Wedekind, Ber. Dtsch. Chem. Ges., 1903, 36, 3793 Search PubMed.
- A. Fock, Z. Kristallogr. Mineral., 1902, 35, 399 Search PubMed.
- R. G. Kostyanovsky, V. R. Kostyanovsky, G. K. Kadorkina and L. A. Lyssenko, Mendeleev Commun., 2001, 11, 1–6 CrossRef.
- R. E. Pincock and K. R. Wilson, J. Am. Chem. Soc., 1971, 93, 1291–1292 CrossRef CAS.
- K. R. Wilson and R. E. Pincock, J. Am. Chem. Soc., 1975, 97, 1474–1478 CrossRef CAS.
- M. A. Sephton, C. R. Emerson, L. N. Zakharov and P. R. Blakemore, Chem. Commun., 2010, 46, 2094–2096 RSC.
- W.-C. Shieh and J. A. Carlson, J. Org. Chem., 1994, 59, 5463–5465 CrossRef CAS.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–977 CAS.
- D. K. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946–954 CrossRef CAS PubMed.
- W. S. Kipping and W. J. Pope, J. Chem. Soc., Trans., 1898, 73, 606–617 RSC.
- J. M. McBride and R. L. Carter, Angew. Chem., Int. Ed. Engl., 1991, 30, 293–295 CrossRef.
- W. J. Boyle Jr., S. Sifniades and J. F. Van Peppen, J. Org. Chem., 1979, 44, 4841–4847 CrossRef.
- M. Sakamoto, M. Kato, Y. Aida, K. Fujita, T. Mino and T. Fujita, J. Am. Chem. Soc., 2008, 130, 1132–1133 CrossRef CAS PubMed.
- F. Yagishita, H. Ishikawa, T. Onuki, S. Hachiya, T. Mino and M. Sakamoto, Angew. Chem., Int. Ed., 2012, 51, 13023–13025 CrossRef CAS PubMed.
- D. K. Kondepudi, J. Laudadio and K. Asakura, J. Am. Chem. Soc., 1999, 121, 1448–1451 CrossRef CAS.
- K. Ziach and J. Jurczak, Chem. Commun., 2015, 51, 4306–4309 RSC.
- P. M. Björemark, S. Olson, T. Kokoli and M. Håkansson, Chem. – Eur. J., 2015, 21, 8750–8753 CrossRef PubMed.
- S.-T. Wu, Y.-R. Wu, Q.-Q. Kang, H. Zhang, L.-S. Long, Z. Zheng, R.-B. Huang and L.-S. Zheng, Angew. Chem., Int. Ed., 2007, 46, 8475–8479 CrossRef CAS PubMed.
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2010, 3, 68–73 CrossRef PubMed.
- C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed.
- C. Viedma, Cryst. Growth Des., 2007, 7, 553–556 CAS.
- D. G. Blackmond, Chem. – Eur. J., 2007, 13, 3290–3295 CrossRef CAS PubMed.
- C. Viedma, Astrobiology, 2007, 7, 312–318 CrossRef CAS PubMed.
- P. S. M. Cheung, J. Gagnon, J. Surprenant, Y. Tao, H. Xu and L. A. Cuccia, Chem. Commun., 2008, 987–999 RSC.
- W. L. Noorduin, T. Izumi, A. Millemaggi, M. Leeman, H. Meekes, W. J. P. Van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158–1159 CrossRef CAS PubMed.
- L. Addadi, Z. Berkovitch-Yellin, N. Domb, E. Gati, M. Lahav and L. Leiserowitz, Nature, 1982, 296, 21–26 CrossRef CAS.
- W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, A. Millemaggi, M. Leeman, B. Kaptein, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 6445–6447 CrossRef CAS PubMed.
- W. L. Noorduin, H. Meekes, A. A. C. Bode, W. J. P. van Enckevort, B. Kaptein, R. M. Kellogg and E. Vlieg, Cryst. Growth Des., 2008, 8, 1675–1681 CAS.
- W. L. Noorduin, W. J. P. van Enckevort, H. Meekes, B. Kaptein, R. M. Kellogg, J. C. Tully, J. M. McBride and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 8435–8438 CrossRef CAS PubMed.
- W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600–9607 CrossRef CAS PubMed.
- W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, B. Kaptein, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2010, 49, 2539–2541 CrossRef CAS PubMed.
- W. L. Noorduin, P. van der Asdonk, H. Meekes, W. J. P. van Enckevort, B. Kaptein, M. Leeman, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2009, 48, 3278–3280 CrossRef CAS PubMed.
- R. R. E. Steendam, B. Harmsen, H. Meekes, W. J. P. van Enckevort, B. Kaptein, R. M. Kellogg, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Cryst. Growth Des., 2013, 13, 4776–4780 CAS.
- B. Kaptein, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 7226–7229 CrossRef CAS PubMed.
- W. L. Noorduin, A. A. C. Bode, M. van der Meijden, H. Meekes, A. F. van Etteger, W. J. P. van Enckevort, P. C. M. Christianen, B. Kaptein, R. M. Kellogg, T. Rasing and E. Vlieg, Nat. Chem., 2009, 1, 729–732 CrossRef CAS PubMed.
- J. E. Hein, B. H. Cao, C. Viedma, R. M. Kellogg and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 12629–12636 CrossRef CAS PubMed.
- B. Kaptein, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, R. D. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2008, 47, 7226–7229 CrossRef CAS PubMed.
- W. L. Noorduin, P. van der Asdonk, H. Meekes, W. J. P. van Enckevort, B. Kaptein, M. Leeman, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2009, 48, 3278–3280 CrossRef CAS PubMed.
- C. Viedma, J. E. Ortiz, T. de Torres, T. Izuma and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274–15275 CrossRef CAS PubMed.
- L. Spix, H. Meekes, R. H. Blaauw, W. J. P. van Enckevort and E. Vlieg, Cryst. Growth Des., 2012, 12, 5796–5799 Search PubMed.
- S. B. Tsogoeva, S. Wei, M. Freund and M. Mauksch, Angew. Chem., Int. Ed., 2009, 48, 590–594 CrossRef CAS PubMed.
- S. Wei, M. Mauksch and S. B. Tsogoeva, Chem. – Eur. J., 2009, 15, 10255–10262 CrossRef CAS PubMed.
- R. R. E. Steendam, T. J. B. van Benthem, E. M. E. Huijs, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Cryst. Growth Des., 2015, 15, 3917–3921 CAS.
- Y. Kaji, N. Uemura, Y. Kasashima, H. Ishikawa, Y. Yoshida, T. Mino and M. Sakamoto, Chem. – Eur. J., 2016, 22, 16429–16432 CrossRef CAS PubMed.
- P. M. Björemark, J. Jönsson and M. Håkansson, Chem. – Eur. J., 2015, 21, 10630–10633 CrossRef PubMed.
- W. L. Noorduin, B. Kaptein, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg and E. Vlieg, Angew. Chem., Int. Ed., 2009, 48, 4581–4583 CrossRef CAS PubMed.
- M. W. van der Meijden, M. Leeman, E. Gelens, W. L. Noorduin, H. Meekes, W. J. P. van Enckevort, B. Kaptein, E. Vlieg and R. M. Kellogg, Org. Process Res. Dev., 2009, 13, 1195–1198 CrossRef CAS.
- P. Wilmink, C. Rougeot, K. Wurst, M. Sanselme, M. van der Meijden, W. Saletra, G. Coquerel and R. M. Kellogg, Org. Process Res. Dev., 2015, 19, 302–308 CrossRef CAS.
- M. Iggland, M. P. Fernández-Ronco, R. Senn, J. Kluge and M. Mazzotti, Chem. Eng. Sci., 2014, 111, 106–111 CrossRef CAS.
- W. H. Pirkle and T. C. Pochapsky, J. Am. Chem. Soc., 1987, 109, 5975–5982 CrossRef CAS.
- C. Wolf, W. A. Koenig and C. Roussel, Chirality, 1995, 7, 610–611 CrossRef CAS.
- E. Tobler, M. Laemmerhofer, G. Mancini and W. Lindner, Chirality, 2001, 13, 641–647 CrossRef CAS PubMed.
- F. Maier and O. Trapp, Angew. Chem., Int. Ed., 2014, 53, 8756–8760 CrossRef CAS PubMed.
- G. Storch and O. Trapp, Angew. Chem., Int. Ed., 2015, 54, 3580–3586 CrossRef CAS PubMed.
- T. Tsunoda, H. Kaku, M. Nagaku and E. Okuyama, Tetrahedron Lett., 1997, 38, 7759–7760 CrossRef CAS.
- H. Kaku, T. Imai, R. Kondo, S. Mamba, Y. Watanabe, M. Inai, T. Nishii, M. Horikawa and T. Tsunoda, Eur. J. Org. Chem., 2013, 8208–8213 CrossRef CAS.
- T. Olszewska, M. J. Milewska, M. Gdaniec, H. Małuszynska and T. Połonski, J. Org. Chem., 2001, 66, 501–506 CrossRef CAS PubMed.
- J. Etxebarria, H. Degenbeck, A.-S. Felten, S. Serres, N. Nieto and A. Vidal-Ferran, J. Org. Chem., 2009, 74, 8794–9797 CrossRef CAS PubMed.
- A. Solladié-Cavallo, O. Sedy, M. Salisova and M. Schmitt, Eur. J. Org. Chem., 2002, 3042–3049 CrossRef.
- H. Park, K. M. Kim, A. Lee, S. Ham, W. Nam and J. Chin, J. Am. Chem. Soc., 2007, 129, 1518–1519 CrossRef CAS PubMed.
- S. M. Soo, K. Moozeh, A. J. Lough and J. Chin, Angew. Chem., Int. Ed., 2014, 53, 829–832 CrossRef PubMed.
- V. A. Soloshonok, T. K. Ellis, H. Ueki and T. Ono, J. Am. Chem. Soc., 2009, 131, 7208–7209 CrossRef CAS PubMed.
- R. Takeda, A. Kawamura, A. Kawashima, T. Sato, H. Moriwaki, K. Izawa, K. Akaji, S. Wang, H. Liu, J. L. Acena and V. A. Soloshonok, Angew. Chem., Int. Ed., 2014, 53, 12214–12217 CrossRef CAS PubMed.
- Y. Nian, J. Wang, S. Zhou, S. Wang, H. Moriwaki, A. Kawashima, V. A. Soloshonok and H. Liu, Angew. Chem., Int. Ed., 2015, 54, 12918–12922 CrossRef CAS PubMed.
- Y. Nian, J. Wang, S. Zhou, W. Dai, S. Wang, H. Moriwaki, A. Kawashima, V. A. Soloshonok and H. Liu, J. Org. Chem., 2015, 80, 9817–9830 CrossRef PubMed.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Wilson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- M. Green, B. A. Garetz, B. Munoz, H. Chang, S. Hoke and R. G. Cooks, J. Am. Chem. Soc., 1995, 117, 4181–4182 CrossRef CAS.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- C. Girard and H. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2923–2959 CrossRef CAS.
- M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138–3154 CrossRef PubMed.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- J. van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490–5494 CrossRef CAS PubMed.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed.
- S. Cantekin, H. M. M. ten Eikelder, A. J. Markvoort, M. A. J. Veld, P. Korevaar, M. M. Green, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2012, 51, 6426–6431 CrossRef CAS PubMed.
- D. J. van Dijken, J. M. Beierle, M. C. A. Stuart, W. Szymanski, W. R. Browne and B. L. Feringa, Angew. Chem., 2014, 126, 5173–5177 CrossRef.
- C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661–673 CrossRef CAS PubMed.
- C. A. Hunter, M. C. Misuraca and S. M. Turega, J. Am. Chem. Soc., 2011, 133, 20416–20425 CrossRef CAS PubMed.
- A. J. Markvoort, H. M. M. ten Eikelder, P. A. J. Hilbers, T. F. A. de Greef and E. W. Meijer, Nat. Commun., 2011, 2, 509 CrossRef PubMed.
- A. J. Markvoort, H. M. M. ten Eikelder, P. A. J. Hilbers and T. F. A. de Greef, ACS Cent. Sci., 2016, 2, 232–241 CrossRef CAS PubMed.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- T. Yamamoto and M. Suginome, Angew. Chem., Int. Ed., 2009, 48, 539–542 CrossRef CAS PubMed.
- T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS PubMed.
- T. Yamada, Y. Nagata and M. Suginome, Chem. Commun., 2010, 46, 4914–4916 RSC.
- (a) Y. Nagata, T. Yamada, T. Adachi, Y. Akai, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2013, 135, 10104–10113 CrossRef CAS PubMed; (b) Y. Nagata, T. Nishikawa and M. Suginome, ACS Macro Lett., 2016, 5, 519–522 CrossRef CAS.
- T. Yamamoto, Y. Akai and M. Suginome, Angew. Chem., Int. Ed., 2014, 53, 12785–12788 CrossRef CAS PubMed.
- T. Yamamoto, Y. Akai, Y. Nagata and M. Suginome, Angew. Chem., Int. Ed., 2011, 50, 8844–8847 CrossRef CAS PubMed.
- Y. Nagata, T. Nishikawa and M. Suginome, ACS Macro Lett., 2016, 5, 519–522 CrossRef CAS.
- Y. Akai, L. Konnert, T. Yamamoto and M. Suginome, Chem. Commun., 2015, 51, 7211–7214 RSC.
- Y. Nagata, T. Nishikawa and M. Suginome, J. Am. Chem. Soc., 2014, 136, 15901–15904 CrossRef CAS PubMed.
- M. Raynal, F. Portier, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2013, 135, 17687–17690 CrossRef CAS PubMed.
- A. Desmarchelier, X. Caumes, M. Raynal, A. Vidal-Ferran, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2016, 138, 4908–4916 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |