
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure-guided optimization of quinoline inhibitors of Plasmodium N-myristoyltransferase†‡
Victor
Goncalves§
*a,
James A.
Brannigan
b,
Alice
Laporte
a,
Andrew S.
Bell
a,
Shirley M.
Roberts
b,
Anthony J.
Wilkinson
b,
Robin J.
Leatherbarrow¶
a and
Edward W.
Tate
*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: victor.goncalves@u-bourgogne.fr; e.tate@imperial.ac.uk
bStructural Biology Laboratory, Department of Chemistry, University of York, York YO10 5DD, UK
First published on 11th November 2016
Abstract
The parasite Plasmodium vivax is the most widely distributed cause of recurring malaria. N-Myristoyltransferase (NMT), an enzyme that catalyses the covalent attachment of myristate to the N-terminal glycine of substrate proteins, has been described as a potential target for the treatment of this disease. Herein, we report the synthesis and the structure-guided optimization of a series of quinolines with balanced activity against both Plasmodium vivax and Plasmodium falciparum N-myristoyltransferase (NMT).
Introduction
N-Myristoyltransferase (NMT, EC 2.3.1.97) catalyses the transfer of the fatty acid chain C14:0 from myristoyl coenzyme A (MyrCoA) to the N-terminal glycine residue of substrate proteins.1 This co- and post-translational modification is ubiquitous in eukaryotic organisms and plays a central role in a variety of cellular processes such as the addressing and reversible anchoring of proteins to membranes.2,3 The N-terminal myristate chain can also participate in the stabilization of the tertiary structure of proteins and form part of recognition elements that establish protein–protein interactions.4,5 Due to the essential functions of myristoylation, the modulation of NMT activity has emerged as an attractive strategy in the treatment of various pathologies. In particular, NMT has been validated as a pharmacological target in fungal infections,6 in a range of parasitic diseases caused by Trypanosoma,7Leishmania8 and Plasmodium9–11 protozoa and filarial nematodes,12 and as a potential target for the treatment of cancer.13The first reported NMT inhibitors were obtained through rational design strategies, by mimicking the structure of peptide substrates,14,15 or by designing non-hydrolysable MyrCoA analogues.16,17
Later, novel families of NMT inhibitors were identified by high throughput screening (HTS) efforts.7,18–20 A selection of different chemotypes are under active development against NMT for numerous indications (Fig. S1‡), and an NMT inhibitor has recently entered the Medicines for Malaria Venture (MMV) development pipeline for the treatment of the lethal form of malaria caused by Plasmodium falciparum. We reported in 2012 the discovery of a Plasmodium vivax NMT (PvNMT) inhibitor based on a quinoline scaffold.21P. vivax is the most frequent parasite responsible for the recurring form of malaria, whose transmission and infection occur through the bite of Anopheles mosquitos.22 Vivax malaria strongly impacts the quality of life of infected patients who experience cyclical fever and weakness episodes and represents a severe burden in endemic countries, due to cost of treatment and loss of productivity.23
Our initial work on the quinoline series resulted in the elucidation of the crystal structure of a hit compound in complex with PvNMT and the identification of an optimized molecule, ethyl 4-((2-cyanoethyl)thio)-6-methoxyquinoline-3-carboxylate 1, displaying micromolar inhibitory potency against PvNMT, some selectivity versus the human NMT isoforms and reasonable physico-chemical properties (Fig. 1). Unfortunately, these compounds showed no activity against the N-myristoyltransferase of Plasmodium falciparum, the species causing the most virulent and deadly form of the disease.
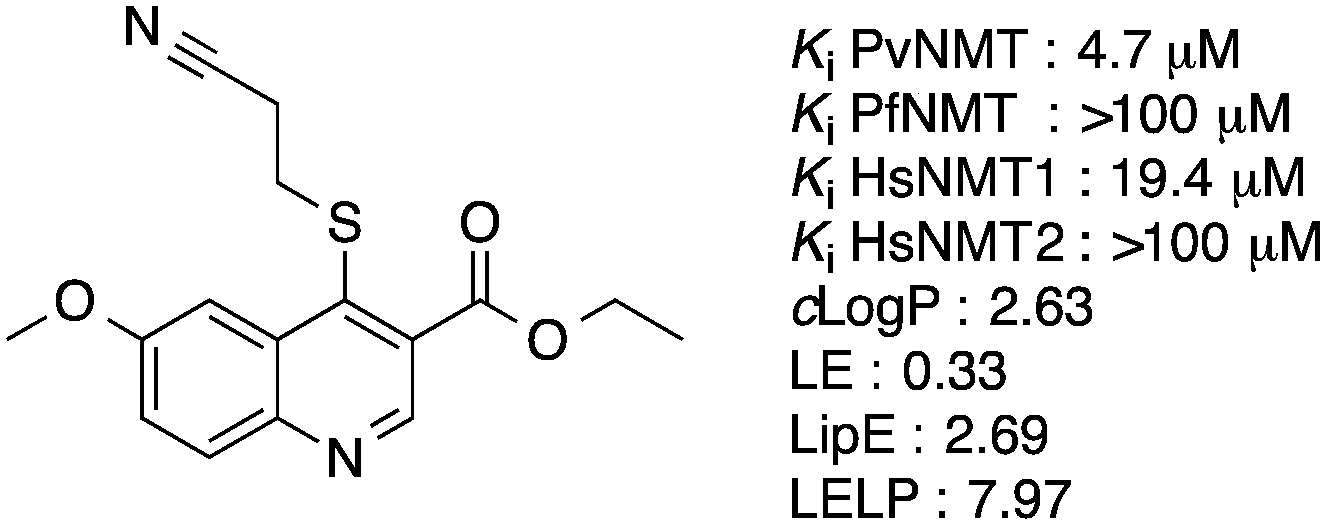 | ||
Fig. 1 Structure of the optimized molecule 1. clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P was determined with ChemAxon, which can be obtained from http://www.chemaxon.com/. LE: ligand efficiency, LE = [−log(Ki)](1.374)/(no. of heavy atoms).24 LipE: lipophilic efficiency, LipE = −log(Ki) − clogP.24 LELP : ligand efficiency dependent lipophilicity, LELP = clogP/LE.25 Here, we report how our attempts to improve the potency of this series of compounds through a structure-guided strategy led to the identification of nanomolar inhibitors of N-myristoyltransferase, active against both PvNMT and PfNMT. P was determined with ChemAxon, which can be obtained from http://www.chemaxon.com/. LE: ligand efficiency, LE = [−log(Ki)](1.374)/(no. of heavy atoms).24 LipE: lipophilic efficiency, LipE = −log(Ki) − clogP.24 LELP : ligand efficiency dependent lipophilicity, LELP = clogP/LE.25 Here, we report how our attempts to improve the potency of this series of compounds through a structure-guided strategy led to the identification of nanomolar inhibitors of N-myristoyltransferase, active against both PvNMT and PfNMT. | ||
Results and discussion
Crystal structure of lead compound 1 in complex with Plasmodium vivax NMT
First, compound 1 was co-crystallized with PvNMT and S-(2-oxo)pentadecyl-coenzyme A, a non-hydrolysable myristoyl-coenzyme A analogue (Fig. 2),26 and the structure of the resulting ternary complex was solved by X-ray crystallography. Overall, 1 adopts a binding mode similar to the original hit compound, 3-butyl-4-((2-cyanoethyl)thio)-6-methoxy-2-methylquinoline (MRT00057965), which we identified in 2012 by high throughput screening.21 The quinoline fits into a hydrophobic pocket, where it establishes parallel-displaced π–π stacking interactions with the adjacent side-chains of Phe103 and Phe105. The binding of the quinoline ring is reinforced by the establishment of an H-bond between its nitrogen and the hydroxyl function of Ser319. Additionally, the nitrile function of the 2-cyanoethylthioether chain appears to interact with the imidazole ring of His213 (distance ∼3.0 Å). The ligand is modelled bound to the protein at half maximal occupancy due to the low affinity of binding. This is mirrored in the two orientations of residues His213 and Tyr211, corresponding to the bound and free forms (Fig. 2). Interestingly, the ester function, introduced to improve the solubility and logP of the hit compound, also establishes polar interactions with PvNMT, through water-mediated H-bonds.
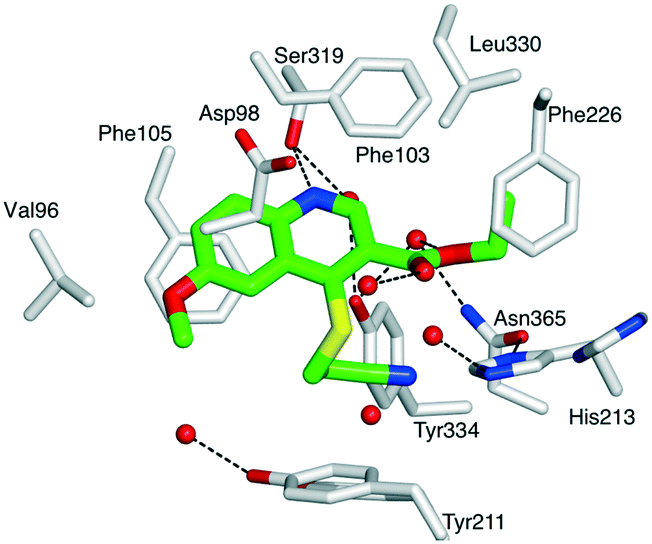 | ||
| Fig. 2 Structure of quinoline 1 in a ternary complex with P. vivax NMT and S-(2-oxo)pentadecylCoA (NHM) (PDB accession code: 5G1Z). View of 1 in chain A of PvNMT in cylinder format and colored by atom: carbon (green), oxygen (red), nitrogen (blue), sulphur (yellow). The side chains of residues located within 4 Å of 1 are displayed as cylinders and labelled. Water molecules are shown as red spheres. Polar interactions with PvNMT are represented as dashed lines. Y211 and H213 are shown in two alternate conformations. For a stereoview, see Fig. S2.‡ | ||
Based on the above structure, the optimization of the 3-, 4- and 6-positions of the quinoline ring was undertaken.
Optimization of the substituent in position 4 of the quinoline ring
It is well known that 2-cyanoethylthioethers can be cleaved in the presence of weak bases such as ammonia through a retro-Michael reaction.27 Although 1 was fully stable in the phosphate buffered solutions used for enzymatic assay, some degradation was observed during its chemical synthesis, suggesting it may represent a hurdle in the future development of this series of compounds. Additionally, structural data indicate that the H-bond formed between the nitrile and His213 may not be critical for the binding of the inhibitor. This prompted us to investigate whether the 2-cyanoethylthioether could be advantageously replaced by a more stable substituent in the 4-position of the quinoline ring.First, the linear alkyl isostere 5, with an n-propyl chain in place of the 2-cyanoethyl, was synthesized. In this series of molecules, the original 6-methoxy group present in the optimized hit 1 was replaced by a benzyloxy. Indeed, the 6-benzyloxy derivative 4, initially prepared as a synthetic intermediate to explore the role of the 6-position, was found to provide an 8-fold improvement in activity compared to the original methoxy compound, probably due to the establishment of edge-to-face π–π interactions with the side chains of residues Phe105 and Tyr211 (Table 1). The intermediate ethyl 6-(benzyloxy)-4-hydroxyquinoline-3-carboxylate 2 was obtained through the thermal condensation and cyclization of 6-benzyloxyaniline and ethyl ethoxymethylenemalonate, and subsequently transformed into its 4-chloro derivative 3 by treatment with POCl3 (Scheme 1).28 Finally, nucleophilic aromatic substitution with thiols or sodium thiolates provided the 4-thioalkyl derivatives 4–8.
| Entry |
|
K i app (μM)a | clogPb |
LipEc | |||||
|---|---|---|---|---|---|---|---|---|---|
| R3 | R4 | R6 | PvNMT | PfNMT | HsNMT1 | HsNMT2 | |||
|
a Apparent Ki values of tested compounds on the enzymatic activity of recombinant P. vivax NMT, P. falciparum NMT and H. sapiens NMT isoforms 1 and 2. Each Ki is the mean ± SD from duplicates. n.d. : not determined.
b clogP were determined with ChemAxon.
c LipE: lipophilic efficiency, LipE = −log(Ki) − clogP.
d No biological data available because of the limited solubility of the compounds in assay buffer.
|
|||||||||
| 1 | COOEt | SCH2CH2CN | Me | 4.74 ± 0.24 (ref. 21) | >100 | 19.43 ± 0.86 (ref. 21) | >100 (ref. 21) | 2.63 | 2.69 |
| 4 | COOEt | SCH2CH2CN | Bn | 0.55 ± 0.06 | >10 | >10 | n.d. | 4.36 | 1.90 |
| 5 | COOEt | SCH2CH2CH3 | Bn | 2.08 ± 0.14 | 1.10 ± 0.08 | 3.36 ± 0.37 | >10 | 5.46 | 0.22 |
| 6 | COOEt | SCH(CH3)2 | Bn | 2.51 ± 0.16 | 1.99 ± 0.30 | 4.84 ± 0.64 | >10 | 5.25 | 0.35 |
| 7 | COOEt | SCH2CH3 | Bn | 0.44 ± 0.05 | 0.67 ± 0.04 | 1.56 ± 0.22 | 4.23 ± 0.53 | 4.94 | 1.42 |
| 8 | COOEt | SCH3 | Bn | 4.14 ± 0.41 | 5.90 ± 0.60 | n.d. | n.d. | 4.69 | 0.69 |
| 9 | COOEt | SOCH2CH3 | Bn | 1.75 ± 0.12 | 5.65 ± 1.24 | 3.60 ± 0.30 | >10 | 3.30 | 2.46 |
| 10 | COOEt | SO2CH2CH3 | Bn | 1.78 ± 0.47 | >10 | >10 | >10 | 3.41 | 2.34 |
| 11 | CH2OH | SCH2CH3 | Bn | 4.19 ± 0.55 | >10 | >10 | >10 | 3.81 | 1.57 |
| 12 | CH2OCH2CH3 | SCH2CH3 | Bn | 2.92 ± 0.15 | 2.69 ± 0.51 | >10 | >10 | 4.81 | 0.72 |
| 13 | CO–NHCH2CH3 | SCH2CH3 | Bn | 0.34 ± 0.05 | 0.96 ± 0.11 | 1.05 ± 0.115 | >10 | 4.01 | 2.46 |
| 14 | CO–N(CH2CH3)2 | SCH2CH3 | Bn | 0.41 ± 0.06 | 0.94 ± 0.09 | 1.02 ± 0.13 | 3.53 ± 0.40 | 4.59 | 1.80 |
| 15 | CO-N-pyrrolidine | SCH2CH3 | Bn | 0.96 ± 0.13 | 1.52 ± 0.29 | 1.36 ± 0.21 | >10 | 4.28 | 1.74 |
| 16 | CO–NH–Ph | SCH2CH3 | Bn | n.d. | n.d. | n.d. | n.d. | 5.67 | |
| 17 | CO–NH-2-thiazole | SCH2CH3 | Bn | n.d. | n.d. | n.d. | n.d. | 5.01 | |
| 18 | CO–NHCH2CH2OH | SCH2CH3 | Bn | 2.84 ± 0.38 | 8.00 ± 3.98 | >10 | >10 | 3.19 | 2.36 |
| 19 | CO-N-morpholine | SCH2CH3 | Bn | 0.20 ± 0.02 | 0.33 ± 0.02 | 0.10 ± 0.01 | 0.59 ± 0.05 | 3.66 | 3.03 |
 | ||
| Scheme 1 Synthetic pathway to compounds 2–8. Reagents and conditions: (i) 140 °C, 1 h; (ii) Ph2O, 240 °C, 2 h; (iii) POCl3, 110 °C, 20 min; (iv) R-SH or R-SNa, K2CO3, THF or DMF, RT, 1.5–24 h. | ||
The inhibitory potency of 4–8 was assessed using a fluorogenic enzymatic assay for PvNMT activity.21 Compounds were also tested on PfNMT, the enzyme present in P. falciparum,29 and on the human isoforms HsNMT1 and HsNMT2.30 Replacing the 2-cyanoethyl thioether with an n-propyl thioether (compound 5) had a limited impact on PvNMT activity, confirming that the nitrile function is not required for high affinity binding to the enzyme. Interestingly, this modification was also associated with a >10-fold improvement in activity versus PfNMT. Indeed, while the original hit compound and compound 4 were totally inactive on PfNMT up to 100 μM, 5 displayed a Ki of 1.1 μM, similar to the value measured with PvNMT. These differences in terms of binding affinity between PvNMT and PfNMT, two enzymes that share 80% identity, are difficult to rationalize. The only residue in the inhibitor-binding pocket that differs between these NMTs is Tyr334 (Pv numbering), which is replaced by a phenylalanine in PfNMT. Based on X-ray data, this residue does not establish polar interactions with the nitrile function of the inhibitor. Therefore, how such a minor modification causes significant changes in binding affinity remains to be defined. Unfortunately, despite substantial crystallisation efforts, no X-ray structure of PfNMT alone, or in complex with inhibitors, has been solved so far.
Subsequent modification of the length and nature of the thioether chain allowed us to identify the ethylthioether derivative 7, as the compound having the best potency against both PvNMT and PfNMT (Table 1).
The X-ray structure of the complex between 1 and PvNMT shows that a cluster of water molecules surrounds the sulfur atom of the thioether chain. We attempted to establish polar contacts with these water molecules through the synthesis of the sulfoxide and sulfone derivatives 9 and 10 respectively, which were obtained by mCPBA-mediated oxidation of 7 at −78 °C and 0 °C respectively (Scheme 2). Unfortunately, both appeared to be less active than the original thioether 7. At this stage, it is difficult to determine whether this reduced activity is due to unfavourable steric clashes/displacement of water molecules or if it originates from the electron-withdrawing effects of the sulfone and sulfoxide on the quinoline ring, which may reduce the strength of the H-bond between the quinoline nitrogen and Ser319 of PvNMT.
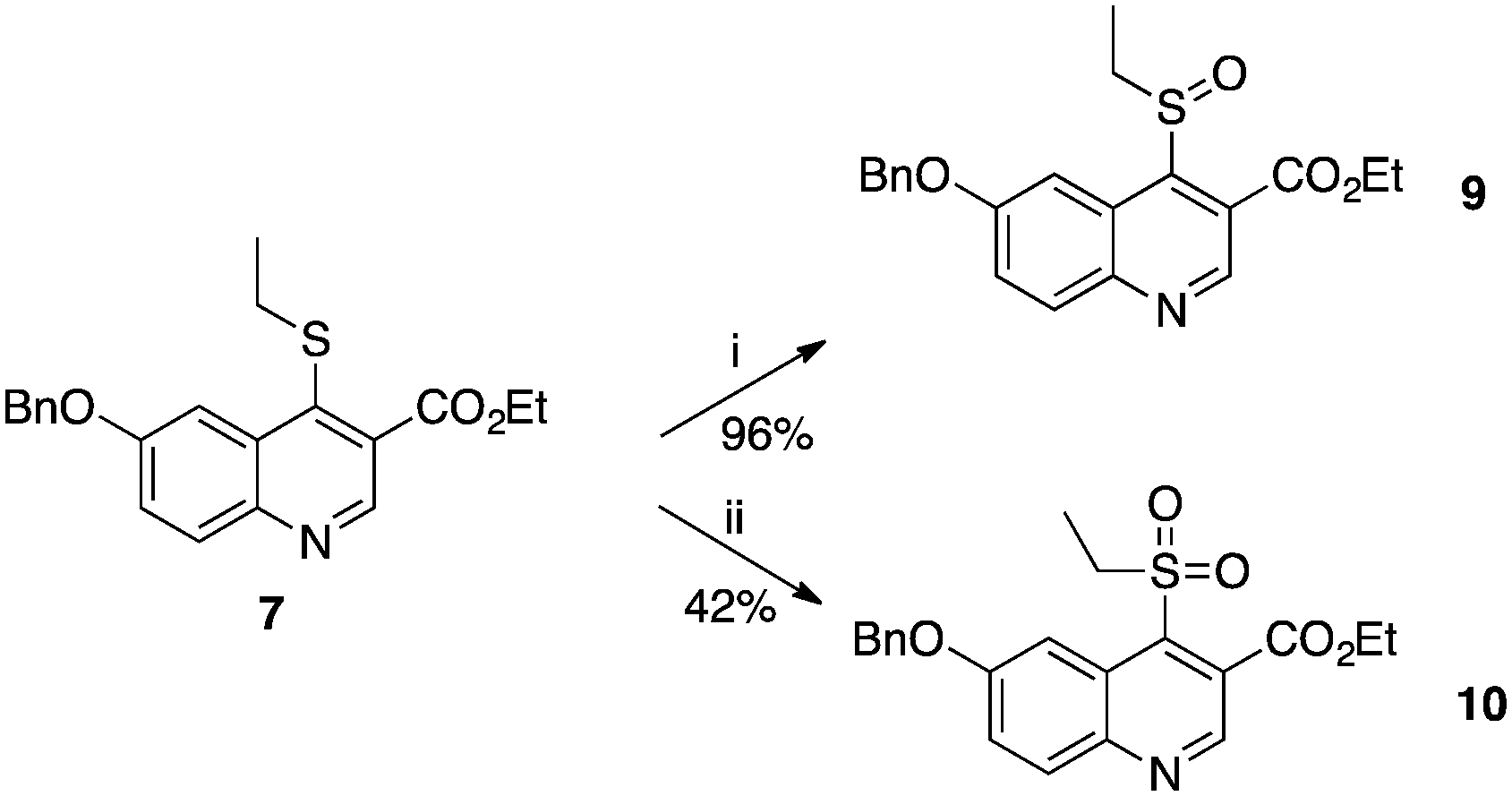 | ||
| Scheme 2 Syntheses of sulfoxide and sulfone derivatives 9 and 10. Reagents and conditions: (i) m-CPBA, DCM, −78 °C, 4 h; (ii) m-CPBA, DCM, 0 °C, 2.5 h. | ||
It is worth noting that, although 9 and 10 were less active than the corresponding alkyl analogue, their LipE values (based on PvNMT data) were significantly higher due to their enhanced hydrophilicity. However, the ethyl thioether was selected as the preferred substituent for the rest of the study because of its better synthetic tractability.
Replacement of the ethyl ester group by amides
The original hit compound, MRT00057965, possessed an n-butyl chain in position 3 of the quinoline ring. In our previous study,21 this group was replaced by an ethyl ester in order to reduce the lipophilicity of the compound. While this modification succeeded in improving the hydrophilicity of the quinoline without significantly impacting the affinity of the molecule for PvNMT, it represents a potential liability with respect to hydrolysis in vivo. In an attempt to identify more suitable substituents, the ester 7 was reduced to form the alcohol 11, which was subsequently alkylated to provide the ethyl ether derivative 12 (Scheme 3). Both compounds showed a 6- to 10-fold reduction in inhibitory activity. Subsequently, the ester was replaced by an amide. In order to allow direct comparison with the ester series, the ethyl amide 13 was synthesized initially, from compound 7 according to Weinreb's procedure (Scheme 3).31 Encouragingly, 13 showed the same inhibitory profile as 7, with a Ki of 0.34 μM and 0.96 μM on PvNMT and PfNMT respectively, and a 3-fold and 30-fold selectivity versus HsNMT1 and HsNMT2 respectively. | ||
| Scheme 3 Syntheses of alcohol, ether and amide derivatives 11–19. Reagents and conditions: (i) LiAlH4, THF, 0 °C, 1 h; (ii) tBuOK, DMSO, RT then EtI, 60 °C, 1 h; (iii) AlMe3, amine, toluene, −15 °C then 7, RT, 24 h. | ||
Superposition of the three quinoline–PvNMT complexes present in the asymmetric unit of the crystal shows that the ethyl ester group is flexible and able to adopt different conformations in a relatively large pocket, suggesting that the ethyl chain is suboptimal and that the enzyme may tolerate larger substituents. This hypothesis was confirmed through the synthesis of the N,N-diethylamide 14 and the N-pyrrolidine amide derivative 15 which showed similar levels of potency as the N-ethylamide 13 across the different enzymes. Substitution of the carboxamide with small aromatic rings (phenyl, 16, or 2-thiazole 17), which could interact with nearby residues Phe226 and His213, led to the formation of poorly soluble compounds that could not be evaluated. Finally, in order to decrease the lipophilicity of the series, and potentially establish novel electrostatic interactions with the enzyme, amides substituted with polar functions were synthesized. While the 2-hydroxyethylamide derivative 18 displayed a significant loss of affinity, the N-morpholine derivative 19 showed an improved potency, as well as a significantly reduced clogP.
A ternary crystal structure of 19 in complex with Leishmania major NMT (LmNMT) and NHM was obtained (Fig. 3).7,32 LmNMT and PvNMT share 39% sequence identity and have been used interchangeably for structural biology studies, with LmNMT offering the more robust platform for rapid crystallography. As before, the ethyl thioether chain in position 4 seems to stack against the phenol group of Tyr217 (Tyr211 in PvNMT) and the position of histidine side chain continues to adopt two orientations. The main difference with compound 1 arises from the (N-morpholine)carboxamide group. The orientation of the amide undergoes a 160° twist compared to the initial orientation of the ester, allowing the carbonyl to H-bond with Asn376 and via a water molecule with Tyr345 (Fig. 3). This change is associated with the full rotation of Phe232 (Phe226 in PvNMT) to accommodate the presence of the morpholine ring (Fig. S3‡). It remains to be determined if these changes are specific to compound 19 or if they occur with all amide derivatives.
 | ||
| Fig. 3 Structure of quinoline 19 in a ternary complex with LmNMT and MyrCoA (PDB accession code: 5G20). 19 is shown in cylinder format and colored by atom: carbon (green), oxygen (red), nitrogen (blue) and sulphur (yellow). A transparent LmNMT surface is shown in grey. The side chains of selected LmNMT residues located within 4 Å of 19 are displayed as grey sticks, and labeled. Water molecules are shown as red spheres. Polar interactions with LmNMT and solvent are represented as black dashed lines. For a stereo view, see Fig. S2.‡ | ||
Investigation of the substituent in position 6 of the quinoline ring
Finally, the role of the substituent located on position 6 of the quinoline ring was explored. The benzyloxy-present in all the above compounds sits at the top of a narrow cavity, which leads to the catalytic C-terminal leucine of N-myristoyltransferase (Fig. 4). Virtually all of the known families of NMT inhibitors interact with the C-terminal residue, which is a leucine in fungi, Plasmodium and Leishmania but is a valine in T. brucei and a glutamine in human NMTs. This interaction is usually formed by an amino group in the inhibitor that establishes a strong ionic bond with the C-terminal NMT carboxylate.7,9,18,33 However, inhibitors containing imidazoles34 or neutral functions35 have also been reported to establish stabilizing interactions with the C-terminus.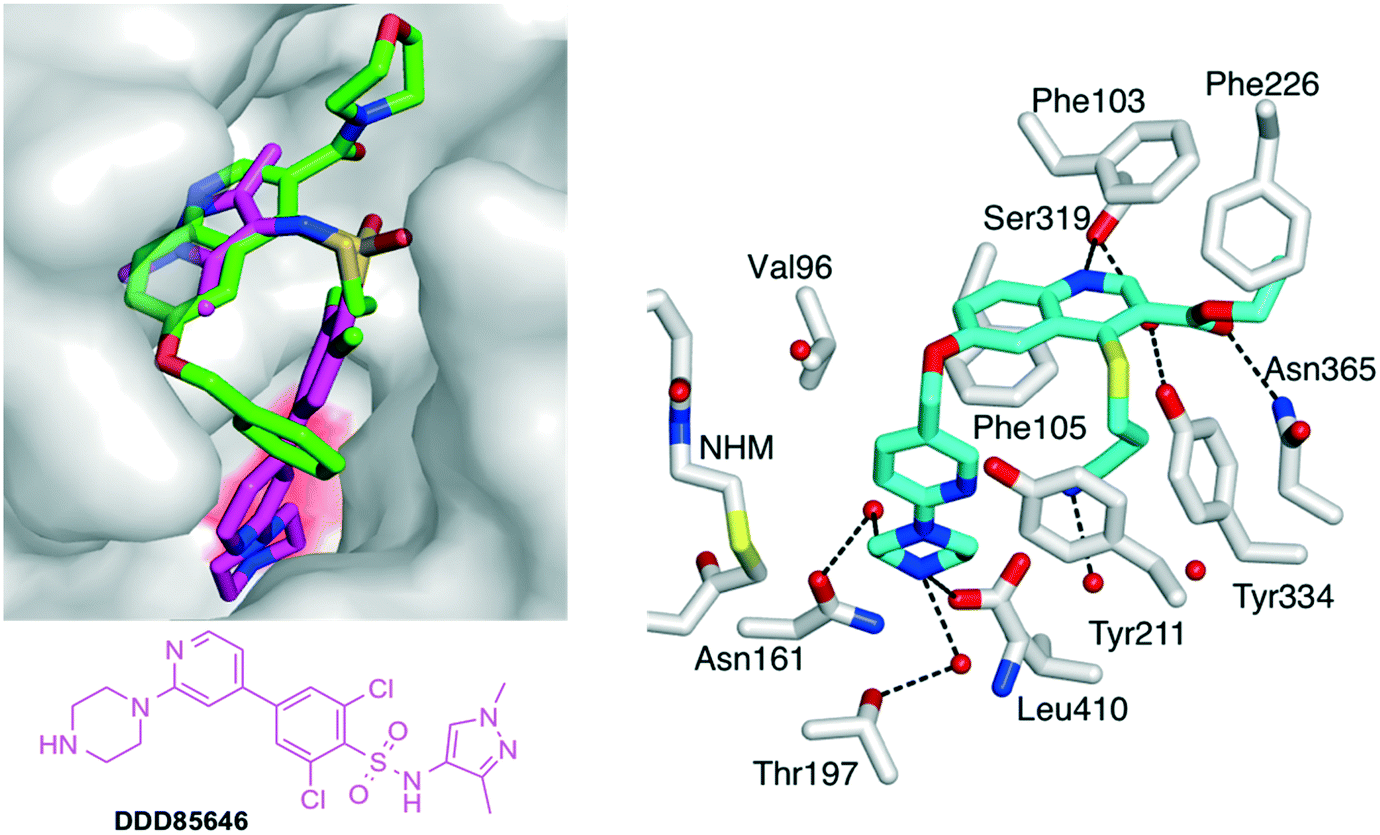 | ||
| Fig. 4 (Left) View of 19 (green carbons) in LmNMT in cylinder format. The surface of LmNMT is shown in grey. 19 is superimposed with DDD85646 (2,6-dichloro-4-(2-piperazin-1-ylpyridin-4-yl)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)-benzenesulfonamide; magenta carbons), a known NMT inhibitor. The surface above the C-terminal leucine of NMT is colored in red. (Right) Crystal structure of 26 (cyan carbons) in complex with PvNMT and non-hydrolizable MyrCoA (pdb accession code: 5G22). Selected side chains of PvNMT are shown in cylinder (format) and labeled according to PvNMT numbering. Dashed lines indicate electrostatic/H-bond interactions. For further views of 26 in complex with PvNMT and LmNMT, see Fig. S4, S5 & S6.‡ | ||
With the objective of further increasing the inhibitory potency of the quinoline series, novel molecules were designed, in which the 6-benzyloxy moiety was replaced by longer substituents elaborating an amine group at their extremity. By analogy with DDD85846, the benzyloxy was first replaced by a (6-(piperazin-1-yl)pyridin-3-yl)methoxy group (Fig. 4).
Compound 21 was prepared from 7 in three steps: 6-hydroxyquinoline intermediate 20 was first formed under acidic conditions, and was subsequently condensed with (6-(4-Boc-piperazin-1-yl)pyridin-3-yl)methanol by a Mitsunobu reaction (Scheme 4). Deprotection of the piperazine afforded 21, with an overall yield after purification of 18%. A similar strategy was employed to synthesize the derivatives 22–25, in which the piperazine was replaced with alternative cyclic or linear diamines (Table 2). These modifications were introduced to identify the most suitable linker and to take into account potential differences between LmNMT and the intended target, PvNMT.
 | ||
| Scheme 4 Synthesis of compound 21. Reagents and conditions: (i) methanesulfonic acid 10% (v/v) in TFA, 60 °C, 4 h; (ii) (6-(4-Boc-piperazin-1-yl)pyridin-3-yl)methanol, DEAD, PPh3, THF, RT, 16 h; (iii) TFA 20% in DCM, RT, 40 min. | ||
| Entry |
|
K i app (μM)a | clogPb |
LipEc | ||||
|---|---|---|---|---|---|---|---|---|
| R4 | R6 | PvNMT | PfNMT | HsNMT1 | HsNMT2 | |||
|
a Apparent Ki values of tested compounds on the enzymatic activity of recombinant P. vivax NMT, P. falciparum NMT and H. sapiens NMT isoforms 1 and 2. Each Ki is the mean ± SD from duplicates.
b clogP was determined with ChemAxon.
c LipE: lipophilic efficiency.
|
||||||||
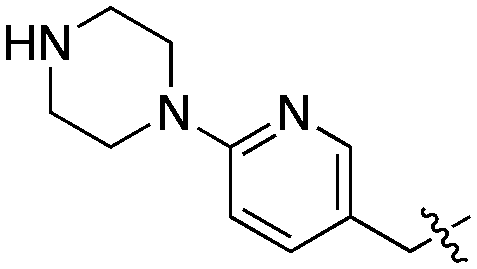
| 7 | SCH2CH3 | Bn- | 0.44 ± 0.05 | 0.67 ± 0.04 | 1.56 ± 0.22 | 4.23 ± 0.53 | 4.94 | 1.42 |
| 21 | SCH2CH3 |
|
0.04 ± 0.01 | 0.24 ± 0.02 | 0.04 ± 0.01 | 0.10 ± 0.01 | 3.89 | 3.53 |
| 22 | SCH2CH3 |
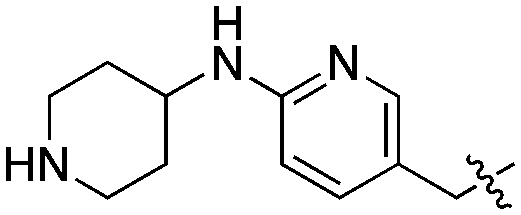
|
0.09 ± 0.01 | 0.40 ± 0.05 | 0.08 ± 0.01 | 0.69 ± 0.08 | 3.43 | 3.64 |
| 23 | SCH2CH3 |

|
0.14 ± 0.01 | 0.96 ± 0.10 | 0.05 ± 0.01 | 0.37 ± 0.06 | 3.37 | 3.49 |
| 24 | SCH2CH3 |
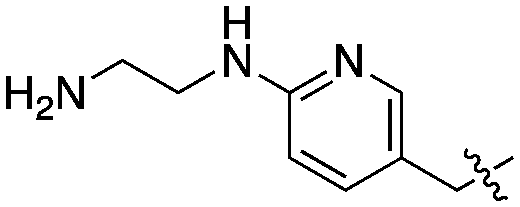
|
0.28 ± 0.02 | 1.25 ± 0.08 | 0.05 ± 0.01 | 0.41 ± 0.07 | 2.99 | 3.57 |
| 25 | SCH2CH3 |
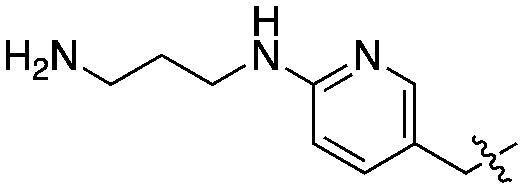
|
0.02 ± 0.01 | 0.15 ± 0.02 | 0.008 ± 0.002 | 0.10 ± 0.01 | 3.05 | 4.69 |
| 26 | SCH2CH2CN |

|
0.10 ± 0.01 | 3.31 ± 1.15 | 0.37 ± 0.03 | 1.35 ± 0.20 | 3.30 | 3.72 |

Compared to the initial benzyloxy 7, the 4-piperazine-pyridin-3-yl derivative 21 showed a dramatic improvement in potency against PvNMT (>10-fold, Ki = 38 nM), and a similar, while more limited, effect on PfNMT (Ki = 242 nM). This increase in potency was also associated with a loss of selectivity versus HsNMT1 and HsNMT2. Replacing the piperazine with a 4-amino-piperazine, a pyrrolidine, or ethylenediamine did not improve activity against PvNMT or PfNMT. The most promising results were obtained with the 1,3-propanediamine derivative 25, which inhibited PvNMT with a Ki of 18 nM.
Crystal structures were determined of 26, in complex with first PvNMT to explore the basis of potency, and then with LmNMT, to allow comparison of its binding mode with that of 19 (Fig. 4, S4, S5 & S6‡). As expected, an ionic interaction is observed between the piperazine and the C-terminal leucine. This complex is further stabilized by direct and water-mediated H-bonds of the piperazine with Thr197 and Asn161. The pyridine ring also establishes a stabilizing polar interaction with the phenol of Tyr211. The structure of 26 bound to LmNMT (PDB code 5G21) is closely superimposable but shows a slightly different orientation of Tyr211, possibly due to multiple orientations of the nitrile tail (Fig. S4‡). It is worth noting that the quinoline plane of compound 26 adopts a slightly tilted orientation compared to the one occupied by 19 in LmNMT (Fig. S5 & S6‡). We assume that this potentially unfavourable change is related to an ionic interaction with LmNMT, which constrains the molecule to reduce the distance between the piperazine nitrogen and Leu421 carboxylate. This may explain the higher potency of the more flexible 1,3-propanediamine derivative 25.
Conclusion
Starting from the crystal structure of compound 1 in PvNMT, the positions 3-, 4- and 6-substituents of the quinoline ring were sequentially modified, with the aim of improving chemical stability, solubility and affinity of this series of compounds for N-myristoyltransferases. Interestingly, while the primary objective of this work was to increase the inhibitory potency of the quinoline series for PvNMT, we observed that the substitution of the 2-cyanoethythioether moiety by an alkyl thioether resulted in a significant improvement in the activity towards Plasmodium falciparum NMT, affording novel lead compounds with balanced activities against both PvNMT and PfNMT. Moreover, controlling the lipophilicity as part of the optimization process allowed us to identify novels inhibitors with significantly improved lipophilic efficiency.The low selectivity over host NMT may limit the potential to progress this series to lead optimisation.36 However, we recently reported that ligands that induce conformational changes in Tyr211 of PvNMT were selective for Plasmodium NMTs over human NMT,9 and further investigation of the 4-position of the quinoline, which is in contact with Tyr211, may be a fruitful area for future exploration.
Finally, we expect that the crystal structures obtained as part of this work, along with information on the quinoline binding mode, will support the development of even more potent N-myristoyltransferase inhibitors, based on a quinoline core alone or hybridized with other series of inhibitors targeting NMT.18,35
Notes
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to Mark Rackham, Zhiyong Yu and Jennie Hutton for valuable discussions. We thank Diamond Light Source for access to beamlines I02 and I04 (proposal numbers mx-1221 and mx-7864). This work was supported by the Medical Research Council (grants G0900278 and U117532067), the EPSRC (grant EP/F500416/1) and the Wellcome Trust (grant 087792).References
- D. R. Johnson, R. S. Bhatnagar, L. J. Knoll and J. I. Gordon, Annu. Rev. Biochem., 1994, 63, 869–914 CrossRef CAS PubMed.
- M. H. Wright, W. P. Heal, D. J. Mann and E. W. Tate, J. Biol. Chem., 2010, 3, 19–35 CrossRef PubMed.
- M. D. Resh, Nat. Chem. Biol., 2006, 2, 584–590 CrossRef CAS PubMed.
- J. Zheng, D. R. Knighton, N. H. Xuong, S. S. Taylor, J. M. Sowadski and L. F. Ten Eyck, Protein Sci., 1993, 2, 1559–1573 CrossRef CAS PubMed.
- M. Chow, J. F. Newman, D. Filman, J. M. Hogle, D. J. Rowlands and F. Brown, Nature, 1987, 327, 482–486 CrossRef CAS PubMed.
- N. H. Georgopapadakou, Expert Opin. Invest. Drugs, 2002, 11, 1117–1125 CrossRef CAS PubMed.
- J. A. Frearson, S. Brand, S. P. McElroy, L. A. T. Cleghorn, O. Smid, L. Stojanovski, H. P. Price, M. L. S. Guther, L. S. Torrie, D. A. Robinson, I. Hallyburton, C. P. Mpamhanga, J. A. Brannigan, A. J. Wilkinson, M. Hodgkinson, R. Hui, W. Qiu, O. G. Raimi, D. M. F. van Aalten, R. Brenk, I. H. Gilbert, K. D. Read, A. H. Fairlamb, M. A. J. Ferguson, D. F. Smith and P. G. Wyatt, Nature, 2010, 464, 728–732 CrossRef CAS PubMed.
- H. P. Price, M. R. Menon, C. Panethymitaki, D. Goulding, P. G. McKean and D. F. Smith, J. Chem. Biol., 2003, 278, 7206–7214 CrossRef CAS PubMed.
- Z. Yu, J. A. Brannigan, D. K. Moss, A. M. Brzozowski, A. J. Wilkinson, A. A. Holder, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2012, 55, 8879–8890 CrossRef CAS PubMed.
- E. W. Tate, A. S. Bell, M. D. Rackham and M. H. Wright, Parasitology, 2014, 141, 37–49 CrossRef CAS PubMed.
- M. H. Wright, B. Clough, M. D. Rackham, K. Rangachari, J. A. Brannigan, M. Grainger, D. K. Moss, A. R. Bottrill, W. P. Heal, M. Broncel, R. A. Serwa, D. Brady, D. J. Mann, R. J. Leatherbarrow, R. Tewari, A. J. Wilkinson, A. A. Holder and E. W. Tate, Nat. Chem., 2014, 6, 112–121 CrossRef CAS PubMed.
- B. D. Galvin, Z. R. Li, E. Villemaine, C. B. Poole, M. S. Chapman, M. P. Pollastri, P. G. Wyatt and C. K. S. Carlow, PLoS Neglected Trop. Dis., 2014, 8 Search PubMed.
- E. Thinon, J. Morales-Sanfrutos, D. J. Mann and E. W. Tate, ACS Chem. Biol., 2016, 11, 2165–2176 CrossRef CAS PubMed.
- D. A. Towler, S. P. Adams, S. R. Eubanks, D. S. Towery, E. Jackson-Machelski, L. Glaser and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2708–2712 CrossRef CAS.
- B. Devadas, M. E. Zupec, S. K. Freeman, D. L. Brown, S. Nagarajan, J. A. Sikorski, C. A. McWherter, D. P. Getman and J. I. Gordon, J. Med. Chem., 1995, 38, 1837–1840 CrossRef CAS PubMed.
- L. A. Paige, G. Q. Zheng, S. A. DeFrees, J. M. Cassady and R. L. Geahlen, J. Med. Chem., 1989, 32, 1665–1667 CrossRef CAS PubMed.
- L. A. Paige, G. Q. Zheng, S. A. DeFrees, J. M. Cassady and R. L. Geahlen, Biochemistry, 1990, 29, 10566–10573 CrossRef CAS PubMed.
- A. S. Bell, J. E. Mills, G. P. Williams, J. A. Brannigan, A. J. Wilkinson, T. Parkinson, R. J. Leatherbarrow, E. W. Tate, A. A. Holder and D. F. Smith, PLoS Negl. Trop. Dis., 2012, 6, e1625 CAS.
- M. Masubuchi, K. Kawasaki, H. Ebiike, Y. Ikeda, S. Tsujii, S. Sogabe, T. Fujii, K. Sakata, Y. Shiratori, Y. Aoki, T. Ohtsuka and N. Shimma, Bioorg. Med. Chem. Lett., 2001, 11, 1833–1837 CrossRef CAS PubMed.
- K. Yamazaki, Y. Kaneko, K. Suwa, S. Ebara, K. Nakazawa and K. Yasuno, Bioorg. Med. Chem., 2005, 13, 2509–2522 CrossRef CAS PubMed.
- V. Goncalves, J. A. Brannigan, D. Whalley, K. H. Ansell, B. Saxty, A. A. Holder, A. J. Wilkinson, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2012, 55, 3578–3582 CrossRef CAS PubMed.
- R. N. Price, E. Tjitra, C. A. Guerra, S. Yeung, N. J. White and N. M. Anstey, Am. J. Trop. Med. Hyg., 2007, 77, 79–87 Search PubMed.
- K. Mendis, B. J. Sina, P. Marchesini and R. Carter, Am. J. Trop. Med. Hyg., 2001, 64, 97–106 CAS.
- A. L. Hopkins, G. M. Keseru, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- G. M. Keseru and G. M. Makara, Nat. Rev. Drug Discovery, 2009, 8, 203–212 CrossRef PubMed.
- J. A. Brannigan, B. A. Smith, Z. Yu, A. M. Brzozowski, M. R. Hodgkinson, A. Maroof, H. P. Price, F. Meier, R. J. Leatherbarrow, E. W. Tate, D. F. Smith and A. J. Wilkinson, J. Mol. Biol., 2010, 396, 985–999 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2006, pp. 647–695, DOI:10.1002/9780470053485.ch6.
- C. G. Wang, T. Langer, P. G. Kamath, Z. Q. Gu, P. Skolnick and R. I. Fryer, J. Med. Chem., 1995, 38, 950–957 CrossRef CAS PubMed.
- M. D. Rackham, J. A. Brannigan, D. K. Moss, Z. Yu, A. J. Wilkinson, A. A. Holder, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2013, 56, 371–375 CrossRef CAS PubMed.
- V. Goncalves, J. A. Brannigan, E. Thinon, T. O. Olaleye, R. Serwa, S. Lanzarone, A. J. Wilkinson, E. W. Tate and R. J. Leatherbarrow, Anal. Biochem., 2012, 421, 342–344 CrossRef CAS PubMed.
- M. F. Lipton, A. Basha and S. M. Weinreb, in Organic Syntheses, John Wiley & Sons, Inc., 2003, DOI:10.1002/0471264180.os059.08.
- J. A. Brannigan, S. M. Roberts, A. S. Bell, J. A. Hutton, M. R. Hodgkinson, E. W. Tate, R. J. Leatherbarrow, D. F. Smith and A. J. Wilkinson, IUCrJ, 2014, 1, 250–260 CAS.
- M. D. Rackham, J. A. Brannigan, D. K. Moss, Z. Yu, A. J. Wilkinson, A. A. Holder, E. W. Tate and R. J. Leatherbarrow, J. Med. Chem., 2012, 56, 371–375 CrossRef PubMed.
- T. O. Olaleye, J. A. Brannigan, S. M. Roberts, R. J. Leatherbarrow, A. J. Wilkinson and E. W. Tate, Org. Biomol. Chem., 2014, 12, 8132–8137 CAS.
- J. A. Hutton, V. Goncalves, J. A. Brannigan, D. Paape, M. H. Wright, T. M. Waugh, S. M. Roberts, A. S. Bell, A. J. Wilkinson, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, J. Med. Chem., 2014, 57, 8664–8670 CrossRef CAS PubMed.
- MMV, Essential information for scientists, http://www.mmv.org/research-development/essential-information-scientists, (accessed 28/08/2015).
Footnotes |
| † The authors declare no competing interests. |
| ‡ Electronic supplementary information (ESI) available: Synthesis details and structural characterization of compounds, X-ray data collection and statistics, supplementary Fig. S1–S6. See DOI: 10.1039/c6md00531d |
| § Current address: ICMUB, Université Bourgogne Franche-Comté, Dijon, 21000, France. |
| ¶ Current address: Liverpool John Moores University, Egerton Court, 2 Rodney Street, Liverpool, L1 2UA, UK. |
| This journal is © The Royal Society of Chemistry 2017 |