
Antimalarial drug discovery targeting apical membrane antigen 1†
Shane M.
Devine
 ,
Christopher A.
MacRaild
,
Raymond S.
Norton
* and
Peter J.
Scammells
*
,
Christopher A.
MacRaild
,
Raymond S.
Norton
* and
Peter J.
Scammells
*
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia. E-mail: ray.norton@monash.edu; peter.scammells@monash.edu
First published on 4th November 2016
Abstract
Malaria continues to frustrate humanity's attempts to eradicate this deadly disease. Although gains have been made over the last 15 years, drug resistance to malaria continues to be a major concern. The lack of new antimalarials with novel mechanisms of action continues to challenge the scientific community to find innovative targets to combat this persistent disease. One such target, apical membrane antigen 1 (AMA1), is an essential protein that helps the parasite invade host erythrocytes. Recently, a number of efforts have focused on the druggability of this target, aiming to block the interactions of AMA1 that mediate invasion of host cells. This review covers recent progress in drug discovery targeting this crucial protein–protein interaction in malaria.
Introduction
Malaria is a complex disease caused by unicellular parasites of the genus Plasmodium. Of the five species of Plasmodium known to infect humans, P. falciparum and P. vivax are the major causes of death and disease. P. ovale, P. malariae and P. knowlesi also infect humans, but represent only a small percentage of infections. Malaria continues to be a critical endemic health problem for much of the world's impoverished and vulnerable societies. Although in recent years the incidence and mortality rates of malaria have decreased in response to increased prevention and treatment strategies, in 2015 alone there were an estimated 214 million new cases of malaria and 438![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.1 Most concerning is the fact that 306000 of these deaths were children under five years of age.
000 deaths.1 Most concerning is the fact that 306000 of these deaths were children under five years of age.
Artemisinin-based therapies have played a central role in recent successes against malaria,2 although resistance to these front-line treatments is now a serious threat, with detection of resistance in five countries in the Greater Mekong region of Asia, and the likelihood of further spread.1 This problem, coupled with the fact that the pipeline of novel antimalarial agents under development suffers from a lack of diversity, underscores the need for new targets to combat malaria.3 Despite recent progress towards a malaria vaccine,4,5 the most advanced of the current candidates possess only modest and short-lived efficacy, and there is no established timetable for widespread clinical roll-out.6 Therefore, there is a clear and present unmet need for novel targets and new antimalarials.
AMA1 – history, importance, function, controversy
The lifecycle of all Plasmodium species involve multiple phases of growth and reproduction within distinct types of host cell. Accordingly, the parasites rely on an elaborate mechanism of host cell invasion, which is conserved across the genus and more broadly across the phylum Apicomplexa.7 One of the best-characterised components of malaria's invasion machinery is the integral membrane protein apical membrane antigen 1 (AMA1). Peterson and co-workers first isolated AMA1 at the Walter and Eliza Hall Institute in 1989 and showed that it was transported to the merozoite surface near the time of schizont rupture.8 AMA1 is one of a range of proteins that is secreted from the microneme and rhoptry organelles,9 many of which are involved in host cell invasion.7 Amongst these proteins are the so-called rhoptry neck proteins (RON), which form a complex that includes RON2, RON4 and RON5. The RON complex is injected into the target host cell, with RON2 integrated into the host cell plasma membrane, where it acts as a receptor for AMA1 by exposing its ectodomain.10,11 Thus, the parasite provides both ligand (AMA1) and receptor (RON2) in forming the AMA1–RON complex.The AMA1–RON complex co-localises with a tight junction that forms between the apical tip of the parasite and its host as the parasite prepares to invade (Fig. 1A).10–12 This junction moves from the apex of the parasite to its posterior end as invasion proceeds, and so is known as the moving junction. By this mechanism, the parasite enters the host cell, forming the parasitophorous vacuole in which it will reside. Once the moving junction reaches the posterior end of the parasite, a membrane fusion event closes the parasitophorous vacuole, separating it from the host cell plasma membrane.
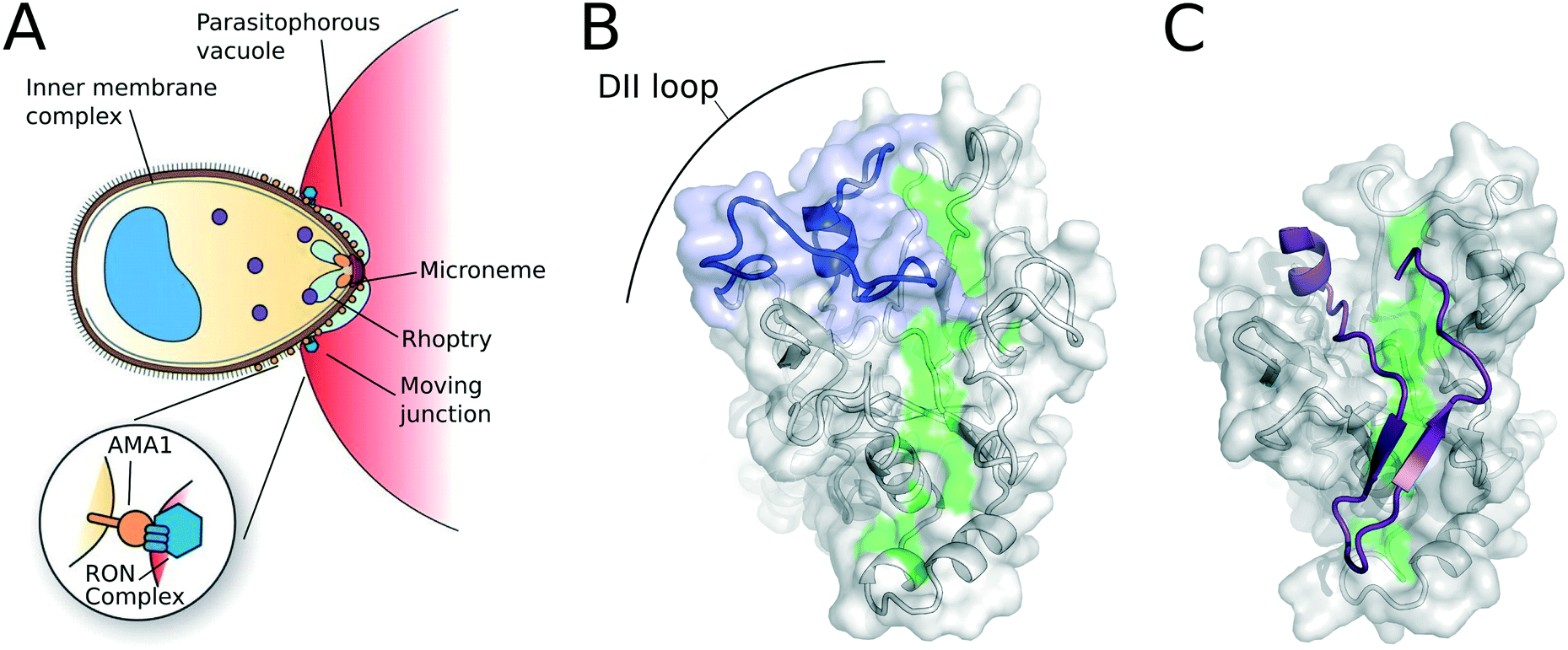 | ||
| Fig. 1 (A) The role of AMA1 in host-cell invasion. Figure modified from Richard et al.21 (B) the conserved hydrophobic cleft (green) and DII loop (blue) on PfAMA1 (PDB: 1Z40).22 (C) The structure of AMA1 bound to RON2 residues 2021–2059 (purple), showing the displacement of the DII loop (top left; PDB: 3ZWZ).23 | ||
Although the molecular details of moving junction formation and the precise role of AMA1 in this process remain unclear, an extensive body of evidence suggests that AMA1 plays an essential role in invasion. Repeated attempts to generate AMA1 knock-outs in several Plasmodium species have failed to yield stable clonal lines, suggesting the gene is essential to parasite growth in blood-stage culture.13–15 Despite clear evidence of strong diversifying pressure acting on AMA1 from the host immune system,16,17 the AMA1–RON2 interaction remains highly conserved across the apicomplexan phylum.18 Furthermore, antibodies targeting AMA1, arising through natural infection or from experimental immunisations, block the AMA1–RON2 interaction, inhibit blood-stage culture of P. falciparum parasites, and are correlated with protection from disease in vivo.19,20
There has been some recent controversy about the essentiality of AMA1 and its role in the moving junction. On the basis of a series of transient AMA1 knock-down and knockout lines in Toxoplasma and P. bergei, Ménard and colleagues posited that AMA1 acts independently of the RON complex and is dispensable in any step of the invasion process.13,15,24 However, several factors complicate the analysis of these lines. In Toxoplasma, paralogues of both AMA1 and RON2 exist, which appear able to complement the loss of AMA1.25 Moreover, no stable clonal knockout lines were established in P. bergei, and the principal assay of host cell invasion used in this work may be unable to distinguish productive invasion from aborted invasion events in which the parasite remains associated with the host cell but is unable to undergo intracellular development.26 Irrespective of the dispensability of AMA1 at the genetic level, a wide range of antibody and peptide inhibitors of the AMA1–RON2 interaction inhibit host cell invasion and parasite growth in culture.27 Accordingly, the potential efficacy of AMA1 as a target for chemical inhibition is now accepted.13
Nonetheless, significant challenges must be overcome if anti-malarials targeting AMA1 are to be developed. The large and relatively flat interface between AMA1 and RON2 is typical of a protein–protein interaction (PPI), and presents a number of challenges shared by this type of target.28,29 Moreover, the formation of the moving junction and the AMA1–RON2 interaction itself is a transient event, occurring only briefly during each ∼48 hour asexual cycle. Together with the fact that AMA1 appears to be present in excess on the parasite surface,26 this may indicate that a saturating amount of inhibitor needs be present at the time of invasion in order to achieve a therapeutic outcome. AMA1 is, however, directly exposed to the host bloodstream prior to invasion, thus removing the difficulties associated with accessing intracellular targets and avoiding the potential for resistance by drug-efflux mechanisms.
Structural insights into the AMA1–RON2 interaction
Mature AMA1 is a 66 kDa type 1 integral membrane protein with a short, well-conserved cytoplasmic region. This cytoplasmic tail has an important functional role in host cell invasion, which appears to involve cAMP-dependent phosphorylation, but other details remain obscure.30,31 A key feature identified by Bai et al. in the first crystal structure of the PfAMA1 ectodomain was a cleft between loops in domains I and II,22 the base of which is lined by conserved hydrophobic residues (Fig. 1B). Initial evidence for the functional importance of this hydrophobic cleft came from the discovery of an inhibitory shark-immunoglobulin new antigen receptor (IgNAR), which binds at one end of the cleft.32 Competitive binding with other inhibitory molecules implied that they too bound at the hydrophobic cleft,27 and subsequent structural studies have confirmed this result.21,23,33 Likewise, the native ligand, RON2, binds along the full length of the hydrophobic cleft.23,34,35 In the case of RON2, as well as the peptide inhibitor R1,27,36 ligand binding displaces the extended DII loop that lines one end of the cleft (Fig. 1C). This opens that end of the cleft, exposing a much greater conserved surface area for the interaction of these ligands.Drug discovery targeting AMA1 inhibitors
Fragment-based approaches
Over the past 20 years, fragment-based drug discovery (FBDD) has emerged as a powerful technique to drive a project from hit to lead. Many campaigns have demonstrated the feasibility of producing clinically useful elaborated molecules from a weak fragment hit.37,38 Significantly, FBDD has been applied successfully to PPIs, a class of target that historically constituted a significant challenge.39 This has culminated in two molecules reaching the clinic: Vemurafenib in 2011 for the treatment of late-stage melanoma40 and Venetoclax earlier this year for the treatment of chronic lymphocytic leukaemia.41 Our group has applied an FBDD approach to AMA1, which commenced with a screen of the MIPS in-house fragment library42 against 3D7 PfAMA1 DI+II.43 We screened 1140 compounds by saturation transfer difference (STD) NMR in cocktails containing six compounds; hits were then re-examined as singletons in competition experiments with R1, an inhibitory peptide with a binding affinity (KD) of 100 nM.27 This experiment served to demonstrate that the fragments were binding along the hydrophobic cleft where R1 has been shown to bind.21,23,27 This secondary screen also exploited an orthogonal ligand-detected NMR technique which detected enhanced NMR relaxation rates upon binding. We thus identified 57 fragments that were positive in both NMR experiments, and which were competed by R1, giving a 5% overall hit rate from our MIPS fragment library. We then assessed these fragments by surface plasmon resonance (SPR) to arrive at a final group of 46 discrete molecules with demonstrable binding to AMA1. These molecules were clustered and assessed for synthetic tractability, solubility and ligand efficiency. A number of relatively conserved scaffolds presented themselves, including 2-aminothiazoles (1), furans (2), pyrazoles (3) and benzimidazoles (4) (Fig. 2A). | ||
| Fig. 2 Small molecule AMA1 inhibitors (A) scaffolds identified via FBDD (1–4);43–46 (B) pyrrolopyrimidines (5–7) identified by Srinivasan et al.;47 (C) and diazepinone 8 identified by Pihan et al.48 | ||
Initially, a series of 4-aryl substituted 2-aminothiazoles was synthesised.43 However, when these were analysed by NMR and SPR, the structure–activity relationships (SAR) obtained were flat and inconsistent. We then reviewed our fragment screening campaigns at MIPS and found that in 14 screens against our fragment library at least one 2-aminothiazole hit all 14 targets by STD NMR.42 This led us to investigate all 2-aminothiazoles, of which there were 28 in our fragment library, for their potential promiscuity by SPR, NMR and computational methods. We also interrogated two high throughput-screening (HTS) libraries, one from academia and one from industry. Although the HTS libraries showed some signs of frequent hitter behaviour, the result was much more pronounced in the fragment set. The 28 fragments in our library were then examined against six unrelated protein targets and, although some patterns emerged, such as the association of the free 2-amino group with increased promiscuity, no clear mechanism of action was evident. Nonetheless, the SAR obtained was confusing and flat against several of the targets, as previously seen in the characterization of pan-assay interference compounds (PAINS) by HTS.49 We therefore concluded that this class of molecule should be excluded from fragment libraries, unless several orthogonal approaches are utilised to avoid these so called promiscuous 2-aminothiazoles (PrATs).45 The remaining scaffolds identified in the original fragment screen are currently under investigation and the outcome of these studies will be reported in due course.
Towards peptidomimetic inhibitors of the AMA1–RON2 interaction
Another potential approach to the development of drugs targeting AMA1 involves peptidomimetic inhibitors based on the endogenous ligand RON2 or peptide inhibitors such as R1. To explore the potential of this approach, we and others have dissected the key interactions responsible for the efficacy of these peptides.23,35,50,51 The results of these studies have demonstrated that RON2 and R1 adopt similar conformations when bound to AMA1, and the key binding interactions of both peptides are similar (Fig. 3). Moreover, these interactions are distributed across the length of the long hydrophobic cleft that serves as their common binding site on AMA1. In the case of R1, key peptide residues include the hydrophobic sequence FLPLF (residues 5–9) and Phe12 and Arg15. The binding sites for these residues on AMA1 are separated by approximately 26 Å. Similar or identical residues on RON2 (including Pro2033, Val2034, Phe2038 and Arg2041) make very similar interactions with AMA1. The minimal high-affinity peptide derived from RON2 is longer than R1, and includes a disulfide-linked 13-residue β-hairpin (residues 2037–2049), which enables additional interactions that contribute to the higher affinity and lack of strain specificity in RON2 binding.50 | ||
| Fig. 3 Peptide ligands R1 and RON2 make similar interactions with AMA1. R1 (orange) (PDB: 3SRJ)23 adopts a similar conformation to residues 2027–2047 of RON2 (blue) (PDB: 3ZWZ).23 Five residues in each peptide are identical (red), and make similar interactions with AMA1. Figure modified from Wang et al.35 | ||
A recent peptide engineering exercise identified two point mutations to RON2 that afford a 1–2 order of magnitude increase in binding affinity for AMA1. On the background of a 29-residue peptide based on residues 2027–2055 of RON2, these mutants give rise to KD values of 2.1 and 0.4 nM, respectively, for 3D7 and FVO AMA1.50 This is the most potent strain-transcending peptide identified to date. Nonetheless, in this peptide, as in the parental RON2 and R1, critical interactions are broadly distributed over the surface of AMA1 in ways that are unlikely to be effectively mimicked in a drug-like peptidomimetic inhibitor.
HTS approaches
Srinivasan and co-workers identified small-molecule inhibitors of the AMA1–RON2 complex, via an AlphaScreen assay of a ∼21000 member library, in which a truncated RON2 peptide was conjugated to the AlphaScreen donor bead, and AMA1 to the AlphaScreen acceptor.47 This screen identified 20 initial hits, 14 of which were tested in blood-stage parasite culture, resulting in three hits that blocked merozoite invasion in vitro with IC50 values in the range 21–29 μM. Re-synthesis of pyrrolopyrimidine 5 (Fig. 2B) and chemical elaboration of this scaffold, produced two molecules (6 and 7) that showed enhanced inhibition, with reported IC50 values of 9.8 μM and 6 μM, respectively. The authors used immunoprecipitation assays to show that these compounds prevented the association of RON2 with AMA1 and electron microscopy of invading parasites to infer that the compounds blocked moving junction formation. Nonetheless, they were unable to quantify a direct interaction between these molecules and AMA1, impeded by poor compound solubility. Moreover, SPR data showed clear evidence of weak, super-stoichiometric binding. In an attempt to address these issues, we synthesized the three active compounds and examined them using our previously-described biophysical techniques based on NMR and SPR.43 Compounds 5–7 showed clear evidence of colloidal aggregation over the concentration range employed for in vitro testing, suggesting these assays may be confounded by the well-known non-specific effects of colloidal aggregates in a broad range of biological assays.52,53 We therefore attempted to improve some of the problematic physicochemical properties of this scaffold, including the high calculated partition co-efficients (clogP),54 a measure of solubility, and the lipophilic ligand efficiency (LLEAT),55 an indicative measure of potency relative to molecular size and lipophilicity. To address these issues, we replaced the 7-cyclopentyl group with a methyl group to reduce the lipophilicity and explored substitutions on the 5-aryl ring, whilst maintaining the 4-amino group of the pyrrolo[2,3-d]pyrimidine scaffold. Although the Srinivasan compounds47 and our novel, more soluble, analogues56 showed modest activity as anti-malarials in a growth inhibition assay (GIA), their affinity for AMA1 (where measurable) was low and did not correlate with GIA activity. Likewise, a third group has been unable to quantify binding of 5 to AMA1, either by SPR or in a fluorescence anisotropy based R1 competition assay (described below).48 We therefore concluded that these compounds were acting in an AMA1-independent manner. As this scaffold is prevalent in the kinase literature, we believe another pathway, possibly the calcium-dependent protein kinase 1 (CDPK), may be the major target in the activity of these compounds.56
Virtual screening approaches
Pihan et al. have approached the inhibition of AMA1 by both computational and biophysical approaches.48 Virtual screening of a collection of 8 million compounds, from sources including the ChEMBL malaria library, MMV Malaria Box57 and approved drugs, was conducted using pharmacophoric models based on the interactions of PfRON2sp1 and R1 with 3D7 AMA1 (PDB: 3ZWZ and 3SRJ).23 Their screening cascade used three ligand-based mapping programs: PHARMER, Align-It and PepMMsMIMIC. From these programs, 2557052 structures were selected from the initial 203738892 structures interrogated. Utilising the crystal structure of the PfAMA1–PfRON2sp1 (PDB: 3ZWZ)23 and the PLANTS program,58in silico docking experiments were conducted. The binding site was a 12 Å sphere centred on the oxygen of the side-chain of Tyr251, a key residue near the centre of the hydrophobic cleft.59 Ten poses were generated per structure and the best-scoring pose was selected for subsequent analysis. The binding poses of these structures were analysed for their interaction with three PfAMA1 residues, Val169, Phe183 and Tyr234. A final visualisation step reduced the structure-based analysis of 7161 down to eight compounds. However, it is unclear how this number was so significantly culled. These compounds included 5 diazepinones, an acetamide and two known drugs: Goserelin, for the treatment of breast and prostate cancer, and Indinavir, a retroviral treatment for HIV/AIDS. They also included the MMV Malaria Box in their computational workflow, resulting in 1 hit, MMV019881.
In order to test these compounds, a fluorescence anisotropy-binding assay was developed. This assay used a fluorescein-labelled peptide probe that was outcompeted by a putative small-molecule inhibitor binding to AMA1. A relatively weak probe is required as the nanomolar binder F*PfRON2sp1 was too potent to permit observation of competitive displacement. A truncated version of the potent binder R1 was utilised featuring a 5-carboxyfluoroscein (5-FAM) linked by an aminohexanoic acid (Ahx) to a 13-residue version with sequence: 5-FAM-Ahx-ALPLFSKFGSRMH, known as F*R1-13A1. The nine hits selected from the in silico screening, together with the Srinivasan compound 5,47 were tested for their ability to disrupt the interaction between PfAMA1 and F*R1-13A1. Diazepinone 8 and pyrrolopyrimidine 5 showed moderate competitive behaviour, with an IC50 determined for diazepinone 8 of 24 μM (Fig. 2C). Limited solubility of pyrrolopyrimidine 5 prevented the quantification of its potency in this assay. The compounds were also tested for their ability to bind AMA1 by SPR, with none showing saturable interaction with the target. Nonetheless, the fluorescence anisotropy assay developed in this study could find greater utility in the future to interrogate putative inhibitory effects at AMA1.
This computational approach followed work that used in silico strategies to identify putative peptidomimetics of AMA1-binding peptides identified by random phage display.60,61 These molecules were then docked into PfAMA1 (PDB: 1Z40)22 along the hydrophobic cleft.44 Then, using the ZINC database,62 small molecules that were similar to the five top scoring results were blindly docked into the surface of PfAMA1. These compounds were not predicted to interact with the polymorphic regions of PfAMA1 and their putative binding site in the hydrophobic cleft may suggest their potential to inhibit the AMA1/RON2 interaction. Nonetheless, experimental validation of this work is lacking, and would be required before the utility of these compounds became evident.
Structural and functional elucidation
One of the key difficulties with drug discovery targeting AMA1 has been the lack of success in obtaining crystal structures of small-molecule inhibitors interacting with AMA1. Many structures are available for PfAMA1, including in complex with the inhibitory monoclonal antibody 1F9,33,63 shark-immunoglobulin domains (IgNARs)32 and the peptide inhibitor R1,23 as well as with the native ligand, RON2.23 Although these structures have enabled much of the foregoing drug-discovery effort, no small-molecules have been crystallised successfully with AMA1 so far, which has significantly impeded the further development of the hits identified to date.The PfAMA1 sequence is relatively polymorphic, with variation being driven by pressure from the host immune system. These polymorphisms are clustered on a single face of PfAMA1. As a result, the potent R1 peptide and inhibitory antibodies are strongly strain-specific; for example R1 binds 3D7 AMA1 with a KD of 100 nM, but to the divergent FVO AMA1 with a KD of ≥500 μM.64 Since our goal was to develop a strain-transcending inhibitor of AMA1, we sought to determine if the sequence divergence between these two strains results in structural change affecting binding. Accordingly the X-ray crystal structure of FVO AMA1 was obtained and compared with previous 3D7 structures.64 Overall the structures were highly similar and suggested that the strain-specificity of R1 and other inhibitors reflects the loss of a few key interactions rather than any significant structural change. Unlike two published structures of 3D7 PfAMA1 (PDB: 1Z40)22 and 1F9–3D7 PfAMA1 complex (PDB: 2Q8A),33 where the DII loop is partially ordered, the DII loop in FVO PfAMA1 is completely disordered in this structure.64 This difference appears to be a consequence of the distinct crystallisation conditions or crystal contacts, rather than any intrinsic difference in the flexibility of the DII loop. Indeed, the flexibility of the DII loop is associated with a conformational change that allows interaction between AMA1 and its endogenous partner RON2,23 and is therefore likely to be essential for AMA1 function.65
19F NMR spectroscopy has proven to be a valuable method for probing ligand-induced conformational changes and has found utility with a number of biologically relevant protein targets.66,67 Our group has utilised 19F NMR spectroscopy to detect ligand-induced movement of the DII loop of AMA1.44 We achieved this by mutating Phe367, near the centre of the DII loop to Trp (Fig. 4A) and replacing the introduced and native Trp residues with 5-fluorotryptophan (5-F-Trp) by expression in the presence of glyphosphate. The resulting 5-F-Trp F367W AMA1 was correctly folded and bound RON2L and R1 with affinities that compared well with those of wild-type AMA1. Additional Trp-to-Phe mutants were used to assign the four native Trp residues in AMA1, each of which lies outside the DII loop (Fig. 4A). The 19F NMR spectrum of the mutant F367W reveals five signals, corresponding to the four native Trp residues in AMA1, and the Trp introduced at residue 367 (Fig. 4B). The linewidth of the signal from Trp367 is more than twice that of the native AMA1 Trp signals, indicating the presence of conformational exchange centred on the DII loop, consistent with evidence from NMR46 and X-ray crystallography64 that the DII loop is quite flexible.
 | ||
| Fig. 4 19F NMR as a probe of ligand binding to AMA1. (A) The structure of AMA1 (domains I and II; PDB: 2Z8V)32 showing the RON2 binding site (green), the DII loop (blue), the site of the introduced 5-F-Trp367 (red) and the four native Trp residues (yellow); (B) 19F NMR spectrum of 5-F-Trp labelled F367W AMA1 (blue) contains signals from the four Trp residues in wt AMA1, plus an additional broad signal attributed to the introduced 5-F-Trp367. Deconvolution of the spectrum as the sum of five Lorentzian signals is shown (grey). Addition of the peptide ligands R1 (green) or RON2L (purple) causes the 5-F-Trp367 resonance to become sharp. A series of aminothiazole fragments at 0 (blue), 1 (purple) and 3 mM (red) bind to AMA1 and cause concentration-dependent sharpening of the 5-F-Trp367 resonance. Samples contained ∼100 μM 5-F-Trp F367W AMA1 in 20 mM sodium phosphate, pH 7.4, and spectra were recorded at 25 °C and a 19F frequency of 564 MHz without proton decoupling. Figure modified from Norton et al.67 | ||
In the presence of the peptide ligands RON2L and R1, a significant change in the 19F NMR of 5-F-Trp-AMA1[F367W] was observed, with the 5-F-Trp367 undergoing a 0.2 ppm change in chemical shift and a 2.5-fold decrease in linewidth, to become the sharpest peak in the spectrum (Fig. 4B). We interpret this as a significant increase in the flexibility of W367 relative to the rest of AMA1, consistent with the DII loop being displaced from the hydrophobic cleft and becoming highly flexible, as inferred from the crystallographic results. We then tested the effect of members of our fragment series on the 5-F-Trp-AMA1[F367W] spectrum.44 Of three series tested, one caused concentration-dependent sharpening of the 5-F-Trp 367 resonance, similar to that caused by the peptide ligands (Fig. 4D). This result suggests that members of this series of elaborated fragments displace the DII loop in much the same way as the peptide ligands do. In contrast, members of two other series caused no change to the 5-F-Trp-AMA1[F367W] spectrum, despite competing with R1 and therefore binding in the hydrophobic cleft. This suggests that these molecules bind at a distinct sub-site within the cleft, distal from the DII loop, and accordingly do not displace the loop. Thus, this approach has the potential to distinguish amongst the several potential binding sites along the length of the cleft, and to reveal the structural mode of action of AMA1–RON2 inhibitors.
To further our understanding of the effects of polymorphism in PfAMA1, the backbone NMR resonance assignments of two P. falciparum strains 3D7 and FVO were obtained by NMR spectroscopy.46 The spectra of FVO PfAMA1 were of higher quality than 3D7, reflecting differences in sample stability and optimal solution conditions for the two proteins. The spectral differences may also reflect differences in conformational dynamics, with 3D7 AMA1 experiencing more extensive line-broadening due to intermediate timescale conformational exchange. Approximately 84% of expected backbone resonances were assigned in FVO AMA1, compared with the less than 50% assignment coverage possible for 3D7. These assignments were used to map the binding site of several small molecules including thiazoles, pyrazoles and benzimidazoles. Of these, three benzimidazoles gave small chemical shift perturbations in 2D [1H–15N]-TROSY experiments and showed binding to a region near the hydrophobic cleft at the tetrapeptide region Lys177, Asp178, Gly179 and Gly180. This information will help guide future development of high-affinity ligands as PfAMA1 inhibitors. Our current research is utilising these findings to further elaborate the 2-phenylbenzimidazole fragment hits.
Conclusion and future prospects
The rise of resistance to all antimalarial drugs creates an urgent need for antimalarials acting on different biological targets such as AMA1. Although, drug discovery targeting AMA1 has its challenges, especially with the lack of structures of small molecule complexes, insights via various biophysical techniques promise to guide the development of small molecules targeting AMA1. Drug discovery efforts have included fragment and high-throughput screening, fragment elaboration and interrogation by SPR and NMR, RON2 peptidomimetic synthesis and computational approaches. The conserved nature of the hydrophobic cleft across AMA1 strains and its essentiality to the parasite continue to provide a strong impetus to develop inhibitors of the AMA1–RON2 interaction in order to block invasion of erythrocytes and help counter the rising burden of resistance to known drugs. The growing wealth of information summarised in this review regarding the structure, function and inhibition of AMA1 will aid in a better understanding of this important drug target.Acknowledgements
This work was supported in part by an Australian National Health and Medical Research Council (NHMRC) project grant (1098884). R. S. N. acknowledges fellowship support from the NHMRC. The authors thank Prof. Robin Anders, Assoc. Prof. Martin Scanlon and Dr Sheena McGowan for numerous helpful discussions.References
- World Health Organization, World Malaria Report 2015, 2015, pp. 1–157 Search PubMed.
- S. Bhatt, D. J. Weiss, E. Cameron, D. Bisanzio, B. Mappin, U. Dalrymple, K. E. Battle, C. L. Moyes, A. Henry, P. A. Eckhoff, E. A. Wenger, O. Briët, M. A. Penny, T. A. Smith, A. Bennett, J. Yukich, T. P. Eisele, J. T. Griffin, C. A. Fergus, M. Lynch, F. Lindgren, J. M. Cohen, C. L. J. Murray, D. L. Smith, S. I. Hay, R. E. Cibulskis and P. W. Gething, Nature, 2015, 526, 207–211 CrossRef CAS PubMed.
- B. Aneja, B. Kumar, M. A. Jairajpuri and M. Abid, RSC Adv., 2016, 6, 18364–18406 RSC.
- RTS, S Clinical Trials Partnership, PLoS Med., 2014, 11, e1001685 Search PubMed.
- RTS, S Clinical Trials Partnership, Lancet, 2015, 386, 31–45 CrossRef.
- R. Gosling and L. von Seidlein, PLoS Med., 2016, 13, e1001994 CrossRef PubMed.
- A. F. Cowman and B. S. Crabb, Cell, 2006, 124, 755–766 CrossRef CAS PubMed.
- M. G. Peterson, V. M. Marshall, J. A. Smythe, P. E. Crewther, A. Lew, A. Silva, R. F. Anders and D. J. Kemp, Mol. Cell. Biol., 1989, 9, 3151–3154 CrossRef CAS PubMed.
- M. J. Blackman, Mol. Biochem. Parasitol., 2001, 117, 11–25 CrossRef CAS PubMed.
- S. Besteiro, J. Poncet, J.-F. Dubremetz and M. Lebrun, PLoS Pathog., 2009, 5, e1000309 Search PubMed.
- P. Srinivasan, W. L. Beatty, A. Diouf, R. Herrera, X. Ambroggio, J. K. Moch, J. S. Tyler, D. L. Narum, S. K. Pierce, J. C. Boothroyd, J. D. Haynes and L. H. Miller, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13275–13280 CrossRef CAS PubMed.
- D. T. Riglar, D. Richard, D. W. Wilson, M. J. Boyle, C. Dekiwadia, L. Turnbull, F. Angrisano, D. S. Marapana, K. L. Rogers, C. B. Whitchurch, J. G. Beeson, A. F. Cowman, S. A. Ralph and J. Baum, Cell Host Microbe, 2011, 9, 9–20 CAS.
- D. Y. Bargieri, N. Andenmatten, V. Lagal, S. Thiberge, J. A. Whitelaw, I. Tardieux, M. Meissner and R. Ménard, Nat. Commun., 2013, 4, 2552 Search PubMed.
- T. Triglia, J. Healer, S. R. Caruana, A. N. Hodder, R. F. Anders, B. S. Crabb and A. F. Cowman, Mol. Microbiol., 2000, 38, 706–718 CrossRef CAS PubMed.
- D. Giovannini, S. Späth, C. Lacroix, A. Perazzi, D. Bargieri, V. Lagal, C. Lebugle, A. Combe, S. Thiberge, P. Baldacci, I. Tardieux and R. Ménard, Cell Host Microbe, 2011, 10, 591–602 CAS.
- J. Healer, V. Murphy, A. N. Hodder, R. Masciantonio, A. W. Gemmill, R. F. Anders, A. F. Cowman and A. Batchelor, Mol. Microbiol., 2004, 52, 159–168 CrossRef CAS PubMed.
- J. Duan, J. Mu, M. A. Thera, D. Joy, S. L. Kosakovsky Pond, D. Diemert, C. Long, H. Zhou, K. Miura, A. Ouattara, A. Dolo, O. Doumbo, X.-Z. Su and L. Miller, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7857–7862 CrossRef CAS PubMed.
- J. S. Tyler, M. Treeck and J. C. Boothroyd, Trends Parasitol., 2011, 27, 410–420 CrossRef CAS PubMed.
- R. F. Anders, C. G. Adda, M. Foley and R. S. Norton, Hum. Vaccines, 2010, 6, 39–53 CrossRef CAS.
- F. J. I. Fowkes, J. S. Richards, J. A. Simpson and J. G. Beeson, PLoS Med., 2010, 7, e1000218 Search PubMed.
- D. Richard, C. A. MacRaild, D. T. Riglar, J.-A. Chan, M. Foley, J. Baum, S. A. Ralph, R. S. Norton and A. F. Cowman, J. Biol. Chem., 2010, 285, 14815–14822 CrossRef CAS PubMed.
- T. Bai, M. Becker, A. Gupta, P. Strike, V. J. Murphy, R. F. Anders and A. H. Batchelor, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12736–12741 CrossRef CAS PubMed.
- B. Vulliez-Le Normand, M. L. Tonkin, M. H. Lamarque, S. Langer, S. Hoos, M. Roques, F. A. Saul, B. W. Faber, G. A. Bentley, M. J. Boulanger and M. Lebrun, PLoS Pathog., 2012, 8, e1002755 CAS.
- D. Bargieri, V. Lagal, N. Andenmatten, I. Tardieux, M. Meissner and R. Ménard, PLoS Pathog., 2014, 10, e1004273 Search PubMed.
- M. H. Lamarque, M. Roques, M. Kong-Hap, M. L. Tonkin, G. Rugarabamu, J.-B. Marq, D. M. Penarete-Vargas, M. J. Boulanger, D. Soldati-Favre and M. Lebrun, Nat. Commun., 2014, 5, 4098 CAS.
- K. L. Harvey, A. Yap, P. R. Gilson, A. F. Cowman and B. S. Crabb, Int. J. Parasitol., 2014, 44, 853–857 CrossRef CAS PubMed.
- K. S. Harris, J. L. Casey, A. M. Coley, R. Masciantonio, J. K. Sabo, D. W. Keizer, E. F. Lee, A. McMahon, R. S. Norton, R. F. Anders and M. Foley, Infect. Immun., 2005, 73, 6981–6989 CrossRef CAS PubMed.
- S. Jones and J. M. Thornton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13–20 CrossRef CAS.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- K. Leykauf, M. Treeck, P. R. Gilson, T. Nebl, T. Braulke, A. F. Cowman, T. W. Gilberger and B. S. Crabb, PLoS Pathog., 2010, 6, e1000941 Search PubMed.
- M. Treeck, S. Zacherl, S. Herrmann, A. Cabrera, M. Kono, N. S. Struck, K. Engelberg, S. Haase, F. Frischknecht, K. Miura, T. Spielmann and T. W. Gilberger, PLoS Pathog., 2009, 5, e1000322 Search PubMed.
- K. A. Henderson, V. A. Streltsov, A. M. Coley, O. Dolezal, P. J. Hudson, A. H. Batchelor, A. Gupta, T. Bai, V. J. Murphy, R. F. Anders, M. Foley and S. D. Nuttall, Structure, 2007, 15, 1452–1466 CrossRef CAS PubMed.
- A. M. Coley, A. Gupta, V. J. Murphy, T. Bai, H. Kim, R. F. Anders, M. Foley and A. H. Batchelor, PLoS Pathog., 2007, 3, 1308–1319 CAS.
- M. L. Tonkin, M. Roques, M. H. Lamarque, M. Pugnière, D. Douguet, J. Crawford, M. Lebrun and M. J. Boulanger, Science, 2011, 333, 463–467 CrossRef CAS PubMed.
- G. Wang, C. A. MacRaild, B. Mohanty, M. Mobli, N. P. Cowieson, R. F. Anders, J. S. Simpson, S. McGowan, R. S. Norton and M. J. Scanlon, PLoS One, 2014, 9, e109674 Search PubMed.
- K. S. Harris, J. L. Casey, A. M. Coley, J. A. Karas, J. K. Sabo, Y. Y. Tan, O. Dolezal, R. S. Norton, A. B. Hughes, D. Scanlon and M. Foley, J. Biol. Chem., 2009, 284, 9361–9371 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- P. J. Hajduk, J. R. Huth and S. W. Fesik, J. Med. Chem., 2005, 48, 2518–2525 CrossRef CAS PubMed.
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Nat. Rev. Drug Discovery, 2012, 11, 873–886 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 147–149 CrossRef CAS PubMed.
- B. C. Doak, C. J. Morton, J. S. Simpson and M. J. Scanlon, Aust. J. Chem., 2013, 66, 1465–1472 CrossRef CAS.
- S. S. Lim, C. O. Debono, C. A. MacRaild, I. R. Chandrashekaran, O. Dolezal, R. F. Anders, J. S. Simpson, M. J. Scanlon, S. M. Devine, P. J. Scammells and R. S. Norton, Aust. J. Chem., 2013, 66, 1530–1536 CrossRef CAS.
- X. Ge, C. A. MacRaild, S. M. Devine, C. O. Debono, G. Wang, P. J. Scammells, M. J. Scanlon, R. F. Anders, M. Foley and R. S. Norton, J. Med. Chem., 2014, 57, 6419–6427 CrossRef CAS PubMed.
- S. M. Devine, M. D. Mulcair, C. O. Debono, E. W. W. Leung, J. W. M. Nissink, S. S. Lim, I. R. Chandrashekaran, M. Vazirani, B. Mohanty, J. S. Simpson, J. B. Baell, P. J. Scammells, R. S. Norton and M. J. Scanlon, J. Med. Chem., 2015, 58, 1205–1214 CrossRef CAS PubMed.
- B. Krishnarjuna, S. S. Lim, S. M. Devine, C. O. Debono, R. Lam, I. R. Chandrashekaran, G. Jaipuria, H. Yagi, H. S. Atreya, M. J. Scanlon, C. A. MacRaild, P. J. Scammells and R. S. Norton, J. Mol. Recognit., 2016, 29, 281–291 CrossRef CAS PubMed.
- P. Srinivasan, A. Yasgar, D. K. Luci, W. L. Beatty, X. Hu, J. Andersen, D. L. Narum, J. K. Moch, H. Sun, J. D. Haynes, D. J. Maloney, A. Jadhav, A. Simeonov and L. H. Miller, Nat. Commun., 2013, 4, 2261 Search PubMed.
- E. Pihan, R. F. Delgadillo, M. L. Tonkin, M. Pugnière, M. Lebrun, M. J. Boulanger and D. Douguet, J. Comput.-Aided Mol. Des., 2015, 29, 525–539 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- G. Wang, N. Drinkwater, D. R. Drew, C. A. MacRaild, D. K. Chalmers, B. Mohanty, S. S. Lim, R. F. Anders, J. G. Beeson, P. E. Thompson, S. McGowan, J. S. Simpson, R. S. Norton and M. J. Scanlon, J. Mol. Biol., 2016, 428, 3986–3998 CrossRef CAS PubMed.
- E. F. Lee, S. Yao, J. K. Sabo, W. D. Fairlie, R. A. Stevenson, K. S. Harris, R. F. Anders, M. Foley and R. S. Norton, Biopolymers, 2011, 95, 354–364 CrossRef CAS PubMed.
- J. Seidler, S. L. McGovern, T. N. Doman and B. K. Shoichet, J. Med. Chem., 2003, 46, 4477–4486 CrossRef CAS PubMed.
- B. J. Davis and D. A. Erlanson, Bioorg. Med. Chem. Lett., 2013, 23, 2844–2852 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- P. N. Mortenson and C. W. Murray, J. Comput.-Aided Mol. Des., 2011, 25, 663–667 CrossRef CAS PubMed.
- S. M. Devine, S. S. Lim, I. R. Chandrashekaran, C. A. MacRaild, D. R. Drew, C. O. Debono, R. Lam, R. F. Anders, J. G. Beeson, M. J. Scanlon, P. J. Scammells and R. S. Norton, Med. Chem. Commun., 2014, 5, 1500–1506 RSC.
- T. Spangenberg, J. N. Burrows, P. Kowalczyk, S. McDonald, T. N. C. Wells and P. Willis, PLoS One, 2013, 8, e62906 CAS.
- O. Korb, T. Stutzle and T. E. Exner, J. Chem. Inf. Model., 2009, 49, 84–96 CrossRef CAS PubMed.
- C. R. Collins, C. Withers-Martinez, F. Hackett and M. J. Blackman, PLoS Pathog., 2009, 5, e1000273 Search PubMed.
- F. Li, A. Dluzewski, A. M. Coley, A. Thomas, L. Tilley, R. F. Anders and M. Foley, J. Biol. Chem., 2002, 277, 50303–50310 CrossRef CAS PubMed.
- A. Alam, Malar. Res. Treat., 2014, 2014, 642391 Search PubMed.
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed.
- A. M. Coley, K. Parisi, R. Masciantonio, J. Hoeck, J. L. Casey, V. J. Murphy, K. S. Harris, A. H. Batchelor, R. F. Anders and M. Foley, Infect. Immun., 2006, 74, 2628–2636 CrossRef CAS PubMed.
- S. S. Lim, W. Yang, B. Krishnarjuna, K. K. Sivaraman, I. R. Chandrashekaran, I. Kass, C. A. MacRaild, S. M. Devine, C. O. Debono, R. F. Anders, M. J. Scanlon, P. J. Scammells, R. S. Norton and S. McGowan, Biochemistry, 2014, 53, 7310–7320 CrossRef CAS PubMed.
- R. F. Delgadillo, M. L. Parker, M. Lebrun, M. J. Boulanger and D. Douguet, PLoS One, 2016, 11, e0144764 Search PubMed.
- K. E. Arntson and W. C. K. Pomerantz, J. Med. Chem., 2016, 59, 5158–5171 CrossRef CAS PubMed.
- R. S. Norton, E. W. W. Leung, I. R. Chandrashekaran and C. A. MacRaild, Molecules, 2016, 21, 860 CrossRef PubMed.
Footnote |
| † The authors declare no competing interests. |
| This journal is © The Royal Society of Chemistry 2017 |