
Hypoxia inducible factor down-regulation, cancer and cancer stem cells (CSCs): ongoing success stories
Anthony R.
Martin
 a,
Cyril
Ronco
a,
Luc
Demange
*abc and
Rachid
Benhida
*a
a,
Cyril
Ronco
a,
Luc
Demange
*abc and
Rachid
Benhida
*a
aUniversité Côte d'Azur, CNRS, Institut de Chimie de Nice UMR 7272 - 06108 Nice, France. E-mail: luc.demange@unice.fr; rachid.benhida@unice.fr; Fax: +33 4 92076189; Tel: +33 4 92076143
bUFR des Sciences Pharmaceutiques, Université Paris Descartes, Sorbonne Paris Cité, 4 avenue de l'Observatoire, Paris Fr-75006, France
cUFR Biomédicale des Saints Pères, 45 rue des Saints Pères, Paris Fr-75006, France
First published on 19th October 2016
Abstract
In cancers, hypoxia inducible factor 1 (HIF-1) is an over-expressed transcription factor, which regulates a large set of genes involved in tumour vascularization, metastases, and cancer stem cells (CSCs) formation and self-renewal. This protein has been identified as a relevant target in oncology and several HIF-1 modulators are now marketed or in advanced clinical trials. The purpose of this review is to summarize the advances in the understanding of its regulation and its inhibition, from the medicinal chemist point of view. To this end, we selected in the recent literature relevant examples of “hit” compounds, including small-sized organic molecules, pseudopeptides and nano-drugs, exhibiting in vitro and/or in vivo both anti-HIF-1 and anti-tumour activities. Whenever possible, a particular emphasis has been dedicated to compounds that selectively target CSCs.
 Anthony R. Martin | Anthony R. Martin received his PhD from the University of Montpellier in 2011 under the supervision of Prof. Michael Smietana and Dr. Jean-Jacques Vasseur, working on the synthesis and self-assembly of borono(oligo)nucleotides. He then joined Prof. Steven Nolan's group (Saint-Andrews University) as a postdoctoral researcher to work on late transition metal coordination and their applications in homogeneous catalysis. In 2013, he moved to the Institute of Chemistry of Nice in Dr. Benhida's group where he was appointed Chargé de Recherche—CNRS in 2014. His researches focus on the design of new chemotherapeutics and encompass various aspects of nucleic acid chemistry. |
 Cyril Ronco | Cyril Ronco received his PhD on cholinesterases ligands under the supervision of Prof. P.-Y. Renard at the University of Rouen. He was then awarded with a Humboldt postdoctoral fellowship to work on the total synthesis of complex natural products in Prof. H.-D. Arndt group at Jena University, Germany. After working on medicinal chemistry projects in Prof. Déprez's group (University of Lille), he joined in 2014 the group of Dr. Benhida as “Maître de Conférences” at the University of Nice and his research focuses currently on the development of bioactive molecules to circumvent resistance mechanisms in the field of oncology. |
 Luc Demange | Luc Demange is a medicinal chemist, graduated from “Ecole Nationale Supérieure de Chimie de Paris”, who completed his PhD working on peptidyl-prolyl isomerases inhibitors (CEA/Saclay, 2001). He was post-doctoral fellow in Sherbrooke (synthesis of cardiac glycosides) then in Montpellier (design of growth hormone secretagogues). In 2006, he was recruited as professor assistant (Université Paris Descartes), and he developed with Prof. Christiane Garbay series CDKs-inhibitors and anti-angiogenic drugs with applications in cancer. In 2013, LD moved to the “Institut de Chimie de Nice” where his research interests are focused on the synthesis of new therapeutic agents able to overcome drug resistances. |
 Rachid Benhida | Rachid Benhida is DR CNRS, Co-Director of the ICN Institute and leader of Bioactive Molecules team. He received his Ph.D. from the University-Paris XI (1992). After two post-doc (Hoechst/Roussel-Uclaf, 1992–93, and “Institut Curie-Paris”, 1994), he joined the “Institut de Chimie des Substances Naturelles” as “ANRS young researcher award” and as “Chargé de Recherche” in the same year (1995) and moved to University of Nice Sophia Antipolis in 2002. His current research is focused on the development of synthetic methods and their applications in nucleic acid chemistry and the development of new strategies to overcome drug resistance in oncology using small molecules. |
1. Introduction
According to the World Health Organization, the worldwide number of new cancer cases will double within the next twenty years, particularly in developing countries. Nevertheless, death rates are decreasing and life expectancy is increasing. Among the factors responsible for this apparent paradox, one can suggest a global better access to early stage diagnosis and the development of new targeted therapies. These therapies are designed to interfere selectively with key molecular targets specific to malignant phenotypes. In this context, the deprivation of oxygen, hypoxia, which is one of the characteristics of the tumour environment, is now commonly targeted by therapeutic agents such as the anti-angiogenic drugs Sutent® and Avastin®, used for the first-line treatment of metastatic renal cell carcinoma. Nevertheless, clinical trials demonstrated a failure to totally eradicate cancers, and the tumours generally relapse within few months. Therefore, there is a real need to identify new drugs and relevant therapeutic targets such as the HIF factors that regulate a variety of cellular processes. The purpose of this review is to summarize the recent advances in HIF regulation and HIF inhibition, from the medicinal chemist point of view.2. Tumour survival and aggressiveness under hypoxia
An apparent paradox is that many regions of growing tumours are poorly vascularized while they have very high needs in oxygen and nutrients due to their anarchic and rapid proliferation. This results in localized areas, known as “hypoxic niches”, where oxygen levels (pO2) are low.1 For example, in pancreatic, prostate and breast cancers, the local concentration of oxygen in these hypoxic niches might be below or close to 2.5 mmHg, hence more than ten fold below the value commonly observed in healthy tissues.2 The presence of these hypoxic niches in tumours is highly relevant for further clinical prognosis: these tissues encompass more aggressive tumour cells which are resistant to chemo- and radio-therapies due to an adaptive response mediated by about 1.0–1.5% of the humane genes (Fig. 1).3 | ||
| Fig. 1 The aggressiveness potential of hypoxic tumors and the cancer stem cells (CSCs) postulate. The tumour growth generates poorly vascularized areas resulting in the induction of an hypoxic stress. If untreated (middle panel), the hypoxic tumor cells (grey) will develop an adaptive response in three points to overcome this stress: (a) metabolic changes (yellow), (b) phenotypic changes resulting in an increase of the metastatic potential of the tumour (the epithelial mesenchymal transition, EMT, resulting in the migration of the mesenchymal cells, blue) and (c) improvement of cell dedifferentiation and promotion of cancer stem cells (CSCs, red). If treated by conventional anti-cancer agents (bottom panel), hypoxic tumour will always relapse within few months, due to the persistence of the untreated CSCs. In this two cases, the relapsed tumours are heterogeneous cells composed by a mixture of mutated cells; this lead to a poor clinical prognosis. By a marked contrast, the combination of a specific anti-CSCs therapy with conventional anti-cancer drugs or surgery, generally leads to a better outcome (top panel). | ||
This adaptive response includes:
- The metabolic reprogramming of tumour cells (yellow cells, Fig. 1). This metabolic switch (the Warburg effect) consists in the induction of ATP production through an anaerobic glycolytic pathway. It is characterized by high levels of glucose uptake, by opposition with the mitochondrial oxidative phosphorylation pathway occurring in healthy tissues.4 In addition, hypoxic tumour cells also induce the pentose phosphate pathway (PPP).5 However, recent evidences revealed the heterogeneity of this metabolic reprogramming in cancers. This induces a ‘metabolic symbiosis’, resulting in the strengthening of the adaptive response to cytotoxic agents.6
- The increase of the metastatic potential of the tumour. On the one hand, hypoxic cells produce pro-angiogenic factors (VEGF, PIGF, HGF)7 and their associated receptors and co-receptors8 leading to the tumour re-vascularization, malignant cells extravasation and dissemination in distant healthy tissues. On the other hand, hypoxia regulates phenotype changes (Epithelial Mesenchymal Transition, EMT, leading to mesenchymal cells) and induces extra-cellular matrix degradations (expression and activation of matrix metalloproteases such as MMP-2, MMP-7, MMP-9, MMP-16).9 The mesenchymal cells (blue, Fig. 1) are characterized by a specific morphology, and their phenotype is characterized by specific markers such as Snail, Slug, twist, vimentin, fibronectin, N-cadherin and also by the accumulation of β-catenin in the nucleus.10 E-cadherin, a potent tumour repressor involved in cell/cell interactions, is downregulated by transcriptional repressors (Snail, Twist-1, ZEB1/2) and post-translational receptors (Hakai).11
- The improvement of the tumour cells dedifferentiation, and the promotion of Cancer Stem Cells (CSCs, red, Fig. 1). The combination of hypoxia and inflammation are responsible for the expression of stat3, the induction of Notch, Jag, Sonic and Hedgehog signalling pathways and for epigenetic changes leading to the phenotypic reversion of cancer cells to CSCs as highlighted in neuroblastoma and breast cancer.12 This dedifferentiation can also be mediated by soluble factors (HGF, in colorectal cancer),13 or receptors (c-Met, in glioblastoma).14 This reversion is dramatic in terms of disease aggressiveness since CSCs have not only the unique capacity to initiate new tumours in distant healthy tissues, but also to develop specific resistances to conventional anti-cancer therapies, as recently reviewed by Sotiropoulou et al.15
In this context, a common denominator involved either in the metabolic and phenotypic changes, in the increase of tumour aggressiveness and in the CSCs promotion and maintenance is the Hypoxia Inducible Factor 1 (HIF-1).16 This protein emerged as a target in oncology several decades ago but remains nowadays a very hot topic. Indeed, approximately 2900 yearly published articles devoted to this transcription factor can be retrieved using the “HIF” keyword on the SciFinder search engine. Nevertheless, although the link between HIF-1 and CSCs have already been evidenced, only a few molecules able to specifically inhibit HIF-1 in this subtype of cancer cells have been reported.17 In this context, our first objective is to briefly summarize the mode of action of HIF-1, and its regulation in hypoxic and normoxic tissues, before reviewing the recent advances in the development of its inhibitors. To this end, we selected ongoing success stories focussed, whenever possible, on molecules with potential activities against CSCs.
3. The hypoxia inducible factor 1: a key target in cancer and cancer stem cells
3.1. The wide scope of the HIF-1 biological activities
In cancer cells, HIF-1 activates the transcription of genes (so-called hypoxia response elements, HRE), and leads as downstream consequence to metabolic switches, EMT induction, ECM changes (which includes the emission of pro-angiogenic factors), and also CSCs formation and maintenance (Fig. 2).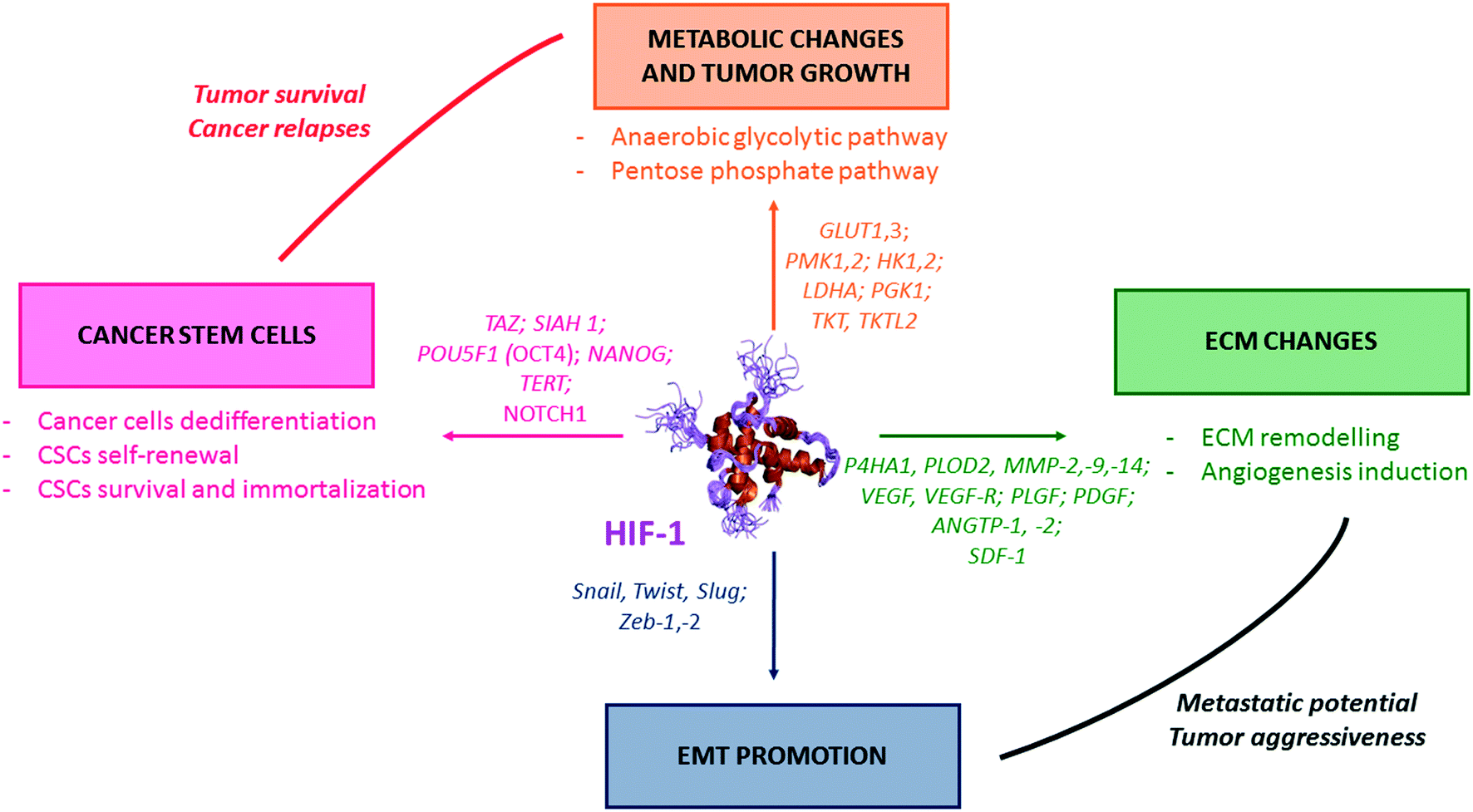 | ||
| Fig. 2 The pivotal role of HIF-1 in tumour survival and aggressiveness. HIF-1 is a transcription factor which induces the transcription a large set of genes, the hypoxia response elements (HRE). Some of them are summarized in this figure. One can divide the HRE in four different classes, and representative examples are depicted herein. (i) HRE promoting metabolic switches (orange); (ii) HRE promoting CSCs formation and self-renewal (pink); (iii) HRE promoting changes in the extra cellular matrix (ECM) (green); and (iv) HRE promoting the epithelial mesenchymal transition (EMT) (blue). | ||
- Among the solid tumours, breast cancer stem cells (BCSCs) have been extensively studied. Indeed, in this type of cancer, the conventional anti-angiogenic drugs, such as bevacizumab and sunitinib, lead to a hypoxia-related formation of BCSCs. This occurs through HIF-1 mediation, and subsequent twist expression, Akt/β-catenin and Wnt signalling pathways induction.30 Moreover, the depletion of HIF-1α gene results in reduced tumour growth and metastasis formation, and is related to the reduction of the BCSCs count.30 More recently, the role of HIF-1 in the expression and activation of TAZ, a transcriptional co-activator of the Hippo signalling pathway, which is required for CSCs maintenance in various subtypes of cancers including breast, esophageal, oral cancers and hepatocellular carcinoma, has been evidenced.31,32 In CSCs, HIF-1 also promotes the transcription of the gene encoding for the Siah E3 ubiquitin protein ligase, which is required for the degradation of large tumour suppressor kinase 2 (LATS2).32
CSCs survival in solid tumours depends, among others, on the expression of telomerases reverse transcriptase (TERT), senescence blockers (such as glucose phosphate isomerase and phosphoglycerate mutase), and specific CSCs factors such as OCT and NANOG involved in multiple signalling pathways; all of them being under the regulation of HIF-1.33,34
The relevance of HIF in improving the CSCs maintenance has been clearly underlined in a large set of cancers, including thyroid, lung, colon, and gastric cancers. Thus, in the particular case of pancreatic carcinomas (Panc-1 and BxPC-3 cell lines), the dedifferentiation of cancer cells to CSCs has been formally associated with both autophagy and high expression levels of HIF-1.34 Additionally, high levels of HIF-1 and upregulation of Akt signalling pathway have also been reported in the case of prostate CSCs. In these specific CSCs, resistance to conventional mTOR inhibitors has been related to HIF-1 expression. Moreover, HIF-1 promotes the downregulation of prostate CSCs metabolism, leading to CSCs quiescence and subsequent maintenance.35 Lastly, in glioblastoma, the pseudopalisading regions are hypoxic and necrotic niches in which CSC-related markers (CD133, CD44, Sox2,…) are overexpressed. In these niches, hypoxia promotes a synergistic effect between HIF-1 and Akt signalling pathway, leading to CSCs maintenance as downstream consequence. Moreover, in this neuronal deadly cancer, the CSCs activity is reduced when HIF-1 encoding genes are silenced by shRNA.36
- In haematological malignancies, HIF-1 stands at the crossroad of different signalling pathways involved in CSCs maintenance. Indeed, the natural antibiotic cyclopeptide echinomycin disrupts HIF-1 binding to specific DNA sequences (read below, section 3.3) and leads to spectacular results on murine models of both Acute Lymphocytic Leukaemia (ALL) and Acute Myeloid Leukaemia (AML). In ALL, this drug cured 100% of mice that have received lethal dose of leukaemia cells, even when echinomycin was used at a low-dose (10 μg kg−1).
In these haematological malignancies, a crosstalk occurs between HIF-1 and Notch signalling pathways. On the one hand, HIF-1 binds the activated NOTCH1 protein and stabilizes it, resulting in the Notch signalling pathway activation; on the other hand, HIF-1 prevents a negative feedback regulation loop by inhibiting HES1 binding to the HES1 promoter, a key Notch target involved in CSCs self-renewal. In the case of AML, echinomycin induces the selective elimination of the AML-stem cells in a xenografted mouse model and also prevents the colony-forming unit with an IC50 in the 100 pm range.37
3.2. The specificities of the three HIF isoforms
The hypoxia inducible factor is active under a heterodimeric form, as illustrated on Fig. 2, which results from the association of an oxygen-regulated subunit (HIF-α) and an oxygen independent subunit (HIF-β, also known as aryl hydrocarbon receptor nuclear translocator ARNT).38Three isoforms of HIF-α (HIF-1α, HIF-2α and HIF-3α) have been reported (Fig. 3). HIF-1α and HIF-2α share 80% of sequence homology, and exhibit close 3D structures. Structurally, they are constituted by 4 domains: a basic helix–loop–helix DNA binding domain (bHLH domain), two per-ARNT-Sim domains (PAS) involved in the HIF-heterodimer stabilization and co-activator recruitment, and the COOH-terminal transactivation domain (C-TAD).39 The latter isoform, HIF-3α, also exhibits a structure divided into four domains, but the C-TAD domain is replaced by an unusual C-terminal leucine zipper (LZIP) involved in protein–protein interactions. In addition, at least ten spliced forms of HIF-3α have been reported to date, resulting in long (among them HIF-3α1, HIF-3α2, and HIF-3α9) and short (among them HIF-3α4) variants.
 | ||
| Fig. 3 Comparison of the 3D structures of the HIF-1α/HIF-β and the HIF-2α/HIF-β heterodimers bound to DNA. Dark blue: HIF-1α; light blue: HIF-2α; red: HIF-β (ARNT); orange: the DNA double helix bound to the dimeric form of HIF. These two heterodimers have very close 3D structures, since HIF-1α and HIF-2α share 80% of sequence homology. HIF-1α/HIF-β: PDB code 4ZPR; HIF-2α/HIF-β: PDB code 4ZPK. | ||
Due to their strong similarities, it has been initially postulated that HIF-1α and HIF-2α share a common mode of regulation and also common functionalities. Nevertheless, this oversimplified model has been balanced in the last decade by growing evidences highlighting that HIF-1α and HIF-2α strongly differ in their expression and are not redundant. First of all, even if these isoforms have common target genes, such as the vascular endothelial growth factor (VEGF) or the interleukine IL-6, HIF-1α and HIF-2α are involved in distinct and specific gene expression. These differences have been exhaustively reviewed by Keith ad co-workers; nevertheless, some relevant specificities will be discussed herein.40 Among these most remarkable specificities, whereas HIF-1α is a specific regulator of the glycolytic enzymes (e.g. hexokinases HK1 and HK2), HIF-2α has been reported to specifically targets the POU transcription factor OCT-4, a stem cell marker, and the growth factor TGF-α, a ligand for the epidermal growth factor receptor. Differences have been also pointed out at the cellular level. Moreover, an in vitro study on SW480 colon cancer cell line revealed that HIF-1α is involved in tumour cell proliferation and migration, while HIF-2α activity is related to cell motility induction.41
Remarkably, in some specific cases, these two isoforms exert opposing effects. This has been demonstrated in the specific regulation of the nitric oxide production by macrophages. Whereas HIF-1α is reported to increase the levels of NO by enhancing the expression of the inducible nitric oxide synthase, HIF-2α induces the expression of arginase 1, as downstream consequence a reduction of NO production.42
By a marked contrast with the two other isoforms, little is known about HIF-3α. An overview of the current advances concerning this isoform has been provided by Ravenna and co-workers in a recent review.43 HIF-3α production is regulated by hypoxia; however, it is to note that its long variants are present in the cytoplasm and nucleus even in normoxia. Interestingly, HIF-1α and HIF-2α are involved in HIF-3α regulation. For example, it has been revealed that HIF-1α, but not HIF-2α, induces the production of two HIF-3α variants (HIF-3α2 and HIF-3α4) in the Caki-1 renal cancer cell line. It is noteworthy that HIF-3α was initially reported as a negative regulator of the transcriptional activities of HIF-1α and HIF-2α. However, HIF-3α is now considered as a mediator for distinct transcriptional hypoxia responses. Additionally, its role in the pro-angiogenesis signalling pathway, which occurs through the VE-cadherin gene repression, has been recently highlighted.43
3.3. The regulation of the HIF-1 translational activity
The regulation of HIF-1 is complex, and depends on distinct features. On the one hand, it has been recently evidenced that the expression of HIF-1 is controlled by a set of growth factors and cytokines, responsible for the induction of two main signalling pathways: PI3K/Akt and MAPK; this induction can occur both in hypoxic and normoxic tissues. On the other hand, HIF-1 levels can be balanced through the local pressure of oxygen (pO2), since the oxygen dependent HIF-1α hydroxylation leads to its degradation by the proteasome. It is noteworthy that HIF-1 stabilization requires its binding to the chaperone protein Hsp90, and also some posttranslational modifications (Fig. 4). | ||
| Fig. 4 The complex regulation of HIF-1. HIF-1 is activated by growth factors and cytokines through two main signalling pathways: PI3K/Akt (orange) and MAPK (blue). Nonetheless, in normoxic tissues, successive hydroxylations lead to (i) HIF-1 degradation by the 26S proteasome or to (ii) the inhibition of its transcriptional activity (blue arrows). Otherwise, interaction with the chaperone protein Hsp90 (for its dimerization) and co-factors such as p300/CEB (for the activation of its transcriptional activity) are also required. | ||
First of all, two prolines belonging to the NH2-terminal transactivation domain (N-TAD) of the HIF-1α subunit (Pro402 and Pro564) are hydroxylated by prolyl hydroxylases domain protein (PHD)1–3. These hydroxylations proceed in combination with the acetylation of the lysine Lys532 performed by the ARD1 acyltransferase. These structural changes allow the recruitment of HIF by the von Hippel–Lindau (VHL) tumour suppressor protein, its subsequent ubiquitination by the E3 ubiquitin protein ligase, and as a downstream consequence, its degradation by the 26S proteasome (Fig. 4 and Table 1).44
| Amino acid residue | Effector | Consequence | Ref. |
|---|---|---|---|
| Hydroxylations | |||
| Pro402, Pro564 | PHD1-3 | Recruitment by VHL and degradation | 44 |
| Asn803 | FIH-1 | Inhibition of the transcriptional activity | 45 |
| Phosphorylations | |||
| Ser641, Ser643 | MAP kinase | Increase the transcriptional activity | 49 |
| Ser668 | CDK1 | Stabilization of HIF-1α | 52 |
| Ser576, Ser657 | Plk-3 | Degradation | 53 |
| Ser551, Ser555, Ser589 | GSK-3 | Degradation | 53 |
| Acetylations | |||
| Lys532 | ARD1 | Recruitment by VHL and degradation | 44 |
| Lys674 | Not determined | Required for transcriptional activity | 113 |
In normoxic tissues, another important hydroxylation exerting a negative control on the activity of HIF-1α occurs at the asparagine Asn803. This residue is located in the COOH-terminal transactivation domain (C-TAD) of HIF-1α, and binds the coactivator p300/Creb binding protein (CBP) required for the interaction of HIF-1 heterodimer with its targeted genes (HRE). The Asn803 hydroxylation is mediated by a Fe(II)-dependant asparginyl oxydase, so-called factor inhibiting HIF-1 (FIH-1), and described as a second oxygen-dependent sensor in the HIF-response to hypoxia. Indeed, FIH-1 is a tumour suppressor which is down-regulated in several cancer tissues and its inhibition can be correlated with a poor clinical outcome. In a recent study on colorectal cancers, FIH-1 has been highlighted as a specific repressor of the HIF-1α-induced transcription of GLUT-1 and VEGF.45
Under hypoxia, the post-translational changes described above do not occur. Consequently, the stability of HIF-α is improved, and its transcriptional activity might be activated.
Several interactions between HIF-1α and partner proteins are also mandatory for its activation. First of all, the conformational changes induced by the chaperone protein Hsp90 are required for the binding of HIF-1α to HIF-β, and for their further dimerization; moreover, inhibition of Hsp90 leads to HIF-1α ubiquitination and subsequent proteasomal degradation.46 Conversely, it is noteworthy that the formation of a ternary complex between HIF-1α, the murine double minute 2 (MDM2) and the tumour suppressor factor p53 induces the ubiquitination of the former and its destruction by the proteasome.47
Specific phosphorylations on HIF-1α serine residues are required for its heterodimerization with HIF-β (Fig. 4 and Table 1). Indeed, a pioneering work suggested that the HIF-1α dephosphorylated form is bound by the tumour repressor p53, and leads consequently to apoptosis.48 Nonetheless, more recent studies evidenced that the stability of HIF-1α depends on a complex balance between the different phosphorylation states of its serine residues (Table 1).
On the one hand, HIF-1α undergoes two p42 and p44 mitogen-activated kinase (MAPKs) mediated phosphorylations on its serine residues Ser641 and Ser643.49 These phosphorylations allow the interaction between HIF-1α and Pin-1, a peptidyl-prolyl isomerase. This interaction induces conformational changes, which result in an enhancement of HIF-1α stabilization and in an increase of its transcriptional activity.50 Moreover, the silencing or the inhibition of Pin-1 reduced in vivo tumour growth and disrupt angiogenesis in correlation with the down-regulation of HIF-1 activity. This pivotal role of Pin-1 has been recently highlighted by Han and co-workers.51 In addition, the cyclin-dependent kinase 1 phosphorylates the serine residue Ser668, and also contributes to the stabilization of HIF-1α.52
Conversely, the phosphorylations mediated by the Polo-like kinase 3 (plk-3), which occur on Ser576 and Ser657, and by the glycogen synthase kinase 3β (GSK-3) (on residues Ser551, Ser555 and Ser589) destabilize HIF-1α and lead to its degradation.53
The PI3K/Akt pathway increases HIF-1α translation through its downstream target, the mammalian target of rapamycin (mTOR). This serine/threonine kinase is one of the main mediators of cellular division and proliferation mechanisms, and its activation is related to tumour growth in several cancer types. mTOR forms two distinct complexes, the rapamycin-sensitive mTOR complex 1 (mTORC1), encompassing the regulatory-associated protein of mTOR (Raptor), and the rapamycin-insensitive mTOR complex 2 (mTORC2), encompassing the rapamycin-insensitive companion of mTOR, (Rictor). These two complexes exert specific functional activities, but HIF-1α production is only activated by mTORC1, through the phosphorylation of the mRNA cap binding protein eukaryotic initiation factor 4E (eIF4E), and of the ribosomal protein S6 kinase (S6K).54
The MAPK (Raf-MEK-ERK) signalling pathway exerts a dual activity on HIF-1. Firstly, ERK enhances HIF-1α biosynthesis. On the one hand, this kinase improves the levels of eIF4E, by phosphorylation of the translational repressor eukaryotic initiation factor 4E binding protein (4E-BP5); one the other hand, ERK phosphorylates the ribosomal protein S6K, a stimulator of HIF-1α translation. Secondly, ERK phosphorylates the transcriptional co-activator p300, and therefore acts in favour of its binding with HIF-1α. This interaction leads to the subsequent activation of HIF-1α transactivation-domain function.55
Interestingly, it has recently been demonstrated that the use of Ginsenoside Rg3, a natural saponin extracted from ginseng, downregulates HIF-1α expression and subsequent angiogenesis through the common inhibition of both PI3K/Akt and Raf-MEK-ERK signalling pathways. In vitro, this extract enhances the radiosensitization of oesophageal cancer cell lines.56
The PI3K signalling pathway has been identified as a main activator of HIF-1 under normoxia. It induces accumulation of HIF-1α through mTOR activation. In normoxic tissues, activation of HIF-1 is also correlated with genetic alterations, such as inactivating mutations in mitochondrial enzymes, which purposes are on the one hand to enhance HIF transcription, and on the other hand to improve HIF-1 stability.58 For example, it has been observed in glioma that specific mutations in PHD and/or its regulators lead to HIF-1 stabilisation; otherwise, it has been evidenced by Nakaota and co-workers that Mit3 destabilizes FIH, thus positively regulates HIF-1. Moreover, VHL expression is downregulated in a specific subset of leukaemia stem cells (c-Kit+Sca-1+).59 Lastly, little is known about the methylation–demethylation cycles of HIF-1α, nevertheless a recent report on this topic underpinned the relevance of the SET7/9 methyltransferase and the lysine-specific demethylase 1 for further development of molecular agents able to tackle HIF-1 activities in cancers.60
To summarize, the activation and stabilization of HIF-1 depends not only on pO2, but also on growth factor-induced signalling pathways; they embody numerous opportunities to the medicinal chemist who aims at designing small-sized molecules able to interact/inhibit them.
4. Strategies to downregulate HIF-1 activity with pharmaceuticals
As discussed above, HIF-1 is an indisputable target in oncology, and therefore remarkable efforts in the discovery of synthetic or natural HIF-1 inhibitors have been sustained in the last decades (Fig. 5). These inhibitors can be divided in three distinct classes, depending on their way-of-action. In fact, these molecules can affect (i) HIF-1 mRNA levels and HIF-1 protein synthesis; (ii) HIF-1 stability and dimerization; (iii) HIF-1 binding to its targeted genes. | ||
| Fig. 5 Three strategies to downregulate HIF-1. These three strategies include (i) the inhibition of HIF-1α biosynthesis (green); (ii) its destabilization leading to its proteasomal degradation (blue); and (iii) the inhibition of its transcriptional activity (orange). | ||
Several therapeutic agents belonging to each class have been approved or are in preclinical stage; representative examples are discussed below. Nevertheless, it is noteworthy that very few HIF inhibitors have been specifically designed and tested on CSCs, in fact many of them have been discovered before the emergence of the CSCs concept. In this context, we decided to particularly highlight the molecules with potential applications against CSCs.
4.1. Therapeutic agents targeting HIF mRNA level and HIF biosynthesis
Blockers of HIF biosynthesis can be divided in five classes: (i) Akt/mTOR signalling pathway inhibitors; (ii) topoisomerase I inhibitors; (iii) molecules targeting HIF mRNA; (iv); molecules targeting the microtubule network and (v) miscellaneous compounds. | ||
| Fig. 6 Representative examples of anti-HIF therapeutic agents targeting the Akt/mTOR signalling pathway: selected PI3K inhibitors. | ||
 | ||
| Fig. 7 Representative examples of anti-HIF therapeutic agents targeting the Akt/mTOR signalling pathway: mTOR inhibitors. | ||
Phosphatidylinositol 3-kinase (PI3K) inhibitors form a large family of Akt/mTOR blockers, which currently includes more than 15 molecules in clinical trials (Fig. 6).62 Wortmannin, a natural product isolated from Penicillium wortmanni, and LY294002 are two “first generation” PI3K inhibitors that have been developed by Eli-Lilly.63 These molecules exert a very potent activity against PI3K, even at a low level (IC50 = 1 nM and 1.4 μM respectively), and affect in a dose dependent manner HIF-1α levels. Moreover, recent studies have highlighted the potential use of LY294002 against BCSCs.64 Indeed, LY294002 has the ability to reverse multidrug resistance in these cells by interfering with the three major ATP-binding cassette transporters: BCRP-1 (breast cancer resistance protein-1), P-glycoprotein and MRP-1 (multidrug resistance-associated protein-1). In addition, LY294002 inhibits the proliferation of BCSCs in hypoxic microspheres and downregulates the biosynthesis of HIF-2α. Nevertheless, these two pioneering molecules exhibit a strong toxicity in mice. For instance, at the daily dose of 1 mg kg−1, Wortmannin kills 50% of the treated mice. This toxicity has strongly restrained the potential clinical use of these two PI3K inhibitors in humans.65
Among the PI3K inhibitors that emerged in the last decade, one should mention NVP-BEZ235 (Dactolisib), an ATP competitive inhibitor. It interacts with different PI3K isoforms with IC50 ranging from 4–7 nM in cell-free assays, suppresses HIF-1α expression, induces autophagy in tumour cells and exhibits strong anti-proliferative activities characterized by cell cycle arrest in G1 phase. Importantly, NVP-BEZ235 is also an inhibitor of the downstream kinase mTOR (IC50 = 6 nM).
This drug is active on different models of mice xenografted with human tumours (gliomas, pancreatic, sarcoma, ovarian, renal, breast, hepatocellular carcinoma), when used alone or in combination with other conventional anti-cancer agents.66 NVP-BEZ235 reverses resistance to other anti-cancer agents in pancreatic neuroendocrine and non-small cell lung cancers.67 Moreover, this drug enhances in vitro the radio-sensitivity of human glioma stem cells, and also inhibits CSCs proliferation in the CD133+ and CD44+ HTC-116 colon cancer lines. Lastly, NVP-BEZ235 also improves the cytotoxic effect of paclitaxel on colorectal CSCs (human SW620 cancer cell line).68
This drug entered for the first time in clinical trials phase I in December 2006 for the treatment of advanced solid tumors (NCT00620594), and in phase II in October 2011. To date, at least ten phase II trials have been launched for the use of NVP-BEZ235 in monotherapy or in combination with other therapeutic agents against solid tumors (including advanced renal cell carcinoma, NCT01453595, advanced breast cancers, NCT01471847, metastatic and castration-resistant prostate cancers, NCT01717898, advanced pancreatic neuroendocrine tumors, NCT01628913). However, most of their results remain unpublished, yet (https://clinicaltrials.gov).
Nevertheless, it is noteworthy that the first-in-class PI3K inhibitor remains to date Idelalisib (CAL-101). The use of this orally bioavailable drug has been approved in 2014 in United States and European Union for the treatment of various leukaemia (Chronic lymphocytic leukaemia, small lymphocytic leukaemia, and follicular lymphoma).62
Another target of interest in the Akt/mTOR cascade is the final serine/threonine kinase mTOR (mammalian target of rapamycin). Its natural ligand, rapamycin (sirolimus), downregulates HIF-1α levels by interfering with Akt/mTOR (Fig. 7). In vitro studies have demonstrated the cytotoxic potential of rapamycin against several cancer cell lines, including glioblastoma CSCs;69 nevertheless, rapamycin used alone in vivo leads to poor clinical benefits, and should be combined with other anti-cancer agents. In this case, very potent effects have been reported: for example, its combined use with an inhibitor of the Sonic Hedgehog pathway leads to the complete CSCs eradication in mice xenografted with human pancreatic tumour in vitro and in vivo. In breast cancers, the combination of tamoxifen and rapamycin reduces the mammosphere formation and counteracts formation of CSCs.70
Among the rapamycin analogues, two molecules of interest are temsirolimus (Torisel®) and everolimus. Temsirolimus was approved in 2007 by the FDA for the treatment of advanced renal carcinomas. Indeed, the phase III clinical trials demonstrated longer overall and progression-free survival for patients treated with this molecule compared with patients treated with interferon-α, which is the standardized drug used in kidney cancers.71 Additionally, the FDA approved in 2011, the use of everolimus for the treatment of progressive and advanced pancreatic neuroendocrine tumours. This drug affects either angiogenesis, cell proliferation and survival. A phase III trial (RADIAN-4) performed on a cohort of 302 patients coming from 25 countries and suffering neuroendocrine tumours of lung or gastrointestinal origin, outlined an increased (11 months) median progression-free survival when using everolimus at a daily dose of 10 mg. Moreover, at this dose, the adverse event observed are infrequent, and include stomatitis (9% of patients), diarrhoea (7%) and infection (7%). These could be managed by adapting the dose of the drug, without changing the duration of the treatment. The long term overall survival results of this trial have not yet been reported.72 Everolimus has also been studied in other clinical trials for the treatment of a set of cancers, including of castration-resistant prostate cancer, advance cervix cancer and breast cancers.73
 | ||
| Fig. 8 Representative examples of anti-HIF therapeutic agents targeting the topoisomerases. | ||
Among the marketed topoisomerase I inhibitors, topotecan is a water soluble camptothecin analogue active against a large scope of human tumours, including resistant ovarian carcinoma, small cell and non-small cell bronchogenic carcinomas and myeloid leukaemia. Moreover, a recent study revealed that topotecan enhances the effect of cisplatin in platinum-resistant ovarian cancers. Topotecan inhibits HIF-1α expression in cell culture at low concentrations (EC50 = 54 nM); in vivo, its daily administration to U251 human glioblastoma xenografted mice results in HIF-1α inhibition, tumour growth inhibition and a marked decrease of HIF-1 target gene expression (VEGF, GLUT1…). Lastly, in a recent clinical trial involving a cohort of patients suffering advanced solid tumours, topotecan decreased HIF-1α expression.75
This molecule has been reformulated for its use as therapeutic agent for humans and vectorization by nano-objects, such as lipidic-, albumin- or micelle-based structures has been considered. For example, camptothecin has been covalently bound to a cyclodextrin-poly(ethyleneglycol) copolymer to form the nanoparticle-drug conjugate: CRLX101. It has a neutral surface charge and the poly(ethyleneglycol) moiety contributes to the improvement of solubility and stealth. This results in a better stability, prolonged circulation time in the organism, and accumulation in the malignant tissues through the enhanced permeability and retention of the tumour (also known as the passive vectorisation effect).76 Pharmacological studies have demonstrated that the release of camptothecin from this nano-conjugate leads to stronger HIF-1α inhibition and as downstream consequence to improved anti-tumor potency compared to other marketed camptothecin derivatives such as topotecan.
CRLX101 reached phase II clinical trials for the treatment of renal, ovarian and rectal cancers.77 Interestingly, its use leads to the reversion of tumour resistance to bevacizumab (conventional anti-angiogenic agent) in mice xenografted with human breast cancer cell line SUM159. In models of mice xenografted with human SUM159 cancer cells, the combined bevacizumab/CRLX101 therapy affects predominantly the BCSCs population hence resulting in tumour regression and delayed recurrence. Moreover, the administration of CRLX101 impedes the BCSCs formation induced by the conventional anti-angiogenic therapeutic agent bevacizumab. Indeed, the tumour size regresses from 200 mm3 to 35 mm3 when the mice are treated for 6 weeks with a combination therapy of 6 mg kg−1 CRLX101 + bevacizumab.78
 | ||
| Fig. 9 Representative examples therapeutic agents targeting HIF-1α expression. | ||
Another molecule of interest is an aminoflavone, issued from the prodrug AFP-464, which recently entered in phase II clinical trial. This anti-cancer agent blocks HIF-1α expression and exhibits anti-proliferative effects against MCF-7 breast tumour cell line (but does not exert any cytotoxicity against the aggressive breast cancer cell line MDA-MB-231). It is also active in neoplastic cells of renal origin. A synergistic effect has been observed in vitro on MCF-7 cell line, when this molecule is used in combination with other conventional anti-cancer agents, such as paclitaxel or camptothecin.81
It is noteworthy that there are growing evidences of the cytotoxicity of flavonoids structurally related to aminoflavone against breast and prostate CSCs.82 For example, 8-chloroflavanone and 2′,3′-dimethoxyflavanone emerged from a screening of 42 flavonoids.83 Additionally, flavopiridol, a molecule that is currently in clinical trials, has been evidenced as an apoptosis inducer (stimulation of caspase-3, caspase-8 and p53) and as an inducer of the cell cycle arrest in CSCs isolated from the DU145 human prostate CSCs. Despite these promising results obtained on structurally-related flavonoids, this aminoflavone has not been studied for its specific effects on breast CSCs, yet.84
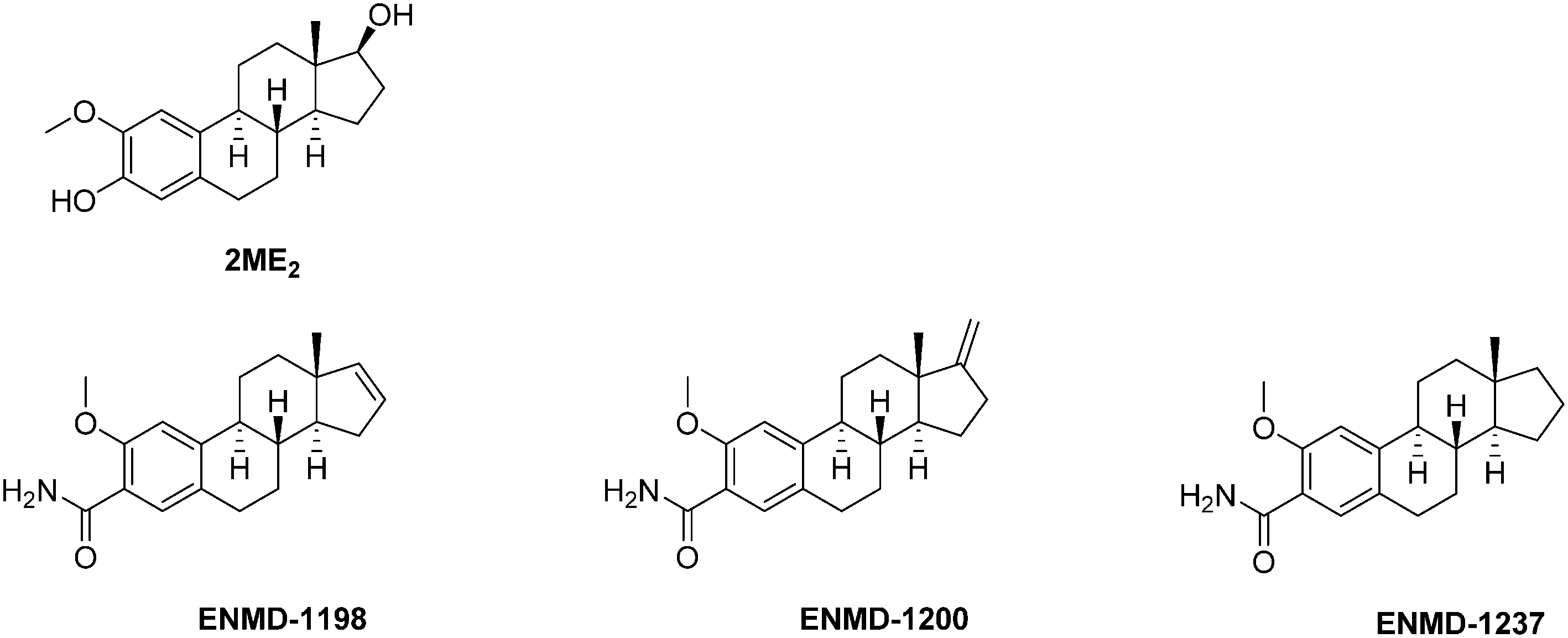 | ||
| Fig. 10 Representative examples therapeutic agents targeting the microtubule network and consequently affecting HIF-1α expression. | ||
Mechanistically, 2ME2 binds the colchicine site of tubulin. Therefore, it prevents the microtubule assembly and inhibits the tubulin polymerization, thus leading to a cell cycle arrest in metaphase. Interestingly, these effects are not correlated with the expression of the estrogen receptor at the surface of the tumour cells. It is noteworthy that 2ME2 also induces ROS formation and activates the caspase-3. As a first consequence, the combination of these different effects induce a pro-apoptotic activity against a panel of different carcinomas (gastric, nasopharyngeal and breast), and consequently, 2ME2 exerts in vitro anti-proliferative effects against a broad scope of human cancer cell lines. It includes solid tumours such as breast carcinoma (MDA-MB-23: IC50 = 1.0–1.4 μM), lung (IC50 = 0.7–5.0 μM), renal (SN12C: IC50 = 0.95 μM), melanoma (UACC62, B16BL6 murine, B16F10: IC50 = 0.3–0.4 μM), prostate (LNCaP, DU145: IC50 = 0.033–10.0 μM), pancreatic (PaTu8902, PaTu8988t, PaTu8988s: IC50 = 1.5–10.0 μM) gastric (IC50 = 1.0–10 μM and cervix cancers (HeLa, HeLaS3: IC50 = 0.001–5.0 μM), and haematological malignancies such as lymphoblast (Jurkat, TK6, WTK1: IC50 = 0.3–2.0 μM).85
The 2ME2-mediated downregulation of HIF-1α and its subsequent effects in vivo on angiogenesis were reported a decade ago. This effect proceeds downstream the binding of 2ME2 to the tubulin colchicine binding site, and results both in the disruption of HIF-1α translation and in the blockade of its nuclear translocation; 2ME2 also inhibits the synthesis of the HIF-1α induced protein.
Currently, 2ME2 is being evaluated, through several clinical trials (phase I & II), for the treatment of solid advanced and metastatic tumours; either alone or in combination with other therapeutic agents including nano-objects).85,86 This drug candidate is well tolerated by patients and exhibits a moderate toxicity at the dose of 1 g per day. Significant clinical benefits have been reported, among them the regression or the stabilization of the tumours.
In this context, the use of 2ME2 for the specific treatment of CSCs in haematological malignancies has been patented in 2011.87
Series of 2ME2 structurally-related compounds have been synthesized to optimize the durability and to unravel the structure–activity relationships. Briefly, while structural changes in the steroid rings B and C are unfavourable in terms of cytotoxicity and binding to tubulin, derivatives with improved stabilities and biological activities have been obtained by modification in rings A and D.85 Among the latter series, three 2ME2 analogs (ENMD1198, ENMD 1200 and ENMD 1237) emerged as potential drug candidates and reached the clinical trial stage against advanced and/or refractory tumours (phases I and II). The assessment of ENMD1198 in a phase I trial revealed a limited drug-related toxicity at a dose of 425 mg m−2 d−1; several patients enrolled in this trial achieved stabilisation of disease after only two treatment cycles.88
 | ||
| Fig. 11 Cardiac glycosides affecting HIF-1α expression. | ||
 | ||
| Fig. 12 PX-478 and its metabolite (melphalan). | ||
In 2008, Zhang and co-workers demonstrated, through the screening of compounds in clinical trials, that these cardiac glycosides inhibit HIF-1α and HIF-2α protein expression in human prostate cancer cell PC-3. The authors observed that, at the dose of 100 nM, digoxin reduces selectively the HIF-1α synthesis by 73%. Moreover, the same authors demonstrated that cardiac glycosides do not reduce the mRNA HIF-1α levels. However, these drugs disrupt the translation of HIF-1α mRNA by a mechanism that is independent of mTOR. Lastly, on mice xenografted with human PC-3 cells, digoxin stops the tumour growth after 7 days of treatment when administrated at a dose of 2 mg kg−1. Importantly, the authors do not report any loss of weight on treated animals.89 More recently, it has been evidenced in pancreatic ductal adenocarcinoma cells, that digoxin is a negative regulator of the stem cell factor (SCF-1). SCF-1 is a multifunctional cytokine involved in tumour growth, and expressed under HIF-1 stimulation.90 The precise inhibition mechanism remains elusive although there are some evidences that it occurs independently from the pVHL/proteasome pathway. Nevertheless, Xie and co-workers recently evidenced that these cardiac glycosides downregulate the polo-like kinase 1,91 which is involved in the phosphorylation of HIF-1α as described above (section 2.3).53
Despite their ancestral use to threat cardiac malignancies, these molecules are now outdated due to their very poor therapeutic index, and they cannot be administrated at a dose greater than 0.012 mg kg−1 (digoxin, iv. administration). Therefore, the pharmacologic profile of these agents has to be strongly optimized for their use in oncology.
PX-478 is another interesting drug candidate. This molecule exerts antitumour and pro-apoptotic effects that are correlated with low HIF-1α levels and the blockade of its targeted genes (VEGF and GLUT). Moreover, when administrated at a dose of 100 mg kg−1 per day for 5 days on mice xenografted with a panel of human tumours (SHP-77, HT-29, OvCar-3, PC-3, DU-145, MCF-7, Caki-1 and Panc-1), PX-478 shows a potent anticancer effect, in vivo. In particular, very large tumour regression (up to 70% in PC-3 prostate cancer; close to 50% in MCF-7 breast cancer) has been observed. This drug leads to a 99% tumour regression and a tumour growth delay of 57 days in the case of OvCar-3 ovarian cancer. A complete cure has been reported with SHP-77 small-cell lung cancers. In this latter case, no tumour cells were detected, at the xenograft site, 100 days after the completion of the protocol.92
Interestingly, PX-478 inhibits HIF-1α both in normoxia and hypoxia by several distinct mechanisms; however, this does not occur through the pVHL/proteasome pathway. Indeed, PX-478 mainly down-regulates levels of HIF-1α mRNA and inhibits it translation. Its second mechanism of action consists in the inhibition of HIF-1α deubiquitination.93
PX-478 completed a phase I clinical trial on 41 patients in 2010 (NCT00522652) for its use against advanced solid tumours and lymphoma. It was administrated at doses ranging from 1 mg m−2 to 88.3 mg m−2, and 35% of treated patients achieved a stable disease and a dose dependent HIF-1α inhibition. However, the pharmacokinetic studies evidenced the rapid conversion of PX-478 into melphalan, a metabolite which is an alkylating agent but that does not inhibit HIF-1α. This result has undermined the clinical development of this molecule.94
The use of nanomaterials opened a new avenue in the medicinal landscape. These new objects are characterized by specific size and surface which act in favour of their interaction with complex biological systems. Indeed, nano-sized objects are mainly used as carrier for therapeutic agents such as small-sized organic compounds or oligonucleotides.
Among these nanomaterials, the fullerene-based Gd@C82(OH)22 is characterized by the encapsulation of a gadolinium atom into an organic cage constituted by 82 carbon atoms and 22 hydroxyl groups.95 This 1 nm nanoparticle exhibits a very low toxicity for healthy tissues but exerts a potent anti-cancer activity. In fact, Gd@C82(OH)22 brought one of the first evidences of the intrinsic anti-tumour effect of certain nanodrugs.96 Recently, Liu and co-workers demonstrated that Gd@C82(OH)22 is a specific and non-toxic inhibitor of HIF-1α expression in breast cancer stem cells. Indeed, Gd@C82(OH)22 reduces EMT, eliminates CSCs under normoxia, and also inhibits, in vivo, HIF-1α expression in hypoxic CSCs. Moreover, the authors evidenced, by means of inductively coupled mass spectrometry analyses, that hypoxia promotes the Gd@C82(OH)22 uptake, thus preventing the EMT. This nanodrug reduces cell migration and invasion, and down-regulates the expression of a set of pro-angiogenic factors at the mRNA and the protein levels (VEGF, IL-6, IL-8, MMP-2 and MMP-9). It is noteworthy that, Gd@C82(OH)22 does affect neither apoptosis and necrosis, nor cell proliferation in the bulk of the tumour. Altogether, these results underline a specific cytotoxic effect towards the CSCs population.97 Due to its novelty and selectivity, one can consider that this therapeutic agent paves the way to a new area for medicine.
4.2. Therapeutic agents affecting HIF stability and its dimerization
Drug candidates able to induce the destabilization of HIF-1α and to prevent its dimerization with HIF-1β might be divided into the four following classes: (i) molecules targeting the chaperone protein Hsp90; (ii) inhibitors of histone deacetylases (HDAC); (iii) inhibitors of thioredoxin-1; and (iv) miscellaneous compounds. | ||
| Fig. 13 Representative examples of Hsp90 inhibitors exhibiting an anti-HIF-1α activity. | ||
As described above (section 2.3),46 HIF-1α binding to Hsp90 results in conformational changes which are mandatory for its subsequent interaction with HIF-1β. Moreover, antagonizing the binding of HIF-1α to Hsp90 leads to the degradation of the former in a pVHL-independant manner. Among the most recently published Hsp90 inhibitors exhibiting a demonstrated effect on HIF-1α level, one can mention small sized organic molecules such as wogonin or degueulin and curcumin derivatives,99 naturals products (e.g. glyceollinin),100 and also macromolecules such as porphyrins derivatives (e.g. protoporphyrin IX).101
The geldanamycin is a natural benzoquinone belonging to the class of the ansamycin antibiotics. It has been isolated from the bacteria Streptomyces hygroscopicus. In renal cell carcinoma and prostate cancer cells, this molecule binds the ATP binding pocket of Hsp90, and subsequently induces HIF-1α degradation.102 However, preclinical studies in dog have highlighted severe side effects. Indeed, at a dose of 2.0 mg kg−1, the geldanamycin induces acute hepatic necrosis, nephro- and gastrointestinal toxicities. Moreover, a randomized phase III study, including patients suffering gastrointestinal stromal tumours, was interrupted due to 4 deaths attributed to liver failure. This toxicity restricted the geldanamycin use in humans.98
Conversely, two structurally-related analogs of galdanamycin exerting marked cytotoxic activities against CSCs are currently in clinical trials (17-N-allylamino-17-demetoxygeldanamycin, so-called 17-AAG or tanespimycin, and 17-dimethylaminoethylamino-17-demethoxygeldanamycin, so-called 17-DMAG or alvespimycin). These two molecules, 17-AAG and 17-DMAG, are potent Hsp90 inhibitors in cell free assays, with IC50 values of 5 nM and 62 nM, respectively.
On haematological malignancies, 17-AAG selectively eliminates lymphoma CSCs both in vitro and in vivo, and supresses HIF-1α transcriptional activity in a dose dependent manner. Indeed, in vitro, 17-AAC is about 50-fold more potent against the lymphoma CSCs (IC50 = 5.6 nM) than against the non-CSCs (IC50 = 238 nM). Moreover, Annexin V immunostaining demonstrated that at the dose of 10 nM, 17-AAG exerts a pro-apoptotic effect on CSCs, while non-CSCs remains unaffected.103 On solid tumours, 17-DMAG potentiates in vitro the cytotoxicity of cisplatin against the bladder CSCs that survived to a conventional chemotherapy. Likewise, 17-AAG improves the therapeutic outcome of a systemic cisplatin-based combination chemotherapy on mice xenografted with bladder CSCs.104 It is to note that 17-AAG reached the phase III clinical trial in 2007 (BMS TIME 1, NCT00546780) for its use in monotherapy for the treatment of patients with multiple myeloma in first relapse; otherwise 17-DMAG entered in phase II clinical trial in 2008 for the treatment of breast cancer (NCT00780000).
However, 17-AAG and 17-DMAG are poorly soluble in water and are usually administrated to patients by intravenous injection in an organic solvent. Recently, while searching for alternative formulations of these drugs, Desale and co-workers reported an efficient process for the dual co-encapsulation of 17-AAG and doxorubicin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 molar ratio) into a biodegradable and biocompatible polypeptide-based nanogel. In mice xenografted with human ErbB2 BT-474 breast cancer cells, the 17-AAG/doxorubicin combination is more efficient to reduce tumour growth and to enhance animal survival when administrated in the nanogel formulation than when the two drugs are administrated separately.105 Otherwise, a recent study on mice xenografted with human hepatocellular carcinoma demonstrated the ability of 17-AAG, when encapsulated into magneto-liposomes bound to an monoclonal antibody directed toward a specific CSCs marker (CD90), to sensitize CSCs to magnetic hyperthermia through the inhibition of Hsp90.106
:6 molar ratio) into a biodegradable and biocompatible polypeptide-based nanogel. In mice xenografted with human ErbB2 BT-474 breast cancer cells, the 17-AAG/doxorubicin combination is more efficient to reduce tumour growth and to enhance animal survival when administrated in the nanogel formulation than when the two drugs are administrated separately.105 Otherwise, a recent study on mice xenografted with human hepatocellular carcinoma demonstrated the ability of 17-AAG, when encapsulated into magneto-liposomes bound to an monoclonal antibody directed toward a specific CSCs marker (CD90), to sensitize CSCs to magnetic hyperthermia through the inhibition of Hsp90.106
Ganetespib is a second generation inhibitor of Hsp90 that encompasses a resorcinol-linked triazolone structural motif (Fig. 14). In vitro cytotoxic assays demonstrated that ganetespib is 20-fold more potent than 17-AAG against a broad scope of human tumours (median IC50 values of 14 nM vs. 280 nM). This molecule binds the N-Terminal ATP-binding domain of Hsp90, and the corresponding X-ray structure has been resolved (PDB code: 3TUH, Fig. 14). The analysis of this crystal reveals at least four H-bond (involving the residues: Lys58, Asp93, Gly97 and Thr184) and a set of hydrophobic interactions (Fig. 14).
 | ||
| Fig. 14 Ganetespib: structure and binding mode. Structure of the molecule (left panel), X-ray crystal of ganetespib bound to the N-Terminal ATP binding pocket of Hsp90 (middle panel, PDB code: 3TUH) and structural features highlighting the H-bond network and the hydrophobic interactions (right panel). | ||
The most dramatic cytotoxicities have been reported against haematological malignancies (e.g. acute myeloid leukaemia, MOLM-13 cell line, IC50 = 5 nM; chronic myeloid leukaemia, KU812 cell line, IC50 = 6 nM; T cell leukaemia, CCRF-HSB-2 cell line, IC50 = 2 nM); nonetheless, melanoma (e.g. A375 cell line, IC50 = 4 nM), prostate (e.g. DU cell line, IC50 = 7 nM) and colon (e.g. HCT-15 cell line, IC50 = 8 nM) cancer cell lines are also highly sensitive to this drug.107 An in vivo study on mice xenografted with the human non-small cell lung cancer, NCI-H1975, revealed that ganetespib is maintained in tumour with t1/2 = 58.3 h, and totally supresses HIF-1α expression only 24 h after injection of a single dose (125 mg kg−1).108 Otherwise, in a human triple-negative breast cancer model (MDA-MB-231 and MDA-MB-435 xenografts), ganetespib administered at the weekly dose of 150 mg kg−1 inhibits selectively HIF-1α expression and the primary tumour growth. It also blocks the vascular invasion and the metastasis formation. Overall, in this in vivo assay, ganetespib reduced dramatically the specific population of breast CSCs from 3.5% to 1% (MDA-MB-231) and from 2.5% to less than 1% (MDA-MB-435).109
To summarize, ganetespib exhibits a better pharmacologic profile than 17-AAG. It affects angiogenesis, tumour metabolism, stem cell self-renewal, invasion and metastasis processes. Moreover, recent clinical trials on prostate cancer, hepatocellular carcinoma and non-small cell lung cancer (phase I and II) revealed that this agent is well tolerated and does not exert any acute toxicity (maximum tolerated dose of 200 mg m−2).110
Altogether, these results promote ganetespib as one of the most promising anti-cancer agent, and as consequence, this drug was assayed in a first phase III study in April 2013. This study was performed on 700 people suffering advanced NSCL cancers. Its purpose was to compare the use of ganetespib in combination with docetaxel versus docetaxel alone (NCT01798485, Galaxy 2). However, the combination therapy did not bring any clinical benefit for patients as demonstrated by the overall survival and progression-free survival, which were equivalent in the two groups of patients. It is to note that another phase III study has been launched in October 2014 for the treatment of haematological malignancies (acute myeloid leukaemia and myelodysplastic syndrome, NCT02272478).
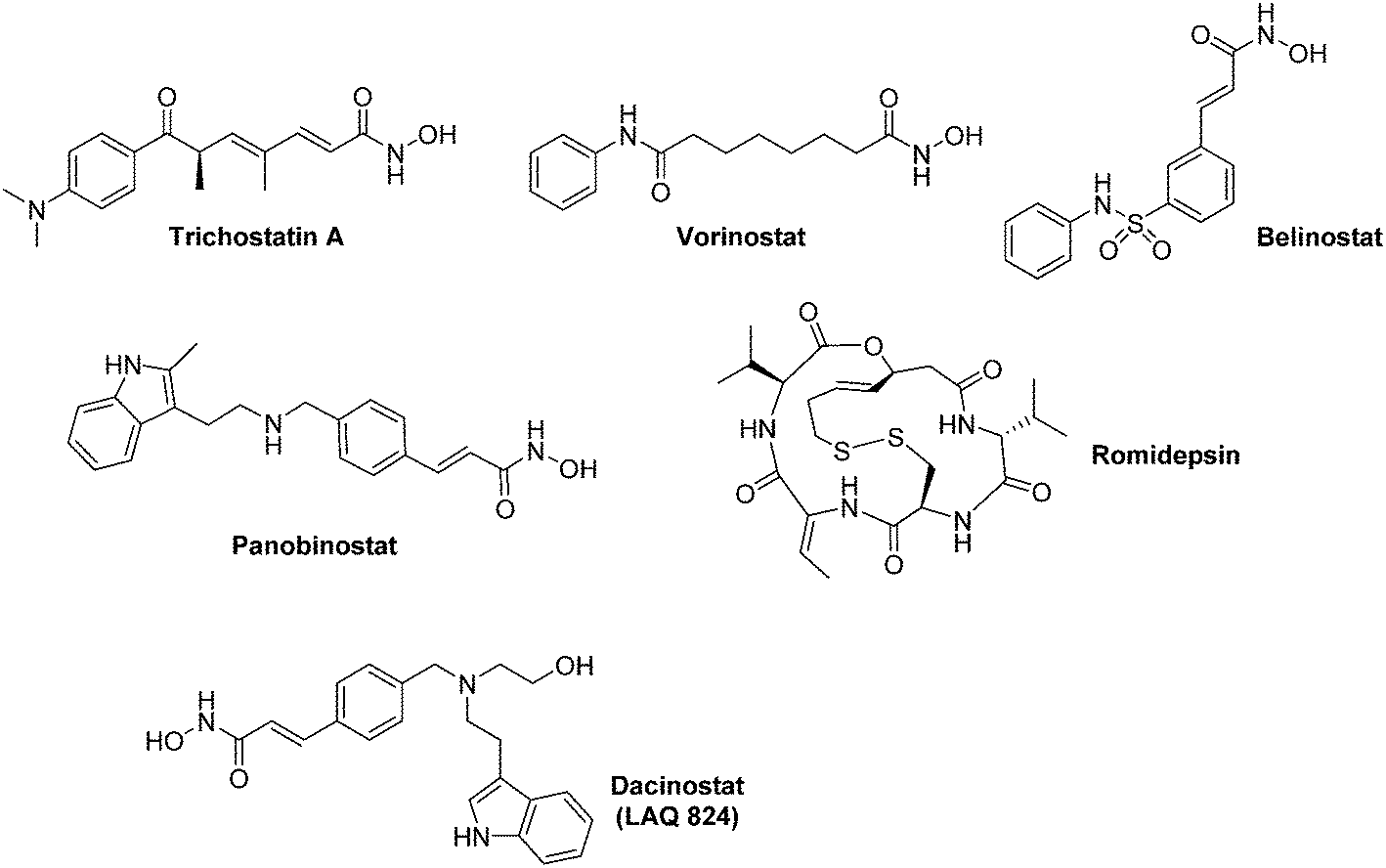 | ||
| Fig. 15 Representative examples of HDAC inhibitors exhibiting an anti-HIF-1α activity. All the compounds represented herein are marketed for their therapeutic uses, excepted Dacinostat. | ||
 | ||
| Fig. 16 Representative examples of sirtuin inhibitors exhibiting an anti-HIF-1α activity. | ||
| HDAC “Classical family” | Sirtuins | |||
|---|---|---|---|---|
| Class I | Class IIa and IIB | Class IV | Class III | |
| Name | HDAC 1, 2, 3, 8. | HDAC 4, 5, 6, 7, 9, 10. | HDAC 11 | SIRT 1, 2, 3, 4, 5, 6, 7. |
| Localization | nucleus (mainly) | IIa: nucleus and cytoplasm IIb: cytoplasm only | Nucleus (mainly) | SIRT 1, 2: cytoplasm SIRT 3, 4, 5: mitochondria SIRT 6, 7: nucleus |
| Mechanism | Zn2+ dependent | NAD+ dependent | ||
| Remarks | Trichostatin A as common inhibitor | — | ||
| Reference | 111 | 111 | 111 | 118 |
Inhibitors of the ‘classical HDAC family’ (HDACis) have been recently reviewed by Manal and co-worker.111 Among them, three hydoxamic derivatives (Vorinostat, Belinostat, and Panobinostat) and one cyclic pseudopeptide (Romidepsin) have recently been approved by the FDA for their use against haematological malignancies (Myeloma, cutaneous T cell lymphoma) (Fig. 15). HDACis exert a large scope of biological effects, including cell cycle arrest, DNA damage through oxidative stress, apoptosis induction, downregulation of tumour cell motility, up regulation of pVHL and repression of pro-angiogenic factors.
HDACis are also involved in the regulation of translational factors, including HIF-1α. To date, the precise mechanism of HIF-1α down-regulation mediated by HDACis remains elusive. However, several studies suggested that class II HDACis lead to a pVHL-independent degradation of HIF-1α.112 This can be related to abnormalities in the acetylation of both HIF-1α (Lys532, Lys674 and Lys709) and Hsp90.113 Nonetheless, a recent study on liver cancer cell lines revealed that Vorinostat down-regulates HIF-1α translation, independently from P53, prolyl hydrolases, autophagy and proteasomal degradation.114 It is noteworthy that synergistic effects have been reported in the case of solid tumours, such as non-small cell lung carcinoma and prostate cancer, when is combined with a conventional cytotoxic agent (cisplatin) or a radiotherapy with HDACi.115
In haematological malignancies, the therapeutic potential of the HDACis against CSCs has been widely evidenced. For example, Romanski and co-workers demonstrated, in a model of acute leukaemia involving primary tumour cells from patients, that the use of dacinostat (10 nM) or vorinostat (1 μM) inhibits the proliferation of leukaemia stem and progenitor cells by interfering with the transcription factor, c-MYC, and with the Polycomb group protein BMI1.116
In solid tumours such as prostate cancers, triple negative breast cancers and head and neck cancers, glioblastoma and thyroid, HDACis exhibit cytotoxic effect against CSCs when used alone or in combination with other therapeutic agents. Nonetheless, these indisputable anti-CSCs effects have not been fully related to their action on HIF-1α, yet.115,117
The sirtuin family, which has been recently reviewed by Kozako and co-workers, encompasses seven isoforms.118 Sirtuins are localized in different cellular compartments, including nucleus (SIRT 1, SIRT 2, SIRT 6 and SIRT 7), cytoplasm (SIRT 1 and SIRT 2) and mitochondria (SIRT 3, SIRT 4 and SIRT 5). Their role in cancer is still controversial. In fact, SIRT 2 and SIRT 6 are considered as tumour suppressors and are downregulated in some malignancies, whereas SIRT 4, SIRT 5 and SIRT 7 are upregulated in cancers. Importantly, depending on the cellular context and tissues, SIRT 1, SIRT 2 and SIRT 3 act either as tumour suppressors or oncogenic factors. Therefore, several sirtuin modulators are currently in clinical trials; nevertheless, to the best of our knowledge, none of them have been marketed, yet (Fig. 16).
A survey of the recent literature evidenced the interactions between sirtuins and HIF-1α expression. Indeed, inhibition of SIRT 1 leads to an anarchic HIF-1α acetylation, impairs its accumulation and consequently downregulates HIF-1α targeted genes expression.119 Interestingly, SIRT 1 is also involved in the regulation of HIF-2α.120 In this context, Dai and co-workers have recently demonstrated that Tenovin-6, a dual SIRT 1/SIRT 2 inhibitor induces apoptosis in uveal melanoma cell lines by activating tumour suppressor genes and by increasing ROS levels. This molecule specifically kills CSCs in the 92.1 and Mel270 uveal melanoma cell lines.121
AK-7 is a specific SIRT 2 inhibitor that has been reported by Kim and co-workers.122 It increases the p-VHL-dependent ubiquitination of HIF-1α under hypoxia, and therefore reduces in a dose dependent manner HIF-1α levels in at least four different cancer cell lines (A549, HeLa, HEK293 and HEK293T). In the HCT116 and HT29 colon cancer cell lines, this inhibitor also induces the proteasomal degradation of Snail, and leads to cell-cycle arrest in phase G1.
Otherwise, SIRT 3 inhibition mediates the metabolic reprogramming of tumour cells through ROS generation. It leads to HIF-1α stabilization and subsequent expression of glycolytic genes.123 Oroxylin A, a flavonoid derivative, activates SIRT 3 in breast cancer cells and promotes the destabilization of HIF-1α. Consequently, oroxylin A inhibits the glycolysis-dependent metabolism and the cellular proliferation.124 Another molecule of interest that targets SIRT 3 is an indole derivative, MIAM. This molecule exerts a large scope of anti-cancer activities both in vitro and in vivo. On mice xenografted with human HCC cancer cells, it triggers the up-regulation of SIRT 3 and consequently destabilizes HIF-1α. Interestingly, MIAM does not exert significant toxicity on threated animals.125
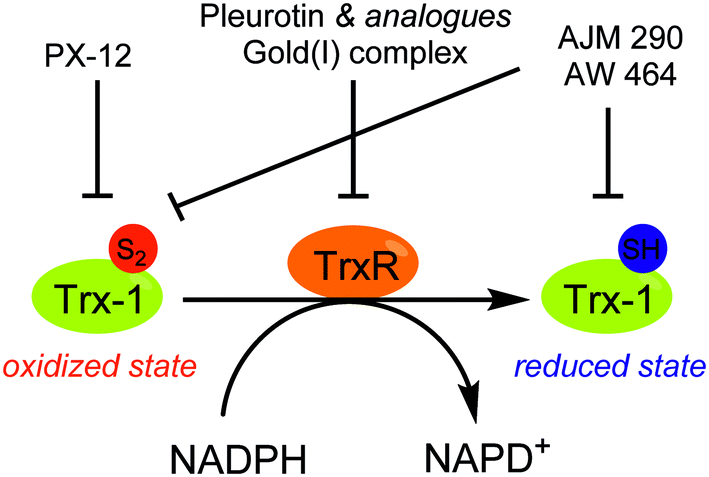 | ||
| Fig. 17 Schematic overview of Thioredoxin reductase (TrxR)-mediated thioredoxin-1 (Trx-1) reduction and the mode of action of some selected inhibitors. The NADPH-dependent reduction of Trx-1, a protein overexpressed in a set of cancer tissues, is mediated by TrxR. Inhibitors of either TrxR and Trx-1 have been developed and exert an action on the activation and stabilization of HIF-1α. See text for details. | ||
In this context some Trx-1/TrxR inhibitors with an attested effect on HIF-1α levels have been reported (Fig. 18). The most studied molecule is the compound PX-12. It exerts a dual activity: on the one hand, PX-12 induces an irreversible thioalkylation of a Trx-1 non-catalytic cysteine residue (Cys73), and on the other hand it disrupts the tubulin polymerisation. Consequently, PX-12 inhibits tumour cell growth with an IC50 in the micromolar range (MCF-7, HT629, MCF-7 tumour cell lines), and decreases in a dose dependent manner HIF-1α levels. Interestingly, Welsh and co-workers evidenced that this degradation occurs independently from the pVHL pathway.129 PX-12 was the first Trx-1 inhibitor to reach clinical development; a phase II study has been launched in December 2006 (NCT00417287) for its use in monotherapy in the treatment of pancreatic neoplasms, but has been interrupted by investigator decision and no results have been published. Indeed, a more recent report (2013) dealing with a phase I trial performed on patients suffering gastrointestinal malignancies highlights that the maximum tolerated dose of PX-12 was estimated at 300 mg m−2 d−1. These trials did not evidence any significant clinical activity of PX-12, since no changes in the levels of Trx-1 and VEGF in plasma of the patients were observed.130 Therefore, the further development of this molecule appears questionable.
 | ||
| Fig. 18 Representative examples of thioredoxin-1 (Trx-1) inhibitors exhibiting an anti-HIF-1α activity. | ||
Nonetheless, two Trx-1 inhibitors (AJM 290 and AW 464) structurally-related to PX-12, increase HIF-1α expression in several human cancer cell lines (e.g. MDA-MB-438) while they paradoxically inhibit the expression of HIF-1 targeted genes. Indeed, AJM 290 and AW 464 act as inhibitors of the HIF-1α COOH-terminal transactivation domain and block HIF-1 binding to DNA. Lastly, AW 464 exerts sub-micromolar anti-proliferative activities against several cancer cell lines, including MCF-7, MDA-MB-468, HCT-116 and HT-29.131
The natural antibiotic pleurotin, isolated from Pleurotus griseus is a naphthoquinone that exerts a large scope of potential therapeutic applications, including antibacterial, antifungal and anti-cancer (Fig. 19). It is a sub-micromolar irreversible inhibitor of TrxR (IC50 = 0.17 μM; Ki = 0.28 μM), which also downregulates HIF-1α in a dose-dependent manner.129 Pleurotin has recently been included in a screening, aiming to select natural products with specific anti-CSCs activity in melanoma. In this study, Sztiller-Sikorska and co-workers demonstrated that it exhibits a more potent anti-proliferative activity against clonogenic cells (IC50 in the 0.1 μm–1 μM range) than against fast-cycling cells.132 Lastly, new structurally-related pleurotin-derivatives, encompassing a benzo [1,2,4]triazin-7-one scaffold have been synthesized. In a preliminary report, Sweeney and co-workers pointed out that these derivatives exhibit interesting cytotoxic effects on selected cancer cell lines including GM00637, DU-145 and MCF-7.133
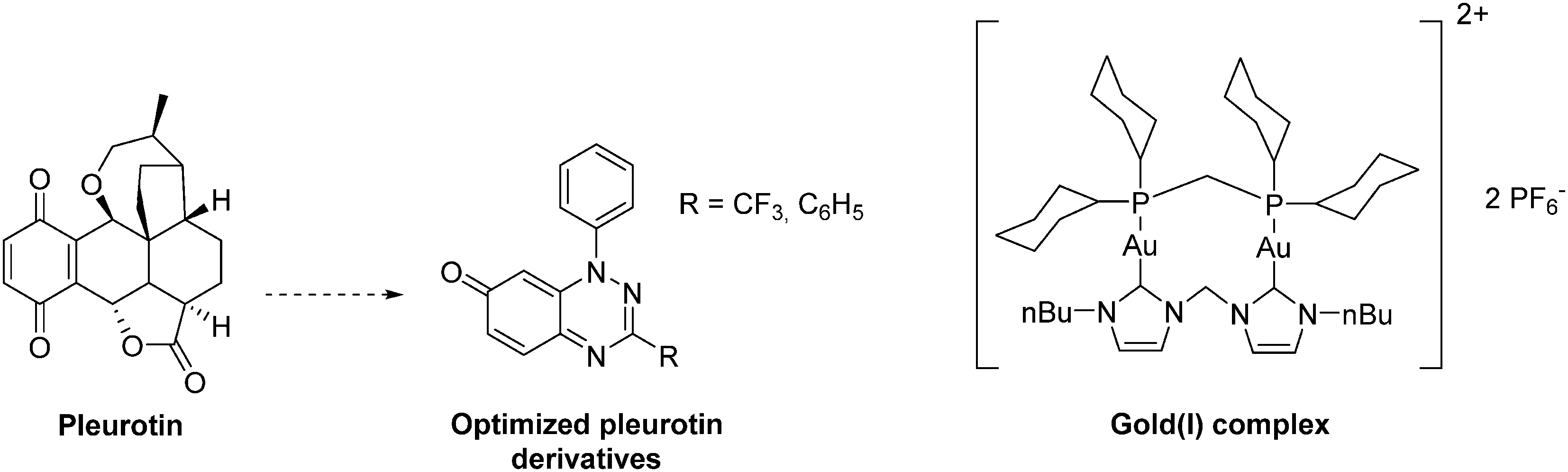 | ||
| Fig. 19 Representative examples of thioredoxin reductase (TrxR) inhibitors exhibiting an anti-HIF-1α activity. | ||
Another TrxR inhibitor of interest is a binuclear gold(I) complex that contains mixed diphosphine and bis N-heterocyclic carbenes (NHCs) ligands (Fig. 19). This drug inhibits in vitro TrxR in a dose dependent manner (IC50 = 38 nM), and exerts significant cytotoxicity toward breast (MCF-7), nasopharyngeal carcinoma (SUNE1), lung adenocarcinoma (H1975) and mouse melanoma (B16-F10) with IC50 values in the 1.3–3.2 μM range. On mice xenografted with HeLa cervical cancer cells, this gold complex reduces the tumour growth by more than 79% in 9 days at a dose of 5 mg kg−1. It does not induce any body weight loss, and exerts an anti-angiogenic activity. Overall, this agent inhibits CSCs self-renewal in human glioblastoma cells (U-87) and cervical cancers (Hela). Therefore, this complex can be seen as a pioneering molecule in the development of new specific anti-CSCs agents.134
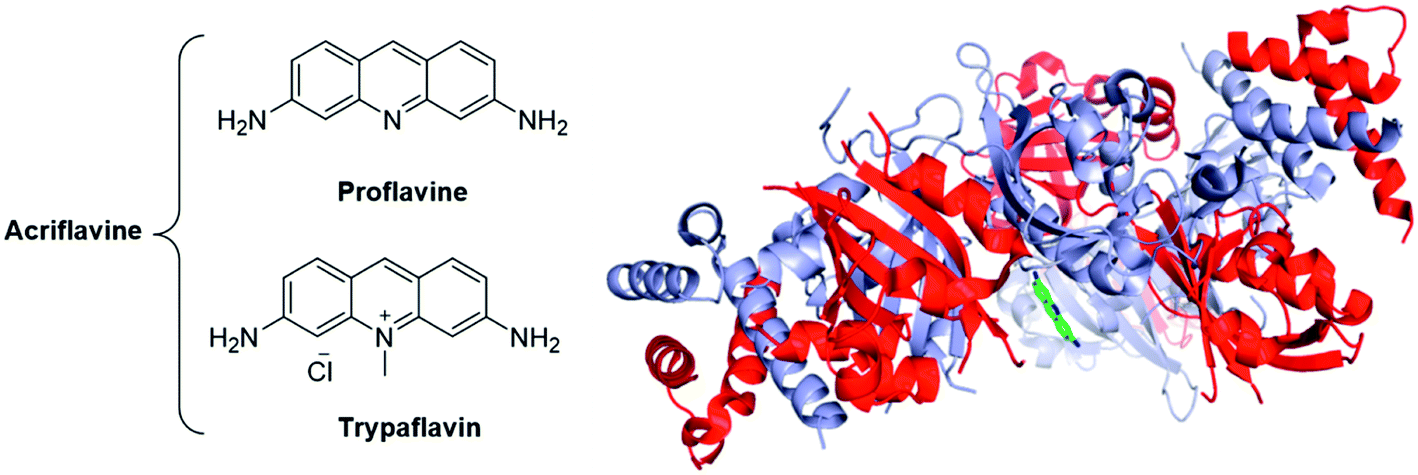 | ||
| Fig. 20 The acridines mixture of Acriflavine is an inhibitor of the HIF dimerization. Left panel: Structure of the two components of acriflavine; Right panel: crystallographic structure of proflavine bound to HIF-2α/HIF-β (PDB code: 4ZPH; blue: HIF-2α ; red: HIF-β ; green: proflavine). | ||
 | ||
| Fig. 21 cyclo-CCLVFY, a cyclopentapeptide inhibitor of HIF-1 dimerization. | ||
The crystallographic structure of proflavine bound to HIF-2α/HIF-β has recently been elucidated by Wu and co-workers.137 It is noteworthy that the binding of acriflavine to HIF-1α or HIF-2α monomers (Kd in the 50 μM range) appears weaker than its binding to the corresponding heterodimeric complexes (Kd in the 40 nM range).
Cellular assays on PC-3 and Hep3B-c1 human cancer cells highlighted that acriflavine affects neither HIF-1α and HIF-2α mRNA levels nor the protein levels of Hsp90, c-Myc, β-actin. Moreover, cell cycle, proliferation or survival are not inhibited by acriflavine. However, this therapeutic agent downregulates the expression of the HIF-1-activated genes (HRE), among them VEGF, stromal derived factor 1 (SDF-1) and SCF-1. In vivo, assays on mice xenografted with PC-3 cells evidenced that acriflavine prevents tumour growth.136
Interestingly, acriflavine sensitization of hypoxic cells to photodynamic therapy (PDT) has recently been suggested by Broekgaarden and co-workers. PDT consists in the local excitation of a photosensitizer (usually a cyanine or a porphyrin derivative), leading to generation of ROS and to the subsequent destruction of the local tumour vasculature. However, tumours may escape from these treatments by activating HIF-1. In this context, acriflavine has been co-encapsulated with zinc phtalocycanines into cationic liposomes and its effect on human epidermoid carcinoma (A431 cells) has been studied. As main result, the authors reported synergistic effects between acriflavine and the PDT-agent when cancer cells were cultured in a hypoxic media.138
Otherwise, the hexapeptide cyclo-CLLFVY (Fig. 21) inhibits HIF-α/HIF-β heterodimerization in vitro with an IC50 = 1.3 μM. It induces a dose-dependent reduction of HIF-1 activity in both human osteosarcoma cells (U2OS) and breast cancer cells (MCF-7). As downstream consequence, this cyclic peptide leads to the inhibition of VEGF production by tumour cells (in U2OS and MCF-7 cells treated at a dose of 50 μM). In addition, it disrupts the HUVEC microtubules formation under hypoxia, which is an evidence of its anti-angiogenic potency. Despite the very high degree of homology between HIF-1α and HIF-2α, cyclo-CLLFVY is reported to only antagonize the HIF-1α/HIF-1β interaction.135
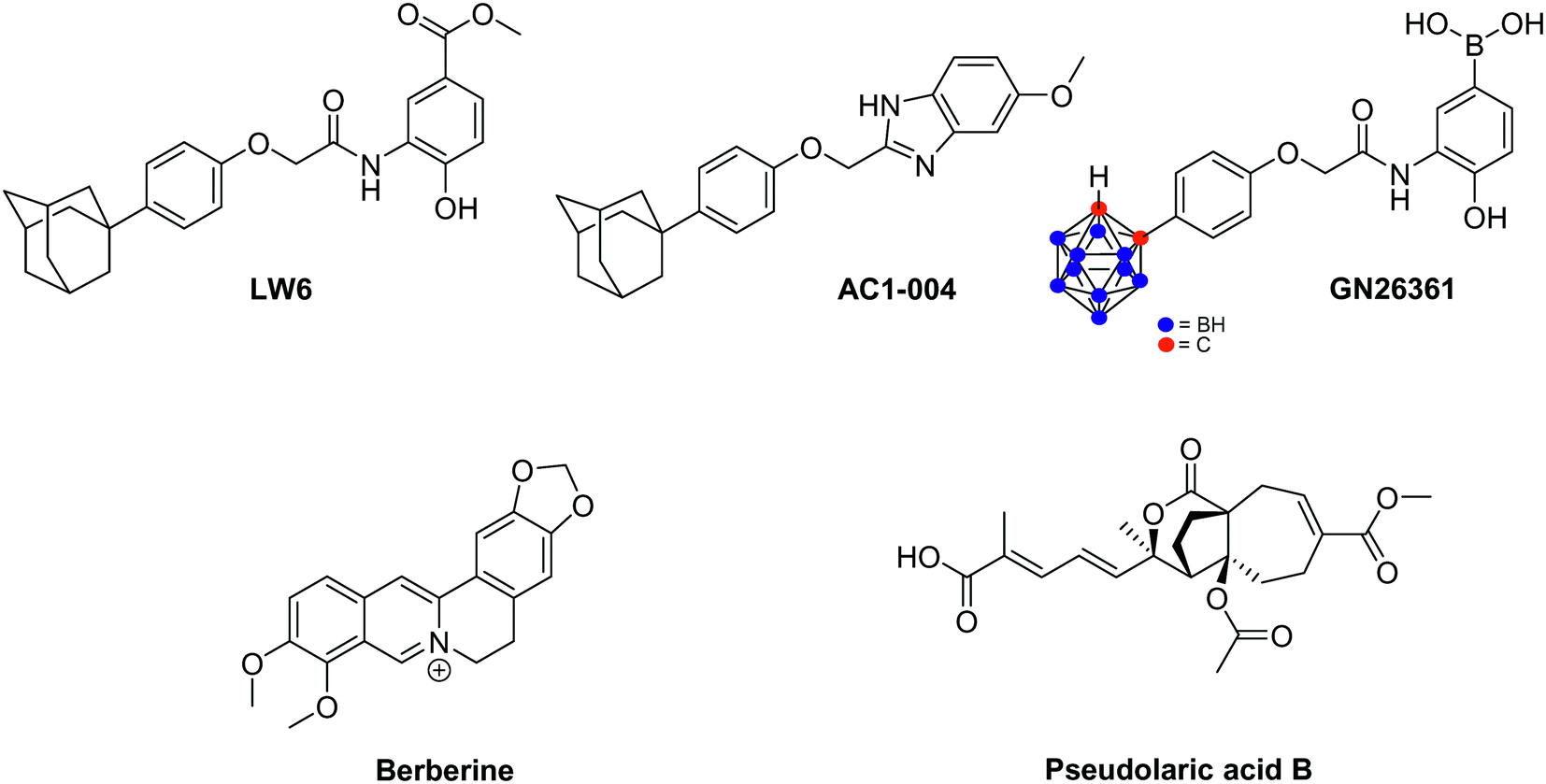 | ||
| Fig. 22 Miscellaneous compounds destabilizing HIF-1α. | ||
Starting from LW6, new series of small-sized organic inhibitors of HIF-1α have been designed. For example, AC1-004, a benzimidazole derivative, inhibits HIF-1α accumulation in cancer cell lines including HCT-116, MDA-MB-435, SK-HEP1 and caki. In vitro, AC1-004 also inhibits microtubule formation, thus underlining its potential anti-angiogenic effects.141 In addition to AC1-004, the carboranylphenoxyacetanilide derivative GN26361 has been reported. It inhibits HIF-1 transcriptional activity in HeLa cells (IC50 = 0.74 μM), supresses HIF-1α accumulation and VEGF mRNA expression. However, it does not affect HIF-1α mRNA levels.142
Berberine is an alkaloid extracted from Huanglian, a plant used in traditional Chinese medicine for the treatment of gastric discomforts such as gastroenteritis. In 2004, Lin and co-workers reported that, in vitro on SC-M1, this alkaloid eradicates angiogenesis at a 7.5 μM dose, and reduces the VEGF expression. These effects are mediated by HIF-1α destabilisation and not by a downregulation of its mRNA. In fact, berberine triggers the acetylation of Lys532 which is catalyzed by the acetyltransferase ARD1, and subsequently leads to the degradation of HIF-1α by the 26S proteasome.143 Currently, growing evidences underline that berberine could potentially be used as radiosensitizer agent against various cancer cell lines, including prostates cancers and esophageal squamous cancer, both in vitro and in vivo.144
Pseudolaric acids A and B, extracted from the root bark of Pseudolarix kaempferi Gordon tree, are other natural compounds that lead to the proteasomal degradation of HIF-1α. Consequently, the downregulation of VEGF expression and the inhibition of angiogenesis are observed. Remarkably, these molecules do not affect MAPK and PI3K/Akt signalling pathways.145 Recently, it has been evidenced that pseudolaric acid B triggers the phosphorylation of two serine residues of c-Jun (Ser63 and Ser73), a transcription factor activated by the activator protein AP-1, which in turn loses its capacity to stabilize HIF-1α.146
4.3. Therapeutic agents affecting DNA binding and gene transcription
 | ||
| Fig. 23 3D-structure of HIF-1α bound to p300 (PDB: 1L3E). The C-TAD domain of HIF-1α (blue ribbon) is characterized by three short helices, wrapping around the CH1 domain of p300 (pink ribbon). CH1 adopts a triangular geometry, and encompasses three cations Zn2+, responsible for its structural stability. Importantly, unbounded C-TAD is not structured. | ||
The HIF-α/p300 protein/protein interaction is characterized by a large surface and a low Kd (7 nM). It involves the C-terminal domain of HIF-1α (C-TAD) and the cystein/histidine-rich 1 domain (CH1) of p300/CBP (also known as a transcriptional adapter zinc-binding domain, TAZ).147 The 3D structure of this interaction has been resolved using high resolution multidimensional NMR spectroscopy. The resulting binding model highlights that three Zn2+ cations are incorporated in the CH1 domain of p300 to ensure its structural stability (PDB: IL3E, 1L8C, Fig. 23).148 Interestingly, unbound CAD is disordered, but its binding to CH1 stabilize its 3D structure, notably by means of hydrophobic and polar interactions. When bounded to p300, C-TAD is structurally characterized by three short helixes (Fig. 23, blue ribbon), wrapping entirely CH1 (Fig. 23, pink ribbon). The carboxamide group of the C-TAD asparagine residue Asn803, located in one of these helixes, is involved in an important H-bond network which stabilizes the protein/protein interaction. However, this asparagine also acts as an hypoxic sensor. Indeed, its hydroxylation occurs in normoxia and is mediated by a tumour-suppressor, the Fe(II)-dependant asparginyl oxydase FIH. As downstream consequence, this hydroxylation breaks the hydrogen-bonding network, and destabilizes the protein/protein interface.148
In this context two rationale approaches have been suggested to design protein/protein interaction inhibitors (PPIis) able to antagonize the interaction between HIF-1 and p300. The first strategy consists in the use of molecule able to destabilize p300 by means of a Zn2+ ejection mechanism. On the other hand, α-helix mimics with high affinities for p300 have been designed to directly compete with its binding to HIF-1α. In addition, natural compounds are also continuously screened in order to identify potent HIF-1/p300 antagonists.
- Studies on PPIis involving a Zn2+ ejection mechanism
Among the series of known inhibitors for the HIF-1α/p300 interaction, the epidithiodiketopiperazines form a class of interesting fungal secondary metabolites, characterized by a diketopiperazine scaffold bridged through two sulfur atoms (Fig. 24). These derivatives specifically destabilize the structure of p300 by a Zn2+ ejection mechanism. The resulting misfolded cofactor loses its capacity to bind HIF-1, whose transcriptional activity is in turn inhibited. Their leading member is chetomin, a metabolite produced by Chaetomim coclides and Chaetomium seminudum.149
 | ||
| Fig. 24 Chetomin and its derivatives. | ||
Indeed, in a model of mice xenografted with HT116 human colon cancer cells, chetomin represses the expression of the HIF-1-mediated genes (HRE) at a dose of 2 mg kg−1. At this dose, chetomin significantly reduces tumour growth without affecting the weight of the treated animals. The histological examination of tissues harvested from tumours revealed significant necrosis in treated animals. However, the repeated intravenous injections cause an unexplained local toxicity.149 More recently, its use in the treatment of incurable multiple myeloma has been suggested by Viziteu and co-workers.150
Chetomin and two derivatives, chaetocin and gliotoxin, exhibit potent anti-tumour activities against several cancer cell lines in vitro, including multiple myeloma. Moreover, in mice xenografted with human prostate tumours (PC-3 and DU-145), administration of chetomin at a dose of 0.25 mg kg−1 reduces the tumour growth by more than 50%, within 15 days.151
Recently, Dubey and co-workers synthesized dimeric chetomin-based derivatives. These new molecules have been designed to act as high-affinity divalent ligands for binding concomitantly two zinc cations from p300. In a panel of human breast (MCF-7 and MDA-MB-231) and lung (A549) cancer cell lines, the same authors reported an inhibition of HIF-induced gene transcription (e.g. VEGF-A, LOX, Glut1 and c-met). Moreover, they reported the strong antitumour effect of these compounds when administrated to mice xenografted with breast and lung tumours, at a dose of 1 mg kg−1.152
Jayatunga and co-workers reported a high throughput screening performed on a library of 10000 natural products. This allowed the mergence of a series of small-sized natural quinones and indandiones which antagonize the HIF-1/p300. Mechanistically, indandione derivatives are unstable and lead to ninhydrin, the actual reactive agent. It is able to bind to Zn2+ cations and thus to trigger the decomplextion of these metal atoms from p300. These quinone and indandione derivatives inhibit HIF-1/p300 interaction with an IC50 in the 5–15 μM range (Fig. 25).153
 | ||
| Fig. 25 Quinone and indandione derivatives which antagonize the HIF-1/p300 interaction. | ||
- α-turn mimetics as PPIis
An interesting alternative to antagonize the HIF-1/p300 interaction is based on the α-helical conformation of C-TAD which surrounds p300 CH1 domain. Indeed, two teams hypothesized that α-helix mimics, designed to exhibit high affinity for p300 CH1, will compete its binding to C-TAD (Fig. 26).
 | ||
| Fig. 26 α-Helix mimics as antagonists of the HIF-1α/p300 interaction. A: Structure of HIF-1α bound to p300 (PDB: 1L8C), highlighting two relevant α-helix involved in the binding of the two partner proteins and their corresponding sequences (blue: HIF-1α; pink: p300; grey: Zn atoms); B: peptidomimetic designed by Henchey (ref. 154), encompassing a hydrogen bon surrogate motive (red); C: oligoamides designed by Burslem (ref. 155) to specifically mimic helix 2 or helix 3. See text for details. | ||
Firtsly, Henchey and co-workers synthesized series of α-helical peptides structurally related to HIF-1α/C-TAD domain and including a hydrogen bond surrogate (HBS) motif.154 Indeed, the HBS strategy consists in the replacement of one of the main chain intramolecular H-bond by a covalent linkage, and more precisely, in the case of this study, the introduction of a C–C linkage (Fig. 26B). Among the different peptides studied, the most potent one exhibits 53% of helicity, and at the dose of 1 μM and after 12 h of incubation reduces the transcription of the VEGF gene by 45% in cervical cancers (HeLa cell line). The authors determined by isothermal microcalorimetry analysis that this peptide binds p300 with a Kd of 0.4 μM.
On the other hand, Burslem and co-workers designed and synthesized trimeric and tetrameric aromatic oligoamides as α-helix mimics (Fig. 26C).155 Among these fully non-peptidic foldamers, several molecules designed as mimics of the HIF-1α helix 2 inhibit the HIF-1/p300 binding with IC50 in the 10–50 μM range (the most potent one depicted in Fig. 26C, exhibits an IC50 = 9 μM). However, conversely to the results described by Henchey, these authors observed that specific mimics of HIF-1α helix 2 are unable to antagonize the HIF-1α/p300 binding. Lastly, these foldamers have not been evaluated in cellular assays, yet.
- Natural products
Classes of natural compounds with the ability to antagonize HIF-1/p300, and subsequently to exert a potential anti-tumour activity, are continuously emerging in the literature (Fig. 27). Among representative examples, one can cite Eudistidine A, a polycyclic marine alkaloid, which inhibits HIF-1/p300 with an IC50 of 75 μM,156 and also a series of pyrroloiminoquinone alkaloids extracted from the marine sponge Latrunculia sp. (including discorhabdin A–W and makaluvamine F) which inhibits the HIF-1 transcriptional activity in HCT 116 and LNCaP cancer cell lines with IC50 in the 0.1–10.0 μM range.157 In addition, several naturally occurring sulfonamide derivatives with IC50 in the 5 μM range, as well as menadione and ethacrynic acid, have been reported to antagonize HIF-1/p300.158
 | ||
| Fig. 27 Series of natural products, antagonists of the HIF-1/p300 interaction. | ||
 | ||
| Fig. 28 Anthracyclines as inhibitors of the HIF-1 transcriptional activity. | ||
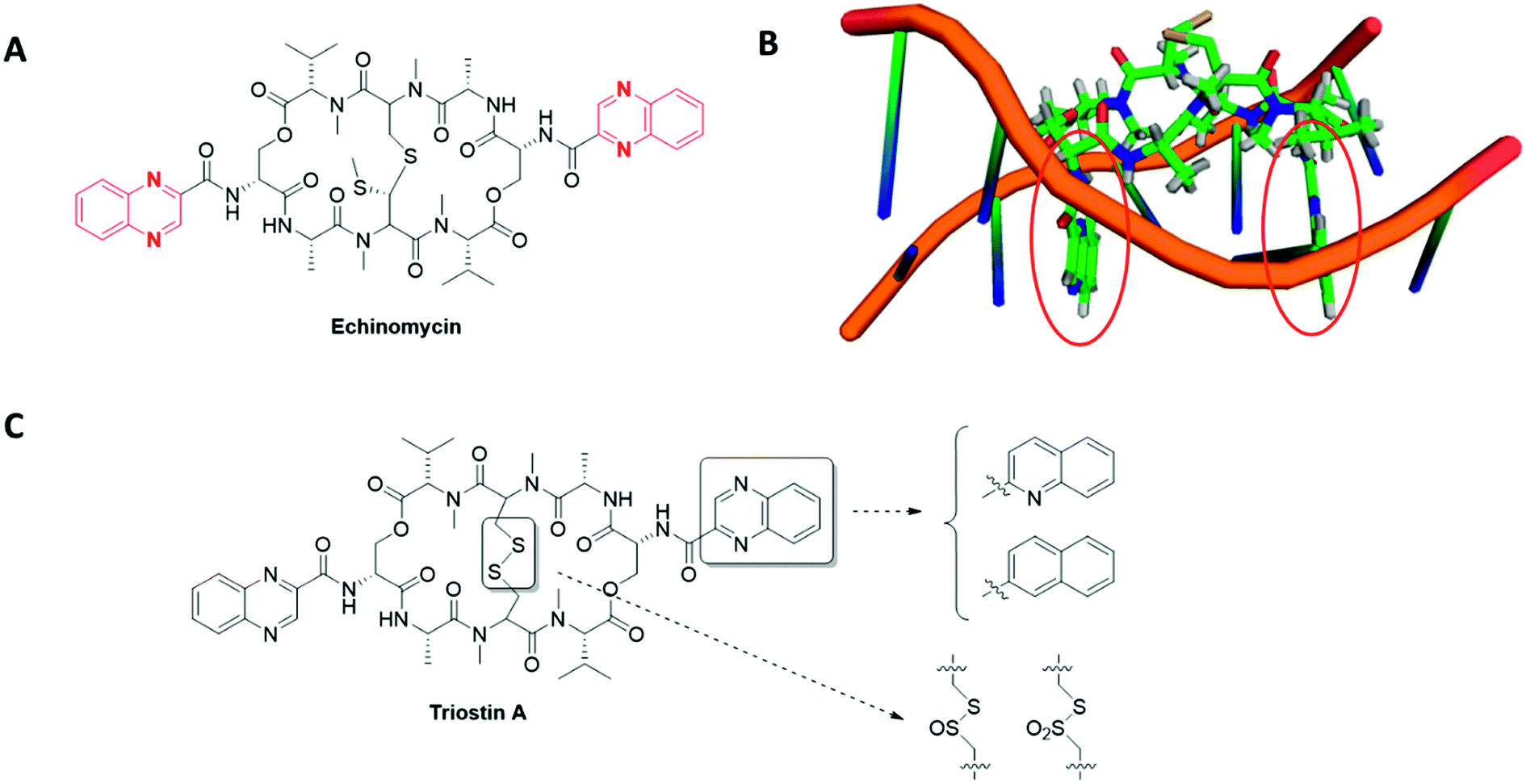 | ||
| Fig. 29 Two cyclopeptides inhibitors of the HIF-1 transcriptional activity. A: Echinomycin; B: crystallographic structure of echinomycin bound to HRE DNA sequence (PDB: 2ADW) featuring the intercalated carboxyquinoxaline cores (red circle); C: triostin A. | ||
Echinomycin, a cyclic pseudopeptide derived from Streptomyces echinatus, belonging to the quinoxalin antibiotics and has been identified as a specific antagonist of HIF binding to a DNA sequence containing the VEGF-A promoter gene (Fig. 29A).162 Indeed, echinomycin binds the HIF-1 recognition sequence 5′-A/TCGT-3′, and the crystallographic data underlines an interesting binding mode in which the two quinoxicarboxaline cores are intercalated between the bases (Fig. 29B).163 Consequently, this molecule downregulates, under hypoxia, the HIF activity in several cancer cell lines (solid and haematological) affecting different type of malignant tissues, such as HepG2, MCF-7, U251.
However, echinomycin has an additional counter productive effect on HIF. Indeed, in normoxic conditions, low concentration levels of echinomycin can enhance the Sp1 protein activity, which lead to an increased HIF gene transcription.164 Therefore, the use of this drug in cancer therapy remains very questionable. The phase II trials devoted to echinomycin did not bring any evidence of significant clinical benefits for patients, and therefore are currently suspended.165 In this context, Hattori and co-workers have recently synthesized derivatives of triostin A, an echinomycin precursor (Fig. 29C). In human breast cancer cells (MCF-7 cell line), several of these pseudopeptides, not only inhibit DNA binding of HIF-1, but also prevent HIF-1α accumulation. This latter result suggests that the cytotoxic effect is not only HIF-1-dependent, and paves the way to the design and development of a new anti-cancer agents.166
5. Perspectives: HIF-1 inhibition and CSCs, what's next?
HIF-1 is a key player in CSCs formation, maintenance and self-renewal and is at the crosstalk of signalling pathways involved in tumour aggressiveness and metastasis formation. It is also responsible for several evolutions of the tumour microenvironment. Therefore, this target is of utmost interest in current medicinal chemistry, and decades of research allowed the emergence of a wide variety of HIF-1 inhibitors/modulators. Several molecules have successfully passed the clinical trial stage and are currently administered to patients. Nonetheless, the survey of the literature presented herein points out that the problem of the HIF-1 inhibition in the specific CSCs context has not been fully addressed, yet. New therapeutic strategies are needed to tackle the adaptive response of this subtype of aggressive cells. In this context, several ongoing research axes can be underlined.Hence, targeting hypoxic region within a tumour by means of hypoxia-activated agents has been envisioned about 30 years ago.167 This strategy consists in the delivery of a prodrug which is first activated by a reductase; the generated reduced radical then undergoes a fragmentation to yield the active substrate in case of oxygen deprivation (hypoxia). In normoxic tissues, oxygen allows the regeneration of the starting products.168 (Fig. 30).
 | ||
| Fig. 30 A. Chemical bases of the hypoxia-activated agents; B. structure of Q39. | ||
In addition to this strategy, the selective delivery of chemotherapeutics to hypoxic niches using hypoxia-sensitive (nano)vectors is also an area of intense research.169 Combining one of these approaches with drugs that directly target HIF or indirectly disrupt its functions could yield doubly specific drugs and may exhibit enhanced selectivity and efficacy towards hypoxic tumours.
In this context, several hypoxia-activated prodrugs (HAPs) have been designed and evaluated in vitro and in vivo.168,170 Nonetheless, Q39 is the only example of HAP specifically directed towards HIF-1 activity (Fig. 30). All the other HAPs do not affect HIF itself or its function and are therefore beyond the scope of this review.
Q39 is a quinoxaline di-N-oxide that was first synthesised and evaluated, in several hypoxic and normoxic tumour cell lines, by Hu and Yang in 2007.171,172 It induces apoptosis of both hypoxic and normoxic tumours through the mitochondria apoptotic pathway. This was corroborated to the increased levels of Bax and p38, and accompanied by the decrease of HIF-1α level. Rather than acting on the degradation rate of HIF-1α or its corresponding mRNA levels, Q39 induces mTOR and 4E-BP1 dephosphorylation and thus perturbs HIF-1α expression at the translational level.172
In addition to the aforementioned approaches, a complementary alternative is the use of antibody drug conjugates (ADCs).173 These therapeutic agents are hybrid macromolecules, which combines a cytotoxic agent with a monoclonal antibody (mAbs) directed toward a specific extracellular marker of cancer cells. These two entities are linked through a spacer (usually disulfide bridges or peptides). In the recent years, this ADC technology has been used to address drugs to CSCs. To this end, mAbs have been designed to specific target CDCs markers such as CD20, CD44, CD133, DLL3 and LGR5, which have been linked to various cytotoxic agents including DNA poisoning molecules, microtubule-targeting drug (paclitaxel), topoisomerase inhibitors and anthracyclines.174–178 Interestingly, no ADCs have been designed to specifically downregulates HIF-1 in CSCs. Moreover, the effects of the above mentioned ADCs on HIF-1 stability have not been assayed. Nonetheless, among them, one encompasses a doxorubicine derivative (the anthracycline PNU 159682),178 and another encompasses a topoisomerase inhibitor (SN-38, a compound in clinical trial phase I, and which is the main metabolite of irinotecan)176 known to downregulate HIF-1α and downstream genes expression when administrated alone (Fig. 31).179
 | ||
| Fig. 31 Structure of two HIF-1 inhibitors used to design ADC directed towards CSCs. | ||
An interesting complementary ADC approach has been suggested by Cui and co-workers in 2010. Owing to the overexpression of HIF-1α in hypoxic regions, its targeting may dramatically enhance drug selectivity. In this context this team reported the preparation and assessment of an antibody anti-HIF-1α conjugated to nano-micelles filled with paclitaxel.180 They describe that the presence of the anti-HIF-1α antibody dramatically increases (from 18% to 81%) the nano-micelles uptake by adherent MGC-803 cells (stomach cancer). In terms of cytotoxicity, the antibody conjugate exhibited moderately higher activities than the untagged nano-micelles, the latter being active due to the EPR effect. Nevertheless, the authors confirmed that the designed nano-micelles could partly discriminate tumour cells from healthy cells. In fact, this antibody drug conjugate did not display obvious toxicity towards HDF fibroblast cells that do not express HIF-1α.
6. Conclusion
To conclude, we summarized in this review some relevant aspects of the current researches devoted to HIF-1α, with a particular emphasis on CSCs. Thus, we underline the pivotal role of this transcription factor in CSCs formation and maintenance. We pointed out selected success stories in the development of HIF-1 inhibitors. Despite indisputable advances, as demonstrated by the continuous emergence of marketed therapeutic agents targeting HIF-1, the molecules able to tackle this transcription factor in the specific context of the CSCs remain rare. This paves the way for new approaches (Fig. 32) in order to identify original therapeutic agents, or a new drug formulation/nanomedecine, able to become the “golden standard” for tackling this aggressive and deadly subtype of cancer cells in a specific way. | ||
| Fig. 32 The three main axes for the development of new anti-HIF compounds. | ||
Acknowledgements
The authors are indebted to Prof. Daniele Passarella, Prof. Ling Peng and Dr. Panagiota Sotiropoulou for their invitation to submit this review to the special issue “Chemical approaches targeting drug resistance in cancer stem cell” in the Royal Society of Chemistry journal MedChemComm.This work was partly funded by the European COST Action CM1106 (http://www.stemchem.org).
References
- G. L. Semenza, Biochim. Biophys. Acta, 2016, 1863, 382 CrossRef CAS PubMed; W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393 CrossRef PubMed.
- J. Liu, Y. Liu, W. Bu, J. Bu, Y. Sun, J. Du and J. Shi, J. Am. Chem. Soc., 2014, 136, 9701 CrossRef CAS PubMed; P. Vaupel, M. Höckel and A. Mayer, Antioxid. Redox Signaling, 2007, 9, 1221 CrossRef PubMed.
- A. N. Bhatt, A. Chauhan, S. Khanna, Y. Rai, S. Singh, R. Soni, N. Kalra and B. S. Dwarakanath, BMC Cancer, 2015, 15, 335 CrossRef CAS PubMed; M. Emara and J. Allalunis-Turner, Oncol. Rep., 2014, 31, 1947 Search PubMed; P. Camaj, J. Carsten, S. Krebs, E. N. DeToni, H. Blum, K.-W. Jauch, P. J. Nelson and C. J. Bruns, Mol. Cancer Res., 2014, 12, 421 CrossRef PubMed; B. Bao, A. Aamir, D. Kong, S. Ali, A. S. Azmi, Y. Li, S. Banerjee, S. Padhye and F. H. Sarkar, PLoS One, 2012, 7, e43726 Search PubMed; R. P. Hill, D. T. Marie-Egyptienne and D. W. Hedley, Semin. Radiat. Oncol., 2009, 19, 106 CrossRef PubMed; H. Axelson, E. Fredlund, M. Ovenberger, G. Landberg and S. Påhlman, Semin. Cell Dev. Biol., 2005, 16, 554 CrossRef PubMed; A. Jögi, I. Øra, H. Nilsson, L. Poellinger, H. Axelson and S. Påhlman, Cancer Lett., 2003, 197, 145 CrossRef.
- Y. Asgari, Z. Zabihinpour, A. Salehzadeh-Yazdi, F. Schreiber and A. Masoudi-Nejad, Genomics, 2015, 105, 275 CrossRef CAS PubMed; X. Chen, Y. Qian and S. Wu, Free Radical Biol. Med., 2015, 79, 253 CrossRef PubMed; W. Zhang, S.-L. Zhang, X. Hu and K. Y. Tam, Int. J. Biol. Sci., 2015, 11, 1390 CrossRef PubMed.
- P. Sancho, D. Barneda and C. Heeschen, Br. J. Cancer, 2016, 114, 1305 CrossRef CAS PubMed; S. Elf, R. Li, S. Xia, Y. Pan, C. Shan, S. Wu, S. Lonial, M. Gaddh, M. L. Arellano, H. J. Khoury, F. R. Khuri, B. H. Lee, T. J. Boggon, J. Fan and J. Chen, Oncogene, 2016 DOI:10.1038/onc.2016.196; Y. Chen, Q. Xu, D. Ji, Y. Wei, H. Chen, T. Li, B. Wan, L. Yuan, R. Huang and G. Chen, Tumour Biol., 2016, 37, 6027 CrossRef PubMed.
- G. J. Yoshida, J. Exp. Clin. Cancer Res., 2015, 34, 111 CrossRef PubMed.
- C. S. Gonsalves, C. Li, M.-S. Eiymo Mwa Mpollo, V. Pullarkat, P. Malik, S. M. Tahara and V. K. Kalra, Biochem. J., 2015, 468, 409 CrossRef CAS PubMed; F. Yu, Y. Lin, T. Zhan, L. Chen and S. Guo, Cell Biol. Int., 2015, 39, 310 CrossRef PubMed; M. Emara and J. Allalunis-Turner, Oncol. Rep., 2014, 31, 1947 Search PubMed; S. Giuliano and G. Pagès, Biochimie, 2013, 95, 1110 CrossRef PubMed; B. Bao, S. Ali, A. Ahmad, A. S. Azmi, Y. Li, S. Banerjee, D. Kong, S. Sethi, A. Aboukameel, S. B. Padhye and F. H. Sarkar, PLoS One, 2012, 7, e50165 Search PubMed; A. Ferrero, D. Dompè, N. Ravarino, A. Ramella, L. Fuso, F. Maggiorotto, E. Tripodi and P. Zola, Gynecol. Oncol., 2011, 123, 301 CrossRef PubMed.
- S. Srinivasan, V. Chitalia, R. D. Meyer, E. Hartsough, M. Mehta, I. Harrold, N. Anderson, H. Feng, L. E. Smith, Y. Jiang, C. E. Costello and N. Rahimi, Angiogenesis, 2015, 18, 449 CrossRef CAS PubMed; R. M. Misra, M. S. Bajbaj and V. P. Kale, PLoS One, 2012, 7, e50153 Search PubMed.
- J. P. Thiery, H. Acloque, R. Y. J. Huang and M. A. Nieto, Cell, 2009, 139, 87 CrossRef CAS PubMed; R. Kalluri and R. A. Weinberg, J. Clin. Invest., 2009, 119, 1420 CrossRef PubMed.
- L. A. Aparicio, V. Abella, M. Valladares and A. Figueroa, Cell. Mol. Life Sci., 2013, 70, 4463 CrossRef CAS PubMed.
- Y. Inoue, Y. Itoh, K. Sato, F. Kawasaki, C. Sumita, T. Tanaka, D. Morishita and H. Hayashi, Curr. Cancer Drug Targets, 2016, 16, 110 CrossRef CAS PubMed; M. Mukherjee, J.-S. Fan, S. Ramachandran, G. R. Guy and J. Sivaraman, J. Biolumin. Chemilumin., 2014, 289, 25611 Search PubMed; L. A. Aparicio, M. Valladares, M. Blanco, G. Alonso and A. Figueroa, Cancer Metastasis Rev., 2012, 31, 375 CrossRef PubMed; A. K. Perl, P. Wilgenbus, U. Dahl, H. Semb and G. Christofori, Nature, 1998, 392, 190 CrossRef PubMed.
- A. Papi, S. DeCarolis, S. Bertoni, G. Storci, V. Sceberras, D. Santini, C. Ceccarelli, M. Taffurelli, M. Orlandi and M. Bonafé, J. Cell. Physiol., 2014, 229, 1595 CrossRef CAS PubMed; D. Illiopoulos, H. A. Hirsch, G. Wang and K. Struhl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1397 CrossRef PubMed; Y. Kim, Q. Lin, D. Zelterman and Z. Yun, Cancer Res., 2009, 69, 9271 CrossRef PubMed.
- C. Boccaccio, P. Luraghi and P. M. Comoglio, Cancer Res., 2014, 74, 3647 CrossRef CAS PubMed; M. Todaro, M. Gaggianesi, V. Catalano, A. Benfante, F. Iovino, M. Biffoni, T. Apuzzo, I. Sperduti, S. Volpe, G. Cocorullo, G. Gulotta, F. Dieli, R. De Maria and G. Stassi, Cell Stem Cell, 2014, 14, 342 CrossRef PubMed.
- K. H. Kim, H. J. Seol, H. Eun, J. Rheey, H. J. Jin, Y. Lee, K. M. Joo, J. Lee and D.-H. Nam, Neuro-Oncology, 2013, 15, 161 CrossRef CAS PubMed; Y. Li, A. Li, M. Glas, B. Lal, M. Ying, Y. Sang, S. Xia, D. Trageser, H. Guerrero-Cazares, C. G. Eberhart, A. Quinones-Hinojosa, B. Scheffer and J. Laterra, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9951 CrossRef PubMed.
- P. A. Sotiropoulou, M. S. Christodoulou, A. Silvani, C. Herold-Mende and D. Passarella, Drug Discovery Today, 2014, 19, 1547 CrossRef CAS PubMed.
- G. Peng and Y. Liu, Trends Pharmacol. Sci., 2015, 36, 374 CrossRef CAS PubMed; B. Keith and M. C. Simon, Cell, 2007, 129, 465 CrossRef PubMed.
- G. L. Semenza, Trends Pharmacol. Sci., 2012, 33, 207 CrossRef CAS PubMed; Y. Xia, H.-K. Choi and K. Lee, Eur. J. Med. Chem., 2012, 49, 21 CrossRef PubMed.
- A. Marin-Hernandez, J. C. Gallardo-Pérez, S. J. Ralph, S. Rodríguez-Enríquez and R. Moreno-Sánchez, Mini-Rev. Med. Chem., 2009, 9, 1084 CrossRef CAS PubMed; S. S. Hong, H. S. Lee and K. W. Kim, Cancer Res. Treat., 2004, 36, 343–353 CrossRef PubMed.
- W. Luo, H. Hu, R. Chang, J. Zhong, M. Knabel, R. O'Meally, R. N. Cole, A. Pandey and G. L. Semenza, Cell, 2011, 145, 732 CrossRef CAS PubMed; H. R. Christofk, M. G. Van der Heiden, M. H. Harris, A. Ramanathan, R. E. Gerszten, R. Wei, M. D. Fleming, S. L. Schreiber and L. C. Cantley, Nature, 2008, 452, 230 CrossRef PubMed.
- G. Bellot, R. Garcia-Medina, P. Gounon, J. Chiche, D. Roux, J. Pouysségur and N. M. Mazure, Mol. Cell. Biol., 2009, 29, 2570 CrossRef CAS PubMed; H. Zhang, J. Biolumin. Chemilumin., 2008, 283, 10892 Search PubMed.
- F. Zhao, Oncogene, 2010, 29, 2962 CrossRef CAS PubMed.
- L. Schito, S. Rey, M. Tafani, H. Zhang, C. C. Wong, A. Russo, M. A. Russo and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2707 CrossRef CAS PubMed; S. Rey and G. L. Semenza, Cardiovasc. Res., 2010, 86, 236 CrossRef PubMed; M. P. Simon, R. Tournaire and J. Pouyssegur, J. Cell. Physiol., 2008, 217, 809 CrossRef PubMed; B. F. Jonhson, T. M. Clay, A. C. Hobeika, H. K. Lyerly and M. A. Morse, Expert Opin. Biol. Ther., 2007, 7, 449 CrossRef PubMed; D. J. Ceradini, A. R. Kulkarni, M. J. Callaghan, O. M. Tepper, N. Bastidas, M. E. Kleinman, J. M. Capla, R. D. Galiano, J. P. Levine and G. C. Gurtner, Nat. Med., 2004, 10, 858 CrossRef PubMed; J. A. Forsythe, B. H. Jiang, N. V. Iyer, F. Agani, S. W. Leung, R. D. Koos and G. L. Semenza, Mol. Cell. Biol., 1996, 16, 4604 CrossRef PubMed.
- J. Zhang, Q. Cheng, Y. Zhou, Y. Wang and X. Chen, Oral. Oncol., 2013, 49, 1043 CrossRef CAS PubMed; G. H. Zhu, C. Huang, Z. Z. Feng, X.-H. Lv and Z.-J. Qiu, Dig. Dis. Sci., 2013, 58, 3502 Search PubMed; M. A. Esteban, M. G. Tran, S. K. Harten, P. Hill, M. C. Castellanos, A. Chandra, R. Ravel, T. S. O'brien and P. H. Maxwell, Cancer Res., 2006, 66, 3567 CrossRef PubMed; B. Krishnamachary, D. Zagzag, H. Nagasawa, K. Rainey, H. Okuyama, J. H. Baek and G. L. Semenza, Cancer Res., 2006, 66, 2725 CrossRef PubMed; M. H. Yang, M. Z. Wu, S. H. Chiou, P. M. Chen, S. Y. Chang, C. J. Liu, S. C. Teng and K. J. Wu, Nat. Cell Biol., 2008, 10, 295 CrossRef PubMed.
- L. Gunaratnam, M. Morley, A. Franovic, N. de Paulsen, K. Mekhail, D. A. Parolin, E. Nakamura, I. A. Lorimer and S. Lee, J. Biolumin. Chemilumin., 2003, 278, 44966 CAS.
- Q. Zhang, X. Bai, W. Chen, T. Ma, Q. Hu, C. Liang, S. Xie, C. Chen, L. Hu, S. Xu and T. Liang, Carcinogenesis, 2013, 34, 962 CrossRef CAS PubMed; Y. G. Jiang, Y. Luo, D. L. He, X. Li, L. L. Zhang, T. Peng, M. C. Li and Y. H. Lin, Int. J. Urol., 2007, 14, 1034 CrossRef PubMed.
- Z. X. Chen, B. Sun, S. J. Wang, Y. Gao, Y. M. Zhang, H. X. Zhou, G. Jia, Y. W. Wang, R. Kong, S. H. Pan, D. B. Xue, H. C. Jiang and X. W. Bai, PLoS One, 2011, 6, 23752 Search PubMed.
- D. M. Gilkes, S. Bajpai, P. Chaturvedi, D. Wirtz and G. L. Semenza, J. Biolumin. Chemilumin., 2013, 288, 10819 Search PubMed; D. M. Gilkes, S. Bajpai, C. C. Wong, P. Chaturvedi, M. E. Hubbi, D. Wirtz and G. L. Semenza, Mol. Cancer Res., 2013, 11, 456 CrossRef CAS PubMed.
- Y. Luo, D.-L. He, L. Ning, S.-L. Shen, L. Li, X. Li, H. E. Zhau and L. W. K. Chung, BJU Int., 2006, 98, 1315 CrossRef CAS PubMed.
- J. M. Heddleston, Z. Li, J. D. Lathia, S. Bao, A. B. Hjelmeland and J. N. Rich, Br. J. Cancer, 2010, 102, 789 CrossRef CAS PubMed.
- E. Chinchar, K. L. Makey, J. Gibson, F. Chen, S. A. Cole, G. C. Megason, S. Vijayakumar, L. Miele and J.-W. Gu, Vasc. Cell, 2014, 6, 12 CrossRef CAS PubMed; D. Zhang, B. Sun, X. Zhao, Y. Ma, R. Ji, Q. Gu, X. Dong, J. Li, F. Liu, X. Jia, X. Leng, C. Zhang, R. Sun and J. Chi, Mol. Cancer Ther., 2014, 13, 207 CrossRef PubMed; S. J. Conley, E. Gheordunescu, P. Kakarala, N. Bryan, H. Korkaya, A. N. Heath, S. G. Clouthier and M. S. Wicha, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2784 CrossRef PubMed; L. P. Schwab, D. L. Peacock, D. Majumdar, J. F. Ingels, L. C. Jensen, K. D. Smith, R. C. Cushing and T. N. Seagroves, Breast Cancer Res., 2012, 14, R6 CrossRef PubMed.
- F. Zanconato, M. Cordenonsi and S. Piccolo, Cancer Cell, 2016, 29, 783 CrossRef CAS PubMed; H. Hayashi, T. Higashi, N. Yokoama, T. Kaida, K. Sakamoto, Y. Fukushima, T. Ishimoto, H. Kuroki, H. Nitta, D. Hashimoto, A. Chikamoto, E. Oki, T. Beppu and H. Baba, Cancer Res., 2015, 75, 4985 CrossRef PubMed.
- L. Xiang, D. Gilkes, H. Hu, W. Luo, H. Lu, J. W. Bullen, D. Samanta, N. Takano and G. L. Semenza, Oncotarget, 2014, 5, 12509 CrossRef PubMed.
- C. Zhang, D. Samanta, H. Lu, J. W. Bullen, H. Zhang, I. Chen, X. He and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2047 CrossRef CAS PubMed; X.-Q. Wu, C. Huang, X. He, Y.-Y. Tian, D.-X. Zhou, Y. He, X.-H. Liu and J. Li, Cell. Signalling, 2013, 25, 2462 CrossRef PubMed; J. Mathieu, Z. Zhang, W. Zhou, A. J. Wang, J. M. Heddleston, C. M. A. Pinna, A. Hubaud, B. Stadler, M. Choi, M. Bar, M. Tewari, A. Liu, R. Vessella, R. Rostomily, D. Born, M. Horwitz, C. Ware, C. A. Blau, M. A. Cleary, J. N. Rich and H. Ruohola-Baker, Cancer Res., 2011, 71, 4640 CrossRef PubMed.
- H. Zhu, D. Wang, Y. Liu, Z. Su, L. Zhang, F. Chen, Y. Zhou, Y. Wu, M. Yu, Z. Zhan and G. Shao, Cancer Cell Int., 2013, 13, 119 CrossRef PubMed.
- R. Diaz, P. A. Nguewa, M. Redrado, I. Manrique and A. Calvo, Prostate, 2015, 75, 1137 CrossRef CAS PubMed; M. Marhold, E. Tomasich, A. El-Gazzar, G. Heller, A. Spittler, R. Horvat, M. Krainer and P. Horak, Mol. Cancer Res., 2015, 13, 556 CrossRef PubMed.
- M. Inukai, A. Hara, Y. Yasui, T. Kumabe, T. Matsumoto and M. Saegusa, Human Pathol., 2015, 46, 1496 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, F. Tang, R. Schore, Y. Liu, K. M. Bernot, G. Marcucci, M. A. Caligiuri and P. Zheng, Blood, 2014, 124, 1127 CrossRef CAS PubMed; S. Yonekura, M. Itoh, Y. Okuhashi, Y. Takahashi, A. Ono, N. Nara and S. Tohsa, Anticancer Res., 2013, 33, 3099 Search PubMed; Y. Wang, Y. Liu, S. N. Malek, P. Zheng and Y. Liu, Cell Stem Cell, 2011, 8, 501 Search PubMed.
- G. L. Semenza, N. Engl. J. Med., 2011, 365, 537 CrossRef CAS PubMed; G. L. Wang, B. H. Jiang, E. A. Rue and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5510 CrossRef.
- M. Ema, S. Taya, N. Yokotani, K. Sogawa, Y. Matsuda and Y. Fujii-Kuriyama, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4273 CrossRef CAS; H. Tian, S. L. Mc Knight and D. W. Russell, Genes Dev., 1997, 11, 72 CrossRef PubMed; Y. Z. Gu, S. M. Moran, J. B. Hogenesch, L. Wartman and C. A. Bradfield, Gene Expression, 1998, 7, 205 Search PubMed.
- B. Keith, R. S. Jonhson and M. Celeste Simon, Nat. Rev. Cancer, 2012, 12, 9–22 Search PubMed; A. Loboda, A. Jozkowicz and J. Dulak, Vasc. Pharmacol., 2012, 56, 245 CrossRef CAS PubMed; B. Keith, R. S. Johnson and M. C. Simon, Nat. Rev. Cancer, 2012, 12, 9 Search PubMed; A. Siddiq, L. R. Aminova, C. M. Troy, K. Suh, Z. Messer, G. L. Semenza and R. R. Ratan, J. Neurosci., 2009, 29, 8838 CrossRef PubMed.
- M. Bhagat, J. K. Palanichamy, K. Jayanth, P. Ramalingal, M. Mudassir, K. Irshad, K. Chosdol, C. Sarkar, P. Seth, S. Goswami, S. Sinha and P. Chattopadhyay, Int. J. Biochem. Cell. Biol., 2016, 74, 60 CrossRef CAS PubMed; T. Imamura, H. Kikuchi, M.-T. Herraiz, D.-Y. Park, Y. Mitzukami, M. Mino-Kenduson, M. P. Lynch, B. R. Rueda, Y. Benita, R. J. Xavier and D. C. Chung, Int. J. Cancer, 2009, 124, 763 CrossRef PubMed.
- N. Takeda, E. L. O'Dea, A. Doedens, J.-W. Kim, A. Weidemann, C. Stockmann, M. Asagiri, M. C. Simon, A. Hoffmann and R. S. Johnson, Genes Dev., 2010, 24, 491 CrossRef CAS PubMed.
- L. Ravenna, L. Salvatori and M. A. Russo, FEBS J., 2016, 283, 993 CrossRef CAS PubMed; S. Kobayashi, T. Yamashita, K. Ohneda, M. Nagano, K. Kimura, H. Nakai, L. Poellinger and O. Ohneda, Genes Cells, 2015, 20, 224 CrossRef PubMed; P. Zhang, Q. Yao, L. Lu, Y. Li, P.-J. Chen and C. Duan, Cell Rep., 2014, 6, 1110 CrossRef PubMed; H. Ando, A. Natsume, K. Iwami, F. Ohka, T. Kuchimaru, S. Kizaka-Kondoh, K. Ito, K. Saito, S. Sugita, T. Hoshino and T. Wakabayashi, Biochem. Biophys. Res. Commun., 2013, 433, 139 CrossRef PubMed; T. Takana, M. Wiesner, W. Bernhardt, K. U. Eckardt and C. Warnecke, Biochem. J., 2009, 424, 143–153 CrossRef PubMed.
- G. L. Semenza, Annu. Rev. Phytopathol., 2014, 9, 47 CrossRef CAS PubMed; R. H. Wenger, D. P. Stiehl and G. Camenisch, Sci. STKE, 2005, 306, re12 Search PubMed; G. L. Semenza, Nat. Rev. Cancer, 2003, 3, 721 CrossRef PubMed.
- T. Chen, Z. Ren, L.-C. Ye, P.-H. Zhou, J.-M. Xu, Q. Shi, L.-Q. Yao and Y.-S. Zhong, Cancer Biol. Ther., 2015, 16, 244 CrossRef CAS PubMed; P. C. Mahon, K. Hirota and G. L. Semenza, Genes Dev., 2001, 15, 2675 CrossRef PubMed; H. Tarhonskaya, A. P. Hardy, E. A. Howe, N. D. Loik, H. B. Kramer, J. S. O. McCullagh, C. J. Schofield and E. Flashman, J. Biolumin. Chemilumin., 2015, 290, 19726 Search PubMed; E. Wang, C. Zhang, N. Polavaram, F. Liu, G. Wu, M. A. Schroeder, J. S. Lau, D. Mukhopadhyay, S.-W. Jiang, B. P. O'Neil, K. Datta and J. Li, PLoS One, 2014, 9, e86102 Search PubMed.
- G. Purnachandra Nagaraju, T.-E. Long, W. Park, J. C. Landry, L. T. Taliaferro-Smith, A. B. Farris, R. Diaz and B. F. El-Rayes, Mol. Carcinog., 2015, 54, 1147 CrossRef CAS PubMed; J. S. Isaacs, Y. J. Jung, E. G. Mimnaugh, A. Martinez, F. Cuttitta and L. M. Neckers, J. Biolumin. Chemilumin., 2002, 277, 29936 Search PubMed.
- R. Ravi, B. Mookerjee, Z. Bhujwalla, C. H. Sutter, D. Artemov, Q. Zeng, L. E. Dillehay, A. Madan, G. L. Semenza and A. Bedi, Genes Dev., 2000, 14, 34 CAS.
- H. Suzuki, A. Tomida and T. Tsuro, Oncogene, 2001, 20, 5779 CrossRef CAS PubMed.
- I. Mylonis, G. Chachami, M. Samiotaki, G. Panayotou, E. Paraskeva, A. Kalousi, E. Georgatsou, S. Bonanou and G. Simos, J. Biolumin. Chemilumin., 2006, 281, 33095 CAS.
- M. Jalouli, M.-A. C. Déry, V. N. Lafleur, L. Lamalice, X. Z. Zhou, K. P. Lu and D. E. Richard, Cell. Signalling, 2014, 26, 1649 CrossRef CAS PubMed.
- H. J. Han, N. Kwon, M.-A. Choi, K. O. Jung, J.-Y. Piao, H. K. C. Ngo, S.-J. Kim, D.-H. Kim, J.-K. Chung, Y.-N. Cha, H. Youn, B. Y. Choi, S.-H. Min and Y.-J. Surh, PLoS One, 2016, 11, e147038 Search PubMed.
- N. A. Warfel, N. G. Dolloff, D. T. Dicker, J. Malysz and W. S. El-Deiry, Cell Cycle, 2013, 12, 3689 CrossRef CAS PubMed.
- D. Xu, Y. Yao, L. Lu, M. Costa and W. Dai, J. Biolumin. Chemilumin., 2010, 50, 38944 Search PubMed; D. Flugel, A. Gorlach, C. Michiels and T. Kietzmann, Mol. Cell. Biol., 2007, 27, 3253 CrossRef PubMed.
- M. Santoni, F. Pantano, C. Amantini, M. Nabissi, A. Conti, L. Burattini, A. Zoccoli, R. Berardi, G. Santoni, G. Tonini, D. Santini and S. Cascinu, Biochim. Biophys. Acta, 2014, 1845, 221 Search PubMed; D. A. Guertini and D. M. Sabatini, Cancer Cell, 2007, 12, 9 CrossRef CAS PubMed; C. C. Hudson, M. Liu, G. G. Chiang, D. M. Otterness, D. C. Loomis, F. Kaper, A. J. Giaccia and R. T. Abraham, Mol. Cell. Biol., 2002, 22, 7004 CrossRef PubMed; M. Alvarez-Tejado, S. Naranjo-Suarez, C. Jimenez, A. C. Carrera, M. O. Landzuri and L. Del Peso, J. Biolumin. Chemilumin., 2001, 276, 22368 Search PubMed.
- R. Fukuda, K. Hirota, F. Fan, Y. D. Jung, L. M. Ellis and G. L. Semenza, J. Biolumin. Chemilumin., 2002, 277, 22368 CAS; N. Sang, D. P. Stiehl, J. Bohensky, I. Leshchinsky, V. Srinivas and J. Caro, J. Biolumin. Chemilumin., 2003, 278, 14013 Search PubMed.
- D. Zeng, J. Wang, P. Kong, C. Chang, J. Li and J. Li, Int. J. Clin. Exp. Pathol., 2014, 7, 2172 Search PubMed; X. Ge, F. Zhen, B. Yang, X. Yang, J. Cai, C. Chang, S. Zhang, Y. Cao, J. Ma, H. Cheng and X. Sun, J. Int. Med. Res., 2014, 42, 628 CrossRef CAS PubMed.
- S. Movafagh, S. Crook and V. Kim, J. Cell. Biol., 2015, 116, 696 CAS.
- A. J. Majmundar, W. J. Kong and M. C. Simon, Mol. Cell, 2010, 40, 294 CrossRef CAS PubMed.
- H. J. Nakaoka, T. Hara, S. Yoshino, A. Kanamori, Y. Matsui, T. Shimamura, H. Sato, Y. Murakami, M. Seiki and T. Sakamoto, Sci. Rep., 2016, 6, 22784 CrossRef CAS PubMed; Y.-H. Kim, K.-C. Yoo, Y.-H. Cui, N. Uddin, E.-J. Lim, M.-J. Kim, S.-Y. Nam, I.-G. Kim, Y. Suh and S.-J. Lee, Cancer Lett., 2014, 354, 132 CrossRef PubMed.
- Y. Kim, H. J. Nam, J. Lee, D. Y. Park, C. Kim, Y. S. Yu, D. Kim, S. W. Park, J. Bhin, D. Hwang, H. Lee, G. Y. Koh and S. H. Baek, Nat. Commun., 2016, 7, 10347 CrossRef CAS PubMed.
- P. Xia and X.-Y. Xu, Am. J. Cancer Res., 2015, 5, 1602 CAS.
- A. Akintunde, P. Avvaru, M. Furqan, Y. Song and D. Liu, J. Hematol. Oncol., 2013, 6, 88 CrossRef PubMed.
- B.-H. Jiang, G. Jiang, J. Z. Zheng, Z. Lu, T. Hunter and P. K. Vogt, Cell Growth Differ., 2001, 12, 363 CAS.
- Y. Imai, H. Yamagishi, Y. Ono and Y. Ueda, Linchuang Shuxue Yu Jianyan, 2012, 1, 24 Search PubMed.
- R. Marone, V. Cmiljanovic, B. Giese and M. P. Wymann, Biochim. Biophys. Acta, 2008, 1784, 159 CrossRef CAS PubMed.
- J. Chen, R. Shao, F. Li, M. Monteiro, J.-P. Liu, Z. P. Xu and W. Gu, Clin. Exp. Pharmacol. Physiol., 2015, 42, 1317 CrossRef CAS PubMed; J. Karar, G. J. Cerniglia, T. Lindsten, C. Koumenis and A. Maity, Cancer Biol. Ther., 2012, 13, 1102 CrossRef PubMed; S.-M. Maira, F. Stauffer, J. Brueggen, P. Furet, C. Schnell, C. Fritsch, S. Brachmann, P. Chène, A. DePover, K. Schoemaker, D. Fabbro, D. Gabriel, M. Simonen, L. Murphy, P. Finan, W. Sellers and C. Garcia-Echeverria, Mol. Cancer Ther., 2008, 7, 1851 CrossRef PubMed.
- T. Vandamme, M. Beyens, K. De Beeck, F. Dogan, P. M. Van Koetsvel, P. Pauwels, G. Mortier, C. Vangestel, W. De Herder, G. Van Camp, M. Peeters and L. J. Hofland, Br. J. Cancer, 2016, 114, 650 CrossRef CAS PubMed; Z. Sun, Q. Li, S. Zhang, J. Chen, L. Huang, J. Ren, Y. Chang, Y. Liang and G. Wu, OncoTargets Ther., 2015, 8, 269 Search PubMed.
- M.-J. Kim, J.-E. Koo, G.-Y. Han, B. Kim, Y.-S. Lee, C. Ahn and C.-W. Kim, J. Korean Med. Sci., 2016, 31, 360 CrossRef CAS PubMed; J. Chen, R. Shao, F. Li, M. Monteiro, J.-P. Liu, Z. P. Xu and W. Gu, Clin. Exp. Pharmacol. Physiol, 2015, 42, 1317 CrossRef PubMed; W.-J. Wang, L.-M. Long, N. Yang, Q.-Q. Zhang, W. J. Ji, J.-H. Zhao, Z.-H. Qin, Z. Wang, G. Chen and Z.-Q. Liang, Acta Pharmacol. Sin., 2013, 34, 681 CrossRef PubMed.
- M. Mendiburu-Eliçabe, J. Gil-Ranedo and M. Izquierdo, Clin. Transl. Oncol., 2014, 16, 495 CrossRef CAS PubMed; Y. Wang, Y. Liu, J. Lu, P. Zhang, Y. Wang, Y. Xu, Z. Wang, J. H. Mao and G. Wei, Biochem. Biophys. Res. Commun., 2013, 434, 352 CrossRef PubMed.
- G.-M. Karthik, R. Ma, J. LÖvrot, L. L. Kis, C. Lindh, L. Blomquist, I. Fredriksson, J. Bergh and J. Hartman, Cancer Lett., 2015, 367, 76 CrossRef CAS PubMed; M.-T. Mueller, P. C. Hermann, J. Witthauer, B. Rubio-Viqueira, S. F. Leicht, S. Huber, J. W. Ellwart, M. Mustafa, P. Barntenstein, J. G. d'Haese, M. H. Schoenberg, F. Berger, K.-W. Jauch, M. Hidalgo and C. Heeschen, Gastroenterology, 2009, 137, 1102 CrossRef PubMed.
- R. A. Figlin, P. De Souza, D. McDermott, J. P. Dutcher, A. Berkenblit, A. Thiele, M. Krygowski, A. Strahs, J. Feingold, J. Boni and G. Hudes, Cancer, 2009, 115, 3651 CrossRef CAS PubMed.
- J. C. Yao, N. Fazio, S. Singh, R. Buzzoni, C. Carnaghi, E. Wolin, J. Tomasek, M. Raderer, H. Lahner, M. Voi, L. B. Pacaud, N. Rouyrre, C. Sachs, J. W. Juan, G. D. Fave, E. Van Cutsem, M. Tesselaar, Y. Margot, Y. Shimada, D.-Y. Oh, J. Strosberg, M. H. Kulke and M. E. Pavel, Lancet, 2016, 387, 968 CrossRef CAS.
- H. Chow, P. M. Ghosh, R. De Vere White, C. P. Evans, M. A. Dall'Era, S. A. Yap, Y. Li, L. A. Beckett, P. N. Lara and C.-X. Pan, Cancer, 2016, 122, 1897 CrossRef CAS PubMed; A. C. De Melo, R. Grazziotin-Reisner, F. Erlich, M. S. Fontes Dias, G. Moralez, M. Carneiro, A. H. Ingles Garces, A. Guerra, V. Flavia, B. Novaes Neto, M. Fuchshuber-Moraes, J. Morando, G. Suarez-Kurtz and C. G. Ferreira, Cancer Chemother. Pharmacol., 2016, 78, 101 CrossRef PubMed; G. N. Hortobagyi, D. Chen, M. Piccart, H. S. Rugo, H. A. Burris, K. I. Pritchard, M. Campone, S. Noguchi, A. T. Perez, I. Deleu, M. Shtivelband, N. Mazuda, S. Dakhil, I. Anderson, D. M. Robinson, W. He, A. Garg, E. R. Mc Donald, H. Bitter, A. Huang, T. Taran, T. Bachelot, F. Lebrun, D. Lebwohl and J. Baselga, J. Clin. Oncol., 2016, 34, 419 CrossRef PubMed.
- Y.-Q. Liu, W.-Q. Li, S. L. Morris-Natschke, K. Qian, L. Yang, G.-X. Zhu, X.-B. Wu, A.-L. Chen, S.-Y. Zhang, X. Nan and K.-H. Lee, Med. Res. Rev., 2015, 35, 735 Search PubMed.
- S. Kummar, M. Raffeld, L. Juwara, Y. Horneffer, A. Strassberger, D. Allen, S. M. Steinberg, A. Rapisarda, S. D. Spencer, W. D. Figg, X. Chen, I. B. Turkbey, P. Choyke, A. J. Murgo, J. H. Doroshow and G. Melillo, Clin. Cancer Res., 2011, 17, 5123 CrossRef CAS PubMed; S. Tsunetoh, Y. Terai, H. Sasaki, A. Tanabe, Y. Tanaka, T. Sekijima, S. Fujiwara, H. Kawaguchi, M. Kanemura, Y. Yamashita and M. Ohmichi, Cancer Biol. Ther., 2010, 11, 1137 CrossRef PubMed; A. Rapisarda, J. Zalek, M. Hollingshead, T. Braunschweig, B. Uranchimeg, C. A. Bonomi, S. D. Borgel, J. P. Carter, S. M. Hewitt, R. H. Shoemaker and G. Melillo, Cancer Res., 2004, 64, 6845 CrossRef PubMed.
- S. Svenson, M. Wolfgang, J. Hwang, J. Ryan and S. Eliasof, J. Controlled Release, 2011, 153, 49 CrossRef CAS PubMed.
- G. J. Weiss, J. Chao, J. D. Neidhart, R. K. Ramanathan, D. Bassett, J. A. Neidhart, C. H. J. Choi, W. Chow, V. Chung, S. J. Forman, E. Garmey, J. Hwang, D. L. Kalinoski, M. Koczywas, J. Longmate, R. J. Melton, R. Morgan, J. Oliver, J. J. Peterkin, J. L. Ryan, T. Schulep, T. W. Synold, P. Twardowski, M. E. Davis and Y. Yen, Invest. New Drugs, 2013, 31, 986 CrossRef CAS PubMed.
- S. J. Conley, T. L. Baker, J. P. Burnett, R. L. Theisen, D. Lazarus, C. G. Peters, S. G. Clouthier, S. Eliasof and M. S. Wicha, Breast Cancer Res. Treat., 2015, 150, 559 CrossRef CAS PubMed; S. Gaur, Y. Wang, L. Kretzner, L. Chen, T. Yen, X. Wu, Y.-C. Yuan, M. Davis and Y. Yen, Nanomedicine, 2014, 10, 1477 Search PubMed; E. Pham, M. J. Birrer, S. Eliasof, E. G. Garmey, D. Lazarus, C. R. Lee, S. Man, U. A. Matulonis, C. G. Peters, P. Xu, C. Krasner and R. S. Kerbel, Clin. Cancer Res., 2014, 21, 808 CrossRef PubMed; S. Gaur, L. Chen, T. Yen, Y. Wang, B. Zhou, M. Davis and Y. Yen, Nanomedicine, 2012, 8, 721 Search PubMed.
- L. M. Greeberger, I. D. Horak, D. Filpula, P. Sapra, M. Westergaad, H. F. Frydenlund, C. Albæk, H. Schrøder and H. Ørum, Mol. Cancer Ther., 2008, 7, 3598 CrossRef PubMed.
- W. Jeong, A. Rapisarda, S. R. Park, R. J. Kinders, A. Chen, G. Melillo, B. Turkbey, S. M. Steinberg, P. Choyke, J. H. Doroshow and S. Kummar, Cancer Chemother. Pharmacol., 2014, 73, 343 CrossRef CAS PubMed.
- M. A. Callero, G. V. Suarez, G. Luzzani, B. Itkin, B. Nguyen and A. I. Loaiza-Perez, Int. J. Oncol., 2012, 41, 125 Search PubMed; E. Terzuoli, M. Puppo, A. Rapisarda, B. Uranchimeg, L. Cao, A. M. Burger, M. Ziche and G. Melillo, Cancer Res., 2010, 70, 6837 CrossRef CAS PubMed.
- H. Jung, S. Y. Shin, Y. Jung, T. A. Tran, H. O. Lee, K.-Y. Jung, D. Koh, S. K. Cho and Y. Lim, Chem. Biol. Drug Des., 2015, 86, 496 CAS.
- T. A. Tran, K. S. Ahn, Y. W. Song, J. Y. Moon, M. Cho, Y. Lim and S. K. Cho, Arch. Biochem. Biophys., 2014, 562, 92 CrossRef CAS PubMed.
- B. C. Soner, H. Aktug, E. Acikgoz, F. Duzagac, U. Guven, S. Ayla, C. Cal and G. Oktem, Int. J. Mol. Med., 2014, 34, 1249 CAS.
- B. S. Kumar, D. S. Raghuvanshi, M. Hasanain, S. Alam, J. Sarkar, K. Mitra, F. Khan and A. S. Negi, Steroids, 2016, 110, 9 CrossRef CAS PubMed.
- M. R. Harrison, N. M. Hahn, R. Pili, W. K. Oh, H. Hammers, C. Sweeney, K. M. Kim, S. Perlman, J. Arnott, C. Sidor, G. Wilding and G. Liu, Invest. New Drugs, 2011, 29, 1465 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, Y. Liu, S. N. Malek and P. Zeng, WO2011/068563A1, 2011.
- Q. Zhou, D. Gustafson, S. Nallapareddy, S. Diab, S. Leong, K. Lewis, L. Gore, W. A. Messersmith, A. M. Treston, S. G. Eckhardt, C. Sidor and D. R. Camidge, Invest. New Drugs, 2011, 29, 340 CrossRef CAS PubMed.
- H. Zhang, D. Z. Qian, Y. S. Tan, K. A. Lee, P. Gao, Y. R. Ren, S. Rey, H. Hammers, D. Chang, R. Pili, C. V. Dang, J. O. Liu and G. S. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19579 CrossRef CAS PubMed.
- C. Gao, S. Li, T. Zhao, J. Chen, H. Ren, H. Zhang, X. Wang, M. Lang, J. Liu, S. Gao, X. Zhao, J. Sheng, Z. Yuan and J. Hao, PLoS One, 2015, 10, e0121338 Search PubMed.
- C.-M. Xie, X.-Y. Liu, S. Yu and H. K. Christopher, Carcinogenesis, 2013, 34, 1870 CrossRef CAS PubMed.
- S. Welsh, R. Williams, L. Kirkpatrick, G. Paine-Murrieta and G. Powis, Mol. Cancer Ther., 2004, 3, 233 CAS.
- M. Y. Koh, T. Spivak-Kroizman, S. Venturini, S. Welsh, R. R. Williams, D. L. Kirkpatrick and G. Powis, Mol. Cancer Ther., 2008, 7, 90 CrossRef CAS PubMed.
- According to the Oncothyreon press release available on the web: http://ir.cascadianrx.com.
- A. Miyamoto, H. Okimoto, H. Shinohara and Y. Shibamoto, Eur. J. Radiol., 2006, 16, 1050 CrossRef CAS PubMed; L. Xu, Y. Liu, Z. Chen, W. Li, Y. Liu, L. Wang, L. Ma, Y. Shao, Y. Zhao and C. Chen, Adv. Mater., 2013, 25, 5928 CrossRef PubMed.
- H. Peng, G. Xing, E. Blanco, Y. Song, L. Zhao, B. Sun, X. Li, P. C. Wang, A. Korotcov, W. Li, X.-J. Liang, C. Chen, H. Yuan, F. Zhao, Z. Chen, T. Sun, Z. Chai, M. Ferrari and Y. Zhao, Nanomedicine, 2012, 8, 136 Search PubMed; C. Chen, G. Xing, J. Wang, Y. Zhao, B. Li, J. Tang, G. Jia, T. Wang, J. Sun, L. Xing, H. Yuan, Y. Gao, H. Meng, Z. Chen, F. Zhao, Z. Chai and X. Fang, Nano Lett., 2005, 5, 2050 CrossRef CAS PubMed.
- Y. Liu, C. Chen, P. Qian, X. Lu, B. Sun, X. Zhang, L. Wang, X. Gao, H. Li, Z. Chen, J. Tang, W. Zhang, J. Dong, R. Bai, P. E. Lobie, Q. Wu, S. Liu, H. Zhang, F. Zhao, M. S. Wicha, T. Zhu and Y. Zhao, Nat. Commun., 2015, 6, 5988 CrossRef CAS PubMed.
- D. S. Hong, U. Banerji, B. Tavana, G. C. George, J. Aaron and R. Kurzrock, Cancer Treat. Rev., 2013, 39, 375 CrossRef CAS PubMed.
- H. S. Kim, M. Hong, S. C. Lee, H.-Y. Lee, Y.-G. Suh, D.-C. Oh, J. H. Seo, H. Choi, J. Y. Kim, K.-W. Kim, J. H. Kim, J. Kim, Y.-M. Kim, S.-J. Park, H.-J. Park and J. Lee, Eur. J. Med. Chem., 2015, 104, 157 CrossRef CAS PubMed; G. P. Nagaraju, S. Zhu, J. E. Ko, N. Ashritha, R. Kandimalla, J. P. Snyder, M. Shoji and B. F. El-Rayes, Cancer Lett., 2015, 357, 557 CrossRef PubMed; X. Song, J. Yao, F. Wang, M. Zhou, Y. Zhou, H. Wang, L. Wei, L. Zhao, Z. Li, N. Lu and Q. Guo, Toxicol. Appl. Pharmacol., 2013, 271, 144 CrossRef PubMed.
- S.-H. Lee, J.-G. Jee, J. S. Bae, K.-H. Liu and Y. M. Lee, J. Cell. Physiol., 2015, 230, 853 CrossRef CAS PubMed.
- W. H. Lee, J. M. Lee, C. Lim, S. Kim and S. G. Kim, Chem.-Biol. Interact., 2013, 204, 49 CrossRef CAS PubMed.
- N. J. Mabjeesh, P. D. E. Ost, M. T. Willard, B. Kaur, E. G. Van Meir, J. W. Simons and H. Zhong, Cancer Res., 2002, 62, 2478 CAS.
- B. Newman, Y. Liu, H.-F. Lee, D. Sun and Y. Wang, Cancer Res., 2012, 72, 4551 CrossRef CAS PubMed.
- M. Tatokoro, F. Koga, S. Yoshida, S. Kawakami, Y. Fujii, L. Neckers and K. Kihara, Int. J. Cancer, 2012, 131, 987 CrossRef CAS PubMed.
- S. S. Desale, S. M. Raja, J. O. Kim, B. Mohapatra, K. S. Soni, H. Luan, S. H. Williams, T. A. Bielecki, D. Feng, M. Storck, V. Band, S. M. Cohan, H. Band and T. K. Bronich, J. Controlled Release, 2015, 208, 59 CrossRef CAS PubMed.
- R. Yang, Q. Tang, F. Miao, Y. An, M. Li, Y. Han, X. Wang, J. Wang, P. Liu and R. Chen, Int. J. Nanomed., 2015, 10, 7345 CrossRef CAS PubMed.
- W. Ying, Z. Du, L. Sun, K. P. Foley, D. A. Proia, R. K. Blackman, D. Zhou, T. Inoue, N. Tatsuta, J. Sang, S. Ye, J. Acquaviva, L. S. Ogawa, Y. Wada, J. Barsoum and K. Koya, Mol. Cancer Ther., 2011, 11, 475 CrossRef PubMed.
- T. Shimamura, S. A. Perera, K. P. Foley, J. Sang, S. J. Rodig, T. Inoue, L. Chen, D. Li, J. Carretero, Y.-C. Li, P. Sinha, C. D. Carey, C. L. Borgman, J.-P. Jimenez, M. Meyerson, W. Ying, J. Barsoum, K.-K. Wong and G. I. Shapiro, Clin. Cancer Res., 2012, 18, 4973 CrossRef CAS PubMed.
- L. Xiang, D. M. Gilkes, P. Chaturvedi, W. Luo, H. Hu, N. Takano, H. Liang and G. L. Semenza, J. Mol. Med., 2014, 92, 151 CrossRef CAS PubMed.
- S. Ramalingam, G. Goss, R. Rosel, G. Schmidt-Bindert, B. Zaric, Z. Andric, I. Bondarenko, D. Komov, T. Ceric, F. Khuri, M. Samarzija, E. Felip, T. Ciuleanu, V. Hirsh, T. Wehler, J. Spicer, R. Salgia, G. Shapiro, E. Sheldon, F. Teofilovici, V. Vukovic and D. Fennell, Ann. Oncol., 2015, 26, 1741 CrossRef CAS PubMed; J. W. Goldman, R. N. Raju, G. A. Gordon, I. El-Hariry, F. Teofilivici, V. M. Vukovic, R. Bradley, M. D. Karol, Y. Chen, W. Guo, T. Inoue and L. S. Rosen, BMC Cancer, 2013, 13, 152 CrossRef PubMed.
- M. Manal, M. J. N. Chandrasekar, J. G. Priya and M. J. Nanjan, Bioorg. Chem., 2016, 67, 18 CrossRef CAS PubMed.
- D. Z. Qian, S. K. Kachhlap, S. J. Collins, H. M. W. Verheul, M. A. Carducci, P. Atadja and R. Pili, Cancer Res., 2006, 66, 8814 CrossRef CAS PubMed.
- H. Geng, Q. Liu, C. Xue, L. L. David, T. M. Beer, G. V. Thomas, M.-S. Dai and D. Z. Qian, J. Biolumin. Chemilumin., 2012, 287, 35496 CAS; H. Geng, C. T. Harvey, J. Pittsenberger, Q. Liu, T. M. Beer, C. Xue and D. Z. Qian, J. Biolumin. Chemilumin., 2011, 286, 38095 Search PubMed.
- D. M. Hutt, D. M. Roth, H. Vignaud, C. Cullin and M. Bouchecareilh, PLoS One, 2014, 9, e106224 Search PubMed.
- C. Fischer, K. Leithner, C. Wohlkoenig, F. Quehenberger, A. Bertsch, A. Olschewski, H. Olschewski and A. Hrzenjak, Mol. Cancer Ther., 2015, 14, 4 CrossRef CAS PubMed; F. M. Frame, D. Pellacani, A. T. Collins, M. S. Simms, V. M. Mann, G. D. D. Jones, M. Meuth, R. G. Bristow and N. J. Maitland, Br. J. Cancer, 2013, 109, 3023 CrossRef PubMed.
- A. Romanski, K. Schwartz, M. Keller, S. Wietbrauk, A. Vogel, J. Roos, C. Oancea, B. Brill, O. H. Krämer, H. Serve, M. Ruthardt and G. Bug, Cell Cycle, 2012, 11, 3219 CrossRef CAS PubMed.
- R. Pathania, S. Ramachandran, G. Mariappan, P. Thakur, H. Shi, J.-H. Choi, S. Manicassamy, R. Kolhe, P. D. Prasad, S. Sharma, B. L. Lokeshwar, V. Ganapathy and M. Thangaraju, Cancer Res., 2016, 76, 3224 CrossRef CAS PubMed; A. Schech, A. Kazi, S. Yu, P. Shah and G. Sabnis, Mol. Cancer Ther., 2015, 14, 1848 CrossRef PubMed; M. Kai, N. Kanaya, S. V. Wu, C. Mendez, D. Nguyen, T. Luu and S. Chen, Breast Cancer Res. Treat., 2015, 151, 281 CrossRef PubMed; A. A. Alvarez, M. Field, S. Bushnev, M. S. Longo and K. Sugaya, J. Mol. Neurosci., 2015, 55, 7 CrossRef PubMed; V. Haghpanah, M. Malehmir, B. Larijani, S. Ahmadian, K. Alimoghaddam and R. Heshmat, J. Thyroid Res., 2014, 218763 Search PubMed; F. S. Giudice, D. S. Pinto Jr., J. E. Nör, C. H. Squarize and R. M. Castilho, PLoS One, 2013, 8, e58672 Search PubMed.
- T. Kozako, T. Suzuki, M. Yoshimitsu, N. Arima, S.-I. Honda and S. Soeda, Molecules, 2014, 19, 20295 CrossRef PubMed.
- A. Laemmle, A. Lechleiter, V. Roh, C. Schwarz, S. Portmann, C. Furer, A. Keogh, M. P. Tschan, D. Candinas, S. A. Vorburger and D. Storka, PLoS One, 2012, 7, e33433 CAS.
- R. Chen, M. Xu, R. T. Hogg, J. Li, B. Little, R. D. Gerard and J. A. Garcia, J. Biolumin. Chemilumin., 2012, 287, 30800 CAS.
- W. Dai, J. Zhou, B. Jin and J. Pan, Nature, 2016, 6, 22622 CAS.
- S. D. Lee, W. Kim, J.-W. Jeong, J.-W. Park and J.-E. Kim, Cancer Lett., 2016, 373, 138 CrossRef CAS PubMed; M. G. Cheon, W. Kim, M. Choi and J.-E. Kim, Cancer Lett., 2015, 356, 637 CrossRef PubMed.
- L. W. S. Finley, A. Carracedo, J. Lee, A. Souza, A. Egia, J. Zhang, J. Teruya-Feldstein, P. I. Moreira, S. M. Cardoso, C. B. Clish, P. P. Pandofi and M. C. Haigis, Cancer Cell, 2011, 19, 416 CrossRef CAS PubMed; P. T. Schumacker, Cancer Cell, 2011, 19, 299 CrossRef PubMed.
- L. Wei, Y. Zhou, C. Qiao, T. Ni, Z. Li, Q. You, Q. Guo and N. Lu, Cell Death Dis., 2015, 6, e1714 CrossRef CAS PubMed.
- Y. Li, W. Wang, X. Xu, S. Sun and X.-J. Qu, Biochem. Pharmacol., 2015, 69, 125 CAS.
- S. J. Welsh, W. T. Bellamy, M. M. Briehl and G. Powis, Cancer Res., 2002, 62, 5089 CAS.
- J. Zhou, A. E. Damdimopoulos, G. Spyrou and B. Brüne, J. Biolumin. Chemilumin., 2007, 287, 7482 Search PubMed.
- S. Naranjo-Suarez, B. A. Carlson, R. Tobe, M.-H. Yoo, P. A. Tsuji, V. N. Gladyshev and D. L. Hatfield, Mol. Cells, 2013, 36, 151 CrossRef CAS PubMed.
- S. J. Welsh, R. R. Williams, A. Birmingham, D. J. Newman, D. L. Kirkpatrick and G. Powis, Mol. Cancer Ther., 2003, 2, 235 Search PubMed; D. L. Kirkpatrick, M. Kuperus, M. Dowdeswell, N. Potier, L. J. Donald, M. Kunkel, M. Berggren, M. Angulo and G. Powis, Biochem. Pharmacol., 1998, 55, 987 CrossRef CAS PubMed.
- A. F. Baker, K. N. Adab, N. Raghunand, H. H. S. Chow, S. P. Stratton, S. W. Squire, M. Boice, L. A. Pestano, D. L. Kirkpatrick and T. Dragovich, Invest. New Drugs, 2013, 31, 631 CrossRef CAS PubMed; R. K. Ramanathan, J. Abbruzzese, T. Dragovich, L. Kirkpatrick, J. M. Guillen, A. F. Baker, L. A. Pestano, S. Green and D. D. Von Hoff, Cancer Chemother. Pharmacol., 2011, 67, 503 CrossRef PubMed.
- D. T. Jones, C. W. Pugh, S. Wingfield, M. F. G. Stevens and A. L. Harris, Clin. Cancer Res., 2006, 12, 5384 CrossRef CAS PubMed.
- M. Sztiller-Sikorska, K. Koprowska, K. Majchrzak, M. Hartman and M. Czyz, PLoS One, 2014, 9, e90783 Search PubMed.
- M. Sweeney, R. Coyle, P. Kavanagh, A. A. Berezin, D. L. Re, G. A. Zissimou, P. A. Koutentis, M. P. Carty and F. Aldabbagh, Bioorg. Med. Chem., 2016, 24, 3665 CrossRef PubMed.
- T. Zou, C. T. Lum, C.-N. Lok, W.-P. To, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810 CrossRef CAS PubMed.
- E. Miranda, I. K. Nordgren, A. L. Male, C. E. Lawrence, F. Hoakwie, F. Cuda, W. Court, K. R. Fox, P. A. Townsend, G. K. Packham, S. A. Eccles and A. Tavassoli, J. Am. Chem. Soc., 2013, 135, 10418 CrossRef CAS PubMed.
- K. A. Lee, H. Zhang, D. Z. Qian, S. Rey, J. O. Liu and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17910 CrossRef CAS PubMed.
- D. Wu, N. Potluri, J. Lu, Y. Kim and F. Rastinejad, Nature, 2015, 524, 303 CrossRef CAS PubMed.
- M. Broekgaarden, R. Weijer, M. Krekorian, B. Van den Ijssel, M. Kos, L. K. Alles, A. C. Van Wijk, Z. Bikadi, E. Hazai, T. M. Van Gulik and M. Heger, Nano Res., 2016, 9, 1639 CrossRef CAS.
- K. Lee, J. H. Lee, S. K. Boovanahalli, Y. Jin, M. Lee, X. Jin, J. H. Kim, Y.-S. Hong and J. J. Lee, J. Med. Chem., 2007, 50, 1675 CrossRef CAS PubMed.
- K. Lee, J. E. Kang, S.-K. Park, Y. Jin, K.-S. Chung, H.-M. Kim, K. Lee, M. R. Kang, M. K. Lee, K. B. Song, E.-G. Yang, J.-J. Lee and M. Won, Biochem. Pharmacol., 2010, 80, 982 CrossRef CAS PubMed.
- M.-S. Won, N. Im, S. Park, S. K. Boovanahalli, Y. Jin, X. Jin, K.-S. Chung, M. Kang, K. Lee, S.-K. Park, H. M. Kim, B. M. Kwon, J. J. Lee and K. Lee, Biochem. Biophys. Res. Commun., 2009, 385, 16 CrossRef CAS PubMed.
- K. Shimizu, M. Maruyama, Y. Yasui, H. Minegishi, H. S. Ban and H. Nakamura, Bioorg. Med. Chem. Lett., 2010, 20, 1453 CrossRef CAS PubMed.
- S. Lin, S.-C. Tsai, C. C. Lee, B.-W. Wang, J. Y. Liou and K.-G. Shyu, Mol. Pharmacol., 2004, 66, 612 CAS.
- Q. Zhang, C. Zhang, X. Yang, B. Yang, J. Wang, Y. Kang, Z. Wang, D. Li, G. Huang, Z. Ma, X. Sun, J. Cai, G. Tao, S. Dai, W. Mo and J. Ma, Diagn. Pathol., 2014, 9, 98 CrossRef CAS PubMed; X. Yang, B. Yang, J. Cai, C. Zhang, Q. Zhang, L. Xu, Q. Qin, H. Zhu, J. Ma, G. Tao, H. Cheng and X. Sun, Cancer Biol. Ther., 2013, 14, 1068 CrossRef PubMed.
- M.-H. Li, Z.-H. Miao, W.-F. Tan, J.-M. Yue, C. Zhang, L.-P. Lin, X.-W. Zhang and J. Ding, Clin. Cancer Res., 2004, 10, 8266 CrossRef CAS PubMed.
- B. Yu, M.-H. Li, W. Wang, Y.-Q. Wang, Y. Jiang, S.-P. Yang, J.-M. Yue, J. Ding and Z.-H. Miao, J. Mol. Med., 2012, 90, 971 CrossRef CAS PubMed.
- G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11570 CrossRef CAS PubMed.
- S. A. Dames, M. Martinez-Yamout, R. N. De Guzman, H. J. Dyson and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5271 CrossRef CAS PubMed; S. J. Freedman, Z.-Y. J. Sun, F. Poy, A. L. Kung, D. M. Livingston, G. Wagner and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5367 CrossRef PubMed.
- K. M. Cook, S. T. Hilton, J. Mecinovic, W. B. Motherwell, W. D. Figg and C. J. Schofield, J. Biolumin. Chemilumin., 2009, 284, 26831 Search PubMed; A. L. Kung, S. D. Zabludoff, D. S. France, S. J. Freedman, E. A. Tanner, A. Viera, S. Cornell-Kennon, J. Lee, B. Wang, K. Memmert, H. U. Naegeli, F. Petersen, M. J. Eck, K. W. Bair, A. W. Wood and D. M. Livingston, Cancer Cell, 2004, 6, 33 CrossRef CAS PubMed.
- E. Viziteu, C. Grandmougin, H. Goldschmidt, A. Seckinger, D. Hose, B. Klein and J. Moreaux, Br. J. Cancer, 2016, 114, 519 CrossRef CAS PubMed.
- K. M. Reece, E. D. Richardson, K. M. Cook, T. J. Campbell, S. T. Pisle, A. J. Holly, D. J. Venson, D. J. Liewehr, C. H. Chau, D. K. Price and W. D. Figg, Mol. Cancer Ther., 2014, 13, 91 CrossRef PubMed.
- R. Dubey, M. D. Levin, L. Z. Szabo, C. F. Laszlo, S. Kushal, J. B. Singh, P. Oh, J. E. Schnitzer and B. Z. Olenyuk, J. Am. Chem. Soc., 2013, 135, 4537 CrossRef CAS PubMed.
- M. K. P. Jayatunga, S. Thompson, T. C. McKee, M. C. Chan, K. M. Reece, A. P. Hardy, R. Sekirnik, P. T. Seden, K. M. Cook, J. B. McMahon, W. D. Figg, C. J. Schofield and A. D. Hamilton, Eur. J. Med. Chem., 2015, 94, 509 CrossRef CAS PubMed.
- L. K. Henchey, S. Kushal, R. Dubey, R. N. Chapman, B. Z. Olenyuk and P. S. Arora, J. Am. Chem. Soc., 2010, 132, 941 CrossRef CAS PubMed.
- G. M. Burslem, H. F. Kyle, P. Prabhakaran, A. L. Breeze, T. A. Edwards, S. L. Warriner, A. Nelson and A. J. Wilson, Org. Biomol. Chem., 2016, 14, 3782 Search PubMed; G. M. Burslem, H. F. Kyle, A. L. Breeze, T. A. Edwards, A. Nelson, S. L. Warriner and A. J. Wilson, ChemBioChem, 2014, 15, 1083 CrossRef CAS PubMed.
- S. T. S. Chan, P. R. Patel, T. R. Ransom, C. J. Henrich, T. C. McKee, A. K. L. Goey, K. M. Cook, W. D. Figg, J. B. McMahon, M. J. Schnermann and K. R. Gustafson, J. Am. Chem. Soc., 2015, 137, 5569 CrossRef CAS PubMed.
- A. K. L. Goey, C. H. Chau, T. M. Sissung, K. M. Cook, D. J. Venzon, A. Castro, T. R. Ransom, C. J. Henrich, T. C. McKee, J. B. McMahon, T. Grkovic, M. M. Cadelis, B. R. Copp, K. R. Gustafson and W. D. Figg, J. Nat. Prod., 2016, 79, 1267 CrossRef CAS PubMed.
- Y.-R. Na, K.-C. Han, H. Park and E. G. Yang, Biochem. Biophys. Res. Commun., 2013, 434, 879 CrossRef CAS PubMed; C. Tan, R. G. De Noronha, N. S. Devi, A. A. Jabbar, S. Kaluz, Y. Liu, S. R. Mooring, K. C. Nicolaou, B. Wang and E. G. Van Meir, Bioorg. Med. Chem. Lett., 2011, 21, 5528 CrossRef PubMed.
- Y. Yamazaki, Y. Hasebe, K. Egawa, K. Nose, S. Kunimoto and D. Ikeda, Biol. Pharm. Bull., 2006, 29, 1999 CAS.
- K. A. Lee, D. Z. Qian, S. Rey, H. Wei, J. O. Liu and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2353 CrossRef CAS PubMed.
- T. Tanaka, J. Yamaguchi, K. Shoji and M. Nangaku, J. Biolumin. Chemilumin., 2012, 287, 34866 CAS.
- D. Kong, E. J. Park, A. G. Stephen, M. Calvani, J. H. Cardellina, A. Monks, R. J. Fisher, R. H. Shoemaker and G. Melillo, Cancer Res., 2005, 65, 9047 CrossRef CAS PubMed.
- M. M. Van Dyke and P. B. Dervan, Science, 1984, 225, 1122 CAS.
- B. Vlaminck, S. Toffoli, B. Ghislain, C. Demazy, M. Raes and C. Michiels, FEBS J., 2007, 274, 5533 CrossRef CAS PubMed.
- A. Y. Chang, K. M. Kim, H. Boucher, P. Bonomi, J. A. Stewart, D. D. Karp and R. H. Blum, Cancer, 1998, 82, 292 CrossRef CAS PubMed.
- K. Hattori, K. Koike, K. Okuda, T. Hirayama, M. Ebihara, M. Takenaka and H. Nagasawa, Org. Biomol. Chem., 2016, 14, 2090 CAS.
- F. W. Hunter, B. G. Wouters and W. R. Wilson, Br. J. Cancer, 2016, 10, 1071 CrossRef CAS PubMed; D. Liang, G. H. Miller and G. K. Tranmer, Curr. Med. Chem., 2015, 22, 4313 CrossRef PubMed.
- R. M. Phillips, Cancer Chemother. Pharmacol., 2016, 77, 441 CrossRef CAS PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388 CrossRef CAS PubMed; T. Thambi, V. G. Deepagan, H. Y. Yoon, H. S. Han, S.-H. Kim, S. Son, D.-G. Jo, C.-H. Ahn, Y. D. Suh, K. Kim, I. C. Kwon, D. S. Lee and J. H. Park, Biomaterials, 2014, 35, 1735 CrossRef PubMed.
- F. W. Hunter, B. G. Wouters and W. R. Wilson, Br. J. Cancer, 2016, 114, 1071 CrossRef CAS PubMed.
- R. Sheng, Y. Xu, Q. J. Weng, Q. Xia, Q. J. He, B. Zhang and Y. Z. Hu, Drug Discoveries Ther., 2007, 1, 119 CAS.
- Q. J. Weng, J. Zhang, J. Cao, Q. Xia, D. D. Wang, Y. Z. Hu, R. Sheng, H. H. Wu, D. F. Zhu, H. Zhu, Q. He and B. Yang, Invest. New Drugs, 2011, 29, 1177 CrossRef CAS PubMed.
- P. L. Ross and J. L. Wolfe, J. Pharm. Sci., 2016, 105, 391 CrossRef CAS PubMed.
- H. Song, X. Su, K. Yang, F. Niu, J. Li, J. Song, H. Chen, B. Li, W. Li, W. Qian, X. Cao, S. Guo, J. Dai, S.-S. Feng, Y. Guo, C. Yin and J. Gao, J. Biomed. Nanotechnol., 2015, 11, 1927 CrossRef CAS PubMed.
- A. Al Faraj, A. S. Sjaik, E. Ratemi and R. Halwani, J. Controlled Release, 2016, 225, 240 CrossRef CAS PubMed.
- S.-T. Ning, S.-Y. Lee, M.-F. Wei, C.-L. Peng, S. Y.-F. Lin, M.-H. Tsai, P.-C. Lee, Y.-H. Shih, C.-Y. Lin, T.-Y. Luo and M.-J. Shieh, ACS Appl. Mater. Interfaces, 2016, 8, 17793 CAS.
- L. R. Saunders, A. J. Bankovich, W. C. Anderson, M. A. Aujay, S. Bheddah, K. A. Black, R. Desai, P. A. Escarpe, J. Hampl, A. Laysang, D. Liu, J. Lopez-Molina, M. Milton, A. Park, M. A. Pysz, H. Shao, B. Slingerland, M. Torgov, S. A. Williams, O. Foord, P. Howard, J. Jassem, A. Badzio, P. Czapiewski, D. H. Harpole, A. Dowlati, P. P. Massion, W. D. Travis, M. C. Pietanza, J. T. Poirier, C. M. Rudin, R. A. Stull and S. J. Dylla, Sci. Transl. Med., 2015, 7, 302ra136 CrossRef PubMed.
- X. Gong, A. Azhdarinia, S. C. Ghosh, W. Xiong, Z. An, Q. Liu and K. S. Carmon, Mol. Cancer Ther., 2016, 15, 1580 CrossRef CAS PubMed; M. R. Junttila, W. Mao, X. Wang, B.-E. Wang, T. Pham, J. Flygare, S.-F. Yu, S. Yee, D. Goldenberg, C. Fields, J. Eastham-Anderson, M. Singh, R. Vij, J.-A. Hongo, R. Firestein, M. Schutten, K. Flagella, P. Polakis and A. G. Polson, Sci. Transl. Med., 2015, 7, 314ra186 CrossRef PubMed; S. Mazzini, L. Scaglioni, R. Mondelli, M. Caruso and F. R. Sirtori, Bioorg. Med. Chem., 2012, 20, 6979 CrossRef PubMed.
- E. Guérin, W. Raffelsberger, E. Pencreach, A. Maier, A. Neuville, A. Schneider, Ph. Bachellier, S. Rohr, A. Petitprez, O. Poch, D. Moras, P. Oudet, A. K. Larsen, M. P. Gaub and D. Guenot, Mol. Med., 2012, 18, 83 Search PubMed.
- H. Song, R. He, K. Wang, J. Ruan, C. Bao, N. Li, J. Ji and D. Cui, Biomaterials, 2010, 31, 2302 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |