DOI:
10.1039/C6MD00466K
(Research Article)
Med. Chem. Commun., 2017,
8, 88-95
Phenotype-based variation as a biomarker of sensitivity to molecularly targeted therapy in melanoma†
Received
15th August 2016
, Accepted 11th October 2016
First published on 27th October 2016
Abstract
Transcriptomic phenotypes defined for melanoma have been reported to correlate with sensitivity to various drugs. In this study, we aimed to define a minimal signature that could be used to distinguish melanoma sub-types in vitro, and to determine suitable drugs by which these sub-types can be targeted. By using primary melanoma cell lines, as well as commercially available melanoma cell lines, we find that the evaluation of MLANA and INHBA expression is as capable as one based on a combined analysis performed with genes for stemness, EMT and invasion/proliferation, in identifying melanoma subtypes that differ in their sensitivity to molecularly targeted drugs. Using this approach, we find that 75% of melanoma cell lines can be treated with either the MEK inhibitor AZD6244 or the HSP90 inhibitor 17AAG.
Introduction
Melanoma is a heterogeneous tumor, as can be defined either by mutations or by transcriptomic analyses. More than 80% of all melanoma cells harbor a mutated gene within the MAPK pathway, all which are mutually exclusive.1,2 Patients with BRAF mutations are candidates for Vemurafenib, a selective BRAF inhibitor,3 and the administration of a MEK inhibitor (Trametinib) in these patients has shown to increase survival time,4 demonstrating the critical function of the MAPK pathway. In addition, close to 60% of melanomas contain mutations within the CDKN2A pathway-related genes. Although these gene mutations are also mutually exclusive within the pathway, they overlap with MAPK-related mutations, indicating the presence of at least two distinct pathways that are deregulated in this tumor type. However, the mutational landscape as defined by these and other mutations has been reported not to correlate with phenotypes that are based on the transcriptome of the same tumors.1,2 That gene expression-based phenotypes reveal the existence of heterogeneity among melanoma tumors is a well-established fact.1,2,5–7 Current data indicates the existence of at least 3 melanoma sub-groups as can be defined by transcriptomic analyses, with a preferential up-regulation of either differentiation, proliferation, or immune-related genes.1,2 Personalized treatment, such as CTLA4 and PD-1 blockade, a most promising treatment for advanced melanoma,8 is likely to benefit from these classifications as they might help design such therapy on an individual basis.9 However, melanoma cells per se show transcriptome-based heterogeneity when studied in vitro as well, and this is important as it has been shown to correlate with drug sensitivity.10 Thus, complete cure of melanoma might have to be pursued from two directions, one based on in vivo effects, and therefore the microenvironment of the tumor cells, and the other based on in vitro defined mechanisms related to drug sensitivity. Switching between phenotypes has been observed for melanoma in vivo, as well as in vitro, and can result in altered drug sensitivity, irrespective of the mutational status of the melanoma cells,5,11 suggesting that a combination treatment consisting of two drugs affecting each one of the two phenotypes might be ultimately necessary for efficient tumor eradication. Drug resistance has traditionally been associated with a stem-cell-like phenotype and a more mesenchymal state, as compared to an epithelial one in almost all tumors.12,13 The “stemness” of melanoma cells is thought to exist in a dynamic state, as melanoma cells with stem-cell markers have been reported to arise from cells lacking these markers, stem-cell like melanoma cells might not be required for tumor initiation, and the stem- and non-stem-cell phenotypes can switch dynamically.7,14 Similar to the plasticity reported for stemness in melanoma, epithelial-to-mesenchymal transition (EMT) has also been implied as a factor driving metastasis and causing drug resistance in melanoma.15,16 Thirdly, melanoma specific in vitro analyses that have aimed to explain metastatic potential have generated gene lists that could robustly classify melanoma cell lines into either a “proliferative” or an “invasive” character;6,17,18 switching among these phenotypes was also shown.5,14 Metastasis, drug resistance and invasion have all been attributed both to the stem cell phenotype and to EMT and have been observed in those classes defined by various researchers for melanoma. In this study, we asked if there was overlap between melanoma cells classified by these various signatures and if such melanoma subtypes would have unique responses to targeted therapy. We also aimed to identify a minimum number of biomarkers that could help classify melanoma subtypes with differing drug sensitivity profiles.
Materials and methods
Cell lines and tissue culture methods
Primary melanoma cell lines used in this study were previously described.19,20 All cell lines (primary and commercial) were incubated in humidified incubators supplied with 5% CO2 and cultured in RPMI1640 medium supplemented with 10% FBS, 1% 10 K/10 K penicillin–streptomycin (P/S Solution, Lonza, Basel, Switzerland) and 1% 200 mM L-glutamine (L-Glu, Lonza, Basel, Switzerland). Two exceptions were commercial cell lines Mewo and SK-MEL-28, which were cultured in DMEM with the same supplements. Melanocytic progeny of primary melanoma cell lines (PrMCLs) were investigated by staining for S100, MART-1, gp100 and GD2 expression using polyclonal rabbit anti-S100, monoclonal A-103, HMB-45 (Invitrogen, Carlsbad, CA, USA) and R-24 (a kind gift of Dr. Aliza Adler) antibodies, and flow cytometry-based (FACS) detection of HMW-MMA (CSPG4) (R&D Systems Minneapolis, MN, USA) (Table S1†). Samples were analyzed by FACS on a Becton Dickinson LSR II cytometer. Data analysis was performed using FCS express V.3 (De Novo Software).
Expression profiling of INHBA and MLANA by q-RT-PCR
Quantitative profiling of INHBA and MLANA by q-RT-PCR was done using a 7500 Real-Time PCR System (Applied Bio-systems, Carlsbad, CA, USA) and SYBR® Green PCR Master Mix (catalog number: 4309155, Applied Biosystems, Carlsbad, CA). PCR reactions were run under cycling conditions according to the manufacturer's instructions. GAPDH was used as the endogenous control in all reactions. PCR primers were designed using the NCBI Primer-Blast Tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primer sequences were: MLANA_F1-ATTAAGGAAGGTGTCCTGTGCC, MLANA_R1-CAGCCGTGGTGTAAGAGTGG, INHBA_F1-GTTTGCCGAGTCAGGAACAGC, INHBA_R1-GAAGAGCCAGACTTCTGCACG, GAPDH_F1-AAAGGGTCATCATCTCTGCCCCC and GAPDH_R1-GGCATTGCTGATGATCTTGAGGCTG. Relative expression values of INHBA and MLANA were calculated by the ddCt method as previously described.21
In vitro drug screening analyses
AZD0530, 17AAG and AZD6244 were purchased from Selleck Chemicals. Drugs were screened on transparent 96-well-plates in which cancer cell lines were seeded at 1000 cells per well 96 hours before cytotoxicity measurements. Drugs were screened using 10 different doses ranging from 0.001 μM to 50 μM, except 10a for which 6 different doses ranging from 0.625 μM to 20 μM were tested. All stock solutions were prepared in 0.1% DMSO. IC50 values were calculated as previously described.22 Cytotoxicity was measured with CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison WI, USA) after 72 hours of drug treatment.
In silico drug screening analyses
The normalized activity area or IC50 values of 24 different commercial drugs were downloaded from the CCLE database (https://portals.broadinstitute.org/ccle/home) for in silico drug screening analyses.
EMT, CSC and PRO/INT/INV score calculation
EMT and CSC scores were calculated based on an R script23 (http://www.R-project.org/) which runs a principal component analysis (PCA) with the following probeset expression values: EMT scoring was based on probeset expression values of E-CAD (201131_s_at) and VIM (201426_s_at); CSC scoring used CD271 (205858_at), CD20 (228599_at, 228592_at, 217418_x_at, 210356_x_at), CD133 (204304_s_at) and ALDH1 (212224_at), all of which were previously published melanoma stem-cell markers.24 CCLE and primary melanoma cell lines were classified as PRO (proliferative), INT (intermediate) and INV (invasive) according to Pro (r) and Inv (r) values generated using the online HOPP tool and the associated gene list.18 CCLE cell lines with a Pro (r) value of more than 0.652 and an Inv (r) value of less than 0.171 were designated as “PRO”, those with a Pro (r) value between 0.041 to 0.630 and an Inv (r) value between 0.254 to 0.600 were designated as “INT” and cells with a Pro (r) value of less than −0.200 and an Inv (r) value of more than 0.445 were designated as “INV” cells. For primary cell lines, designations were as follows: PRO: Pro (r) > 0.533 and Inv (r) < 0.162; INT: Pro (r): 0.186 to 0.588 and Inv (r): 0.221 to 0.581; INV: Pro (r) < 0.03 and Inv (r) > 0.554.
Melanoma gene expression signature analyses
E-MTAB-783 and GSE36133 commercial cancer cell line datasets were normalized separately using RMA normalization without background correction. Differentially expressed probesets were determined between melanoma cell lines and other solid tumor cell lines separately in both datasets according to p-value and fold change (FC) cutoffs, 0.0001 (Bonferroni corrected) and 4 (in either direction), respectively. Significant probesets common to both datasets were determined using VENNY, “Oliveros, J. C. (2007–15), an interactive tool for comparing lists with Venn diagrams (http://bioinfogp.cnb.csic.es/tools/venny/index.html).” These 223 probesets constitute “gene list #1”. Standardized expression values of these probesets obtained from the C-MAP microarray data of primary melanoma cell lines20 were used for hierarchical clustering analysis using Euclidean distance as the similarity metric and complete linkage as the clustering method in Cluster3.0 software. Results were visualized in JavaTreeView. Two main clusters were generated with this approach. One of the clusters showed upregulation of genes which were also upregulated in commercial melanoma cell lines compared with other solid cancer cell lines. The second cluster of primary cell lines showed upregulation of genes that were downregulated in commercial melanoma cell lines, when compared to other solid cancer cell lines. In the second round of analyses, the most differentially expressed genes between two main clusters were determined using the same significance cutoffs used in generating “gene list#1”. This resulted in “gene list#2”. INHBA and MLANA were selected among the most significantly differentially expressed genes from gene list#2 for further validation of phenotypic features of the two main clusters.
Results & discussion
1. Characterizing melanoma cell lines established from primary tumor tissue
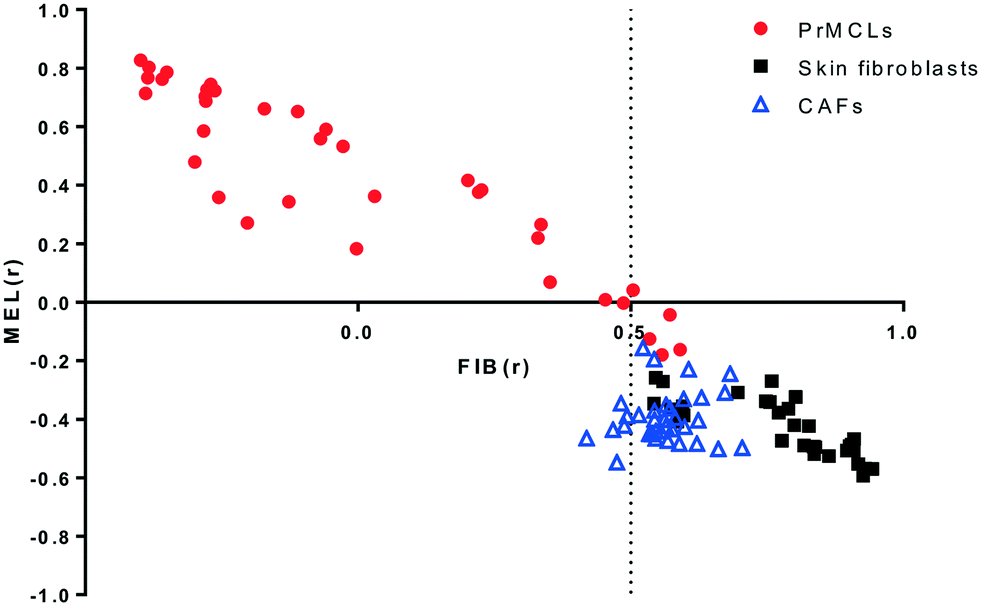
Melanoma cell lines derived from primary tumor tissues were used throughout this study. Cells were initially tested for melanoma markers S100, MART-1 and gp100 to differentiate them from non-melanoma cells.20 As a further measure of specificity, gene expression data obtained from all cell lines via C-MAP technology was used to generate two gene lists representing “melanoma” (MEL) and “fibroblast” (FIB) like qualities using a previously defined algorithm.18 Gene expression-based correlation measures obtained for melanoma cell lines, as well as for normal skin fibroblasts and cancer-associated fibroblasts, show that this approach is an effective means for separating melanoma from fibroblasts. As shown in Fig. 1, gene expression-based analysis revealed that only 4 out of 36 melanoma cell lines show an inverse correlation with the MEL signature, while only 6 out of 65 fibroblasts have a FIBr value lower than 0.5. The 4 melanoma cell lines that can be thus designated “fibroblast-like” are, however, clearly melanoma cell lines as judged by morphology. Other primary cell lines which were negative for S100, MART-1 or gp100, and which lacked a melanoma-like morphology, were also identified as non-melanoma and fibroblast-like by this analysis and were excluded from our studies (not shown). Similar to our analyses, “fibroblast-like” cells, as defined by low MELr and high FIBr values, are present in two out of three datasets generated from melanoma primary cell lines (Fig. S1a†). We consistently observed that such cells grow much slower, compared to those with high MELr scores, explaining why they are extremely rare among commercially available melanoma cell lines (CMCL, Fig. S1b†). The average daily proliferation rate of cells with low INVMELr scores (<0.4) was 18%, compared to 65% for cells with high INVMELr scores (>0.4); similarly, cells with low FIBr scores (<0), had an average proliferation rate of 67%, compared to 20% for those with high FIBr scores (>0).
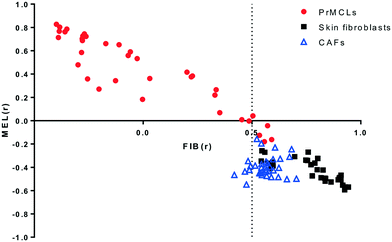 |
| | Fig. 1 Fibroblast/melanoma correlation plot of primary melanoma cell lines (PrMCLs), skin fibroblasts and cancer-associated fibroblasts (CAFs). C-MAP-based gene expression data for PrMCLs and publically available data from skin fibroblasts (GSE11919, GSE16715, GSE21648, GSE34309, GSE35551) and CAFs (GSE46824) were used to calculate gene expression-based correlations to a fibroblast (FIB) and a melanoma (MEL) gene list. Correlation measures (Pearson's r) for each sample are shown for FIB on the x-axis and INVMEL on the y-axis (see Materials and methods for details). | |
2. Classification of melanoma cell lines based on a combination of three gene signatures and their relation to drug sensitivity
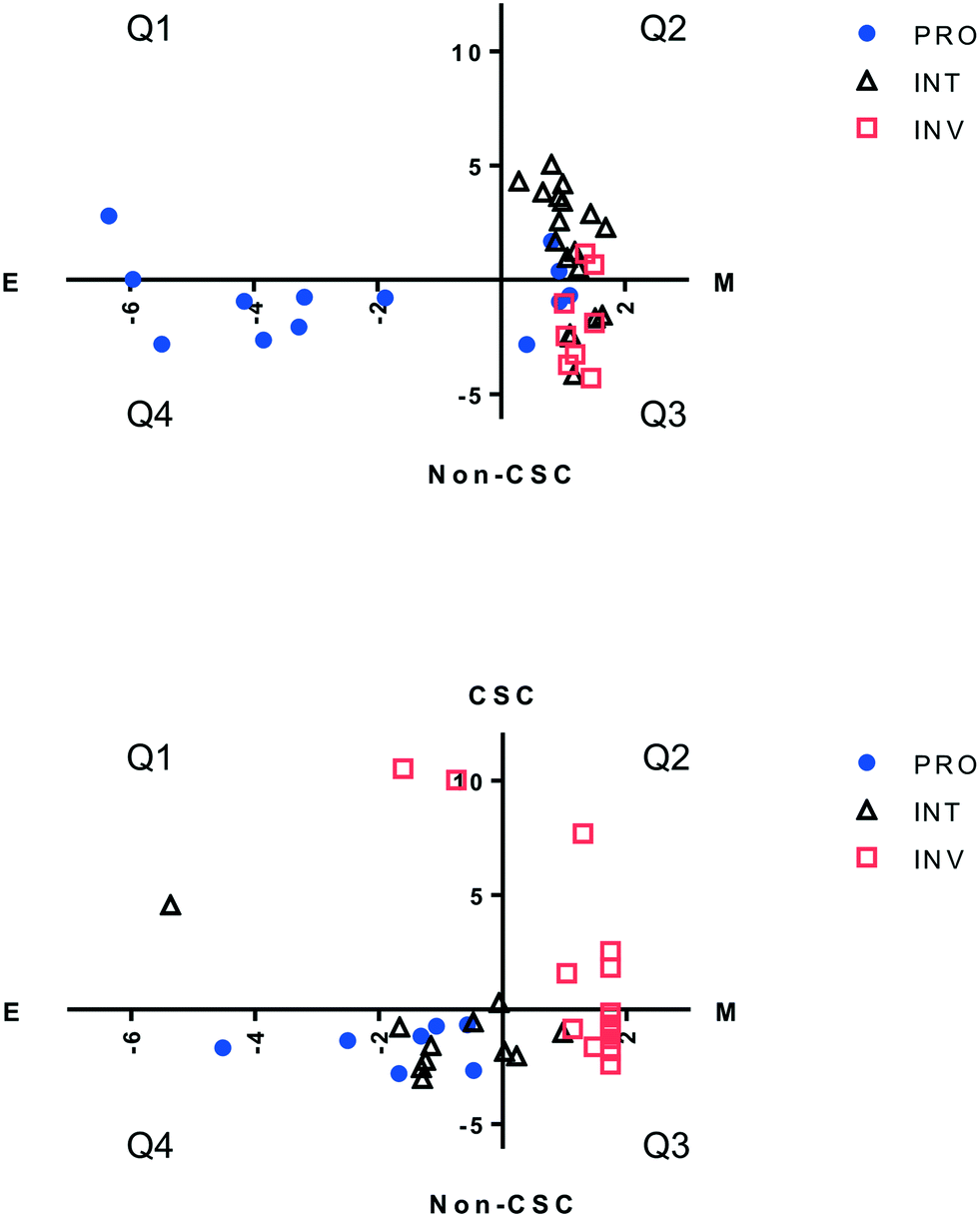
Drug resistance in melanoma has been reported to associate with qualities determined by phenotypes such as EMT, stemness, and measures pertaining to the cell's proliferative and invasive capacity. We evaluated each primary melanoma cell line, as well as CMCLs, for each of these 3 qualities in silico, to determine any overlap between these definitions, and if one or a combination of these could be used to better explain sensitivity to a molecularly targeted drug. For this purpose, cells were first classified into proliferative (PRO), invasive (INV) or intermediate (INT) categories using an online algorithm based on a previously published gene list.18 Each cell was then assigned a score corresponding to its epithelial/mesenchymal state (EMT score), in addition to a score reflecting how stem-like the cell was (CSC score; see methods). Not surprisingly, only one CMCL and 3 PrMCLs had both epithelial and CSC-like qualities, as these two phenotypes are expected to show mutual exclusivity (quadrant 1, Fig. 2). Similarly, most INV cells were more mesenchymal than epithelial, while PRO cells were more epithelial. However, while the INT group among CMCLs was mostly of a CSC-like and mesenchymal phenotype (Q3, Fig. 2, top), the INT group among primary melanoma cell lines had more non-CSC-like and epithelial features (Q4, Fig. 2, bottom). We analyzed if cells harboring BRAF (V600) mutations were preferentially of a particular transcriptomic subtype. CSC and EMT scores were not significantly different between mutant and WT cells. However, the PRO scores of mutant cells were significantly larger than that of WT cells, and the inverse was observed for INV scores; p < 0.01 and p = 0.03, respectively. This suggested that BRAF mutations were not a primary determinant of stemness or the EMT state of cells, but were related to PRO and INV scores.
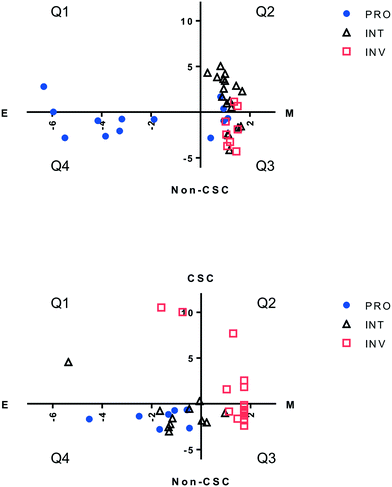 |
| | Fig. 2 Clustering melanoma cell lines based on different molecular signatures. Distribution of CMCLs (top) and PrMCLs (bottom), categorized into PRO, INV or INT groups according to an EMT score (X axis) and stemness score (Y axis). E: epithelial, M: mesenchymal phenotype. CSC: cancer stem-cell-like phenotype, non-CSC: non-cancer-stem-cell-like phenotype. Cells are shown distributed into 4 quadrants: Q1: E and CSC-like, Q2: M and CSC-like, Q3: M and non-CSC-like, and Q4: E and non-CSC-like. | |
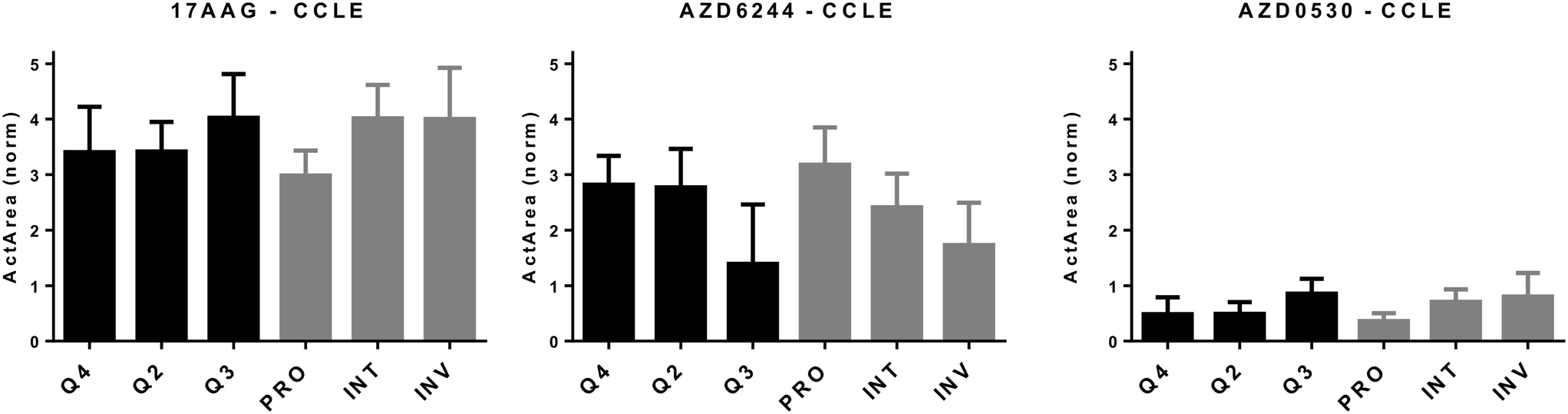
We then asked if the classification based on the combination of these three transcriptomic groups could predict drug sensitivity better than that which could be predicted by using the “PRO/INT/INV” classification by itself. For this purpose, we correlated INV and PRO values obtained for melanoma cell lines for which there was cytotoxicity data in the CCLE database.22 The strongest direct correlation between PRO values and simultaneous negative correlation with INV values was observed for cytotoxicity data from two MEK inhibitors, AZD6244 and PD0325901, in line with the correlation of these phenotypes with BRAF mutation status. The exact opposite pattern was observed most prominently for three other drugs, the HSP90 inhibitor 17-AAG, the SRC-inhibitor AZD0530, and a cMET inhibitor PF-2341066 (Table S2†). As HSP90 and MEK inhibitors have previously been shown to affect opposite phenotypes,25 and similar findings exist for SRC and MEK inhibitor combinations,10 we decided to focus on 17AAG, AZD6244 and AZD0530. We first determined in silico how melanoma cells, as defined by the combination of 3 scores, behaved in response to these drugs and whether this classification would help improve on the PRO/INT/INV classification. As shown in Fig. 3, PRO cells were primarily sensitive to the MEK inhibitor AZD6244, while INV cells were to 17AAG (HSP90 inhibitor) and AZD0530 (SRK inhibitor). By further classifying cells into quadrants based on the added EMT and stemness score, we were unable to identify a subpopulation that was more sensitive to any of the three drugs tested, with the most resistant and sensitive groups identified based on quarters showing similar values to those obtained for PRO, INT and INV groups (Fig. 3). This indicates that classifying cells based on a combination of EMT and CSC markers is able to identify distinct cell populations that differ in their chemosensitivity profiles, but does not help identify cells that are more sensitive to these drugs, compared to the classification generated by the PRO/INV score.
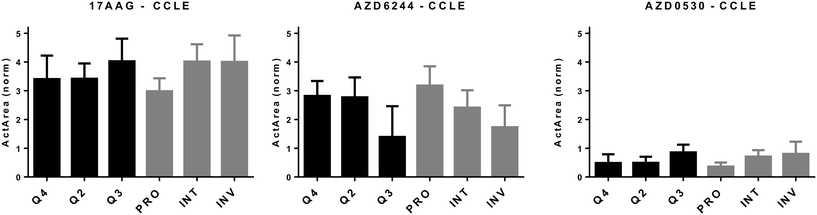 |
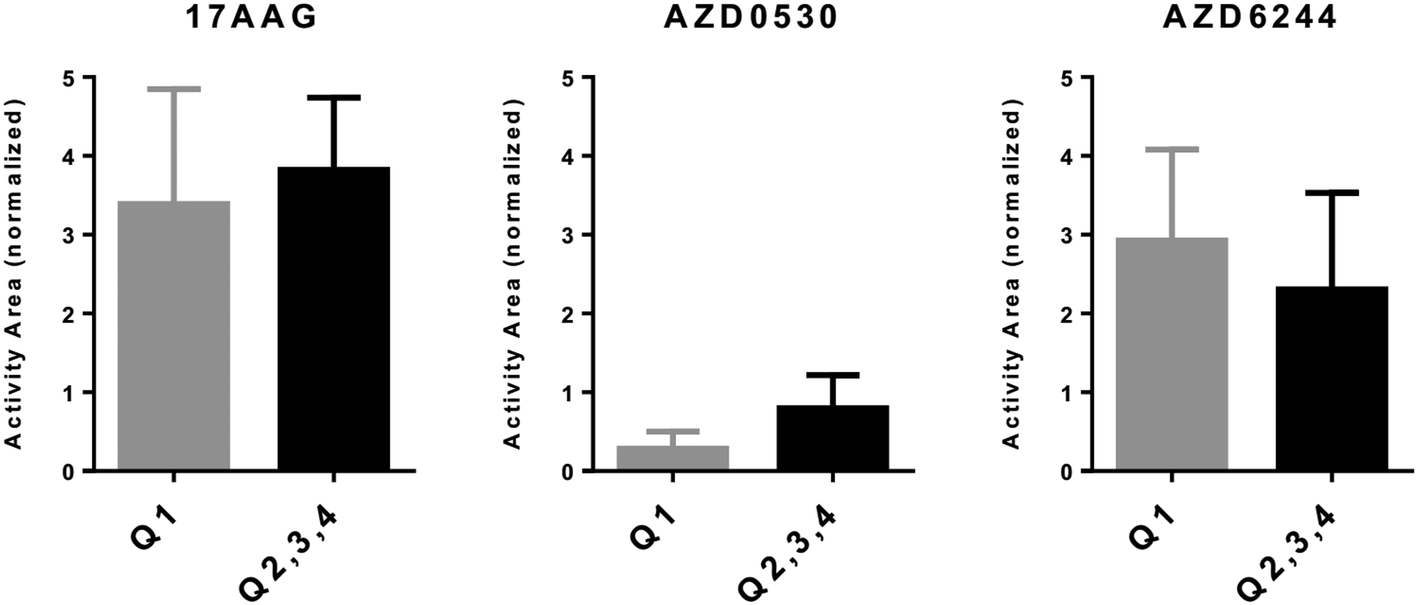
| | Fig. 3
In silico cytotoxicity analyses of molecularly targeted drugs on CMCLs. CMCLs clustered in quadrants 2, 3, and 4 (as indicated in Fig. 2) as well as according to the PRO/INV classification were analyzed for sensitivity against the shown drugs. Cytotoxicity is shown as normalized activity area (Y axis). 1-Way ANOVA-based analysis of quadrant-based classification of cells gave p values of 0.07, 0.034 and 0.05 for 17AAG, AZD6244 and AZD0530, respectively, while the same statistical analysis for PRO, INT and INV groups generated p values of 0.025, 0.023 and 0.03 for 17AAG, AZD6244 and AZD0530, respectively. Q2: M and CSC-like, Q3: M and non-CSC-like, and Q4: E and non-CSC-like. | |
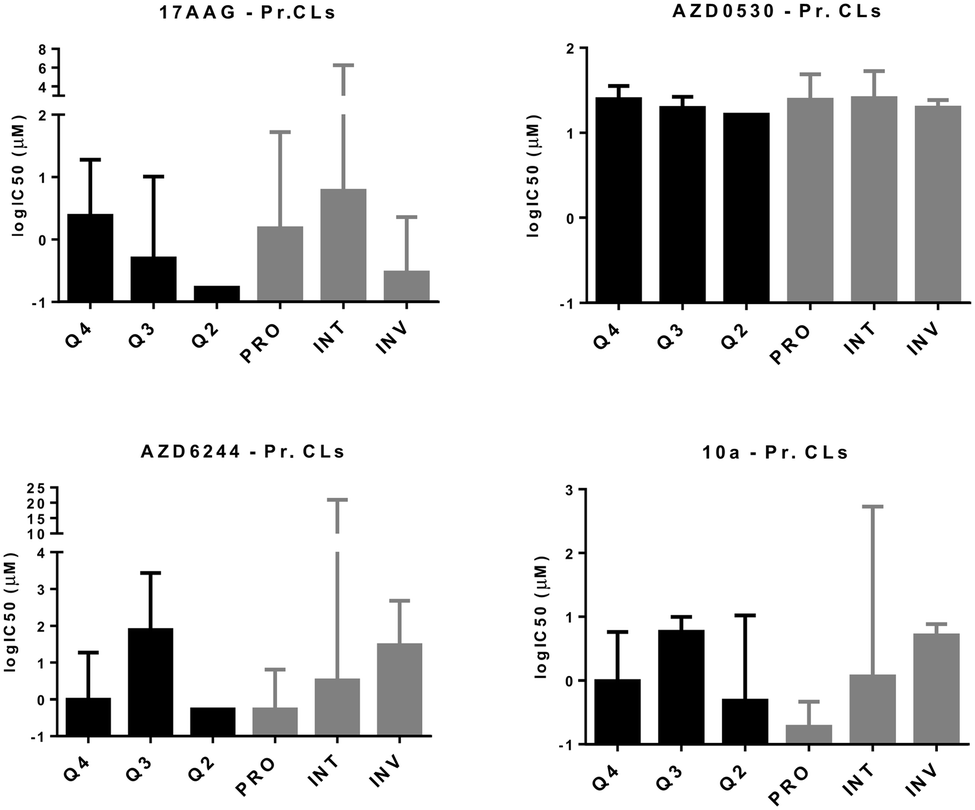
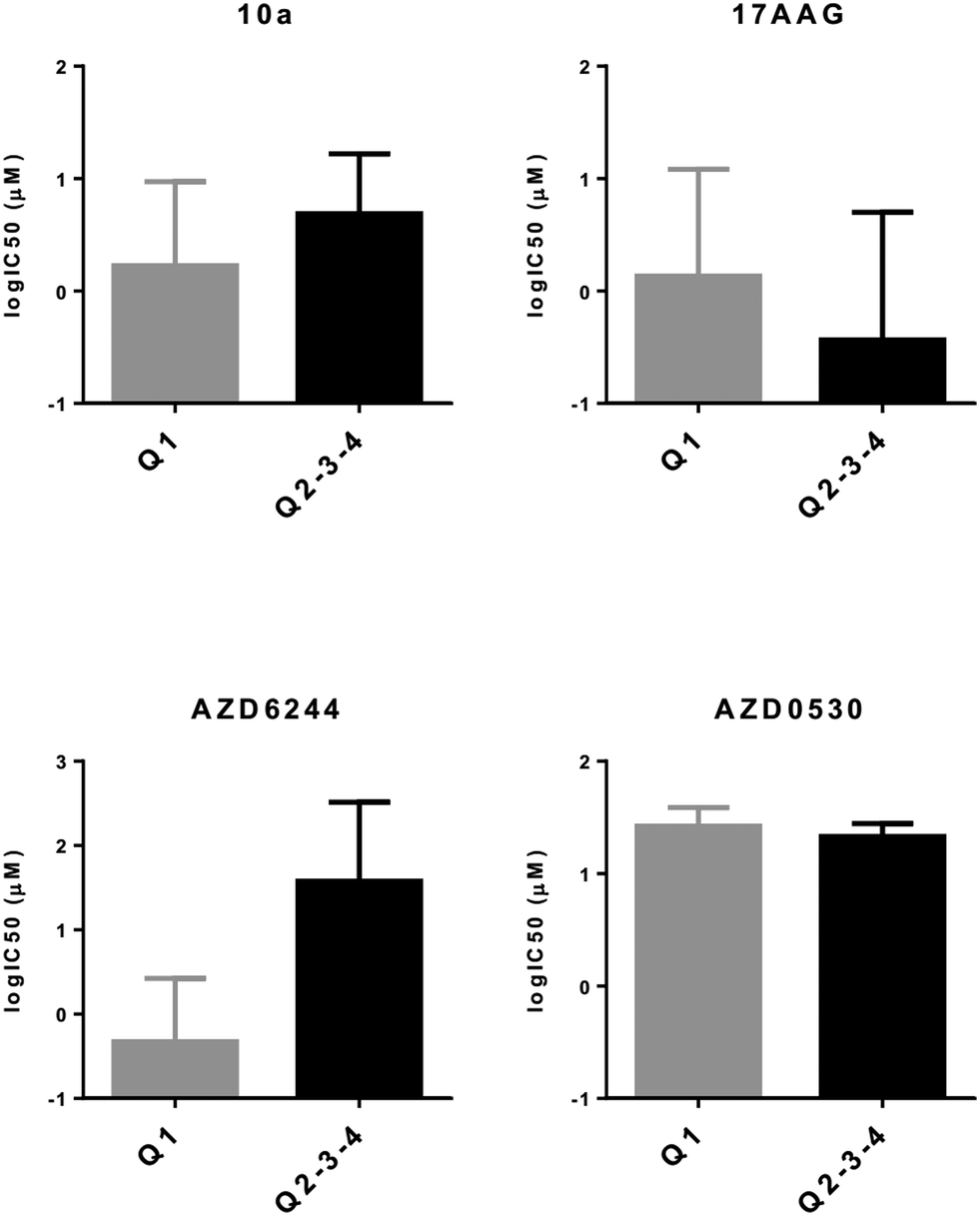
Next, we wanted to validate the in silico cytotoxicity correlations in vitro using PrMCLs. In contrast to what we observed for CMCLs, PRO, INT and INV groups were not as clearly separated into quadrants in analyses of PrMCLs (Fig. 2). Most PRO cells were observed in Q4 (epithelial and non-CSC-like), together with INT cells, while INV cells were present in quarters 1, 2 and 3, which is a different distribution compared to the classification we observed for CMCLs. Cytotoxicity profiles of PrMCLs, however, were quite different between Q-based and PRO/INV-based classes (Fig. 4). For 17-AAG, although INV cells were the most sensitive, INT cells were generally more resistant than PRO cells. A single cell tested in Q2 was the most sensitive, supporting this observation. For AZD6244, PRO and Q4 cells were relatively sensitive. Although a single cell in Q2 was also sensitive, as this is a sole observation, we do not think it indicates an exception. AZD0530, similar to its in silico data, did not show much variation among groups, although INV cells and that in Q2 were somewhat more sensitive. We then tested a novel drug 10a, that has been shown to be a specific SRC inhibitor.26 10a was effective in particular on PRO cells and those in Q4 (epithelial, non-CSC-like), but also on cells in Q2 (mesenchymal, CSC-like) (Fig. 4). There were no statistically significant differences among either quadrant or PRO/INV-based classes for 17AAG and AZD0530 IC50 values. However, there were significant differences for AZD6244 between Q3 vs. Q4 (p = 0.03, t-test), and for 10a, in quadrant based (p = 0.026) or PRO/INT/INV based (p < 0.001) classifications as identified by 1-way ANOVA. This observation reveals that PRO/INV-based and quadrant-based classifications are distinct, in that the cells categorized by both differ in their responses to targeted therapy, especially to the SRC inhibitor 10a. Interestingly, the sensitivity profile of 10a is the complete inverse of that observed for AZD0530. Our data shows that SRC inhibition by 10a exhibits a profile almost identical to that of AZD6244 for PrMCLs (Fig. 4). As MEK and SRC are shared in most pathways, this suggests the SRC-MAPK pathway is active and responsible for growth primarily in PRO cells and those in quarters 2 and 4. AZD0530 shows minimal activity in both CMCLs and PrMCLs at the concentrations tested and does not happen to have distinctly different effects on subgroups of melanoma. That its activity profile is different from 10a suggests either that AZD0530 is unable to inhibit SRC in these cells or that 10a is inhibiting a non-SRC protein.
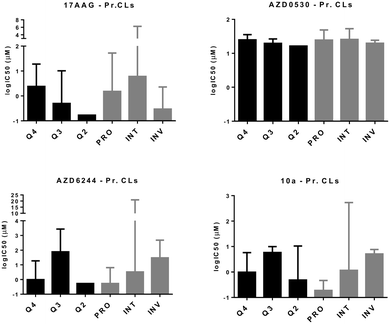 |
| | Fig. 4
In vitro cytotoxicity analyses of molecularly targeted drugs on melanoma cell lines. CMCLs and PrMCLs clustered in quadrants 2, 3, and 4 (Fig. 2) as well as according to the PRO/INV classification were analyzed for sensitivity against the shown drugs. Cytotoxicity is shown as log IC50 (Y axis). Q2: M and CSC-like, Q3: M and non-CSC-like, and Q4: E and non-CSC-like. | |
We also compared cytotoxicity responses among cells with and without BRAF mutations. We did not observe any significant differences in IC50 values for any of the drugs tested between the two groups, although BRAF mutant cells were generally more sensitive to the MEK inhibitor AZD6244 (data not shown).
3. MLANA and INHBA as biomarkers of molecularly targeted drug sensitivity in melanoma
All phenotypical categorizations described thus far are based on the evaluation of a relatively large number of genes. However, as the various categories defined by the three different phenotypical markers generate clusters with similar drug sensitivity profiles, we argued that such profiles could be reflected by a much smaller number of gene expression patterns. As a number of genes used in these classifiers are related to differentiation markers of melanoma, as a discovery strategy, we sought to identify genes whose expression could differentiate melanoma from non-melanoma tumors by comparing gene expression profiles of tumors within E-MTAB-783 and GSE36133 datasets. 223 probesets with a Bonferroni corrected p value of 0.0001 or below and a difference of more than 4 fold that were common to both datasets were used to cluster PrMCLs in silico (Fig. S2†). This analysis revealed two distinct clusters (A and B) which were then used to discover genes that were most differentially expressed between these two clusters (Fig. S2†). MLANA and INHBA were selected as representatives of the most upregulated genes in each of the two distinct clusters, especially since they have been reported to identify opposite phenotypes in melanoma: MLANA is critical for the regulation of mammalian pigmentation,27 while INHBA is reported to decrease melanin content of B16 murine melanoma cell lines.28 Furthermore, INHBA is a member of the transforming growth factor superfamily which induces a mesenchymal phenotype by increasing N-cadherin expression in esophageal carcinoma cell lines.29 These two genes are inversely expressed in both commercial and PrMCLs (Fig. S3†). Although most INHBA expressing cells are low expressors of MLANA and vice versa (thus in quadrants 1 and 3, Fig. S4†), there are cells that express both or neither. Both genes have been reported to show a strong correlation between mRNA and protein levels;17,30 we therefore believe the mRNA-based phenotype should be reflected at the protein level as well. As expected, MLANA and INHBA mRNA expression correlated tightly with MITF and AXL, two genes that are known to play critical roles in phenotype switching in melanoma31,32 (Fig. S5†). In line with BRAF mutations not correlating with previously reported transcriptomic signatures for melanoma, the BRAF mutation status of cells does not seem to correlate with MLANA or INHBA either (Fig. S4†).
We observed that MLANA and INHBA expression-based classification was successful in revealing drug sensitivity differences for both CMCLs and PrMCLs: we combined cells in Q2, 3 and 4 as their sensitivity profiles were similar. We then compared their sensitivity to cells within Q1 (MLANA-high, INHBA-low). This revealed the expected trends for all three drugs. In silico analysis showed that Q1 cells (MLANA-high, INHBA-low) were relatively sensitive to AZD6244 (MEK inhibitor), while they were relatively resistant to 17AAG (HSP90 inhibitor) and AZD0530 (SRC inhibitor), compared to cells in Q2, 3, 4 (Fig. 5). We then validated these findings in vitro for PrMCLs and included the SRC inhibitor 10a in our analysis. As shown in Fig. 6, AZD6244 and 10a are both more effective on Q1 (MLANA-high, INHBA-low) cells, while 17AAG and AZD0530 showed the exact opposite trend. The differential effect of AZD6244 was statistically significant. To test if the cells that were resistant to AZD6244 were also sensitive to 17-AAG or 10a, we plotted the drug sensitivity profiles of cells for either of the two drugs. This analysis, however, revealed that while half of either CMCLs or PrMCLs followed this pattern, about a quarter were sensitive to both, and another quarter of the cells were resistant to both drugs analyzed (Fig. S6†). As previously observed, the sensitivity profiles of 10a and AZD6244 were highly similar (Fig. S6c†). We therefore conclude that phenotyping based on MLANA and INHBA will be able to identify cells that are sensitive to either AZD6244/10a or 17AAG, in about 50% of the cells tested. 25% of cells will be sensitive to both drugs, and a quarter might be resistant to both.
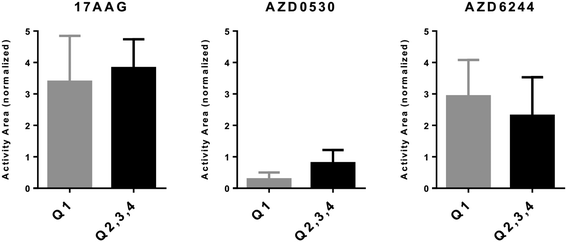 |
| | Fig. 5 MLANA–INHBA-based drug sensitivity analysis of CMCLs. In silico cytotoxiciy analysis of CMCLs against 3 molecularly targeted drugs. Cells contained within quadrants identified in Fig. S3† are indicated. P values (t-test) for 17AAG, AZD0530 and AZD6244 are 0.26, 0.0002 and 0.13, respectively. Q1: E and CSC-like, Q2: M and CSC-like, Q3: M and non-CSC-like, and Q4: E and non-CSC-like. | |
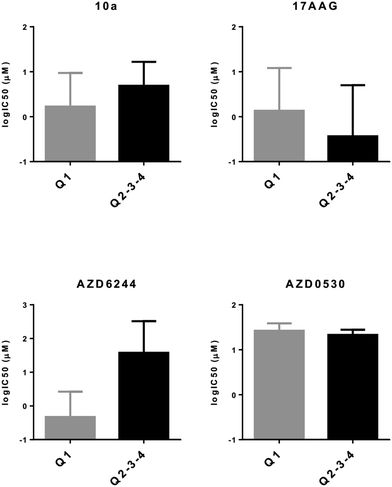 |
| | Fig. 6 MLANA–INHBA-based drug sensitivity analysis of PrMCLs. In vitro cytotoxiciy analysis of PrMCLs against 4 molecularly targeted drugs. Cells contained within quadrants identified in Fig. S3† are indicated. P values (t-test) for 10a, 17AAG, AZD6244, and AZD0530 are 0.1, 0.43, 0.01 and 0.32, respectively. Q1: E and CSC-like, Q2: M and CSC-like, Q3: M and non-CSC-like, and Q4: E and non-CSC-like. | |
Discussion
Plasticity of stemness, EMT and INV/PRO phenotypes in melanoma are clearly related to changes in the signaling pathway choices of these cells. We hypothesized that different melanoma cells, as characterized by gene expression profiles reported to relate to distinct phenotypes, would primarily rely on one particular signaling pathway which could be blocked by one of the drugs generated to target specific signaling pathways. We also hypothesized that the various phenotypes defined for melanoma could overlap. Despite the limited number of cell lines used here, we have been able to support our second hypothesis, as we were able to show that cells grouped by utilizing multiple gene lists are not necessarily better characterized in terms of their drug sensitivity profiles, compared to using any given one. If each of these different phenotypes arises due to the utilization of a distinct pathway, then the phenotypic switch should relate to a shift in the signaling pathways the cell uses, resulting in altered drug sensitivity to a molecularly targeted drug. In line with this argument, we were able to generate partial support for our first hypothesis: although we were able to identify distinct drugs that were more effective on cells as defined by a given phenotype, and that drugs that target different molecules within the same pathway show similar outcomes (shown for 10a and AZD6244), we observed that drugs showing opposite correlations with a given phenotype (AZD6244 or 10a versus 17AAG) did not necessarily have mutually exclusive actions on cells. This is in contrast to the fact that the biomarkers identifying these cells are almost mutually exclusive (MLANA and INHBA). The explanation for this could be that mRNA-based biomarkers, however good they may be in identifying cells with homogenous transcriptomic phenotypes, are not necessarily identifying cells that utilize the same survival pathways. This discrepancy could then likely be due to the fact that simultaneous but distinct pathways are active in a given cell and they have to be targeted at the same time for effective control of cell growth. Despite not being statistically significant, the similarity in sensitivity profiles we observe for 17AAG and AZD0530, two drugs that target non-overlapping signal pathways, support this line of thought. It is possible that the ultimate aim of the cell is to alter its transcriptomic profile, generating a given functional phenotype. If various pathways can be utilized for this purpose, then the cell will use one out of the several ones possible. On the other hand, the fact that melanoma cells that develop resistance to BRAF inhibitors re-activate the MAPK pathway suggests there are situations where the cell relies solely on one pathway.33
Targeted therapy via agents like Dabrafenib (BRAF inhibitor) and Trametinib (MEK inhibitor) significantly increase overall survival of metastatic melanoma patients with BRAF V600E or V600K mutations.34,35 That MEK and RAF inhibitor cytotoxicity profiles are similar according to our in silico analysis is also supportive of this. One reason we could not correlate BRAF mutations to sensitivity to the drugs tested here could be the limited number of mutant cells we studied. This is supported by the fact that BRAF V600 mutant cells have much higher PRO scores compared to INV scores, and the fact that PRO/INV scores do correlate with drug sensitivity, especially to that of the MEK inhibitor AZD6244. However, melanoma subclasses as defined by transcriptomic analyses cannot be explained based on their mutational profile.1,2 It is therefore likely that a simple classification based on MLANA and INHBA can help identify tumors that will benefit from one of the drugs analyzed in this study more by itself or in combination with mutation information.
Conclusions
Our results indicate that melanoma cells are possibly utilizing multiple survival pathways and that the best personalized treatment approach will be one that first reveals the signaling pathways critical for the survival in each cell, followed by the utilization of a drug or a combination of drugs that can target all of these pathways at once. Our data shows that a profiling based on gene expression testing of only MLANA and INHBA is as capable of classifying in vitro responses to molecularly targeted drugs. Larger validation experiments in vivo and tests of drug synergy will need to be performed to confirm this observation.
Abbreviations
| CCLE | Cancer Cell Line Encyclopedia |
| C-MAP | Connectivity map |
| CMCL | Commercial melanoma cell line |
| CSC | Cancer stem cell |
| EMT | Epithelilal to mesenchymal transition |
| FIB | Fibroblast |
| INHBA | Inhibin beta A |
| INT | Intermediate |
| INV | Invasive |
| MEL | Melanoma |
| MLANA | Melan-A |
| PrMCL | Primary melanoma cell line |
| PRO | Proliferative |
| Q | Quadrant |
| RMA | Robust multi-array average |
References
- N. Cancer Genome Atlas, Cell, 2015, 161, 1681–1696 CrossRef PubMed.
- H. Cirenajwis, H. Ekedahl, M. Lauss, K. Harbst, A. Carneiro, J. Enoksson, F. Rosengren, L. Werner-Hartman, T. Torngren, A. Kvist, E. Fredlund, P. O. Bendahl, K. Jirstrom, L. Lundgren, J. Howlin, A. Borg, S. K. Gruvberger-Saal, L. H. Saal, K. Nielsen, M. Ringner, H. Tsao, H. Olsson, C. Ingvar, J. Staaf and G. Jonsson, Oncotarget, 2015, 6, 12297–12309 CrossRef PubMed.
- P. B. Chapman, A. Hauschild, C. Robert, J. B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S. J. O'Day, J. A. Sosman, J. M. Kirkwood, A. M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R. J. Lee, K. T. Flaherty, G. A. McArthur and B.-S. Group, N. Engl. J. Med., 2011, 364, 2507–2516 CrossRef CAS PubMed.
- K. T. Flaherty, C. Robert, P. Hersey, P. Nathan, C. Garbe, M. Milhem, L. V. Demidov, J. C. Hassel, P. Rutkowski, P. Mohr, R. Dummer, U. Trefzer, J. M. Larkin, J. Utikal, B. Dreno, M. Nyakas, M. R. Middleton, J. C. Becker, M. Casey, L. J. Sherman, F. S. Wu, D. Ouellet, A. M. Martin, K. Patel, D. Schadendorf and M. S. Group, N. Engl. J. Med., 2012, 367, 107–114 CrossRef CAS PubMed.
- K. S. Hoek, O. M. Eichhoff, N. C. Schlegel, U. Dobbeling, N. Kobert, L. Schaerer, S. Hemmi and R. Dummer, Cancer Res., 2008, 68, 650–656 CrossRef CAS PubMed.
- A. R. Jeffs, A. C. Glover, L. J. Slobbe, L. Wang, S. He, J. A. Hazlett, A. Awasthi, A. G. Woolley, E. S. Marshall, W. R. Joseph, C. G. Print, B. C. Baguley and M. R. Eccles, PLoS One, 2009, 4, e8461 Search PubMed.
- A. Roesch, M. Fukunaga-Kalabis, E. C. Schmidt, S. E. Zabierowski, P. A. Brafford, A. Vultur, D. Basu, P. Gimotty, T. Vogt and M. Herlyn, Cell, 2010, 141, 583–594 CrossRef CAS PubMed.
- J. D. Wolchok, H. Kluger, M. K. Callahan, M. A. Postow, N. A. Rizvi, A. M. Lesokhin, N. H. Segal, C. E. Ariyan, R. A. Gordon, K. Reed, M. M. Burke, A. Caldwell, S. A. Kronenberg, B. U. Agunwamba, X. Zhang, I. Lowy, H. D. Inzunza, W. Feely, C. E. Horak, Q. Hong, A. J. Korman, J. M. Wigginton, A. Gupta and M. Sznol, N. Engl. J. Med., 2013, 369, 122–133 CrossRef PubMed.
- M. Lauss, J. Nsengimana, J. Staaf, J. Newton-Bishop and G. Jönsson, J. Invest. Dermatol., 2016 DOI:10.1016/j.jid.2016.05.119 , in press.
- J. Ferguson, I. Arozarena, M. Ehrhardt and C. Wellbrock, Oncogene, 2013, 32, 86–96 CrossRef CAS PubMed.
- M. Saez-Ayala, M. F. Montenegro, L. Sanchez-Del-Campo, M. P. Fernandez-Perez, S. Chazarra, R. Freter, M. Middleton, A. Pinero-Madrona, J. Cabezas-Herrera, C. R. Goding and J. N. Rodriguez-Lopez, Cancer Cell, 2013, 24, 105–119 CrossRef CAS PubMed.
- A. Singh and J. Settleman, Oncogene, 2010, 29, 4741–4751 CrossRef CAS PubMed.
- A. Mitra, L. Mishra and S. Li, Oncotarget, 2015, 6, 10697–10711 CrossRef PubMed.
- K. S. Hoek and C. R. Goding, Pigm. Cell Melanoma Res., 2010, 23, 746–759 CrossRef CAS PubMed.
- A. Jayachandran, M. Anaka, P. Prithviraj, C. Hudson, S. J. McKeown, P. H. Lo, L. J. Vella, C. R. Goding, J. Cebon and A. Behren, Oncotarget, 2014, 5, 5782–5797 CrossRef PubMed.
- B. Kreiseder, Y. M. Holper-Schichl, B. Muellauer, N. Jacobi, A. Pretsch, J. A. Schmid, R. de Martin, H. Hundsberger, A. Eger and C. Wiesner, PLoS One, 2015, 10, e0119402 Search PubMed.
- K. S. Hoek, N. C. Schlegel, P. Brafford, A. Sucker, S. Ugurel, R. Kumar, B. L. Weber, K. L. Nathanson, D. J. Phillips, M. Herlyn, D. Schadendorf and R. Dummer, Pigm. Cell Res., 2006, 19, 290–302 CrossRef CAS PubMed.
- D. S. Widmer, P. F. Cheng, O. M. Eichhoff, B. C. Belloni, M. C. Zipser, N. C. Schlegel, D. Javelaud, A. Mauviel, R. Dummer and K. S. Hoek, Pigm. Cell Melanoma Res., 2012, 25, 343–353 CrossRef CAS PubMed.
- M. Lotem, T. Peretz, O. Drize, Z. Gimmon, D. Ad El, R. Weitzen, H. Goldberg, I. Ben David, D. Prus, T. Hamburger and E. Shiloni, Br. J. Cancer, 2002, 86, 1534–1539 CrossRef CAS PubMed.
- M. Lotem, S. Merims, S. Frank, T. Hamburger, A. Nissan, L. Kadouri, J. Cohen, R. Straussman, G. Eisenberg, S. Frankenburg, E. Carmon, B. Alaiyan, S. Shneibaum, Z. Ozge Ayyildiz, M. Isbilen, K. Mert Senses, I. Ron, H. Steinberg, Y. Smith, E. Shiloni, A. O. Gure and T. Peretz, J. Immunol. Res., 2016, 2016, 8121985 Search PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- J. Barretina, G. Caponigro, N. Stransky, K. Venkatesan, A. A. Margolin, S. Kim, C. J. Wilson, J. Lehar, G. V. Kryukov, D. Sonkin, A. Reddy, M. Liu, L. Murray, M. F. Berger, J. E. Monahan, P. Morais, J. Meltzer, A. Korejwa, J. Jane-Valbuena, F. A. Mapa, J. Thibault, E. Bric-Furlong, P. Raman, A. Shipway, I. H. Engels, J. Cheng, G. K. Yu, J. Yu, P. Aspesi Jr., M. de Silva, K. Jagtap, M. D. Jones, L. Wang, C. Hatton, E. Palescandolo, S. Gupta, S. Mahan, C. Sougnez, R. C. Onofrio, T. Liefeld, L. MacConaill, W. Winckler, M. Reich, N. Li, J. P. Mesirov, S. B. Gabriel, G. Getz, K. Ardlie, V. Chan, V. E. Myer, B. L. Weber, J. Porter, M. Warmuth, P. Finan, J. L. Harris, M. Meyerson, T. R. Golub, M. P. Morrissey, W. R. Sellers, R. Schlegel and L. A. Garraway, Nature, 2012, 483, 603–607 CrossRef CAS PubMed.
-
T. R. D. C. Team, R: A Language and Environment for Statistical Computing, 2010 Search PubMed.
- J. P. Medema, Nat. Cell Biol., 2013, 15, 338–344 CrossRef CAS PubMed.
- M. Zajac, G. Gomez, J. Benitez and B. Martinez-Delgado, BMC Med. Genomics, 2010, 3, 44 CrossRef PubMed.
- T. Kruewel, S. Schenone, M. Radi, G. Maga, A. Rohrbeck, M. Botta and J. Borlak, PLoS One, 2010, 5, e14143 CAS.
- T. Hoashi, H. Watabe, J. Muller, Y. Yamaguchi, W. D. Vieira and V. J. Hearing, J. Biol. Chem., 2005, 280, 14006–14016 CrossRef CAS PubMed.
- M. Murakami, F. Matsuzaki and M. Funaba, Mol. Biol. Rep., 2008, 36, 1247–1250 CrossRef PubMed.
- K. Yoshinaga, H. Inoue, T. Utsunomiya, H. Sonoda, T. Masuda, K. Mimori, Y. Tanaka and M. Mori, Clin. Cancer Res., 2004, 10, 5702–5707 CrossRef CAS PubMed.
- J. Du, A. J. Miller, H. R. Widlund, M. A. Horstmann, S. Ramaswamy and D. E. Fisher, Am. J. Pathol., 2003, 163, 333–343 CrossRef CAS PubMed.
- K. Kemper, P. L. de Goeje, D. S. Peeper and R. van Amerongen, Cancer Res., 2014, 74, 5937–5941 CrossRef CAS PubMed.
- D. J. Konieczkowski, C. M. Johannessen, O. Abudayyeh, J. W. Kim, Z. A. Cooper, A. Piris, D. T. Frederick, M. Barzily-Rokni, R. Straussman, R. Haq, D. E. Fisher, J. P. Mesirov, W. C. Hahn, K. T. Flaherty, J. A. Wargo, P. Tamayo and L. A. Garraway, Cancer Discovery, 2014, 4, 816–827 CrossRef CAS PubMed.
- H. Shi, W. Hugo, X. Kong, A. Hong, R. C. Koya, G. Moriceau, T. Chodon, R. Guo, D. B. Johnson, K. B. Dahlman, M. C. Kelley, R. F. Kefford, B. Chmielowski, J. A. Glaspy, J. A. Sosman, N. van Baren, G. V. Long, A. Ribas and R. S. Lo, Cancer Discovery, 2014, 4, 80–93 CrossRef CAS PubMed.
- K. Margolin, Curr Treat Options Oncol, 2016, 17, 48 CrossRef PubMed.
- C. Robert, B. Karaszewska, J. Schachter, P. Rutkowski, A. Mackiewicz, D. Stroiakovski, M. Lichinitser, R. Dummer, F. Grange, L. Mortier, V. Chiarion-Sileni, K. Drucis, I. Krajsova, A. Hauschild, P. Lorigan, P. Wolter, G. V. Long, K. Flaherty, P. Nathan, A. Ribas, A. M. Martin, P. Sun, W. Crist, J. Legos, S. D. Rubin, S. M. Little and D. Schadendorf, N. Engl. J. Med., 2015, 372, 30–39 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00466k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Mehdi
Ghasemi
a,
Muhammad W.
Akbar
a,
Murat
Isbilen
a,
Anna L.
Fallacara
b,
Shoshana
Frankenburg
c,
Silvia
Schenone
d,
Michal
Lotem
c,
Maurizio
Botta
b and
Ali O.
Gure
*a
a,
Mehdi
Ghasemi
a,
Muhammad W.
Akbar
a,
Murat
Isbilen
a,
Anna L.
Fallacara
b,
Shoshana
Frankenburg
c,
Silvia
Schenone
d,
Michal
Lotem
c,
Maurizio
Botta
b and
Ali O.
Gure
*a