
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTumor cell resistance against targeted therapeutics: the density of cultured glioma tumor cells enhances Stat3 activity and offers protection against the tyrosine kinase inhibitor canertinib†
V.
von Manstein
and
B.
Groner
*
Georg Speyer Haus, Institute for Tumor Biology and Experimental Therapy, Paul Ehrlich Str. 42, 60596 Frankfurt am Main, Germany. E-mail: groner@em.uni-frankfurt.de; Tel: +49 6963395180
First published on 14th October 2016
Abstract
Tumor cell resistance to drug treatment severely limits the therapeutic success of treatment. Tumor cells, exposed to chemotherapeutic drugs, have developed intricate strategies to escape the cytotoxic effects and adapt to adverse conditions. The molecular mechanisms causing drug resistance can be based upon modifications of drug transport or metabolism, structural alterations of drug targets or adaptation of cellular signaling. An important component in the transformation of cells and the emergence of drug resistance is the activation of the transcription factor Stat3. The persistent, inappropriate activation of Stat3 causes the expression of target genes which promote tumor cell proliferation, survival, invasion and immune suppression, and it is instrumental in the process of the emergence of resistance to both conventional chemotherapeutic agents and novel targeted compounds. For these reasons, Stat3 inhibition is being pursued as a promising therapeutic strategy. We have investigated the effects of the tyrosine kinase inhibitor canertinib on the glioma cell line Tu-2449. In these cells Stat3 is persistently phosphorylated and activated downstream of the oncogenic driver v-Src and its effector, the cytoplasmic tyrosine kinase Bmx. Canertinib exposure of Tu-2449 cells rapidly caused the inhibition of the Bmx kinase and the deactivation of Stat3. Prolonged exposure of the cells to canertinib caused the death of the large majority of the cells. Only a few cells became resistant to canertinib and survived in tight clusters. These cells have become drug resistant. When the canertinib resistant cells were expanded and cultured at lower cell densities, they regained their sensitivity towards canertinib. We measured the extent of Stat3 activation as a function of cell density and found that higher cell densities are accompanied by increased Stat3 activation and a higher expression of Stat3 target genes. We suggest that Stat3 induction through tight cell–cell interactions, most likely through the engagement of cadherins, can counteract the inhibitory effects exerted by canertinib on Bmx. Cell–cell interactions induced Stat3 and compensated for the suppression of Stat3 by canertinib, thus transiently protecting the cells from the cytotoxic effects of the inhibitor.
Introduction
Targeted drugs have become valuable new therapeutics in the treatment of cancer. They have proven beneficial for patients with diverse cancer diseases. These drugs are usually administered to patients in which a characteristic driver mutation has led to the enhanced activity of a particular oncogene and the activation of a defined signaling cascade. The combination of molecular diagnostics and targeted drugs has allowed the definition of subpopulations of patients, suffering from a particular indication, with a high probability of responsiveness.1,2 The identification of driver mutations provides valuable information for the choice of first line treatment regimens and contributes to the cost effectiveness of cancer therapies.3 However, the responsiveness towards targeted drugs, even within a molecularly eligible subpopulation of patients, is not uniform. In addition, even if a favorable treatment response can be achieved, the duration of such a response is usually limited. Resistant tumor cells emerge, a situation comparable to treatment with conventional, non-specifically cytotoxic chemotherapeutic agents.4Intrinsic and acquired resistance limits drug efficacy. The mechanisms of resistance based upon drug transport and metabolism have been well studied.5 For example, P-glycoprotein (P-gp) acts as a drug efflux transporter protein and limits the intracellular concentration of multiple therapeutic compounds causing multidrug resistance. MDR can also involve changes in the level of target proteins, mutations that diminish drug binding, trapping of drugs in acidic vesicles, enhanced metabolism of drugs by cytochrome P450 mixed function oxidases, increased tolerance of cellular DNA damage and diminished apoptotic signaling.6
More recently, additional mechanisms have been discovered which contribute to drug resistance. These discoveries extend to “targeted drugs”, compounds which are causal in transformation and tumor formation. Resistance to tyrosine kinase inhibitors can e.g., be caused by mutations in the drug target structure which prevent the interaction of the drugs with the ATP-binding pocket. Other mechanisms of resistance can be elicited by the activation of compensatory signaling pathways. They can be based not only on gene mutations or amplifications, but also on non-genomic mechanisms. Factors present in the tumor microenvironment can induce proliferative or anti-apoptotic signal responses and can thus contribute to drug resistance.7–9 Adaptive responses can be triggered which counteract the initial dependence of tumor cells upon a particular signaling molecule and allow only a transient inhibition of tumor cell growth. These compensatory signaling mechanisms are often based upon the relief of repression of regulatory feedback loops.10,11 They might involve cell autonomous, intracellular events12,13 or they can be mediated via the secretion of growth factor receptor ligands into the tumor microenvironment and signal induction in an auto- or paracrine fashion.14
Drug resistance is often associated with the activation of the transcription factor Stat3.10,11,15 Here we describe a mechanism of drug resistance which relies upon the activation Stat3 mediated by high cell density in cultured cells. Stat3 is regulated by cytokines, interleukins and growth factors and acts as an endpoint of multiple signaling pathways.16,17 In normal cells this activation is transient and the Stat3 molecules return to their non-phosphorylated, latent state within a short time period. In tumor cells the balance between activating and de-activating signals is disturbed, resulting in the persistent activation of Stat3.18 The hyper activation of Stat3 induces the expression of target genes, which enhance the proliferation and survival potential of cancer cells, and thus contribute to drug resistance.15,19 Activating components of the Jak–Stat pathway have been recognized as potentially valuable drug targets and important principles of compensatory signaling circuit induction during targeted drug treatment have been discovered.20 Our experiments suggest that the effects on Stat3 activation in the Tu-2449 glioma cell line through the inhibition of the Bmx tyrosine kinase can be compensated by an alternate Stat3 activation mechanism. It has been demonstrated that engagement of E-cadherin as a function of cell density activates Stat3.21 Classical cadherins like N-cadherin and cadherin-11 can trigger an increase in Stat3 activity as well.22 Unlike other resistance mechanisms, this mechanism is not dependent upon stable genomic alterations and is only transiently active as a function of the cell growth conditions.
Results
Canertinib inhibits the growth of Tu-2449 glioma cells and strongly impedes their viability
We have investigated the effects of the tyrosine kinase inhibitor canertinib on the glioma cell line Tu-2449. Tu-2449 is an established cell line derived from a spontaneous tumor in transgenic mice, induced by a glial fibrillary acidic protein (Gfap)-v-Src transgene.23 The cells can be transplanted into the brains of mice. There they form tumors with high mitotic activity, focal necrosis and diffuse infiltration into the adjacent neuroepithelial tissue.24 These characteristics can also be observed in human gliomas. The tyrosine kinase inhibitor canertinib has shown activity towards Bmx (bone marrow X-linked kinase), an intracellular, non-receptor tyrosine kinase. Its inhibitory activity is not strictly specific. It also functions as a pan-ErbB receptor tyrosine kinase inhibitor, but does not inhibit v-Src.25–27Stat3 is a transcription factor which is persistently activated by the v-Src-Bmx axis in Tu-2449 cells, through phosphorylation on tyrosine residue 705. Throughout the text we refer to the tyrosine phosphate activated form of Stat3 as P-Stat3. We treated Tu-2449 cells with the indicated concentrations of canertinib for 24, 48 and 72 hours and supplemented the cells every 24 h with fresh medium and inhibitor. The exposure to 5 μM canertinib caused the inhibition of Bmx and Stat3 phosphorylation and resulted in an arrest of proliferation after 24 hours.28 The cellular viability is reduced after 48 hours and 10 μM canertinib caused massive cell death already after 24 hours (Fig. 1 and ref. 28).
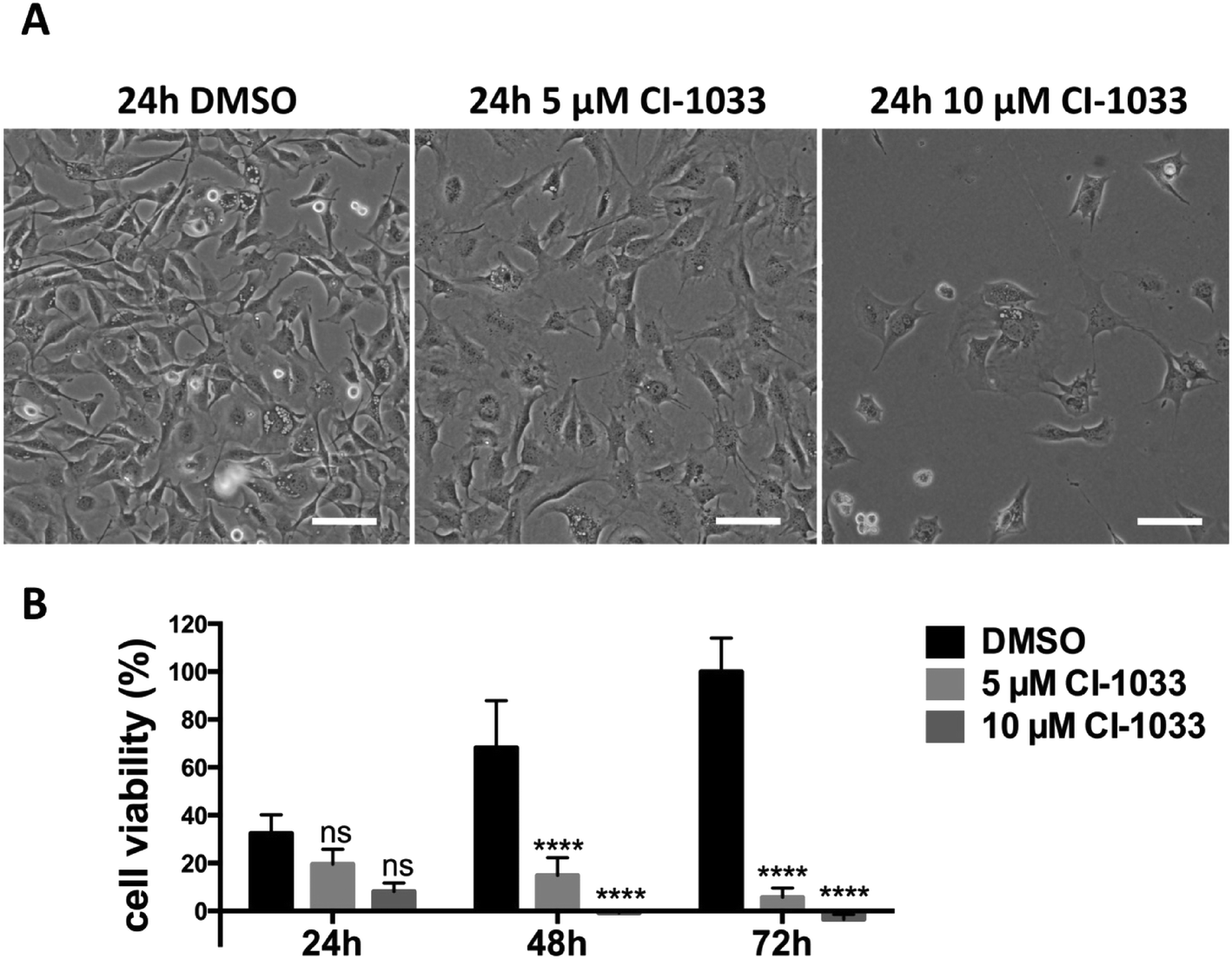 | ||
| Fig. 1 Canertinib treatment of Tu-2449 glioma cells induces the arrest of cell growth after 24 h and prolonged inhibition causes cell death dependent upon exposure time and drug dose. (A) Tu-2449 cells were treated with a single dose of 5 or 10 μM canertinib or DMSO and cultured for 24 h. Microscopic monitoring after 24 h reveals growth arrest in canertinib treated cells; scale bar: 100 μm. (B) Tu-2449 cells were treated with canertinib or DMSO and the drug was replenished in the growth medium in 24 hour intervals. Cell viability was monitored after 24, 48 and 72 h using the AlamarBlue® assay. Repeated addition of canertinib arrests the cells and causes cell death after 24 h (10 μM canertinib) or 48 h (5 μM canertinib). Results are presented as mean ± SD, expressed as a percentage with respect to 72 h DMSO. ns = not significant, ****p < 0.0001. | ||
Canertinib resistant cells can be selected upon prolonged exposure of Tu-2449 cells
When Tu-2449 cells were exposed to 5 μM canertinib for extended periods of time, surviving cells emerged with low frequency (Fig. 2). Canertinib was replenished at 24 hour intervals in the growth medium and small colonies of cells could be observed after 5 to 6 days. These cells are characterized by cell–cell contacts. The surrounding cells, not in contact with neighboring cells, died. Viable cell clusters survived for more than 10 days in the presence of the drug. The cells remain viable, but divided only slowly. When the drug was removed from the medium, proliferation of the cells accelerated again. We expanded the drug resistant cells by culturing them without canertinib for 8 days and observed massive proliferation.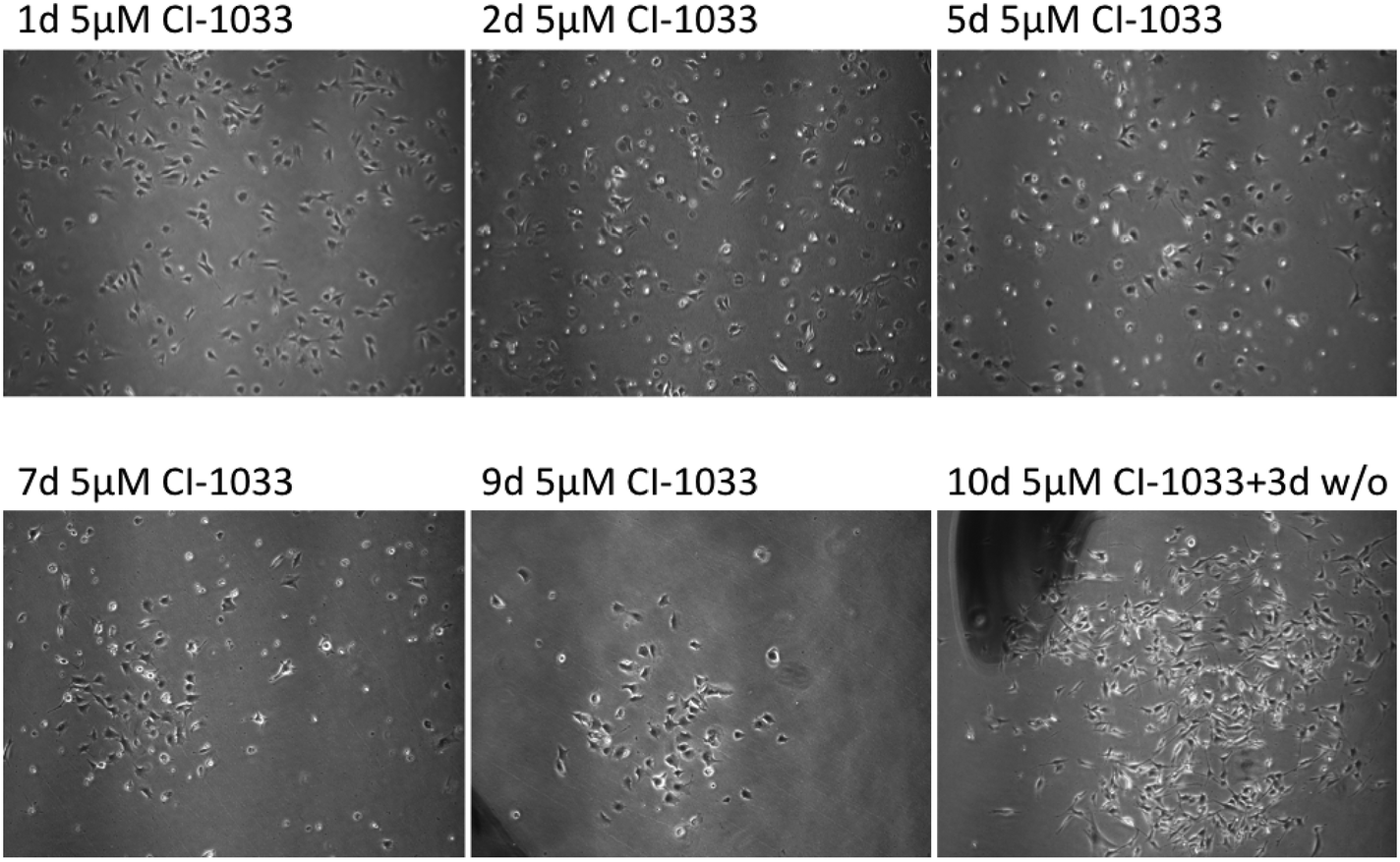 | ||
| Fig. 2 Selection of canertinib resistant cells. Tu-2449 cells were treated with canertinib for 10 days. The drug was replenished in the growth medium in 24 hour intervals by half-medium change. After 10 days, drug treatment was discontinued for three days. Cell growth was microscopically monitored after 1, 3, 5, 7, 9 and 13 days. The upper panel shows different fields while the lower panel shows the same field. Magnification, 40×. Drug treatment selects for small cell colonies with higher cell density. | ||
Canertinib resistance is transient and cell density dependent
We investigated the cells which had grown out from the canertinib resistant cell colonies (Fig. 3). Five individually obtained cell colonies and the parental Tu-2449 cells were trypsinized and replated at 20–40% confluency and treated with 5 μM canertinib for 3 days. Cell viability assays showed no difference in cell viability between the parental Tu-2449 cells and the cells selected as resistant for 9 days. These experiments show that the resistance to canertinib is transient and the cells became sensitive to canertinib again upon expansion and replating. The comparison of the cell morphology between the parental Tu-2449 cells and the selected cell colonies upon exposure to canertinib at low cell density did not show differences. The cells died uniformly within 48 to 72 hours.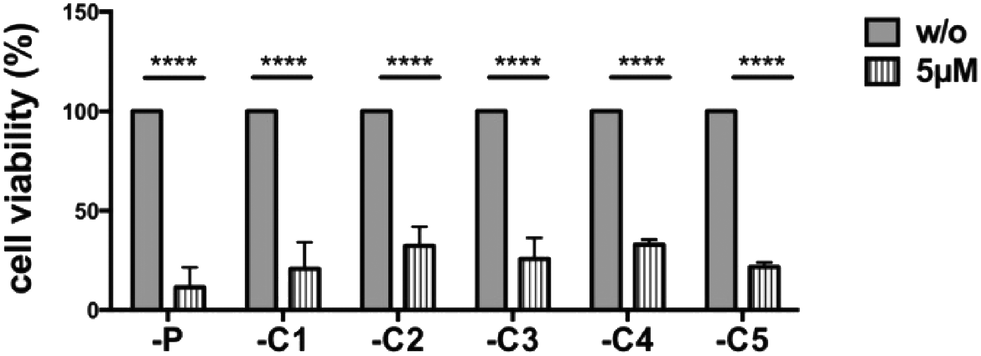 | ||
| Fig. 3 Selected canertinib resistant colonies become sensitive again to drug treatment when cultured at low cell densities. Parental and 5 selected colonies of Tu-2449 glioma cells were plated at low densities (comparable to Fig. 2 panel one) and treated with 5 μM canertinib or only the growth medium. The drug was replenished in the growth medium in 24 hour intervals. Cell viability was monitored after 24, 48 and 72 h using the AlamarBlue® assay. Repeated addition of 5 μM canertinib significantly reduced the cell viability in all analyzed cell clones after 72 h. Results are presented as mean ± SD, with respect to 72 h untreated cells; ****p < 0.0001. | ||
High cell density causes P-Stat3 induction and canertinib resistance of Tu-2449 cell colonies
The resistance to canertinib was seen in cell colonies in which the cells had established cell–cell contacts (Fig. 2). We suspected that these contacts could be a cause of resistance. For these reasons we plated Tu-2449 cells at different densities, 20, 40, 60, 80 and 100% confluency, and measured the extent of Stat3 activation (Fig. 4). Cell lysates were obtained and tyrosine phosphorylated Stat3 was quantitated by Western blotting. Our results show that Stat3 activation can be observed even at the lowest density, 20%, but the extent of activated Stat3 increases steadily with the increasing density of the cells on the plate.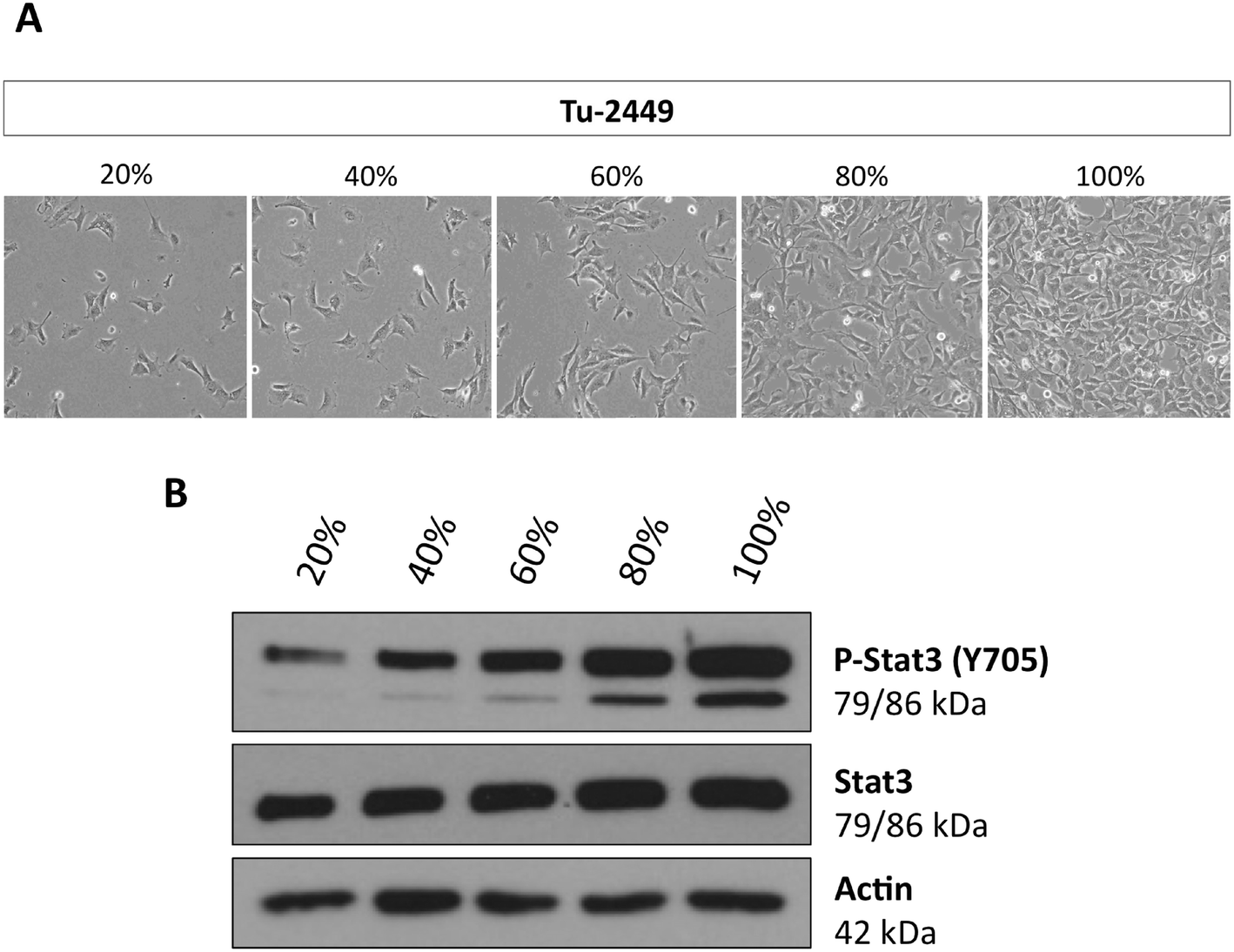 | ||
| Fig. 4 Phosphorylation of Stat3 in Tu-2449 cells increases with cell density. Tu-2449 glioma cells were plated with different cell numbers. (A) Cell density was microscopically monitored after 24 h (20% low, 100% high cell density), magnification: 40×, and (B) protein lysates were analyzed for phosphorylated Stat3 (P-Stat3 Y705) and total Stat3 by Western blotting analyses. Actin served as the loading control. The extent of P-Stat3 increases with cell density. | ||
A number of genes are known to be induced by activated Stat3, e.g. SOCS3, Bcl2 and Stat3 itself. We measured the expression of the mRNA of these genes as a function of the density at which the Tu-2449 cells were grown (Fig. 5). Similar to the increased levels of Stat3 activation in densely grown cells, the mRNA expression levels of these Stat3 target genes increased.
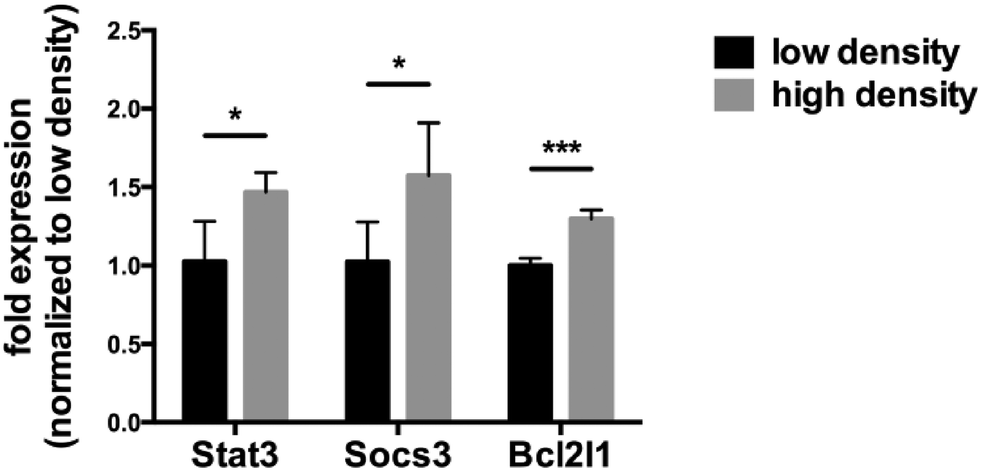 | ||
| Fig. 5 High cell density increases the expression of selected Stat3 target genes. Tu-2449 glioma cells were plated with different cell numbers and cultured for 24 h. RNA was isolated, reverse transcribed and analyzed using q-real-time PCR. Expression of Stat3, Socs3 and Bcl2l1 was significantly increased in cells grown at high density. The values were normalized versus Gapdh expression, which served as an endogenous control. Results are presented as mean ± SD. Statistical significance was calculated compared to cells with low density; *p < 0.05, ***p < 0.001. | ||
Discussion
The resistance of tumor cells to the action of cancer drugs is one of the most important clinical problems in cancer therapy.5 The exposure of cancer cells to a selective pressure, exerted by drug treatment, elicits tumor cell responses and promotes their evolution through adaptation. A multitude of molecular mechanisms are being exploited by the tumor cells to counteract the toxic effects of drugs and to survive their onslaught.13,29In many of these mechanisms the transcription factor Stat3 plays a central role.15 Persistent Stat3 activation not only regulates cell proliferation, differentiation, migration and survival of tumor cells, but also contributes to drug resistance against conventional chemotherapeutic drugs and targeted compounds. Since Stat3 activation is regulated by multiple layers of feedback loops, cancer cells can adjust these regulatory mechanisms to compensate for particular inhibitory influences and thus counterbalance drug effects.18 The relief of negative feedback inhibition and crosstalk between signaling pathways allows the cells to defend themselves against drug effects.16
We provide evidence that a mechanism of Stat3 activation, independent of genetic alterations, can participate in drug resistance. In addition to the endocrine and paracrine mechanisms used by secreted ligands through the activation of cytokine and growth factor receptors, Stat3 induction can be triggered through cell–cell interactions. In v-Src transformed NIH3T3 cells an increase in Stat3 activation was observed as a function of cell to cell adhesion. Src inhibition caused the death of sparsely growing cells.30 Cadherins mediate cell–cell interactions and these interactions have been shown to result in Stat3 activation at high densities of cultured cells through the increased secretion of IL-6 cytokines.31,32 In Tu-2449 glioma cells v-Src is a Stat3 inducer, an effect mediated via the phosphorylation of the Bmx kinase. Canertinib is a tyrosine kinase inhibitor which blocks the enzymatic activity of Bmx and the phosphorylation of Stat3. Cell interaction mediated Stat3 induction can offset the Bmx inhibition and temporarily protect the cells from the cytotoxic effects of canertinib. We corroborated these conclusions in experiments in which we exposed Tu-2449 cells grown at high densities to canertinib. We observed that canertinib was still able to suppress Bmx activation. Under these conditions Stat3 phosphorylation was only slightly affected (data not shown). These protective effects are transient; Tu-2449 cells lose their resistance against canertinib again when they are grown at low cell density. In a clinical setting, high cell densities and tight cell–cell contacts within tumor tissues could limit the efficacy of therapeutics and allow residual tumor cells to survive periods of drug exposure during treatment. Concurrent Stat3 inhibition could be beneficial.33
Experimental
Cells and culture conditions
Tu-2449, a glioma cell line34 derived from a spontaneous tumor occurring in a Gfap-v-Src-transgenic mice (provided by Dr. Kögel, Neuroscience Center, University of Frankfurt), was cultured at 37 °C in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 2 mM L-glutamine, penicillin (100 units per ml) and streptomycin (100 μg ml−1) (all from Life Technologies, Darmstadt, Germany) with 5% CO2. The cells were subcultured 2 or 3 times a week using 0.25% trypsin (Life Technologies, Darmstadt, Germany) as a detaching agent.Chemicals
The tyrosine kinase inhibitor canertinib (CI-1033) was purchased from SelleckChem, Munich, Germany, dissolved in sterile dimethyl sulfoxide (DMSO) and stored at −80 °C. The stock was added to the growth medium while the concentration of DMSO in the medium was <0.03%.Antibodies
The monoclonal antibody specific to P-Stat3 (Y705; #9145) was from Cell Signalling Technology Inc., Frankfurt, Germany. The polyclonal antibody specific to Actin (A2066) was purchased from Sigma-Aldrich. Polyclonal horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (#7074) from Cell Signalling Technology was used as a secondary antibody.Cell viability assay
Tu-2449 cells were cultured in 96-well plates and treated with medium containing the indicated concentrations of canertinib, DMSO or just plain medium for increasing lengths of time. After the treatment, 10% (v/v) AlamarBlue® (Life Technologies, Darmstadt, Germany) was added and the cells were incubated for 4 hours. Absorption at 570 nm was measured by a spectrophotometer (absorption at 600 nm as a reference). The absorption measurements of the treated cells were compared to those obtained upon incubation with DMSO and expressed as percentage values. All values are mean ± SD. Statistical analysis was performed using 2-way Anova with Tukey's post test; an α value of less than 0.05 was considered as significant.Western blot analysis
Cells were lysed for 10 min in RIPA buffer (1% NP40, 0.5% sodium desoxycholate, 0.1% SDS, 150 mM NaCl, 5 mM EDTA, 50 mM Tris–HCl pH 7.5) freshly supplemented with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 protease inhibitor (protease inhibitor cocktail set III, Merck, Darmstadt, Germany) and 1 pill per 10 ml of phosphatase inhibitor (PhosphoStop, Roche Diagnostics, Munich, Germany). The lysates were sonified and then cleared by centrifugation. Protein concentrations were determined spectrophotometrically using the Bradford method. 25 μg of the protein lysates was separated by electrophoresis in 8% SDS-polyacrylamide gels, and transferred to nitrocellulose membranes (Protran, Whatman, Kassel, Germany) by semi-dry blotting. The membranes were blocked for 90 min in TBST (20 mM Tris–HCl, 150 mM NaCl, pH 7.5, 0.1% Tween 20) with 5% non-fat dry milk and then incubated with P-Stat3 (Y705) (5% BSA) or Actin (5% non-fat dry milk) antibodies overnight at 4 °C. The membranes were subsequently incubated with the HRP-conjugated secondary antibody for 1 h at RT. Immunoreactive bands were visualized by enhanced chemiluminescence (Advansta, Menlo Park, USA).
:500 protease inhibitor (protease inhibitor cocktail set III, Merck, Darmstadt, Germany) and 1 pill per 10 ml of phosphatase inhibitor (PhosphoStop, Roche Diagnostics, Munich, Germany). The lysates were sonified and then cleared by centrifugation. Protein concentrations were determined spectrophotometrically using the Bradford method. 25 μg of the protein lysates was separated by electrophoresis in 8% SDS-polyacrylamide gels, and transferred to nitrocellulose membranes (Protran, Whatman, Kassel, Germany) by semi-dry blotting. The membranes were blocked for 90 min in TBST (20 mM Tris–HCl, 150 mM NaCl, pH 7.5, 0.1% Tween 20) with 5% non-fat dry milk and then incubated with P-Stat3 (Y705) (5% BSA) or Actin (5% non-fat dry milk) antibodies overnight at 4 °C. The membranes were subsequently incubated with the HRP-conjugated secondary antibody for 1 h at RT. Immunoreactive bands were visualized by enhanced chemiluminescence (Advansta, Menlo Park, USA).
RNA isolation, cDNA synthesis and quantitative real-time PCR
Total RNA was extracted from cells using the NucleoSpin RNA II Kit (Macherey-Nagel, Düren, Germany). The RNA quality and concentration was measured by spectrophotometry (NanoDrop ND 1000, Nano Drop Technologies, Wilmington, USA). cDNA synthesis with 2 μg of RNA was performed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Darmstadt, Germany). 62.5 ng of the diluted cDNA was used in quantitative real-time-PCR reactions using Maxima SYBR Green qPCR Master Mix (Fermentas, St. Leon-Rot, Germany). All primers were obtained from Sigma-Aldrich and their final concentration was 0.6 μM(Stat3 fwd: AAGAGTCTCGCCTCCTCCAG,
Stat3 rev: GCTTCTCTGTCACTACGGCG;
Socs3 fwd: ATGGTCACCCACAGCAAGTTT,
Socs3 rev: TCCAGTAGAATCCGCTCTCCT;
Bcl2l1 fwd: GACAAGGAGATGCAGGTATTGG,
Bcl2l1 rev: TCCCGTAGAGATCCACAAAAGT).
Cycling conditions comprised an activation step lasting 10 min at 95 °C and 50 cycles of denaturation at 95 °C for 15 s and annealing/amplification at 60 °C for 30 s, followed by melting curve analyses to confirm the quality of the assays. The q-real-time-PCR reactions were performed on a Light Cycler 480 II Real-Time PCR System (Roche Applied Science, Penzberg, Germany). The reactions were carried out in triplicate and the results were standardized. Analysis of relative gene expression was performed employing the 2-ΔΔCt-method with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA expression as an endogenous control. All values are mean ± SD. Statistical analysis was performed using unpaired student's t-test; a p value of less than 0.05 was considered as significant.
Image acquisition and presentation
Microscopy images were acquired with a Nikon Eclipse TE300 or TS100 microscope (Nikon Instruments Europe B.V., Kingston, UK). Image adjustment and composition were generated using PowerPoint (Microsoft, Redmond, USA) software.Conclusions
Tumor cell resistance to drug treatment is a serious clinical problem. To overcome the limitations for cancer therapy, set by the emergence of drug resistant tumor cell variants, it is important to understand the molecular principles underlying this process. Tumor cells have developed diverse strategies to escape the cytotoxic effects of anti-cancer drugs and adapt to adverse conditions. Among the mechanisms identified are modifications of drug transport or metabolism, structural alterations of drug targets and adaptation of cellular signaling. The transcription factor Stat3 plays a central role in the emergence of drug resistance to conventional chemotherapeutic agents and to novel targeted compounds. In a glioma cell model, the tyrosine kinase inhibitor canertinib inhibits the cytoplasmic tyrosine kinase Bmx and the phosphorylation of its substrate, Stat3. It also causes the death of the large majority of the cells. A small fraction of drug resistant cells survive the canertinib treatment, but only at high cell densities. Stat3 activation increases as a function of cell density and we suggest that Stat3 induction through tight cell–cell interactions, most likely through the engagement of E-cadherin, can counteract the inhibitory effects exerted by canertinib on Bmx. Cell–cell interactions induced Stat3 and compensated the suppression of Stat3 by canertinib, thus protecting the cells from the cytotoxic effects of the inhibitor.Acknowledgements
We thank Jakob Weissenberger and Donat Kögel (Neuroscience Center, Goethe University Hospital, Frankfurt) for Tu-2449 cells. This work was supported by the institutional funds of the Georg Speyer Haus. The Georg Speyer Haus is funded jointly by the German Federal Ministry of Health (BMG) and the Ministry of Higher Education, Research and Arts of the State of Hessen (HMWK).References
- D. A. Haber, N. S. Gray and J. Baselga, The evolving war on cancer, Cell, 2011, 145(1), 19–24 CrossRef CAS PubMed.
- F. Iorio, T. A. Knijnenburg, D. J. Vis, G. R. Bignell, M. P. Menden, M. Schubert, N. Aben, E. Gonçalves, S. Barthorpe, H. Lightfoot, T. Cokelaer, P. Greninger, E. van Dyk, H. Chang, H. de Silva, H. Heyn, X. Deng, R. K. Egan, Q. Liu, T. Mironenko, X. Mitropoulos, L. Richardson, J. Wang, T. Zhang, S. Moran, S. Sayols, M. Soleimani, D. Tamborero, N. Lopez-Bigas, P. Ross-Macdonald, M. Esteller, N. S. Gray, D. A. Haber, M. R. Stratton, C. H. Benes, L. F. Wessels, J. Saez-Rodriguez, U. McDermott and M. J. Garnett, A Landscape of Pharmacogenomic Interactions in Cancer, Cell, 2016, 166(3), 740–754 CrossRef CAS PubMed.
- J. Settleman and R. L. Cohen, Communication in Drug Development: “Translating” Scientific Discovery, Cell, 2016, 164(6), 1101–1104 CrossRef CAS PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Cancer drug resistance: an evolving paradigm, Nat. Rev. Cancer., 2013, 13(10), 714–726 CrossRef CAS PubMed.
- B. C. Baguley, Classical and Targeted Anticancer Drugs: An Appraisal of Mechanisms of Multidrug Resistance, Methods Mol. Biol., 2016, 1395, 19–37 Search PubMed.
- J. Rueff and A. S. Rodrigues, Cancer Drug Resistance: A Brief Overview from a Genetic Viewpoint, Methods Mol. Biol., 2016, 1395, 1–18 Search PubMed.
- P. Lito, C. A. Pratilas, E. W. Joseph, M. Tadi, E. Halilovic and M. Zubrowski, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas, Cancer Cell, 2012, 22(5), 668–682 CrossRef CAS PubMed.
- P. Lito, N. Rosen and D. B. Solit, Tumor adaptation and resistance to RAF inhibitors, Nat. Med., 2013, 19(11), 1401–1409 CrossRef CAS PubMed.
- M. Cojoc, K. Mabert, M. H. Muders and A. Dubrovska, A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms, Semin. Cancer Biol., 2015, 31, 16–27 CrossRef CAS PubMed.
- H. J. Lee, G. Zhuang, Y. Cao, P. Du, H. J. Kim and J. Settleman, Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells, Cancer Cell, 2014, 26(2), 207–221 CrossRef CAS PubMed.
- C. Zhao, H. Li, H. J. Lin, S. Yang, J. Lin and G. Liang, Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism, Trends Pharmacol. Sci., 2016, 37(1), 47–61 CrossRef CAS PubMed.
- A. Britschgi, R. Andraos, H. Brinkhaus, I. Klebba, V. Romanet and U. Muller, et al. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer, Cancer Cell, 2012, 22(6), 796–811 CrossRef CAS PubMed.
- P. Ramos and M. Bentires-Alj, Mechanism-based cancer therapy: resistance to therapy, therapy for resistance, Oncogene, 2015, 34(28), 3617–3626 CrossRef CAS PubMed.
- A. Eichten, J. Su, A. P. Adler, L. Zhang, E. Ioffe and A. A. Parveen, et al. Resistance to Anti-VEGF Therapy Mediated by Autocrine IL6/STAT3 Signaling and Overcome by IL6 Blockade, Cancer Res., 2016, 76(8), 2327–2339 CrossRef CAS PubMed.
- F. H. Tan, T. L. Putoczki, S. S. Stylli and R. B. Luwor, The role of STAT3 signaling in mediating tumor resistance to cancer therapy, Curr. Drug Targets, 2014, 15(14), 1341–1353 CrossRef CAS PubMed.
- H. Yu, H. Lee, A. Herrmann, R. Buettner and R. Jove, Revisiting STAT3 signalling in cancer: new and unexpected biological functions, Nat. Rev. Cancer, 2014, 14(11), 736–746 CrossRef CAS PubMed.
- B. Groner and L. Hennighausen, The versatile regulation of cellular events by Jak-Stat signaling: from transcriptional control to microtubule dynamics and energy metabolism, Horm. Mol. Biol. Clin. Invest., 2012, 10(1), 193–200 CAS.
- B. Groner, Determinants of the extent and duration of STAT3 signaling, JAKSTAT, 2012, 1(3), 211–215 Search PubMed.
- V. Poli and A. Camporeale, STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance, Front Oncol., 2015, 5, 121 Search PubMed.
- M. S. Wake and C. J. Watson, STAT3 the oncogene - still eluding therapy?, FEBS J., 2015, 282(14), 2600–2611 CrossRef CAS PubMed.
- R. Arulanandam, M. Geletu, H. Feracci and L. Raptis, Activated Rac1 requires gp130 for Stat3 activation, cell proliferation and migration, Exp. Cell Res., 2010, 316(5), 875–886 CrossRef CAS PubMed.
- M. Geletu, R. Arulanandam, S. Chevalier, B. Saez, L. Larue and H. Feracci, et al. Classical cadherins control survival through the gp130/Stat3 axis, Biochim. Biophys. Acta, 2013, 1833(8), 1947–1959 CrossRef CAS PubMed.
- H. M. Smilowitz, J. Weissenberger, J. Weis, J. D. Brown, R. J. O'Neill and J. A. Laissue, Orthotopic transplantation of v-src-expressing glioma cell lines into immunocompetent mice: establishment of a new transplantable in vivo model for malignant glioma, J. Neurosurg., 2007, 106(4), 652–659 CrossRef PubMed.
- M. Priester, E. Copanaki, V. Vafaizadeh, S. Hensel, C. Bernreuther and M. Glatzel, et al. STAT3 silencing inhibits glioma single cell infiltration and tumor growth, Neuro Oncol., 2013, 15(7), 840–852 CrossRef CAS PubMed.
- J. S. Jarboe, S. Dutta, S. E. Velu and C. D. Willey, Mini-review: bmx kinase inhibitors for cancer therapy, Recent Pat. Anti-Cancer Drug Discovery, 2013, 8(3), 228–238 CrossRef CAS PubMed.
- E. A. Djerf Severinsson, C. Trinks, H. Green, A. Abdiu, A. L. Hallbeck and O. Stal, et al. The pan-ErbB receptor tyrosine kinase inhibitor canertinib promotes apoptosis of malignant melanoma in vitro and displays anti-tumor activity in vivo, Biochem. Biophys. Res. Commun., 2011, 414(3), 563–568 CrossRef CAS PubMed.
- J. Tang, Y. Qian, H. Li, B. J. Kopecky, D. Ding and H. C. Ou, et al. Canertinib induces ototoxicity in three preclinical models, Hear Res., 2015, 328, 59–66 CrossRef CAS PubMed.
- V. von Manstein and B. Groner, The Oncogenic Drivers V-Src in Tu-2449 Glioma Cells and EGFR in MDA-MB-468 Breast Cancer Cells Cause Differential Sensitivity to Stat3 Inhibition, J. Drug Des. Res., 2016, 3(2), 1026 Search PubMed.
- V. von Manstein, C. M. Yang, D. Richter, N. Delis, V. Vafaizadeh and B. Groner, Resistance of Cancer Cells to Targeted Therapies Through the Activation of Compensating Signaling Loops, Curr. Signal Transduction Ther., 2013, 8(3), 193–202 CrossRef CAS PubMed.
- A. Anagnostopoulou, A. Vultur, R. Arulanandam, J. Cao, J. Turkson and R. Jove, et al. Differential effects of Stat3 inhibition in sparse vs confluent normal and breast cancer cells, Cancer Lett., 2006, 242(1), 120–132 CrossRef CAS PubMed.
- L. Raptis, R. Arulanandam, A. Vultur, M. Geletu, S. Chevalier and H. Feracci, Beyond structure, to survival: activation of Stat3 by cadherin engagement, Biochem. Cell Biol., 2009, 87(6), 835–843 CrossRef CAS PubMed.
- L. Raptis, R. Arulanandam, M. Geletu and J. Turkson, The R(h)oads to Stat3: Stat3 activation by the Rho GTPases, Exp. Cell Res., 2011, 317(13), 1787–1795 CrossRef CAS PubMed.
- M. Niit, V. Hoskin, E. Carefoot, M. Geletu, R. Arulanandam and B. Elliott, et al. Cell-cell and cell-matrix adhesion in survival and metastasis: Stat3 versus Akt, Biomol Concepts., 2015, 6(5–6), 383–399 CAS.
- U. Pohl, W. Wick, J. Weissenberger, J. P. Steinbach, J. Dichgans and A. Aguzzi, et al. Characterization of Tu-2449, a glioma cell line derived from a spontaneous tumor in GFAP-v-src-transgenic mice: comparison with established murine glioma cell lines, Int. J. Oncol., 1999, 15(4), 829–834 CAS.
Footnote |
| † The authors declare no conflict of interest. |
| This journal is © The Royal Society of Chemistry 2017 |