
A portable and reconfigurable multi-organ platform for drug development with onboard microfluidic flow control†
J. R.
Coppeta‡
,
M. J.
Mescher‡
,
B. C.
Isenberg
,
A. J.
Spencer
,
E. S.
Kim
,
A. R.
Lever
,
T. J.
Mulhern
,
R.
Prantil-Baun
,
J. C.
Comolli
and
J. T.
Borenstein
*
Materials and Microfabrication Directorate, Draper, Cambridge, MA 02139, USA. E-mail: jborenstein@draper.com
First published on 23rd November 2016
Abstract
The drug development pipeline is severely limited by a lack of reliable tools for prediction of human clinical safety and efficacy profiles for compounds at the pre-clinical stage. Here we present the design and implementation of a platform technology comprising multiple human cell-based tissue models in a portable and reconfigurable format that supports individual organ function and crosstalk for periods of up to several weeks. Organ perfusion and crosstalk are enabled by a precision flow control technology based on electromagnetic actuators embedded in an arrayed format on a microfluidic platform. We demonstrate two parallel circuits of connected airway and liver modules on a platform containing 62 electromagnetic microactuators, with precise and controlled flow rates as well as functional biological metrics over a two week time course. Technical advancements enabled by this platform include the use of non-sorptive construction materials, enhanced scalability, portability, flow control, and usability relative to conventional flow control modes (such as capillary action, pressure heads, or pneumatic air lines), and a reconfigurable and modular organ model format with common fluidic port architecture. We demonstrate stable biological function for multiple pairs of airway–liver models for periods of 2 weeks in the platform, with precise control over fluid levels, temperature, flow rate and oxygenation in order to support relevant use cases involving drug toxicity, efficacy testing, and organ–organ interaction.
Introduction
Therapeutic drug development requires extensive pre-clinical testing and validation of drug candidates to accurately predict human safety and efficacy, which is both costly and time-consuming. However it is estimated that only one in nine drug candidates that enter clinical testing reach the market,1,2 indicating a need for pre-clinical assessments that are more predictive of human responses. In addition, some compounds may fail preclinical testing due to toxic side effects seen only in animal models, such as in drug-induced vascular injury,3 thereby depriving the pipeline of potentially safe and effective therapies.Current pre-clinical testing relies heavily on conventional in vitro laboratory assays that use primary cell cultures or cell lines cultured on a polystyrene surface or semi-permeable plastic membrane. These remain the gold standard because of their robustness, simplicity, and compatibility with high-throughput operation. However, a significant disadvantage of these models is that they often fail to accurately mimic the complexity of human organs, where multiple tissues and cell types are arranged in a complex, 3-dimensional architecture. Pre-clinical animal models are used to more accurately represent this complexity, and to provide data on more comprehensive and systemic responses. However, due to the phylogenetic differences between species, animals can have poor predictive capability for the drug responses of humans.4,5 Animal models are also inherently limited in their ability to support precisely controlled mechanistic studies.
The availability of more versatile, informative, and rapid pre-clinical models would advance drug development through better prediction of the human response. An active area of development is in physiologically relevant cell culture models produced by engineering the in vitro microenvironment of cells and tissues, often using microfluidics.6 These technologies enable researchers to reproduce important cell-to-cell signaling and mechanical cues that induce more realistic tissue phenotypes in vitro.7,8
Drug toxicity, and often efficacy, can be a consequence of a series of events involving several organs, spurring efforts toward advanced platform technologies that enable communication between organs. These systems are designed to more accurately recapitulate human drug absorption, distribution, metabolism, and elimination. Recently developed body-on-a-chip or human-on-a-chip platforms have been used to assess drug metabolism, absorption/barrier function, immune response, and drug interaction.6,7,9–12 In order for these systems to gain widespread use, they must demonstrate predictive power through in vitro–in vivo correlation (IVIVC) while providing ease of use, portability and low cost of ownership.
Adoption of these multi-organ platform technologies will require addressing several significant technical challenges involving appropriate temporal and spatial control of biochemical signaling between organs. One principal challenge involves scaling of the organ model device, media volume and cell number; this has been confronted by several groups in the human-on-a-chip domain.11,13,14
Another major biological challenge for interacting organ models involves the requirement for a common media, or blood substitute, that supports organ function in a connected circuit.6,15 Most existing cell culture models comprise a single or multiplex array of individual models of an organ or tissue bathed in media optimized for the function of the cells comprising a specific model. In interacting systems, development of a media that supports the function of the entire circuit of organ models must be accomplished, balancing critical media components for each element of the interacting system.
A third major challenge is predominantly an engineering obstacle; maintaining precise fluid volumes in each organ model for periods of several weeks while operating at relatively high fluid exchange rates. This challenge is heightened by effects such as evaporation, surface tension, slight variations in pump stroke volume, and the relatively low media volume in the circuit. For microfluidic platforms, this has been attempted using techniques involving capillary action,16 gravity flow,17,18 peristaltic micropumps19,20 and micropipetting with an aspirator/dispenser approach,21 among others. Systems that rely on passive diffusion or gravity flow suffer from limitations in dynamic range, and are not easily reconfigurable given the intertwined nature of the fluid exchange rate and the flow path. Pneumatic systems are more flexible, but do not easily scale, due to the need for individual wall-connected air lines to control each pump. Micropipetting systems are in the early stages of development; however their ability to mimic physiologic exchange processes may ultimately be limited due to the inherently transient nature of the aspiration and dispensing steps.
In response to these challenges and limitations, we have developed a robust, portable and scalable microfluidic platform that uses electromagnetically actuated micropumps to drive fluidic exchange.22–24 A key feature of the platform is the wide dynamic range of fluid exchange enabled by electromagnetic micropumps, which affords the ability to achieve precise temporal and spatial control of flow for organ perfusion, recirculation, concentration gradients, organ crosstalk, and fluid mechanical shear of cultured cell populations. Additional benefits realized by this approach include the portability and scalability enabled by embedding the micropumps within the platform, with only a ribbon cable connecting the platform to a unit providing control signals and power, essentially realizing the first electronically controlled dynamic well plate.
In this work, we present the integration of the programmable electromagnetic micropump technology reported in ref. 23 in an arrayed format that enables on-chip organ model perfusion and crosstalk for microphysiological systems (MPS) applications. We evaluate fluidic performance of the platform in supporting the demonstration of an interconnected circuit comprising four airway modules and one liver module with frequent fluid exchange between modules over the course of 14 days of operation. The airway–liver interaction was chosen because we have established a robust solution for common media between these organ models, as we will later describe. The key demonstration reported here is stable and programmable operation of a microphysiological system that enables dynamic organ perfusion and organ crosstalk in a reconfigurable, convenient and nearly fully portable manner. This provides an initial demonstration of the ability of the platform to support organ model interaction schemes that can be used to mimic key aspects of human metabolism, thus enabling more accurate prediction of human drug responses commonly referred to as IVIVC, in future biological applications.
Methods
Cell culture
Cryopreserved primary normal human bronchial epithelial (NHBE) cells (Lonza; Walkersville, MD) were seeded onto human placental collagen-IV (Sigma, St. Louis, MO) coated 6.5 mm Transwell inserts (Costar, Corning, NY) at a density of 3.0 × 105 cells per cm2. Cultures were submerged for three days and then maintained at an air–liquid interface (ALI) in lung differentiation medium (LDM – Fulcher et al.25) with media replenished every other day. Airway modules were transferred to the platform seven days after their transition to ALI then maintained on the platform for 14 days.Cryopreserved primary human hepatocytes (Thermo Fisher, Waltham MA) were grown in Cryopreserved Hepatocyte Recovery Media (CHRM), centrifuged, then suspended in hepatocyte plating media (supplemented William's E media (WEM) – Life Technologies, NY) to a concentration of 2.0 × 106 cells per ml. Cryopreserved human Kupffer cells (Life Technologies, NY) were grown in advanced Dulbecco's modified Eagle media (DMEM) containing supplements and FBS, centrifuged, then suspended to a concentration of 6.0 × 105 cells per ml. Cells were seeded onto polystyrene scaffolds coated with rat tail collagen I and assembled along with a filter and retaining ring into the liver MPS. The seeding protocol was adapted from one used by CN Bio Innovations (Hertfordshire, UK). Hepatocytes (6.0 × 105) were seeded directly onto each module scaffolds followed by 6.0 × 104 Kupffer cells. Seeded cells were exposed to downward flow of medium for 8 hours to assist cell adhesion to the scaffold, then pumping was reversed. The next day (considered day 1), plating medium was changed to supplemented advanced DMEM without glucocorticoid (Life Technologies). Maintenance media, WEM with Life Technologies cocktail B and 100 nM hydrocortisone, was used after day 3 and was changed every 48 hours. Hydrocortisone was chosen in the media rather than dexamethasone for its ability to maintain hepatocyte function, as is seen with upregulation of hepatocyte nuclear factor, for instance.
Tissue assessment
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 in WEM maintenance medium) to each liver MPS and incubated at 37 °C for 1 hour with self-circulating flow in the upwards direction. Tissue formation was assessed by staining for cell nuclei on scaffolds using Hoescht 33342 (Thermo Fisher, Waltham MA) diluted 1:1000 in WEM for 20 minutes. Following image analysis, scaffolds were incubated in RIPA buffer at 4 °C then scraped to remove cells; total protein was measured using a BCA ELISA kit (Pierce). Human serum albumin (HSA) levels were normalized to the total amount of protein present.
:1000 in WEM maintenance medium) to each liver MPS and incubated at 37 °C for 1 hour with self-circulating flow in the upwards direction. Tissue formation was assessed by staining for cell nuclei on scaffolds using Hoescht 33342 (Thermo Fisher, Waltham MA) diluted 1:1000 in WEM for 20 minutes. Following image analysis, scaffolds were incubated in RIPA buffer at 4 °C then scraped to remove cells; total protein was measured using a BCA ELISA kit (Pierce). Human serum albumin (HSA) levels were normalized to the total amount of protein present.
 | ||
| Fig. 1 Multi-organ direct drive platform overview. A) Photo of assembled platform, B) exploded view of the MPS (microphysiological system) platform, showing the individual layers and components which make up the laminated fluidic layer and the actuator plate, C) graphical layout of single fluidic circuit comprising five organ modules as well as a systemic mixing chamber (top), showing both the cross-flow and through-flow module configurations, along with the pumps (Pm – module mixing, Pd – delivery pump, Pi – mixing chamber intake) and channel-select valves (V) controlling flow between elements of the circuit, D) cross-section schematic of three actuators comprising a single pump (two valves and a pump chamber). The blue arrows trace the fluid path. In the configuration depicted, the upstream valve closes off flow into the pump by depressing the membrane onto the rubber valve seat; the pump chamber actuator displaces fluid forward through the downstream valve, which is open, allowing the membrane to relax and allow fluid flow through the valve seat and onward into the flow path. | ||
The fluidic layer module is typically 3.18 mm thick and is machined from polyetherimide Ultem® 1000 sheets after annealing for 1 h at 175 °C. The channels layers are also polyetherimide, vary in thickness from 0.076 to 0.25 mm, and are patterned via UV laser machining. The elastomer valve seat layer is 0.25 mm thick Viton and is also patterned by UV laser. The membrane layer is Kapton® polyimide, typically of thickness 0.025 mm. The various layers are laminated in a thirty ton press in air at 175 °C using RFlex® 1000 adhesives with thicknesses of 0.025 or 0.125 mm. The elastomer layer is tensioned during lamination to compensate for thermal expansion mismatches. The membrane is also tensioned, in this case to produce repeatable stroke volumes by eliminating the occurrence of buckling which can occur due to the fabrication processes or mechanical creep of the pump membranes from repeated actuation.
As shown in Fig. 1B, the platform is composed of three primary interconnected elements: 1) reconfigurable MPS modules that house the individual tissue models, 2) the fluidic manifold, which contains a network of microchannels connecting the individual modules via a system of pumps and valves, and 3) an actuation layer that contains an array of electromagnetic actuators that operate the valves and pump chambers. The modules attach to the top of the fluidic manifold and interface with the microchannels via a set of standardized ports. The fluidic manifold sits directly on top of the actuator layer such that the actuators contact the valves and pump chambers. Individual actuators are opened and closed via current pulses supplied by an external controller, which is itself is controlled by a user-programmable smart device. The MPS modules possess one or more fluidic compartments, which may be sealed or open to the atmosphere, and may contain sensor hardware supporting measurements such as oxygen tension and TEER. Additionally, the system is compatible with an inverted microscope for moderate magnification (10–20×) in situ tissue inspection. Electromagnetic pumps embedded in the platform pull fluid from or dispense fluid into culture-insert-containing modules. Real time selection of fluid routes are controlled by multiple-outlet-port pumps and/or a set of channel-selector valves. A sample configuration of the fluidic circuit for the platform, comprising five organ modules and a systemic mixing module, is shown in Fig. 1C. For example, fluid can be routed from the topmost module (the systemic mixer module) to the organ module immediately below it by actuating the inlet valve and closing the outlet valve to the delivery pump (labelled P_d). In Fig. 1C, the inlet valve to P_d is the valve between the mixing chamber pump (P_i) and P_d, and the outlet valve to P_d is the valve immediately downstream from P_d. Similarly, fluid can be routed from the mixer module to one of the lower organ modules by closing the inlet valve and actuating the outlet valve to P_d while simultaneously opening one or more of the downstream channel select valves (labelled V) controlling flow to a particular channel. The channel select valves are used (as opposed to incorporating additional pump-port valves) to avoid the pump performance degradation of many-port pumps (valve compliances add and eventually degrade the pump stroke volume).

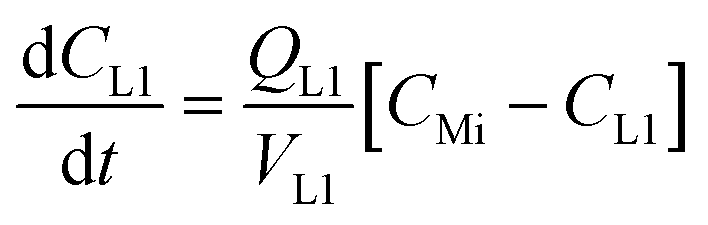



where CMi = drug concentration in mixer [mol L−1], CLn = drug concentration in airway #n compartment [mol L−1] (n = 1 to 4), CLi = drug concentration in liver compartment [mol L−1], VMi = volume in mixer [L], VLn = volume in airway #n compartment [L] (n = 1 to 4), VLi = volume in liver [L], QMi = flow thru mixer [L min−1], QLn = flow thru airway #n compartment [L min−1] (n = 1 to 3), QLi = flow thru HPV (mixer to liver) [L min−1].
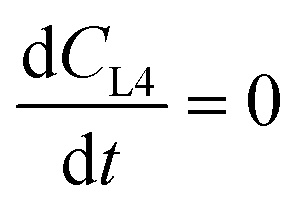
Results and discussion
Plug-and-play platform technology
The platform presented here can be used to recapitulate a myriad of in vivo organ systems, including up to twelve independent organs. For this initial demonstration, we chose a pair of interacting liver–airway systems with open-well MPS modules to demonstrate the functionality of the platform (Fig. 1C). The liver–airway interaction was chosen for two reasons. First, hepatotoxicity resulting from inhaled anaesthetics such as halothane and isoflurane represents one of the most prominent and widespread examples of an unexpected drug safety issue in history27 and remains a significant challenge in the developing world today. Second, the liver and lung are known to interact in the pathogenesis and outcome of sepsis induced acute lung injury.28 For instance, ischemia/reperfusion injury occurring during Gram-negative sepsis has been observed to result in higher rates of acute respiratory distress syndrome (ARDS)29 While this has been extensively studied in rodent models, alteration of the kinetics of circulating TNF-α and endotoxin may be more accurately probed in a human-based in vitro model system.A six-module fluidic circuit, duplicated on each half of the plate, can either create a biological replicate if using the same flow patterns and modules or, alternatively, two different experiments by programming different flow rates in each circuit (Fig. S1†). The figure illustrates how pump chambers, valves and fluid paths are configured to mimic complex multi-organ circulatory interaction, specifically with open-well modules, utilizing both inlet and outlet pumps and a robust means for maintaining fluid volume control within modules; this can be challenging in a multi-pump open module system. A mixing reservoir is connected to inlet and outlet pumps to distribute fluid between modules. This design allows future experiments connecting up to five unique tissues while simultaneously testing mixing rates on the individual airway modules.
The basic electromagnetic actuator, pump chambers and valve architectures have been described previously.23 Briefly, Fig. 1D shows a cross-section of the pump architecture consisting of two valves and a central pump chamber. In this low-power design, pins are biased against a tensioned pump or valve membrane when unpowered (normally closed) via spring force. Applying a current pulse causes the pin to overcome the spring force and retract from the fluid chamber, allowing the tensioned membrane to open the valve or pump chamber. A six-step sequence of opening the first valve, opening the pump chamber, closing the first valve, opening the second valve, closing the pump chamber, and closing the second valve displaces one aliquot of fluid (ca. 0.5–1 μL). Using this sequence, these pumps are able to operate in the pressure range of approximately −30 kPa to 60 kPa and achieve average flow rates ranging from approximately 0 to 13 μL s−1 (see supplementary data). Operational pump rates are set to capture the appropriate interaction and mixing time scales relative to the biological processes of interest. Arranging more than two valves to a central pump chamber can create a variety of multi-directional flow paths (Fig. S6†).
All 20 platform pumps were tested over a two-week period in an incubator maintained at 37 °C without any programmatic flow corrections, and demonstrated stable flow control of 0.58 ± 0.12 μL per stroke. Given the pulsatile flow of the pumps, inline fluidic compliance may be required to reduce ripple flow24 and was incorporated in the current design to maintain shear rates below the damage threshold of hepatocytes.
Platform interaction: liver–airway tissue 14 day study
A 14 day liver–airway interaction study with a high inter-module fluid exchange rate was executed evaluating tissue biology and platform performance (Fig. 2A). The airway tissue is a tracheobronchial model comprised of primary NHBE cells cultured at air–liquid interface. Tracheobronchial epithelial cells are seeded at 100000 per insert, a cell number that rises to a degree during the growth phase. The liver model is comprised of a perfused primary human hepatocytes co-cultured with primary human Kupffer cells in a 10:1 cellular ratio. The hepatocytes are seeded at a target of 540000 per liver module.
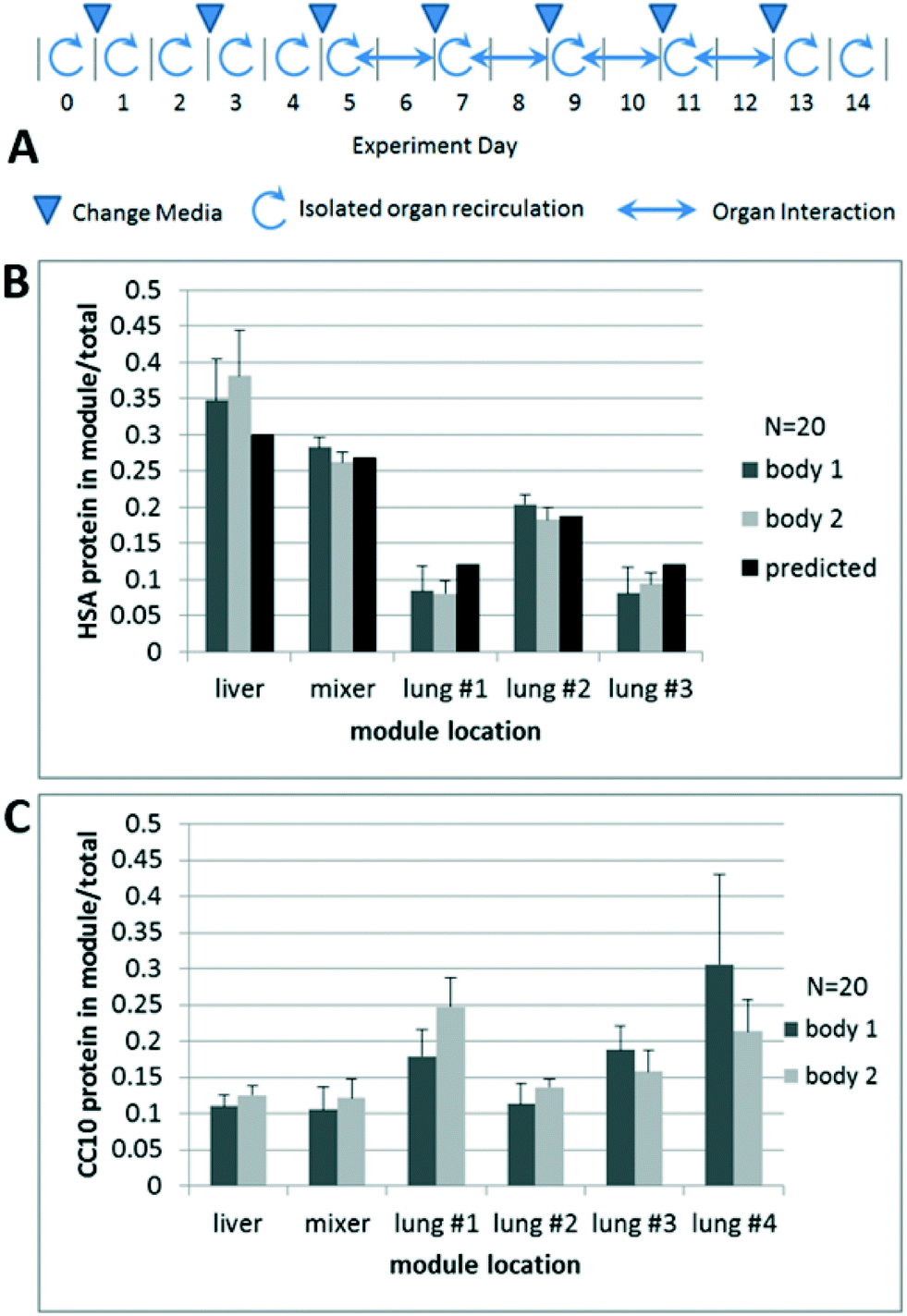 | ||
| Fig. 2 Results from 14 day liver–airway interaction experiment. A) Graphical summary of 14 day experiment interaction details, B) liver HSA (human serum albumin) distribution in each MPS module normalized by total HSA on platform versus pharmacokinetic prediction. C) Lung CC10 (club cell 10 kDa) protein distribution in each MPS (microphysiological system) module normalized against total CC10 summed for all modules. Data is represented as the mean ± SD (standard deviation) for three replicates for each measurement for both bodies 1 and 2. | ||
To demonstrate platform function, duplicate sets of a single liver module and four airway modules, were equilibrated on the platform for five days with re-circulating within-module medium flow followed by four consecutive 48 hour interaction cycles. Each interaction cycle began with a media change followed by a 17 hour conditioning period (see below) with intra-module circulation of medium to help reduce detrimental effects encountered when exposed to the other tissue's maintenance medium. During each 48-cycle, samples were collected at 17 (start of the interaction period), 24, and 48 hours after medium change, after which the medium was replaced and the conditioning period began again for a total of four cycles.
The flow distribution between organs during interactions was based on a five organ system. For this case, we reference Fig. 1C regarding the fluid distribution for each element of the circuit. The fluid distribution included 100% of fluid routed through the mixer (top module), 50% directly through the first airway module (airway #1, right side of second row) to the liver (left side of second row), 30% directly to the liver, 10% through two MPS modules each (airway #2 & #3, third and fourth rows), and 0% through the airway #4 (bottom row of Fig. 1C) as an on-platform static control. In order to demonstrate high exchange rates, the flow rate through the mixer was set at 10 mL per day, and each module's flow rate was set to 1.8 volume changes per hour, resulting in greater than 90% mixing of a tracer at the tissue scaffold to approximate a well-mixed system at the 1 hour time scale. Actual fluid exchange volumes per day can be calculated by multiplying the total mixer flow rate (10 mL per day) by the percentage of fluid exchanged between each organ module, as indicated above. Media volumes for each module are indicated in Table 1s (see ESI†). A comprehensive effort to address organ scaling was not considered for this initial assessment of the platform function, however, for reference, 10 mL per day corresponds to approximately four times the blood perfusion rate of media through the liver MPS model relative to a human liver using the number of hepatocytes as a scale factor.11 As Table 1s† shows, the ratio of media to cell volume in the in vitro platform is far higher for both the liver and the airway models relative to physiologic values, resulting in dilution factors of roughly 200–950× versus in vivo ratios. However, the platform technology presented here can be readily adapted to closed microfluidic organ models, reducing the dilution factors significantly and more closely approximating in vivo levels.
Lung and liver-secreted specific biomarkers (i.e., CC10 (club cell protein 10 kDa) and albumin) were selected to assess medium exchange, organ function, and platform performance. For the lung, CC10 is a specific marker due to the presence of club cells present in culture, and they serve as a marker of tissue health.30 These markers served to validate organ health throughout the experiment, while additional endpoint metrics were developed to further confirm that the platform supports long-term function and physiology. Mucin production ranged from 10–12 μg per day across the platform.
To demonstrate that the platform performed as designed with respect to exchange of medium between organ models, we measured the amount of albumin, secreted by hepatocytes, and CC10, which is secreted by bronchiolar exocrine cells (club cells),31 in each platform module after each of the four 48 hour interaction cycles. Though the total amount of albumin in the platform modules varied over the 14 day study (Fig. 2B), the distribution of albumin after each interaction cycle was comparable, indicating that the platform fluidics reproducibly controlled tissue interactions and the liver behaved predictably over the course of the experiment. In addition, the average albumin distribution on the two individual systems/bodies on the platform were similar, demonstrating consistent operation of both systems. The concentration of albumin in each module after an interaction showed reasonable agreement with multi-compartment pharmacokinetic modeling. Overall distribution of CC10 was also similar over the four interaction runs and between platform bodies (Fig. 2C), although it was not simulated due to variability in airway tissue production on a daily basis. By day 14, CC10 levels ranged from 10–60 ng per mL between the various lung modules, with much lower amounts in the mixer and liver module.
Common media strategy is enabled by platform controlled tissue interactions
Individual components of each media were examined to determine constituents which may compromise one of the MPS tissues. While both mediums were serum free, the differentiation medium required by the lung is rich in supplements and additives relative to that of the liver MPS. From this analysis, epinephrine, bovine pituitary extract, and bovine serum albumin (BSA) were identified as constituents requiring modification in the lung medium because of their effects on liver function.32–34 LDM was modified by reducing the epinephrine concentration to 0.25 μM and lowering the bovine pituitary extract (BPE) concentration to 5 μg per mL, which maintained the airway and liver as measured by the functional and morphological metrics shown below. Bovine serum albumin was increased from 0.5 to 1.25 mg mL−1 to match the concentration in the liver medium and maintain carrier protein concentrations.We hypothesized that if each organ was allowed sufficient time to “condition” its own media – i.e., metabolize critical components to a point that reduces its potency/concentration – then upon medium exchange, the level of damage caused by potentially incompatible compounds could be reduced. The platform allowed this strategy to be implemented by controlling the timing and extent of tissue interactions, however, additional experimentation will be required to verify the effects of this strategy.
Liver MPS model morphology and function over 14 day on-platform experiment
Morphology and health of perfused primary human hepatocyte co-culture tissue scaffolds were assessed after 14 days by staining cell nuclei and monitoring albumin production and oxidative enzyme activity (CYP3A4). Hepatocyte co-cultures were effectively seeded at acceptable densities across most scaffold wells, as observed via visual inspection; some wells exhibit a “half-moon” pattern of cells, partly an artifact of removing and preparing the scaffold for staining (Fig. 3A shows Hoechst nuclear stain and live stain for a representative liver module on the platform), but also indicative of the fact that the pore regions are typically not filled completely with hepatocytes. The morphology is similar to that observed after 4–7 days in other non-interacting perfused hepatocyte bioreactor systems such as the LiverChip™.35 As a control study, we maintained the same lot of primary liver hepatocyte co-cultures used on the multi-organ platform on a LiverChip™, which houses 12 individual liver modules but does not support organ interaction. Albumin production by primary human hepatocytes on the platform peaked on day 6 (24 ± 0.6 μg per day per mg protein) with levels gradually declining until the end of the study to 2.6 ± 0.8 μg per day per mg protein. This was comparable to the range of albumin production, 5.8 ± 0.6 to 19.3 ± 1.5 μg per day per mg protein, from primary liver hepatocytes maintained on the Liverchip™ platform though the rate of production peaked on day 10 in these perfusion cultures. These values were also consistent with published albumin production data from cryopreserved human primary hepatocytes.36 Average total protein production for the two liver tissues maintained on the multi-organ platform was 180 ± 10 μg after 14 days, while those on the LiverChip™ yielded 70 ± 10 μg total protein. Overall, primary human hepatocytes actively interacting with human primary airway cells on the platform maintained viability and albumin production consistent with non-interacting perfusion systems. | ||
| Fig. 3 Liver results from 14 day interacting liver–airway experiment. A) Representative images of liver co-culture formation in scaffold wells of liver MPS. Cell nuclei (blue) are shown via fluorescent microscopy after Hoechst staining (left), with live stain (green) shown for the same field of view on the right. Tissue formation is completely confluent in majority of scaffold wells, exhibiting a “half-moon” appearance in others. B) Evaluation of CYP3A4 (cytochrome P450 3A4) activity normalized to total protein for liver MPS measured at day 14. Data is represented as the mean ± SD (standard deviation). Scale bar = 500 μm. | ||
The activity of CYP3A4, the most highly expressed cytochrome P450 drug-metabolizing enzyme in human hepatocytes, was also measured in the two on-platform liver tissues upon completion of the 14 day interaction study. CYP3A4 activity was 1.15 ± 0.03 and 0.85 ± 0.05 pmol min−1 mg−1 total protein, respectively, which was not significantly different than that of hepatocytes maintained on the LiverChip™ (Fig. 3B). This indicated that the hepatocytes retained their metabolic capability on platform for 14 days and that there was no detrimental effect of the liver–airway interaction on expression of CYP3A4.
Airway MPS model tissue morphology and function over 14 day on-platform experiment
Airway tissue barrier function and morphology was maintained throughout the 14 day experiment on each set of four airway tissues as measured on day 14 by immunohistochemistry and functional metrics including TEER and mucus production rate. The ratio of essential cell subtypes in the pseudostratified airway epithelium was determined by IHC staining with acetylated tubulin for ciliated cells and CK5 for basal cells. This analysis indicated that there was no significant change in the population of ciliated and basal cells in on-platform airway tissues compared to off-platform controls (Fig. 4A). TEER measurements of on-platform airway tissue under dynamic flow were not significantly different from airway tissues maintained in static culture either on-platform (airway #4) or off-platform (also noted in Fig. 4B) and are in the expected range of similar models,36 with a range of roughly 700–1000 Ωcm2. Also, as shown in Fig. 4B, the rate of mucus production from goblet/mucus-producing cells within the primary tracheobronchial tissue was not significantly different in tissues maintained off platform and those interacting with the liver on platform. These metrics indicate that the platform was able to maintain viability of the airway tissues and that interaction with the liver tissue had no significant detrimental effect on airway tissue integrity or function. | ||
| Fig. 4 Airway results from 14 day interacting liver–airway experiment. A) Validation of airway specific cell subtypes. On-platform and off platform airways were stained for ciliated cells (green) and basal cells (red) at day 21 of differentiation with all lungs staining positive for both airway markers. Images were taken for bodies 1 and 2 (n = 2 airways, n = 4 FOV (field of view) per lung MPS) and for static controls (n = 2 airways, n = 4 FOV per airway). Scale bar = 100 μm. (B) Mucus production and TEER (TransEpithelial Electrical Resistance) were evaluated as an endpoint analysis on day 14. No significant change was observed in on-platform airways relative to static controls. Data is represented as the mean ± SD (standard deviation) for bodies 1 and 2. | ||
The airway MPS was comprised of NHBE cells that when differentiated at an air–liquid interface form a pseudo-stratified epithelium that recapitulates in vivo airway physiology and function, such as barrier function, mucus and CC10 secretion as well as the presence of essential cell subtypes. Using metrics to measure these baseline functions and responses developed by Lever et al.,26 we confirmed that the airway MPS maintained physiology following the 14 day on-platform study and interaction with liver MPS. Our results show that the airway MPS constructs maintained epithelial integrity, as indicated by TEER, along with baseline levels of secreted mucus. Mucus is secreted by goblet cells apically, coating the epithelial surface, and adding another level of protection to the epithelial barrier.37 Our imaging results demonstrated that the ciliated cell population was abundant in our airway model following platform studies. Together, ciliated cells and mucus-producing cells trap and clear pathogens from the lung, a process known as mucociliary transport.38 Basal cells and club cells are also essential for airway function and are both required for cell turnover and epithelial repair upon injury.39,40 We confirmed the presence of basal cells via positive staining for Ck5. Club cells secrete CC10,41 a protein specific to the airway MPS, which we measured via ELISA and found to remain stable throughout experiment. Our results demonstrate that the platform supported physiology of the airway MPS and baseline functions representative of the in vivo airway.
Liver function was also maintained on platform following the 14 day study and interaction with the airway MPS. We chose to evaluate albumin secretion, CYP3A4 activity and tissue formation as these are all well-studied metrics for assessing liver performance in vitro.41,42 In humans, albumin synthesis takes place only in the liver at a rate of approximately 194 mg kg−1 per day in healthy individuals.43 We measured the rate of albumin production throughout the experiment and found that albumin peaked on day 6 with levels gradually declining to day 14. This decline is typical of long-term hepatocyte cultures.41,44 We also evaluated CYP3A4 activity, an essential drug metabolizing enzyme that is critical for studying pharmacokinetic drug–drug interactions.45 Both liver MPSs expressed CYP3A4 activity after 14 days, higher than what has been reported in sandwich culture conditions.41,46 Tissue formation corroborated these results, with most scaffold wells exhibiting robust hepatocyte seeding. Together, these results show that the platform maintained liver MPS function throughout the 14 day experiment.
Conclusions
Our platform allows for dynamic multi-organ interaction and medium exchange in a highly controllable manner. The overall design supports an unprecedented level of reconfigurability in contrast to traditional well plates. For instance, the fluidic manifold plate can be redesigned with different fluid routing schemes, MPS modules can be assigned to multiple locations on the fluidic plate and accept multiple tissue formats, and flow schemes can be varied in real time. Unlike many pneumatically actuated systems,47 the present platform technology does not require separate vacuum or air lines for each actuating element, and it operates independently of routine variations in laboratory environmental conditions such as air or vacuum pressure levels or humidity levels in the lines.The open design and capabilities of the platform present limitless opportunities for the addition of complex organ interaction systems, as well as the application of use cases for controlled drug delivery or multi-organ toxicity and efficacy evaluations. We demonstrated that the platform is capable not only of controlled medium exchange and mixing, but also of supporting long-term organ model health and function. The interaction of these two organ models demonstrated successful simultaneous achievement of key historical organ-on-chip challenges including common media development to support tissue function, constant reliable perfusion to support liver metabolic needs, long term tissue interaction, and programmable interactions creating predictable mixing patterns amenable to modeling.
While the platform technology presented here offers a precise, robust and reliable approach that enables crosstalk between multiple organ models, several additional near-term opportunities are made possible by this new capability. Current in vitro ADME-Tox tools are generally limited to static systems that do not adequately recapitulate the organ microenvironment and cannot precisely control drug, nutrient and metabolite gradients in the media, reducing physiologic relevance for many organ systems and disease models. This dynamic platform technology described here can be customized to provide precisely controlled levels of shear flow and tuned to generate specific concentration gradients for soluble factors, while maintaining the simplicity and compatibility of standard well plate technology in pharmaceutical laboratories. Scaling these systems into a multiplexed architecture for individual organ models for diseases and toxicology studies will provide a significant benefit to the drug development process, since the inability to scale emerging microfluidic organ models stands as one of the principal barriers to entry for wider use. Ultimately the reconfigurable and scalable nature of this technology will enable highly multiplexed systems and more complex models involving multiple organ constructs and systems as these in vitro platform technologies gain broader use throughout the drug development process.
Acknowledgements
We would like to thank David Hughes of CN Bio Innovations, Patrick Hayden of MatTek, Micah Sam Brickman Raredon of Yale Medical School and Ujjal Sarkar, Ravi Khodilalli, Dinelia Burgos-Rivera and Steve Tannenbaum of MIT for technical advice and assistance. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the U.S. Government or any other third party. This work was supported in part by NIH/NCATS/NIEHS/NICHD/OWHR/UH3TR001207, and by Draper.References
- J. A. DiMasi, L. Feldman, A. Seckler and A. Wilson, Trends in risks associated with new drug development: success rates for investigational drugs, Clin. Pharmacol. Ther., 2010, 87(3), 272–277 CrossRef CAS PubMed.
- M. Hay, D. W. Thomas, J. L. Craighead, C. Economides and J. Rosenthal, Clinical development success rates for investigational drugs, Nat. Biotechnol., 2014, 32(1), 40–51 CrossRef CAS PubMed.
- W. Kerns, et al., Drug-induced vascular injury--a quest for biomarkers, Toxicol. Appl. Pharmacol., 2005, 203(1), 62–87 CrossRef CAS PubMed.
- J. J. Hornberg, et al., Exploratory toxicology as an integrated part of drug discovery. Part I: Why and how, Drug Discovery Today, 2014, 19(8), 1131–1136 CrossRef CAS PubMed.
- C. Tamaki, et al., Potentials and limitations of nonclinical safety assessment for predicting clinical adverse drug reactions: correlation analysis of 142 approved drugs in Japan, J. Toxicol. Sci., 2013, 38(4), 581–598 CrossRef PubMed.
- J. H. Sung, et al., Microfabricated mammalian organ systems and their integration into models of whole animals and humans, Lab Chip, 2013, 13(7), 1201–1212 RSC.
- A. Polini, et al., Organs-on-a-chip: a new tool for drug discovery, Expert Opin. Drug Discovery, 2014, 9(4), 335–352 CrossRef CAS PubMed.
- T. S. Chan, et al., Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome P450 using a novel hepatocyte model, HepatoPac, Drug Metab. Dispos., 2013, 41(12), 2024–2032 CrossRef CAS PubMed.
- D. Huh, et al., A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice, Sci. Transl. Med., 2012, 4(159), 159ra147 Search PubMed.
- K.-H. Nam, A. S. T. Smith, S. Lone, S. Kwon and D.-H. Kim, Biomimetic 3D Tissue Models for Advanced High-Throughput Drug Screening, J. Lab. Autom., 2015, 20(3), 201–215 CrossRef CAS PubMed.
- J. P. Wikswo, et al., Scaling and systems biology for integrating multiple organs-on-a-chip, Lab Chip, 2013, 13(18), 3496–3511 RSC.
- M. B. Esch, et al., How multi-organ microdevices can help foster drug development, Adv. Drug Delivery Rev., 2014, 69–70, 158–169 CrossRef CAS PubMed.
- C. Moraes, et al., On being the right size: scaling effects in designing a human-on-a-chip, Integr. Biol., 2013, 5(9), 1149–1161 RSC.
- J. P. Wikswo, et al., Engineering challenges for instrumenting and controlling integrated organ-on-chip systems, IEEE Trans. Biomed. Eng., 2013, 60(3), 682–690 CrossRef PubMed.
- S. Li, J. Nickels and A. F. Palmer, Liposome-encapsulated actin-hemoglobin (LEAcHb) artificial blood substitutes, Biomaterials, 2005, 26(17), 3759–3769 CrossRef CAS PubMed.
- F. Trietsch, R. J. Snijder, C. Kloosterziel and D. A. F. van den Heuvel, Pulmonary artery embolization for refractory hypoxemia caused by invasive mucinous adenocarcinoma, J. Thorac. Oncol., 2013, 8(2), e15–e16 CrossRef PubMed.
- J. H. Sung, C. Kam and M. L. Shuler, A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip, Lab Chip, 2010, 10(4), 446–455 RSC.
- P. J. Lee, T. A. Gaige, N. Ghorashian and P. J. Hung, Microfluidic tissue model for live cell screening, Biotechnol. Prog., 2007, 23(4), 946–951 CrossRef CAS PubMed.
- W. Inman, et al., Design, modeling and fabrication of a constant flow pneumatic micropump, J. Micromech. Microeng., 2007, 17(5), 891–899 CrossRef.
- I. Wagner, et al., A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture, Lab Chip, 2013, 13(18), 3538–3547 RSC.
- D. E. Ingber, D. Levner, I. Thompson, J. Fernandez-Alcon and C. D. Hinojosa, Systems and Methods for Cell Culture Device Interconnection and Fluidic Device Interconnection, 2015, Available at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015006751, [Accessed February 16, 2016] Search PubMed.
- M. Mescher, J. Fiering and C. Dube, Electromagnetically-actuated microfluidic flow regulators and related applications, 2005, Available at: http://www.google.com/patents/US20050238506, [Accessed February 16, 2016] Search PubMed.
- V. Tandon, et al., Microfabricated reciprocating micropump for intracochlear drug delivery with integrated drug/fluid storage and electronically controlled dosing, Lab Chip, 2016, 16(5), 829–846 RSC.
- M. J. Mescher, et al., Fabrication Methods and Performance of Low-Permeability Microfluidic Components for a Miniaturized Wearable Drug Delivery System, J. Microelectromech. Syst., 2009, 18(3), 501–510 CrossRef PubMed.
- L. M. Fulcher, et al., Well-differentiated human airway epithelial cell cultures, in Human Cell Culture Protocols, ed. J. Picot, Springer, Available at: http://www.springer.com/us/book/9781588292223, pp. 183–206, [Accessed March 7, 2016] Search PubMed.
- A. R. Lever, et al., Comprehensive evaluation of poly(I:C) induced inflammatory response in an airway epithelial model, Physiol. Rep., 2015, 3(4), e12334 CrossRef PubMed.
- D. K. Spracklin, D. C. Hankins, J. M. Fisher, K. E. Thummel and E. D. Kharasch, Cytochrome P450 2E1 is the principal catalyst of human oxidative halothane metabolism in vitro, J. Pharmacol. Exp. Ther., 1997, 281(1), 400–411 CAS.
- A. M. Siore, Endotoxin-induced acute lung injury requires interaction with the liver, Am. J. Physiol., 2005, 289(5), L769–L776 CAS.
- G. M. Matuschak, K. A. Henry, C. A. Johanns and A. J. Lechner, Liver-lung interactions following Escherichia coli bacteremic sepsis and secondary hepatic ischemia/reperfusion injury, Am. J. Respir. Crit. Care Med., 2001, 163(4), 1002–1009 CrossRef CAS PubMed.
- F. Broeckaert and A. Bernard, Clara cell secretory protein (CC16): characteristics and perspectives as lung peripheral biomarker, Clin. Exp. Allergy, 2000, 30(4), 469–475 CrossRef CAS PubMed.
- G. Singh and S. L. Katyal, Clara Cells and Clara Cell 10 kD Protein (CC10), Am. J. Respir. Cell Mol. Biol., 1997, 17(2), 141–143 CrossRef CAS PubMed.
- L. A. Sachs, W. E. Finkbeiner and J. H. Widdicombe, Effects of media on differentiation of cultured human tracheal epithelium, In Vitro Cell. Dev. Biol.: Anim., 2003, 39(1–2), 56–62 CrossRef CAS.
- J. C. Willey, M. A. Laveck, I. A. McClendon and J. F. Lechner, Relationship of ornithine decarboxylase activity and cAMP metabolism to proliferation of normal human bronchial epithelial cells, J. Cell. Physiol., 1985, 124(2), 207–212 CrossRef CAS PubMed.
- C. Aninat, et al., Catecholamines induce an inflammatory response in human hepatocytes, Crit. Care Med., 2008, 36(3), 848–854 CrossRef CAS PubMed.
- K. Domansky, et al., Perfused multiwell plate for 3D liver tissue engineering, Lab Chip, 2010, 10(1), 51–58 RSC.
- A. Vivares, et al., Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device, Xenobiotica, 2015, 45(1), 29–44 CrossRef CAS PubMed.
- M. C. Rose and J. A. Voynow, Respiratory Tract Mucin Genes and Mucin Glycoproteins in Health and Disease, Physiol. Rev., 2006, 86(1), 245–278 CrossRef CAS PubMed.
- M. Vareille, E. Kieninger, M. R. Edwards and N. Regamey, The Airway Epithelium: Soldier in the Fight against Respiratory Viruses, Clin. Microbiol. Rev., 2011, 24(1), 210–229 CrossRef CAS PubMed.
- J. R. Rock, S. H. Randell and B. L. M. Hogan, Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling, Dis. Models & Mech., 2010, 3(9–10), 545–556 CAS.
- F. Broeckaert, A. Clippe, B. Knoops, C. Hermans and A. Bernard, Clara cell secretory protein (CC16): features as a peripheral lung biomarker, Ann. N. Y. Acad. Sci., 2000, 923, 68–77 CrossRef CAS PubMed.
- Y. Kono, S. Yang and E. A. Roberts, Extended primary culture of human hepatocytes in a collagen gel sandwich system, In Vitro Cell. Dev. Biol.: Anim., 1997, 33(6), 467–472 CrossRef CAS PubMed.
- D. Roymans, et al., Determination of cytochrome P450 1A2 and cytochrome P450 3A4 induction in cryopreserved human hepatocytes, Biochem. Pharmacol., 2004, 67(3), 427–437 CrossRef CAS PubMed.
- J. P. Nicholson, M. R. Wolmarans and G. R. Park, The role of albumin in critical illness, Br. J. Anaesth., 2000, 85(4), 599–610 CrossRef CAS PubMed.
- B. Clement, et al., Long-Term Co-Cultures of Adult Human Hepatocytes with Rat Liver Epithelial Cells: Modulation of Albumin Secretion and Accumulation of Extracellular Material, Hepatology, 1984, 4(3), 373–380 CrossRef CAS PubMed.
- Wiley: Drug-Drug Interactions in Pharmaceutical Development, ed. A. P. Li and B. Wang, Available at: http://www.wiley.com/WileyCDA/WileyTitle/productCd-0471794414.html, [Accessed February 16, 2016] Search PubMed.
- Q. Yang, U. Doshi, N. Li and A. P. Li, Effects of culture duration on gene expression of P450 isoforms, uptake and efflux transporters in primary hepatocytes cultured in the absence and presence of interleukin-6: implications for experimental design for the evaluation of downregulatory effects of biotherapeutics, Curr. Drug Metab., 2012, 13(7), 938–946 CrossRef CAS PubMed.
- M.-H. Wu, S.-B. Huang, Z. Cui, Z. Cui and G.-B. Lee, A high throughput perfusion-based microbioreactor platform integrated with pneumatic micropumps for three-dimensional cell culture, Biomed. Microdevices, 2008, 10(2), 309–319 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6lc01236a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |