
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multidisciplinary investigation of the technical and environmental performances of TAML/peroxide elimination of Bisphenol A compounds from water†‡
Yusuf
Onundi
a,
Bethany A.
Drake
b,
Ryan T.
Malecky
b,
Matthew A.
DeNardo
 *b,
Matthew R.
Mills
b,
Soumen
Kundu
b,
Alexander D.
Ryabov
*b,
Evan S.
Beach
b,
Colin P.
Horwitz
b,
Michael T.
Simonich
c,
Lisa
Truong
c,
Robert L.
Tanguay
*c,
L. James
Wright
*d,
Naresh
Singhal
*a and
Terrence J.
Collins
*b
*b,
Matthew R.
Mills
b,
Soumen
Kundu
b,
Alexander D.
Ryabov
*b,
Evan S.
Beach
b,
Colin P.
Horwitz
b,
Michael T.
Simonich
c,
Lisa
Truong
c,
Robert L.
Tanguay
*c,
L. James
Wright
*d,
Naresh
Singhal
*a and
Terrence J.
Collins
*b
aDepartment of Civil & Environmental Engineering, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
bInstitute for Green Science, Department of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, Pennsylvania 15213, USA
cDepartment of Environmental and Molecular Toxicology, Sinnhuber Aquatic Research Laboratory, Environmental Health Sciences Center, Oregon State University, 28645 East HWY 34, Corvallis, OR 97333, USA
dCentre for Green Chemical Science, School of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: lj.wright@auckland.ac.nz; Tel: +64 9923 8257
First published on 2nd August 2017
Abstract
Designing technologies that mitigate the low-dose adverse effects of exposures to large-volume, everyday-everywhere chemicals such as bisphenol A (BPA, 1a) requires an understanding of the scope of the exposures and the nature of the adverse effects. Therefore, we review the literature of, (i) the occurrences of 1a in humans, waters and products and the effectiveness of widely deployed mitigation methods in 1a stewardship and, (ii) the adverse effects of 1a exposures on human cells and fish. Within this broad context, we present and evaluate experimental results on TAML/H2O2 purification of 1a contaminated waters. TAML/H2O2 catalysis readily oxidizes BPA (1a) and the ring-tetramethyl (1b), tetrachloro (1c), and tetrabromo (1d)-substituted derivatives. At pH 8.5, TAML/H2O2 induces controllable, oxidative oligomerisation of 1a (2-, 3-, 4-, and 5-unit species were identified) with precipitation, establishing a green synthetic pathway to these substances for biological safety characterisation and an easy method for near quantitative removal of 1a from water. TAML/H2O2 (24 nM/4 mM) treatment of 1a (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg L−1) in pH 8.5 (0.01 M, carbonate) lab water effects a >99% reduction (to <100 μg L−11a) within 30 min. Yeast oestrogen screens (YES) of the pH 8.5, TAML/H2O2 treated, catalase quenched, and filtered oxidation solutions show elimination of 1a oestrogenicity. Zebrafish developmental assays of TAML/H2O2 treated, unfiltered, agitated pH 7, 1a solutions showed no significant incidences of abnormality among any of 22 endpoints—treated samples showed an insignificant increase in mortality. At pH 11, the TAML/H2O2 oxidations of 1a–d are fast with second order rate constants for the substrate oxidation process (kII values) of (0.57–8) × 104 M−1 s−1. The 1a oxidation gives CO and CO2 (∼78%), acetone (∼25%) and formate (∼1%). In striking contrast with pH 8.5 treatment, no oligomers were detected. TAML/H2O2 (150 nM/7.5 mM) treatment of 1a (34244 μg L−1) in pH 11 (0.01 M, phosphate) lab water effected a >99.9% reduction (to <23 μg L−11a) within 15 min. The pH dependent behaviour of 1a was examined as a possible origin of the differing outcomes. The 1st and 2nd pKa values of 1a were estimated by fitting the pH dependence of the UV-vis spectra (pKa1 = 9.4 ± 0.3; pKa2 = 10.37 ± 0.07). At pH 8.5, coupling of the radical produced on initial oxidation evidently outcompetes further oxidation. A linear free energy relationship between the logarithm of the pH 11, kII values and the redox potentials of 1a–d as determined by differential pulse voltammetry in CH3CN is consistent with rate-limiting, electron transfer from the dianionic form of 1a at pH 11, followed by a multistep, deep degradation without observation of 4-(prop-1-en-2-yl)phenol 12, a common 1a oxidation product—an improved synthesis of 12 is described. Microtox® analyses of pH 12, TAML/H2O2 treated 1a solutions showed significantly reduced toxicity. The facility and high efficiency by which TAML/H2O2 catalysis eliminates 1a from water, by either mechanism, suggests a new and simple procedure for 1a stewardship.
000 μg L−1) in pH 8.5 (0.01 M, carbonate) lab water effects a >99% reduction (to <100 μg L−11a) within 30 min. Yeast oestrogen screens (YES) of the pH 8.5, TAML/H2O2 treated, catalase quenched, and filtered oxidation solutions show elimination of 1a oestrogenicity. Zebrafish developmental assays of TAML/H2O2 treated, unfiltered, agitated pH 7, 1a solutions showed no significant incidences of abnormality among any of 22 endpoints—treated samples showed an insignificant increase in mortality. At pH 11, the TAML/H2O2 oxidations of 1a–d are fast with second order rate constants for the substrate oxidation process (kII values) of (0.57–8) × 104 M−1 s−1. The 1a oxidation gives CO and CO2 (∼78%), acetone (∼25%) and formate (∼1%). In striking contrast with pH 8.5 treatment, no oligomers were detected. TAML/H2O2 (150 nM/7.5 mM) treatment of 1a (34244 μg L−1) in pH 11 (0.01 M, phosphate) lab water effected a >99.9% reduction (to <23 μg L−11a) within 15 min. The pH dependent behaviour of 1a was examined as a possible origin of the differing outcomes. The 1st and 2nd pKa values of 1a were estimated by fitting the pH dependence of the UV-vis spectra (pKa1 = 9.4 ± 0.3; pKa2 = 10.37 ± 0.07). At pH 8.5, coupling of the radical produced on initial oxidation evidently outcompetes further oxidation. A linear free energy relationship between the logarithm of the pH 11, kII values and the redox potentials of 1a–d as determined by differential pulse voltammetry in CH3CN is consistent with rate-limiting, electron transfer from the dianionic form of 1a at pH 11, followed by a multistep, deep degradation without observation of 4-(prop-1-en-2-yl)phenol 12, a common 1a oxidation product—an improved synthesis of 12 is described. Microtox® analyses of pH 12, TAML/H2O2 treated 1a solutions showed significantly reduced toxicity. The facility and high efficiency by which TAML/H2O2 catalysis eliminates 1a from water, by either mechanism, suggests a new and simple procedure for 1a stewardship.
Introduction
Green Chemistry has much to offer at every stage of the life-cycle of chemical products and processes. Both during and at the end of commercial utility, many chemical products become water contaminants.1,2 These may or may not be persistent. Those that negatively impact flora or fauna at low concentrations (ng L−1–μg L−1) are called “micropollutants” (MPs). Some MPs are endocrine disruptors (EDs). An endocrine disruptor is, “an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action”.3 Minimizing exposures to EDs is a significant sustainability challenge. High volume EDs that sufficiently elude water treatment processes to threaten the environment and human health are among the most difficult MPs to manage.4,5 For example, phenolic compounds are not completely removed by the combined physical, biological and chemical processes in water treatment plants resulting in contamination of released effluent streams.6,7Bisphenol A (2,2-bis(4-hydroxyphenyl)propane, BPA, 1a, Fig. 1) is one such continually emitted, anthropogenic, xenooestrogenic, high volume, commodity, phenolic ED found in multiple products.8–11 Although 1a is often regarded as weakly oestrogenic, its capacity to impact biological processes is modulated by several factors including interactions with plasma oestrogen-binding proteins,12,13 varying potential for metabolism,13–19 and the types and quantity of oestrogen receptors (ERs) present, including those bound to membranes.20,21 In some cases, 1a has been shown to have the same effect as and be as potent as the endogenous, primary, female sex hormone oestradiol (E2, Fig. 1),3,13,20,22,23 which can alter the functioning of cellular proteins at sub-picomolar to nanomolar concentrations.13,24 Oestrogens regulate development25–27 and actions in bone, brain, cardiovascular, liver, and reproductive tissues.28 ERs are targeted by some endogenous hormones and pharmaceuticals. Such drugs include oestrogens, anti-oestrogens and selective oestrogen receptor modulators like 1a,13 which lack the steroid ring structure of oestrogens (see E2, Fig. 1), but retain structural elements necessary to bind to ERs.28,30 Drugs that target ERs include fertility enhancers and contraceptives, as well as menopausal hormone,31 breast32 and prostate cancer33,34 therapeutics. In addition to acting as an oestrogen, 1a can act as a thyroid hormone35,36 and androgen.37 Inappropriate adjustment of oestrogen, thyroid hormone and androgen regulated processes, such that which can result from exposure to xeno-oestrogens like 1a, can have negative effects.20,25–27,31,38–44 Consequently, in 2016 the European union voted to recognize 1a as a presumed human reproductive toxicant.45 In 2017, it voted to also add 1a to the list of substances of very high concern for adverse effects on human mammary gland development, cognitive functions and metabolism, identifying it as a general disruptor of the human endocrine system.46
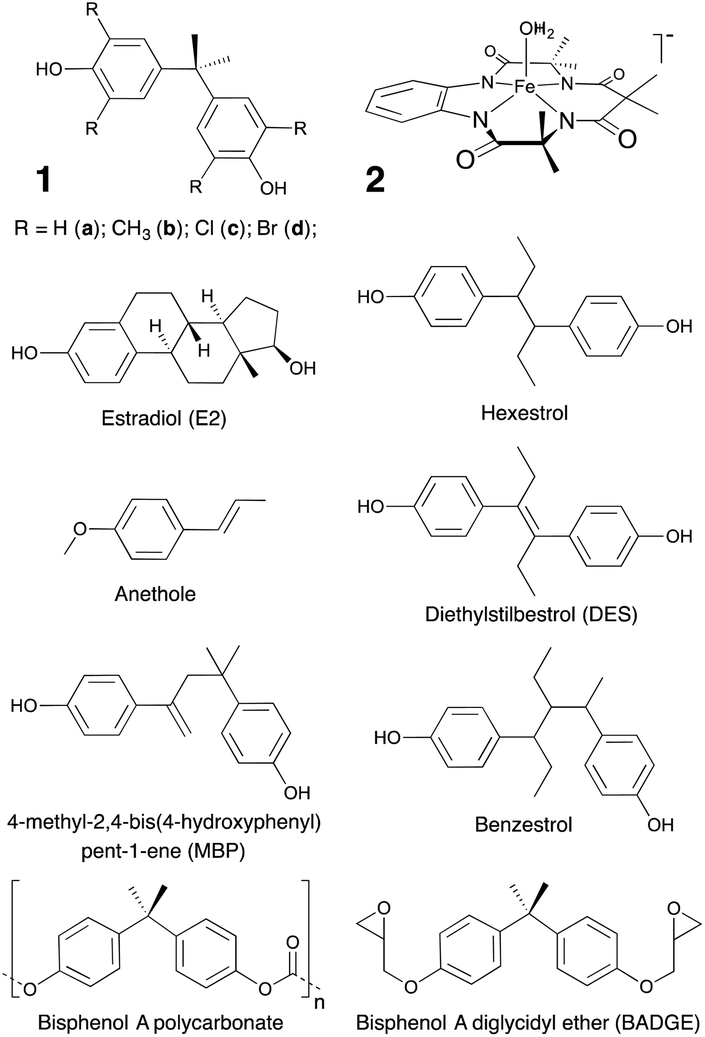 | ||
| Fig. 1 Bisphenol A (BPA, 1a), its derivatives (1b–d) and the TAML activator (2) used in this work, as well as compounds referred to in this work. TAML is a registered trademark of Carnegie Mellon University covering tetra-organic-amido-N macrocyclic ligand complexes.29 | ||
Approximately 15 billion pounds of 1a are produced annually.1 This figure is expected to increase with a compound annual growth rate (CAGR) of almost 6% by 2020.47 About 95% of the 1a produced is incorporated into BPA polycarbonate plastic and bisphenol A diglycidyl ether (BADGE) epoxy resins48 (Fig. 1), including those used to line food and water containers such as metal cans,49,50 metal and concrete drinking water pipes51,52 and residential water storage tanks.52,53 BPA is also added to phenoxy,54 polyacrylate,55,56 polyarylate,57,58 polyetherimide,58 polyester,55,56,58,59 polyester–styrene,55 and polysulfone54,56,58,59 plastic and resins,54 and rubber, polyethylene tetraphthalate,13,54,55,59 and polyvinyl chloride (PVC) products where it can function as a stabilizer,54,60,61 antioxidant62,63 or inhibitor of end polymerization.64
Many of these products leach 1a to contribute to ubiquitous environmental contamination.50,53,65 For example, 7.8 μg L−1 of 1a was detected in the water of an epoxy resin lined drinking water tank,66,67 the contents of canned goods have been estimated to contain 4–23 μg of 1a as a result of leaching from the epoxy resin can linings,50,68 and, at room temperature, polycarbonate food containers have been estimated to be capable of leaching ca. 6.5 μg of 1a into each gram of food stored inside.68,69 Aqueous 1a concentrations of 1.98–139 μg L−1 have been found to result from the room temperature soaking of each of one gram of PVC products and synthetic leather for two weeks in the dark.60,67 Room temperature, 24-hour exposure of pure water to PVC hoses resulted in leaching of 4–1730 μg L−1 of 1a.67,70 Contamination also occurred upon passage through the tube and increased from 8.7–558 μg L−1 as the water residence time increased from 0–24 hours.70 At 20 °C, polycarbonate tubes leach 1a into lab, river, and sea waters at rates of 0.15, 0.2, and 1.6 μg L−1 day−1, respectively—the amount of 1a leached is less than that from PVC hoses.67,70,71
Herein we examine the consequences of widespread BPA use and present an enticing laboratory study of a potential method for BPA stewardship. The impacts of BPA use are presented in two parts, a ‘Mini-Review of the occurrence of BPA’ and a ‘Mini-Review of BPA toxicity’. These draw extensively upon reviews both from within and outside of traditional chemistry.1,10,11,13,25,28,48,49,68,72–109 The BPA occurrence mini-review encompasses the industrial synthesis and associated releases, the diverse product space, the associated contamination of air, water, soils, food crops, and recycled products, and measured human body burdens. The immense scope of BPA use and contamination is detailed to highlight the complexity of the stewardship challenge. Thus, the design and implementation of sustainable, chemical processes for removing BPA from waters is an important goal for green chemists. Therefore, the technical performances of currently deployed BPA water treatment technologies are surveyed. The BPA toxicity mini-review outlines the health and ecological implications of exposures to BPA at the concentrations currently observed in humans and environmental waters. With this understanding, we evaluate the technical and environmental performances of TAML/peroxide (2/H2O2, Fig. 1) oxidation of heavily BPA-contaminated lab water at pHs 8.5 and >11 at similar concentrations to certain processing streams and landfill runoff. The TAML/peroxide processes are found to be remarkably simple and effective warranting further studies in a variety of real-world scenarios.
Mini-review of BPA occurrences
This mini-review demonstrates the panoptic contamination of the ecosphere by BPA (1a) and 1a removal by the water treatment strategies currently deployed. We have largely focused on the sources and surface water occurrences of 1a. However, it is also important to recognize the significance of the reported contamination of oceans110–112 and sediments.11,67,113 The presence of polycarbonate in oceans is of special concern because leaching of 1a from polycarbonate occurs more rapidly in sea than in fresh waters,74,1141a oxidation by radical oxygen species is slower in seawater than in control water,71 and marine bacteria strains have been found to degrade 1a much more slowly than freshwater strains.107,115 Furthermore, 1a has been detected in marine organisms including phytoplankton, zooplankton, mussels, herring, flounder, and cod, and bioamplification between phytoplankton and zooplankton has been documented.110,111,116The reported sediment contamination levels exceed those of both sea and fresh water.67 This assessment is consistent with the observation of higher concentrations of 1a in the lower layer of water columns than in the upper layers and a positive correlation between the low layer and sediment concentrations.117 Since 1a degradation is slower under anaerobic than aerobic conditions,67,109,117,118 and agents in the environment accumulate in the accessible reservoir in which they have the longest half-life and that reservoir then becomes a secondary source of emissions,119 the potential for rerelease of sediment-bound 1a is concerning. A report on the fates of plastics and the need for reforms has recently been released.113
Biomonitoring studies have revealed that 1a is also a pervasive human contaminant—unconjugated 1a has been detected in foetal, child and adult fluids and tissues (central tendency: 0.3–4.4 μg L−1 or 1.3–19 nM).120 Total (unconjugated + bioconjugated) 1a urinary concentrations have been detected in 92.6% of 2517 Americans older than 6 years (mean: 5.2 μg L−1 or 23 nM, range: 0.3–149 μg L−1 or 1.3–653 nM).121 Concentrations have been found in human colostrum (total 1a, all samples, 1–7 μg L−1, mean of 3.41 μg L−1)122 and breast milk (total 1a, 90% of samples, <0.3–6.3 μg L−1, mean of 1.3 μg L−1).123 Breast milk concentrations were found to rise from 6.2 μg L−1 to ca. 30 μg L−1 one hour after consumption of a canned coffee drink containing 37.4 μg,124 indicating that exposure of mothers correlates with that of nursing children. An overview of the available science on the impacts of human exposures to these concentrations is presented later in the ‘Mini-Review of BPA toxicity’.
The concentrations found in humans, colostrum, and breastmilk are comparable to some of the higher surface water concentrations of 1a that have been reported throughout the globe (US: median of 0.14 and maximum of 12 μg L−1;125 Atibaia River watershed of São Paulo, Brazil: weighted average of 4.6 and maximum of 13 μg L−1;126 Dongguan watershed of China: average of 6.5 and maximum of 56 μg L−1;127 Nagara River of Japan: average of 4.8 and maximum of 22.2 μg L−1;117 and Portugal: 0.07–4 μg L−1).128 An overview of the available science on the impacts of fish exposures to these concentrations is also presented in the ‘Mini-Review of BPA Toxicity’.
Human exposure to 1a is continuous and occurs through numerous known and unknown routes.78,79 While municipal drinking water itself is not typically considered a major source of 1a (France 2011: <9–50 ng L−1;129 Germany 2000: 0.003–2 ng L−1;130 Malaysia 2009: 3.5–59.8 ng L−1;131 Spain 2003: <5–25 ng L−1),132 significantly higher drinking water concentrations have been reported (Nigeria 2015: 109.00–882.50 μg L−1).133 Removal of 1a by conversion to transformation products,129 including 1c (Fig. 1),134,135 during disinfection by chlorination prior to release may contribute to the reported, low drinking water 1a concentrations. The potential human health and environmental impacts of exposure to 1a chlorination products are briefly discussed later in this work. The higher reported 1a drinking water concentrations derive from both a higher initial 1a concentration and contributions from plastic storage container leachate.133 Passage through tap mounted filter devices,131 pipes lined with epoxy resin, or polycarbonate or PVC tubing can also raise 1a concentrations.129 Ingestion of leachate50,136 from polycarbonate,137,138 epoxy53,62,136 and organosol62,139 resins such as those made from bisphenol A diglycidyl ether (BADGE, Fig. 1), and 1a stabilizers and antioxidants62,63 in, for example, food contact papers,140 PVC stretch films,141 dental sealants,80,101,142–151 and food and drink containers49,50,52,63,139,152–156 including commercial polycarbonate containers used for microwave ovens,69 adult131,152 and baby52,133,157–162 water bottles and cans50,52,163 is the most authoritatively documented route of human 1a exposure.
Canned goods are perhaps the most well-studied source of dietary 1a. Globally, contamination with 1a has been detected in canned beers,154 decaffeinated and non-decaffeinated coffees,162,164 soft drinks165,166 including diet and regular ginger ales, diet and regular root beers,165 diet and regular colas,154,165 energy drinks,154,165 orange and lemon soft drinks,154 flavoured and unflavoured soda waters,165 teas162,165 and tonic waters,165 infant167–169 and follow up formulas,170 the liquid phases of canned artichokes, green beans, corn, mushrooms, peas, and mixed vegetables,50 the homogenized liquid and solid contents of canned fruit products including coconut cream,154,171 coconut milk,156 lychees, mangoes,154 olives,171 peaches, light pineapples,154 tomatoes,63,155,156,171 tomato juice156 and tomato paste,172 and fruit pieces and cocktails,63 the homogenized liquid and solid contents of canned vegetable products including asparagus,156 baked beans in tomato sauce, green beans,63 beetroot,171 carrots,63 corn,63,171,172 mount elephants, mushrooms,156 peas,63,171 jalapeño peppers,153 potatoes,63 and goulash,154 the homogenized liquid and solid contents of canned soups171,172 including cream of chicken, chicken and white wine,63 potato,154 tomato,63 and Tom Kha,154 the homogenized liquid and solid contents of canned sauces171 of many varieties including demi-glace, fond de volaille, gratin, meat, tomato, and white,156 the homogenized liquid and solid contents of canned pastas in tomato sauce,63 the homogenized liquid and solid contents of canned seafood including Japanese sand lance, mackerel,156 pilchards in tomato sauce,63 salmon,63,156,171 sardine,156 sardine in tomato sauce63 shrimp, squid,156 tuna,156,171,172 and fish and vegetable mixtures,156 the homogenized liquid and solid contents of canned meats171 including chicken, corned beef, fish balls, ham, hot dogs, and pork,63 the homogenized liquid and solid contents of canned quail eggs,156 the homogenized liquid and solid contents of desserts including evaporated milk63,173 and creamed rice,63 the solids of canned crushed tomatoes, young peas,154 corn,154,174 haricot beans, red kidney beans, lentils,154 mushrooms,174 tuna in oil, and sardines in oil,154 mackerel filet in tomato sauce, and canned dinners.166 Contamination of vegetable solids has been found to be greater than that of the liquids.174
Alone, the estimated child and adult dietary intakes (0.008–14.7 μg kg−1 day−1)82 are unlikely to account for the concentrations observed in human bloodstreams.79 The human oral dosage necessary to maintain the average unconjugated 1a blood concentrations observed in healthy adults, adults having diseases, pregnant women, and foetuses (0.5–10 μg L−1, average ca. 1–3 μg L−1, greater than that found in most surface waters) has been estimated at >500 μg kg−1 day−1.81 This estimate agrees with a prediction of an oral dose of 1.43 mg kg−1 day−1 for 10 days as necessary to effect human steady state blood levels of 0.9–1.6 μg L−1.175 Therefore, alternate routes of exposure which obviate the first pass metabolism of the liver that greatly reduces blood concentrations of unconjugated 1a,79 including dermal uptake, which increases when skin is wet and/or greasy,176,177 and sublingual absorption78,79 are thought to contribute to human blood concentrations of 1a. For example, the handling of thermal receipt papers, which are coated in amounts of unbound 1a that have been found to be at least one thousand times greater than that of food can epoxy linings,178 is the most well-known route of dermal exposure.140,177,179–182
Given the myriad of products into which 1a is incorporated or contaminates, and the known sublingual, oral and dermal routes of exposure, other less-discussed sources likely also contribute to human blood concentrations of 1a. While not directly proven to raise blood concentrations of 1a, it is reasonable to expect additional exposures from, inter alia, indoor and outdoor air,183–186 indoor dust,183–185,187–189 5 gallon water carboys,157 polyethylene tetraphthalate13,132,190 and polycarbonate water bottles,129 drinking water generators52 and pipes,51 water main55 and tap131 filters, fungicides,54,55,59 randomly selected fresh foods including fresh cherries, courgettes, eggplants, medlars, oranges, peaches, peppers, and tomatoes,191 white clams, crabs, blood cockles, fish, prawn, and squid,111 buns, flour, hard cheese, minced meat, sausages, hamburgers, sliced salami and turkey, and frozen pizza in plastic packaging, bread in plastic or paper packaging, liver paté in plastic packaging with metal foil, fish pudding in plastic or paper packaging, caviar spread in a metal tube, jam in glass jars with metal or plastic screw caps, whole eggs packaged in cardboard,166 honey packaged in glass or plastic that was imported in epoxy-lined metal drums,192 baby food products in glass193 and high-density polyethylene plastic168 jars52 with metal lids, solid185 and liquid foods served at day care centers,183,184 baby teethers,194 breastpumps,52 children's books and toys,52,195,196 cosmetics,197 training cups,157 dishwasher and laundry detergents,198 protective and general sporting equipment,52,79 inhaler housings, musical instrument mouthpieces,52 plastic plates and utensils52,79,133 including those used at elementary schools,162 nail polish,55,198 pillow protectors,198 outdoor play area soil,183 artificial teeth,55 hand, hard floor surface and food preparation surface wipes,184 personal care-hygiene products198,199 including cleansers, conditioner, shaving cream, lotions, shampoo, bar soap, sunscreen,198 toothbrushes,133,198 toothpaste,133 and face and body washes,198 automotive interiors and exteriors including bumpers, interior light covers, dashboards, radiator grills, fog lamps, headlight lenses, head, brake and tail lights, indicator reflectors, roofs, windows, covers, and coatings,52 building materials52,91 including structural adhesives and fillers, coatings, carport covers,52 flooring,52,55 architectural, conservatory, greenhouse, and bus stop shelter glazings, grouting, mortars, concrete reinforcement, sheets for roofing,52 ceiling tiles,133 and road and train track noise reduction walls, the casings of cameras, computers, copiers, monitors,52 phones,52,133 pens,121 steamirons, suitcases, and TVs,52 paper currencies,200 CDs52,55,60,79 and DVDs,52,79 hair dryers,52 cigarette filters,201 contact lens holders,52 eyeglass frames,79 lenses52,91,133 and nosepads,79 razors,52 cellulose products including chromo board,202 business cards,140 catalogues,202 coatings, dye developers,91 mailing envelopes,140 magazines,140,202 carbonless copy paper,203 free advertising papers, and advertising supplements,202 recycled cellulose fiber (RCF) products including newspapers,140,202,204 napkins,140 paper towels,140,205 toilet tissue,140,202 and paperboard140,202 including the food contact surfaces of confectionary, fried chicken, fried potato,204 pizza,204,206 sandwich,204 and general food storage boxes202 as well as noodle cups,204 virgin paper products including coffee filters, cooking papers, cups, dishes, napkins, tea bags, tissues, and fried chicken wrapping paper,204 the epoxy paints,52,207,208 coatings and composites of aircraft, car, boat,52 and DIY repair adhesives52,91 and fillers, household appliances including vacuum cleaners, dishwashers, dryers, fridges, freezers, and washing machines, windmill blades, engine blocks, concrete and steel bridges, gas bottles, aircraft, boats, buses, canoes, caravans, cars, helicopters, mobile homes, railcars, yachts, general cans including those containing oils and hairsprays, caps, closures and crown corks, general and sea containers, cookers, decking, drums, heat, ventilation and air conditioning equipment, steel frames, furniture and racks, office furnishings, underwater ship hulls, printing inks, cargo and storage tank linings, primed metals, electric motors, engines, machinery, and parts, pails, automotive body, construction cladding, metal roofing, ceiling, and garage door panels, automotive and electronic parts, pipes, valves and fittings, gas pipes, offshore drilling platforms, general plastics, radiators, concrete reinforcing rebars, traffic light reflectors, roadsigns, emissions scrubbers, supporting steel structures including bars, beams, gratings, rods, and shafts, metal and concrete storage tanks, menus, and food trays, gardening tools and equipment, collapsible tubes including those containing creams and toothpastes, paper and board varnish including food packaging, secondary containment walls, and wood, medical equipment including ampoules, i.v. connectors, dialysers, medical packaging film, blood oxygenators and sample reservoirs, cardiotomy reservoirs, respirators,52 single use surgical tools,52 and medical tubing,13 the polycarbonate plastics, resin linings, and printed circuit boards of electronic products52,60,209,210 and electronic waste,60,91,209 distributor boxes, plug connectors, large advertising displays, fuses, lamp globes, inductors, lamp holders, lamps, electrical meters, switch modules, solar panels, sockets, streetlights, battery power stations, switches, transformers, kitchen tools and appliances including front panels for electric cookers,52 refrigerator crisper drawers,55 electrical kettles, coffee makers, microwaves, mixers,52 and food processors,55 and PVC products48,60 including synthetic leather211–213 in, for example, seating materials and clothing,214 shower curtains,198 and cord coverings.60,211 It is important to note that the concentrations found can be high. For example, a Nigerian study recorded in the occurrences above reported 1a contamination ranges in consumer products and food/drink samples of 915.00–1415.50 μg L−1 and 163.00–2785.00 μg L−1, respectively.133
When used, discarded or recycled, 1a-containing goods and the contaminated solutions generated in the manufacture and processing of 1a and 1a-containing goods, become the largest sources of 1a in the environment.60,211,215,216 In 2008, the government of Canada concluded that “bisphenol A is entering or may be entering the environment in a quantity or concentration or under conditions that have or may have an immediate or long-term harmful effect on the environment or its biological diversity or constitute or may constitute a danger in Canada to human life or health”.217 Consequently, in 2009, it recommended 1a releases be reduced to the lowest level technically and economically feasible and proposed an upper limit of 1.75 μg L−1 in emissions.218 This value was chosen because it is an order of magnitude greater than the 2009 partial no effect concentration (PNEC) of 1a218—it has been argued that the PNEC for 1a in surface waters should be lowered to 0.06 μg L−1,219 and a maximum concentration of 0.03 μg L−1 has been proposed to safeguard 95% of exposed species from the chronic toxicity of 1a.74 Therefore, easy to implement, safe technologies like the potential process documented in this contribution are critical to achieving emissions standards that, like the proposed Canadian standard of 1.75 μg L−1, safeguard human health and the environment.
In 2014, the 258 million tons (mt) of municipal solid waste generated in the US before recycling, composting or combustion was composed of 69 mt of paper and paperboard (28% of which was landfilled, 65% was recycled and 7% was combusted), 33 mt of plastics (76% landfilled, 10% recycled and 15% combusted), 16 mt of textiles (65% landfilled, 16% recycled and 19% combusted), and 8 mt of rubber and leather (51% landfilled, 18% recycled and 32% combusted).220 Landfilled postconsumer goods release 1a.211 Consequently, water that has contacted waste, known as landfill leachate221,222 (mostly pH 6.5–8.5),61,89 has been reported to be a major source of 1a in the environment202,215 (Germany 2002: 4200–25000 μg L−1, average of 14067 μg L−1;223 Germany 2003: ca. 500–5000 μg L−1;224 Japan 1999: ca. 500–7500 μg L−1;225 Japan 1996: <0.5–17200 μg L−1, median of 269 μg L−1;211 Norway 2015: 0.7–200 μg L−1, average of 66.5 μg L−1;226 Philippines 2003: ca. 9000 μg L−1;54 and Sweden 2000: 4–136 μg L−1).227 These mixed pollutant streams containing 1a may or may not be treated based on local judgments of the need for such.54 Treatments often include recycling through the landfill, on-site biological treatment,90 coagulation, filtration, and/or activated carbon adsorption,54,89 prior to discharge to surface waters or a municipal WWTP.
For surface water discharges, a higher treatment requirement often applies. One such treatment process entails pumping collected leachate containing ca. 100 μg L−11a into an adjustment tank where it is combined with leachate of unreported 1a concentration from four other landfill blocks and from neighbouring landfill sites and aerated. This reduced the concentration of 1a to ca. 0.15 μg L−1. This combined influent was further treated by phosphate addition and passage through three biological contactors, coagulation with FeCl3 and NaOH, mixing, sedimentation, and sterilization prior to discharge into a river.61 When operated at ca. 25 °C, the stages of the treatment process that followed combination with leachate of undisclosed composition and aeration removed ca. 80% of the influent ca. 0.15 μg L−11a and produced effluents of ca. 0.03 μg L−1. Biological treatment and coagulation followed by sedimentation each account for roughly half of the amount of 1a removed. When the process was operated at ca. 20 °C, the effluents were found to contain higher concentrations of 1a than the post-aeration influents. Both biological treatment and coagulation produce contaminated secondary wastes that must be landfilled, which risks rereleases of 1a, or incinerated.
Another treatment entailed 1a removal through the use of a membrane bioreactor (MBR) consisting of three activated sludge tanks. The first two tanks engaged nitrifying bacterial cultures (NH3 → NO3−). The third engaged a denitrifying culture (NO3− → N2). This was followed by ultrafiltration (UF).224 The influent ca. 500 μg L−11a was reduced to ca. 300 μg L−1 by the biological treatment of the MBR and the subsequent UF of the MBR effluent reduced this to ca. 2 μg L−1. Biological treatment and UF produce secondary wastes in the forms of contaminated sludge and retentate, respectively, that must be landfilled, which risks rerelease of 1a, or dried and incinerated. Further treatment by nanofiltration (NF) increased the MBR effluent concentration of 1a to ca. 3 μg L−1. Ozonation (0.2–0.5 kg O3 per kg COD) of the NF retentate, which contained ca. 4 μg L−11a, removed ca. 83% to give effluent containing ca. 0.7 μg L−1.
For discharge to municipal WWTPs, a lower treatment requirement often applies. One such treatment entailed collection of the raw leachate in a regulating reservoir for pumping through a sequence of adjustment and aeration, mixing and sedimentation tanks before discharge into a sewer.61 About 30% of the influent ca. 90 μg L−11a was removed giving effluents of ca. 65 μg L−1. Another treatment entailed passage through a MBR which is similar to that discussed previously.224 The biological treatment of the MBR reduced the highly contaminated, ca. 5000 μg L−11a-containing leachate to ca. 1300 μg L−1. This was further reduced to ca. 70 μg L−1 by UF. Further treatment by passage through a granular activated charcoal (GAC) column removed ca. 50% of the remaining 1a, resulting in final discharges of ca. 35 μg L−1. GAC treatment produces contaminated GAC which, when removal performance diminishes due to saturation, must be landfilled with risk of 1a rerelease and replaced at cost, or thermally regenerated.
In addition to landfill leachate, other process and waste streams containing 1a in excess of 1.75 μg L−1 have been reported worldwide. Some are treated before release and some are not. For example, 1a has been detected in untreated paper production and recycling solutions (Spain 2004: 3.0–142 μg L−1, mean of 53.6 μg L−1).228 Primary and secondary treatment at the plant reduced these 1a concentrations to 1.6–27 μg L−1 (mean of 13.8 μg L−1).228 At various times, 1a concentrations have been reported to be high in final effluents of the chemical (Austria 2000: 2.5–50 μg L−1, mean of 18 μg L−1),229 paper production and recycling (Austria 2000: 28–72 μg L−1, mean of 41 μg L−1;229 Japan 2002: 0.2–370 μg L−1, mean of 59 μg L−1),230 plastics manufacturing and recycling (Nigeria 2015: 108–163 μg L−1, mean of 130 μg L−1),133 and industrial laundry (US 2008: 21.5 μg L−1)231 industries. Unfortunately, we were unable to locate data on the concentrations of 1a in various other industrial solutions and effluents including washing residue and wastewater generated in the production of 1a itself91 and the sink-float/heavy media separation slurries, chemical etchants, detinning, and chemical delacquering solutions employed in the recycling of, for example, cans and bulk metals.102 As with landfill leachate treatment plant effluent, industrial effluents may or may not be treated before release to the environment or wastewater treatment plants (WWTPs).216
When directed to WWTPs, treated or untreated landfill leachate and industrial effluents may join with domestic wastewater which often also contains 1a. For example, the use of toilet tissue made from recycled paper has been estimated to contribute ca. 36000 pounds of 1a to wastewater per year.202,232 Consequently, at various times, 1a concentrations in WWTP influent have been reported to be high (Austria 2000: 10–37 μg L−1, mean of 21 μg L−1;229 Canada 2004: 0.16–28.1 μg L−1;233 Germany 2008: <0.02–12.2 μg L−1, weighted average of 3.67 μg L−1).234 Though the removals of EDs including 1a have been observed to vary with the operating conditions,216,235–237 conventional wastewater (pH 7–8) treatment processes have been reported to remove 82 ± 12% of influent 1a.87,233 Primarily, this removal occurs through sorption to primary sludge and sorption to activated sludge which may or may not be followed by biological degradation.87,237,238 The final effluents are often discharged to surface waters.233 Reports indicate that ca. 2% of the influent 1a is not degraded by and, as a result, remains in the activated sludge, along with a mixture of other EDs, the mass-normalized concentrations of which are significantly greater than those typically found in effluents and surface waters.5,95,98,216,239–243 As a consequence of lesser degradation in anaerobic than aerobic biological treatment, adsorption of several EDs, including 1a, to sludge occurs to a greater extent in anaerobic treatment.239 Therefore while the use of nitrifying cultures can provide cleaner effluents,244 these can come at the expense of increased sludge contamination.202
The contaminated sludge generated in biological treatment can also become a source of 1a. Sludge concentrations of 0.10–3.2 × 107 μg kg−1 of dry weight have been reported.11,245–248 While incineration is practicable, the high costs, technological demands, low to no energetic gain, concentration of hazardous metal content in the ash produced which is usually landfilled, air emissions, and negative public perception can hinder its deployment.249 Consequently, the contaminated sludge is often disposed of by landfilling or application to agricultural lands and composting.99 For example, in 2004 the US produced 7.18 × 106 dry US tons of sewage sludge solids, 55% (3.95 × 106 tons) of which was land applied,250 and in 2005, the EU-27 countries produced ca. 10.96 × 106 dry tons of sewage sludge solids, 41% (4.49 tons) of which was directed towards agricultural use.99 These percentages are consistent with the 50% land application and 50% incineration reported in 2004 for the Canadian Ashbridges Bay, Humber and North Toronto WWTPs.233 While the practice recycles nutrients and, thus, can enhance the sustainability of societies, land application of sewage sludge can increase the risk of ED rerelease to the environment233 and contributes to ED contamination of soils.5,238,251–256 This has led to a considerable literature of the effects on animals and their offspring that graze on pastureland amended with sludge containing multiple EDs.95,257–267 Since these studies mostly concern multi-ED exposures, we consider this area outside the scope of the current mini-reviews. Sludge is the primary source of 1a soil contamination.238 If practiced for an extended period of time,239,255,256 land application to agricultural soils may contribute to the aforementioned levels of 1a detected in fresh produce.191 Soil 1a can partition into soil water83 which, in turn, can result in contamination of leafy vegetables,238,268 root crops238 and cereal grains.238 Unfortunately, while never applying sewage sludge or only applying decontaminated sewage sludge would limit soil concentrations of 1a, this alone cannot ensure 1a-free soil.
Ongoing global deposition from ubiquitous contamination186 of the atmosphere238 and irrigation with treated and untreated contaminated water also contribute to soil concentrations of 1a.70,268 Sources of atmospheric aerosol concentrations of 1a include volatilisation during processing, handling and transportation of 1a (at least 109 metric tons per year)91 the open burning of municipal wastes (US: estimated to be >75000 kg per year)269 and plastics,186 the thermal degradation of polycarbonate,270–272 waste electrical and electronic equipment recycling, disposal and burning,186,210 waste sorting facilities including metal shredders,226 and landfills.226 For example, 1a is a major product of the depolymerisation of polycarbonate observed on heating to 475 °C in air,272 and flash pyrolysis gas chromatography of polycarbonate at 500–850 °C shows the release of H2O, CO2, 1a, phenol, isopropenyl phenol, and diphenyl carbonate in addition to higher molar mass compounds.273–275 Resuspension of contaminated soil also contributes to atmospheric concentrations.186
In addition to contributing to dietary exposures, cured, epoxy resins made from the 1a-containing prepolymer Bisphenol A diglycidyl ether (BADGE, Fig. 1), such as those used in the metal coatings of canned goods, may contribute to atmospheric concentrations of 1a when thermally degraded. In studies of heating the purified, cured resins made from BADGE prepolymers, stepwise alterations occur as the temperature increases. From ca. 250–350 °C, evaporation occurs of the compounds encaged in the cured polymer, including unreacted modifiers such as diols, and residual prepolymer mono- and bis(hydroxyether)s of 1a.276,277 From 300–340 °C, thermal degradation begins and 1a is a major product released along with smaller amounts of isopropenylphenol, isopropylphenol and phenol.278 Up to 500 °C, there is very little decomposition of the 1a moiety.278 From 500–600 °C, aryl–alkyl ether bonds are further cleaved and 1a remains a major product released.276–279 At >600 °C, the release of high boiling pyrolyzates with epoxide end groups decreases markedly,279 presumably giving a more 1a-rich product mixture.
The heating of cured resins made from BADGE prepolymers occurs in the recycling of metals. In, for example, aluminium recycling, thermal decoating or delacquering of the shredded metal is commonly performed prior to melting.102,103 Approximately 18 metric tons of scrap pass through each delacquering machine per hour.103 There are two major thermal delacquering approaches for Used Beverage Cans (UBCs).103 The first is based on conveying crushed and shredded UBCs through zones of increasing temperature, which may be fixed at 520 °C103 or may progressively rise to ca. 540 °C.280 The furnace temperatures must be maintained below ca. 566 °C to prevent ignition of the surface contaminants.280 The second approach relies upon roasting the scrap in a rotary kiln through various temperature stages, the last of which occurs at near 615 °C, with recirculation of the produced combustion gases.103 Consequently, the gases produced in the delacquering process can be rich in 1a. These then may or may not be treated with activated charcoal281 and/or lime or calcium carbonate282 before or after passage through a baghouse filter followed by release via a high stack.283 These purifications create contaminated materials which must be disposed of by, for example, landfilling or incineration.282 Alternatively, the delacquering gases may be combusted.282 Activated charcoal may, or may not, be added to the resulting flue gases281 which are then purified by passage through a baghouse filter.283 This creates contaminated activated charcoal. In either case, any compounds not sequestered or destroyed are released.
Despite the activated sludge removals, WWTP effluent concentrations of 1a in excess of 1.75 μg L−1, which are often discharged to surface waters, have been reported (Austria 2000: <0.5–2.5 μg L−1, mean of 1.5 μg L−1;229 Canada 2004: 0.01–17.3 μg L−1;233 EU 2008: 3.13–45 μg L−1;284 2008: <0.02–7.6 μg L−1, weighted average of 0.52 μg L−1;234 US 1999: <0.01–2.7 μg L−1).285 While these alone are cause for concern, of perhaps greater concern are the by-products that can be formed at landfill leachate135 and wastewater286 treatment facilities if effluents are disinfected by chlorination233 prior to release.
These disinfection by-products can include mono- and poly-chlorinated forms of 1a including the tetra-chlorinated 1c.134,135 When product mixtures resulting from chlorination of 1a in water were tested in a binding assay employing a recombinant form of the endogenous human classical oestrogen receptor,287 ERα, 24-fold greater activity than the untreated 1a solution was observed.134 The mixtures also induced β-galactosidase activity in a yeast two-hybrid system employing human ERα.134 In addition, 2-ClBPA, 2,2′-diClBPA, 2,6-diClBPA, 2,2′,6-triClBPA and 1c exhibited oestrogenic activity as measured by oestrogen-induced green fluorescent protein expression in transgenic human breast carcinoma MCF7 cells (ERE-GFP-MCF7 cells).288 In addition to being oestrogenic, the multiply chlorinated forms of 1a are more resistant to activated sludge treatment than 1a.203 Notably, 2,2′-diClBPA only undergoes very slow degradation while 2,2′,6-triClBPA and 1c are not degraded. Thus, in addition to emissions resulting from the use of 1c in flame retardants and as an epoxy intermediate,243 chlorination of 1a-containing waters may contribute to the 1c concentrations which have been detected in sewage sludge,243,289–291 river water289 and sediment.289
Further treatment of WWTP effluent with powdered activated carbon (PAC) or ozone has been advanced to enhance removal of MPs from wastewater.292 The energy and resource demands of PAC treatment followed by sand filtration (PAC-SF) to retain the PAC and ozone are significant and similar.293 PAC has been observed to remove 89.5–99% and 33–98.5% of 1a from pH 8.3 lab and pH 8.3–8.4 raw drinking waters, respectively, and these results are expected to transfer to waters containing ≥500 ng l−11a.294 However, difficulties arise from handling of the finely powdered PAC and retention of the PAC which can necessitate employment of ultrafiltration membranes at significantly greater cost than either PAC-SF or ozone.293 Additional problems derive from interference from dissolved organic carbon, a decrease in the performance over time due to saturation and management of the spent PAC, which is expensive to produce for replacement or to regenerate at elevated temperature.293 BPA is readily degraded by ozone.295–299 For example, treatment of 10045 μg L−11a in pH 6.0 lab water with ca. 0.1 mg L−1 O3 (4.05 mg O3 per min) effects a >98% removal in 10 minutes with ca. 20% mineralization.295 The technical performance of ozone decreases as the water matrix becomes more complex296 and despite the extensive deployment of ozone in some countries, it is not widely used globally—the reasons vary by country.88,293 Where it is deployed, staff training, safety measures293 and additional infrastructure are necessary.
Significant amounts of 1a have also been detected in recycled cellulose fiber (RCF) plant process solutions, effluents and products, the last of which comprise roughly 50% of the furnish for worldwide paper and board production.140,202,300 This can result in the previously noted contamination of virgin and RCF products. Thermal receipt papers typically containing low mg g−1 quantities of 1a contribute significantly to RCF contamination.140 Up to 10% of the thermal paper produced is never used and directly enters recycling streams along with an additional 30% of that which is used.284 A phase-out of 1a would remove it from the paper cycle, however a lag period of 10–30 years has been estimated before 1a concentrations would reach the limit of nondetection.301 The contamination of RCF with EDs of any description is a wrench in the sustainability machinery required to move the civilization progressively toward renewable feedstocks. For example, incineration of highly 1a-contaminated waste at state-of-the-art, low-emission facilities has been advanced over recycling as a method for avoiding further contamination of recycled materials.226 However, incineration precludes the energetic gains and decreased production emissions of material reuse, generates emissions including carbon dioxide302 and can also generate PCDDs and PCDFs.104,303,304 Another recently suggested option for dealing with 1a contamination of feedstocks may involve establishing acceptable levels for 1a in recycled products.215 However, any level of 1a in goods exposes the consumer and compromises material streams. Therefore, improving decontamination methods for removing 1a from recycling process solutions and waste streams is an important sustainability research trajectory advanced by the empirical results detailed in this study.
Opportunities for 1a removal vary with whether or not the process includes a deinking stage. In the deinking of paper products, the addition of NaOH, sodium silicate, and surfactants extracts 95% of the contaminating 1a into the aqueous solution (pH 9.5–11) and sludge.284,305 The deinking sludge is then separated from the pulp slurry and dewatered. The process water may be clarified prior to reuse resulting in contaminated sludge and the enrichment of 1a in the process water.306 Effluents from the pulp thickening process have been reported to contain 196–10300 μg L−11a.230 Washing of the pulp also generates contaminated solutions that are reused and may be bleached.305,306 If alkaline paper recycling plant pulping solutions are chlorine-bleached, chlorinated forms of 1a, including 1c, can be generated,203 as noted above. Primary treatment of these process waters transfers 95.9% of the influent 1a to the primary sludge.284 The water is then sent to a WWTP for secondary treatment, often with activated sludge. In a study of 40 Korean WWTPs that receive influent comprised of varied proportions of industrial and domestic effluent, the concentrations of 1a detected in sludge at plants receiving primarily industrial effluent (I-WWTPs, >70% of inflow rate from industrial wastewater) were an order of magnitude greater than those receiving primarily domestic effluents (D-WWTPs, 0–3% of inflow rate from industrial wastewater).216 Of the I-WWTPs, the highest sludge concentrations of 1a were found at plants receiving wastewater from the paper industry.
If deinking is not necessary, such as in the production of corrugated packing materials, the majority of 1a remains in the finished paper products limiting opportunities for removal and creating sources of environmental contamination. However, 10% of the influent 1a is transferred to water from the pulping process which is sent to primary treatment where 50% is removed with 18% incorporated into the primary sludge and 32% unaccounted for.284 Final effluents have been reported to contain significant quantities of 1a (Japan 2002: 0.2–370 μg L−1, average of 59 μg L−1).230 In most plants, this effluent is subjected to secondary treatment with activated sludge.306 The deinking, primary, and secondary sludges are dewatered and dried. These are then incinerated with negative to low net energy production, used in biogas production, landfilled, or applied to agricultural land where allowed by law. Thus, the massive global use of BPA in myriad products and processes further burdens an overstrained water treatment infrastructure where upgrading and maintenance require great public expense. Low cost, high efficiency BPA removal approaches, such as the lab experimental results herein promise, can improve both the technical and cost performances of dealing with this contamination.
TAML/peroxide removal of BPA from water
One green science strategy for reducing exposures to compounds such as 1a is to pursue safer, more efficient and flexible stewardship processes. To this end we have been developing TAML activators (2 is the prototype, Fig. 1) which are highly effective catalysts for H2O2 oxidations. TAML catalysts and TAML/H2O2 processes appear to be environmentally benign based on a diversity of evidence307–310 and, as further shown here, are extremely simple to deploy. TAML/H2O2 processes have been employed to oxidatively destroy numerous targets in water,311–315 including bromo-,310 chloro-309 and nitro-316 phenols, drugs,317 thiophosphate pesticides,318,319 molluscicides,320,321 nitroaromatics,316,322 the B. atrophaeus non-infectious surrogate for pathogenic B. anthracis and protozoa,323 dyes,324–327 coloured paper industry effluents,328 and signature oestrogenic micropollutants.4,329,330 Importantly, 2/H2O2 chemistry is comprised exclusively of biochemically common elements as a foundational strategy for avoiding toxicity.The destructive potency toward micropollutants315,331 suggests that 2 should easily oxidise electron rich 1a and BPA-like compounds. In fact, well over a decade ago at Carnegie Mellon, 1a was one of the first compounds studied in this context. At that time, 2/H2O2 was found to readily catalyse the oxidative elimination of BPA from water.332 However, further studies revealed a pH-dependence of the product distribution. As detailed herein, at pH > 10 2/H2O2 oxidizes 1a rapidly and deeply giving a potential solution for BPA water contamination. At or near neutral pH (optimal for most water treatment processes), 2/H2O2 induces oligomerisation of 1a to form precipitates as the principal products. While this could also represent a solution, the toxicity properties of the aqueous products were unknown, and the intervening years of this long study were primarily focused on developing confidence that no new toxicities were being introduced.
This concern is well-grounded in literature precedent. The unanticipated formation of phenolic oligomers found in the demethylation of anethole (Fig. 1) resulted in the discovery of hexestrol (Fig. 1).94 Hexestrol is a mono-hydrogenated form of diethylstilbestrol (DES, Fig. 1), a compound structurally similar to 1 that, at the time, was one of the most potent known oestrogens with ca. one hundred thousand-fold greater oestrogenic activity than 1a as indicated by the relative minimum total weights of a substance required to induce a full oestrus response in ovariectomized female rats when administered by six injections over three days of a solution in sesame oil.333,334 Additionally, the oxidation of 1a by fungal manganese peroxidase335 has been observed to give a product mixture containing hexestrol. Therefore, in all cases, evidence was clearly needed that the treated solutions do not contain compounds that are also MPs across the domain of endocrine endpoints and beyond. Addressing these concerns necessitated the development of methodologies for analysing the catalysts and post-treatment solutions. In 2008, through the leadership of J. P. Myers and Advancing Green Chemistry, a coalition of environmental health scientists and green chemists formed to develop and eventually publish in this journal the Tiered Protocol for Endocrine Disruption (TiPED).336 The TiPED is an organized suite of mammalian, fish and amphibian, cellular and computational assays designed to detect low dose, adverse effects as a pre-commercial guide to the chemical enterprise for avoiding EDs and MPs. The development of this protocol and the various resulting collaborations,4,307,308,336 as extended herein, have allowed TAML activators and TAML-treated BPA media to be scrutinized for low dose toxicity.
Herein, we present a study of 2/H2O2 treatment of 1a in lab water at near neutral pH and at pH 11 to determine the potential for improved decontamination of 1a-containing waters. This work demonstrates the many levels of complexity that accompany the oxidative degradation of BPA and its derivatives in water treatment. Here we report (i) that 1a–d are all readily decomposed by 2/H2O2 at pH 11 and substantially mineralized and effectively eliminated from water, (ii) that 2/H2O2 treatment of high concentrations of 1a at near neutral pH leads to a green procedure for oligomerising BPA, (iii) on the acid–base and redox properties of 1, (iv) on an improved synthesis of 12 (Scheme 4), a product of enzymatic degradation of BPA, (v) on a kinetic and mechanistic study of the oxidation of 1a–d and, (vi) on the toxicity of 1a samples before and after 2/H2O2 treatment at pHs 8 and 11 via bacterial, oestrogenicity, and zebrafish developmental assays. Given that 1a and 1d are deployed commercially in large quantities, the work also highlights the requirement for further investigation of the degradation profiles of 1b–d and points to the need for expanded studies on the environmental safety of the 1a–d oligomers.
Experimental section
Materials
Bisphenol A (1a, Sigma Aldrich, GC grade >99%) was purified by re-crystallization using a mixture of hot ethanol and water and 2 was obtained from GreenOx Catalysts, Inc. Fresh stock solutions of H2O2 were prepared daily from reagent grade H2O2 (30% w/w, Fluka) and standardized by measuring the absorbance at 230 nm (ε = 72.8 M−1 cm−1).337 All other chemicals and solvents obtained from Sigma-Aldrich or Fischer Chemicals were of ACS reagent grade quality or higher and were used as received. Water was either Fisher HPLC grade or deionized Milli-Q water (Millipore). Regenerated Cellulose (RC) 15 mm syringe filters (0.2 μm pore diameter) were supplied by Phenomenex.Synthesis of 4-(4-(4-hydroxyphenyl)-2-methylpent-4-en-2-yl)-phenol, a precursor of 12 (Scheme 4)
Bisphenol A (15.2 g, 0.07 mol) was dissolved in concentrated H2SO4 (50 mL) and stirred (20 min) at 22 °C. The reaction mixture was quenched by transfer into deionized water (900 mL) in an Erlenmeyer flask with vigorous stirring. The solids were filtered on a medium porosity frit and the filtrate was extracted 3 times with diethyl ether (3 × 200 mL). The organic extracts were washed with aqueous NaHCO3 (5%, 100 mL) and combined with the solids. The resulting solution was dried over MgSO4, filtered, and the solvent was removed by rotary evaporation. The residue was purified by flash chromatography on silica gel with 2:1 hexane:ethyl acetate to yield a white solid (3.5 g, 0.01 mol, 15%). 1H NMR (300 MHz, d6-acetone, J in Hz) δ 8.20 (s, 1H, OH), 7.95 (s, 1H, OH), 7.17 (dd, J 8.8, 7.4, 4H, ArH), 6.72 (t, J 8.8, 4H, ArH), 5.04 (d, J 2.2, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 4.68–4.66 (m, 1H, CH), 2.75 (d, J 0.8, 2H, CH2), 1.16 (s, 9H, CH3). ESI-MS (m/z, negative mode): 267.3 (100), 268.2 (17), 269.2 (3%).
CH), 4.68–4.66 (m, 1H, CH), 2.75 (d, J 0.8, 2H, CH2), 1.16 (s, 9H, CH3). ESI-MS (m/z, negative mode): 267.3 (100), 268.2 (17), 269.2 (3%).
Synthesis of 4-(prop-1-en-2-yl)phenol (12) (Scheme 4)
4-(4-(4-Hydroxyphenyl)-2-methylpent-4-en-2-yl)phenol (3.5 g, 0.01 mol) and NaOH (9 mg) were placed in a vacuum distillation apparatus to which a water aspirator was connected. The product was distilled under vacuum at 200 °C using a silicone oil bath and a fraction boiling in the range of 75–135 °C was collected. The light-yellow solid was dissolved in diethyl ether (5 mL) and was added to deionized water (25 mL). The mixture was stirred rapidly and sparged with nitrogen until a white precipitate was observed. This was isolated by suction filtration on a fine porosity glass frit (2.3 g, 0.020 mol, 86%). An analytical sample was recrystallized by slow evaporation of diethyl ether from heptane. 1H NMR (300 MHz, d6-acetone): δ 8.20 (s, 1H, OH), 7.36 (d, J 8.9, 2H, ArH), 6.80 (d, J 8.9, 2H, ArH), 5.26 (dq, J 1.6, 0.8, 1H, H2), 4.93 (quintet, J 1.5, 1H, H1), 2.09 (dd, J 1.5, 0.8, 3H, CH3). 1H NMR (500 MHz, H2O + D2O) δ 7.41 (d, J 8.8, ArH), 6.67 (d, J 8.8, ArH), 2.11 (s, CH3). 1H NMR (500 MHz, H2O + D2O) δ 7.41 (d, J 8.8, ArH), 6.67 (d, J 8.8, ArH), 2.11 (s, CH3). GC-MS (m/z): 134 (100), 119 (77), 94 (14), 91 (35), 77 (15%).Methods
UV-vis spectra and kinetic data were obtained using a Hewlett Packard 8453 Diode Array spectrophotometer equipped with a thermostatted cell holder and an 8-cell sample positioner. The temperature was maintained at 25 °C using a Thermo digital temperature controller RTE17 with a precision of ±1 °C. Stock solutions of 1 were prepared by dissolving solid (7.50 × 10−5 mol) in water made basic with KOH (5.0 mL). Stock solutions were then diluted with phosphate buffer (usually pH 11). For experiments at pH 8.5, aliquots of BPA stock solutions (10000 ppm in CH3OH) were added to the required volumes of 0.01 M buffer (sodium carbonate/bicarbonate) to give solutions with final concentrations of 10 ppm BPA. Aliquots of a 2 stock solution (40 μM in deionised H2O) were added to the 1 containing buffer solutions to give the required final concentration (4–40 nM). Differential pulse voltammetry was performed with an Autolab PGSTAT100 potentiostat and GPES 4.9 software. The working electrode was a glassy carbon disk, with a saturated calomel reference electrode and platinum wire counter electrode. HPLC measurements were performed using a Waters® 600 system with 717 autosampler and 2996 photodiode array detector. Separations were carried out on a Varian Microsorb-MV 100-5 C18 (250 × 4.6 mm internal diameter, particle size 5 mm) column. The system at 40 °C was run in isocratic mode with an acetonitrile/water (3/1) mobile phase. HPLC measurements for the determination of kII at pH 11 were performed using a Shimadzu HPLC system with a Shimadzu CMB-20A controller, LC-20AB pump, DGU-20A3 degasser, SPD-M20A diode array detector, RF-20A XS fluorimeter detector, CTO-20A column oven, and SIL-20A HT auto sampler. Separations were performed on a Phenomenex EVO C18 column at 40 °C with a mobile phase of 50% methanol:50% water. After H2O2 addition to initiate reactions, aliquots (1 mL) were quenched by addition to an HPLC vial containing a catalase solution—12000 units of bovine liver catalase or 60 times the concentration capable of destroying 2.0 mL of H2O2 (4.0 × 10−3 M) in 1 min with shaking (5 min). 1H NMR spectra (500 MHz Bruker Avance 500) of the reaction mixtures were recorded for reaction mixtures containing 10% D2O and 1a (1.5 × 10−4 M), 2 (1.5 × 10−7 M) and H2O2 (7 × 10−3 M). The Watergate water suppression technique was applied. Ion chromatography (IC) studies were conducted on a Dionex DX500 chromatography system with a GP50 gradient pump, an AS40 automated sampler, an ED40 electrochemical detector, a LC25 chromatography oven, and an ASRS® 300 (P/N 064554) self-regenerating suppressor. Chromatographic data were analysed using Chromeleon chromatography software (Version 6.70 Build 1820, S/N 50398). IonPac® AS9-HC (4 × 250 mm) analytical and IonPac® AG9-HC (4 × 50 mm) guard columns were obtained from Dionex. The IC analysis was performed under isocratic conditions with an aqueous Na2CO3 (9 × 10−3 M) mobile phase at a flow rate of 1 mL min−1 with the oven temperature set at 35 °C and the SRS current set at 100 mA. The injection volume for all IC samples was 100 μL. The IC mobile phase was prepared with water from a Barnstead Nanopure system. Total organic carbon (TOC) analysis was performed by Analytical Laboratory Services, Inc. Middletown, PA—samples consisted of 1a treated with 2/H2O2 for a 15 min at pH 11.
Catalytic oxidation processes
Studies were performed across the pH range of 7–12: pHs 11 and 8.5 were chosen for 1a product characterizations and mechanistic investigations. At pH 11, reactions typically employed 1 (1.50 × 10−4 M), H2O2 (7.5 × 10−3 M) and 2 (1.5 × 10−7 M). An excess (50-fold) of H2O2 relative to 1 was used (36 eq. H2O2 are required for mineralization). Reactions were initiated by addition with mixing of the H2O2 stock solution to a mixture of all other reagents in 0.01 M phosphate buffer in 10.0 mm quartz cells.For the studies at pH 8.5, the appropriate buffer (250 mL, 0.020 M) and aliquots of the 1a and 2 solutions were combined in a volumetric flask (500 mL) and the volume was made up to 500 mL with deionized water to give [1a] = 43.8 × 10−6 M, [2] = (4.0–40) × 10−9 M and [buffer] = 0.01 M. In a typical reaction, an aliquot (120 mL) of this medium was added to a conical flask (500 mL) and the reaction was initiated by adding H2O2 (54 μL of 8.82 M standard) with mixing in a mechanical shaker (IKA KS 260) at 150 rpm for 180 min. Aliquots (2 mL) were withdrawn at appropriate intervals and catalase treated to remove residual peroxide. This medium was then filtered (RC syringe filter) into an HPLC vial (2.0 mL) and analysed by injection (10 μL) into a Shimadzu LC-ESI-MS Model 2020: Phenomenex MAX-RP C12 column (2.0 × 150 mm) at 30 °C with a mobile phase of 80% acetonitrile/methanol (2/3 v/v) and 20% deionized water pumping at 0.2 mL min−1. 1a was monitored at m/z 227 (peak at 5 min) under isocratic elution (20 min) in the negative ion mode.
Reaction solutions at pH 8.5 were also subjected to solid phase extraction (SPE) using 500 mg hydrophilic-lipophilic balance (HLB) cartridges from Waters Corp. The cartridges were preconditioned with methanol (2 mL) followed by Milli-Q water (2 mL) and the sample was passed through the cartridge at a flow rate of 10.0 mL min−1. The SPE cartridges were dried under high vacuum and then eluted with methanol (5.0 mL) at a flow rate of 3 mL min−1. The eluent was collected and dried under a gentle stream of nitrogen gas. The residue was dissolved in methanol (2.0 mL) for analysis by high resolution (HR) mass spectrometry (Bruker micro-ToF-QII, Bruker Daltonics, Germany) coupled with a Dionex Ultimate 3000 HPLC with autosampler (Dionex, Germany) following a previously described procedure.330 Samples were scanned within the range m/z 50–1500 in both the positive and negative ESI modes. Pure samples of 1a and 2 were similarly analyzed. Nitrogen dried methanol eluents were also derivatised for GC-MS analysis by treatment with BSTFA + TMCS (150 μL, 2 h, 60 °C). The samples were then dissolved in benzene (350 μL) and injected (1 μL) onto a column (Restek RXi-5 ms, 30 m long, 0.25 mm ID, 0.25 μm) for GC-MS analysis using an Agilent GC 7890A gas chromatogram equipped with an Agilent 5975C inert XL MSD mass spectrometer with a Triple-Axis detector. 1H NMR analysis (Bruker AVIII-400 MHz spectrometer) was also carried out using a sample obtained by SPE extraction of a BPA oxidation reaction after the methanol eluent had been evaporated to dryness under a gentle stream of nitrogen and the products redissolved in CD3OD.
Kinetic measurements
Monitoring of pH 11 processes was performed at the wavelength of maximal absorbance (λmax) of the corresponding phenol. Initial rates were calculated using the independently measured pH 11 extinction coefficients [ε/M−1 cm−1 (λmax, nm)]; 1a [4.3 × 103 (294)], 1b [3.6 × 103 (294)], 1c [9.0 × 103 (305)], and 1d [10.0 × 103 (310). The data were analysed using Microsoft Excel 2003, UV-Visible ChemStation (Rev. A. 10.01) and Mathematica (version 10.0) software packages. All measurements were performed in triplicate to obtain the mean values and standard deviations.Toxicity measurements
Results and discussion
Degradation at pH 8.5
At pH 8, no degradation of 1a was detected over 1 h in the absence of H2O2, but 1a is vulnerable to H2O2 alone (36% degradation in 60 min). At pH 8.5 in the presence of 2 (2.4 × 10−8 M) and H2O2 (4.0 × 10−3 M), the concentration of 1a (4.38 × 10−5 M or 10000 μg L−1) decreased to 3.97 × 10−7 M (90 μg L−1) within 30 min. Visually obvious differences were found between 2/H2O2 oxidation of 1a at pH ≥11 and at pH ≤8.5. At the lower pHs, white precipitates formed.
The oxidation of 1a was studied using very low concentrations of 2 to achieve gentle oxidising conditions as a way of gaining insight into the formation of insoluble material by slowing its rates of formation and potential further oxidation. Following 2/H2O2 (4 × 10−9 M/4 × 10−6 M) treatment of 1a (4.4 × 10−5 M) at pH 8.5 (0.01 M carbonate buffer) for 180 min, HR-ESI-MS (negative ion mode) of the SPE collected reaction media (ESI, Fig. S1a‡) showed two major ions. One was attributable to 1a (m/z 227.1082 [M − H]−; calcd for C15H15O2 227.1067) and the other to a dimer of 1a apparently formed via oxidative coupling (m/z 453.2076 [2M − H]−; calcd for C30H29O4 453.2060).
To better characterize the dimer, the crude material was TMS-silylated and analysed by GC-MS. Two major ions were observed. The first, at 51.4 min with m/z 742 [M]+, is consistent with a tetrasilylated dimer (ESI, Fig. S2a‡). This suggests the formation of the C–C coupled product 3 (Fig. 3). The second, at 53.79 min with m/z 670 [M]+, is consistent with a trisilylated dimer (ESI, Fig. S2b‡) suggesting the formation of the C–O coupled product 4 (Fig. 3). The 3:4 ratio of the relative integrals was ca. 14:1 i.e. the C–C coupling is a dominant pathway. Since authentic samples of the silylated dimers were not available, standard response curves were not generated. 1H NMR analysis of small amounts of the crude reaction product before silylation gave spectra with poor signal to noise ratios consistent with a similar 3:4 ratio. Signals assigned to the proposed C–C coupled product 3 were clearly visible at δ 6.78 (d, 8.2), 7.05 (dd, 8.2, 2.4), 7.05 (d, 8.6), 6.68 (d, 8.6) and 1.57 (s) (ESI, Fig. S3‡). C–O coupled isomer signals could not be clearly assigned.
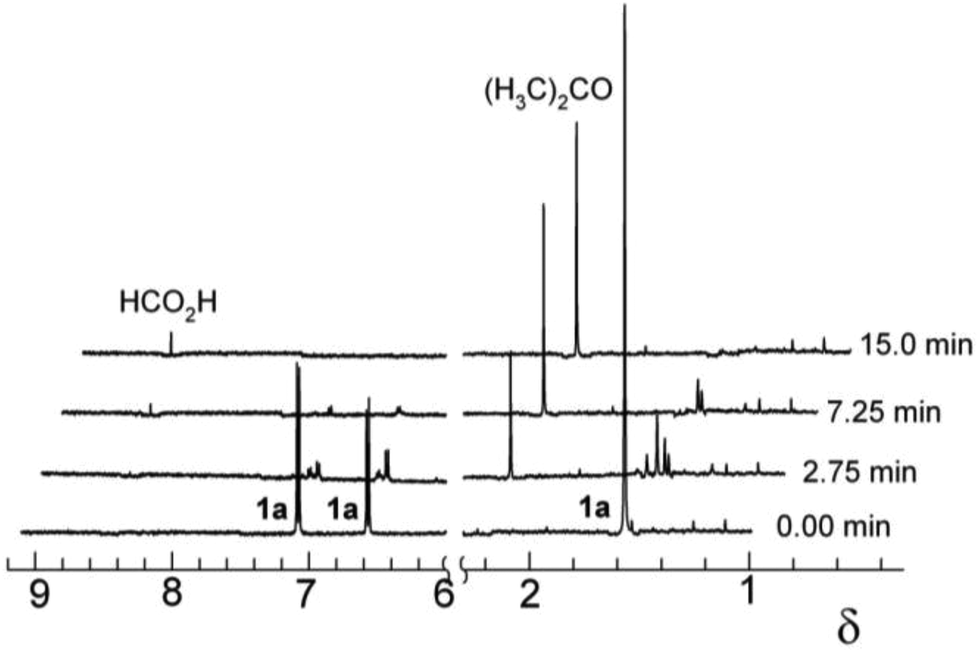 | ||
| Fig. 2 Progress of the oxidation of 1a (1.5 × 10−4 M) by H2O2 (7 × 10−3 M) in the presence of 2 (1.5 × 10−7 M) monitored by 500 MHz 1H NMR spectroscopy (Watergate suppression technique). Conditions: pH 11, 0.01 M phosphate, 10% D2O in H2O, 16 scans per spectrum. | ||
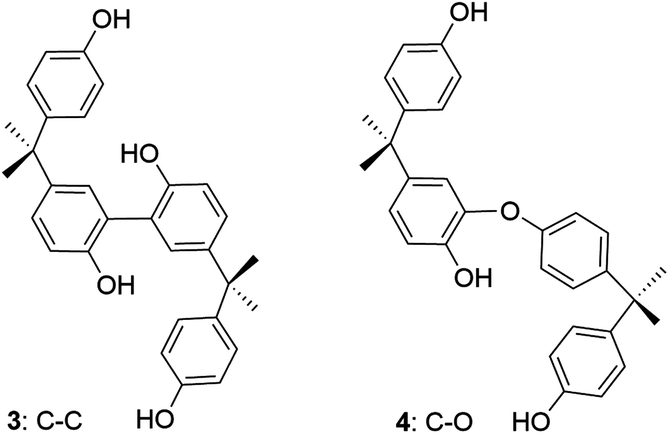 | ||
| Fig. 3 Proposed coupling products in 2/H2O2 oxidation of 1a at pH 8.5. | ||
The influence of the 2 concentration on 3 and 4 formation was investigated (ESI, Fig. S4‡). The reactions were monitored at set time intervals by low resolution electrospray ionization mass spectrometry (LR-ESI-MS, negative ion, selected ion-monitoring mode (SIM), m/z 453) without SPE. With 4 nM 2, the yields of 3 and 4 reached a maximum after 60 min and then declined slightly over the next 120 min. This 2/H2O2 oxidative procedure provides a green chemical synthesis for the oligomers. At 8 and 16 nM 2, approximately the same amounts of 3 and 4 formed more rapidly, but then declined more quickly, ultimately nearing zero.
The fates of 3 and 4 in treatment with 2 (4 and 40 nM) were next investigated under the same general conditions with a reaction volume of 500 mL and SPE product extraction prior to analysis by high resolution electrospray ionization (HR-ESI). The HR-ESI mass spectrum (ESI, Fig. S1b‡) for treatment with 4 nM 2 showed unreacted 1a (m/z 227.1120 [M − H]−; calcd for C15H15O2 227.1067), oxidatively coupled 1a dimers (m/z 453.2095 [2M − H]−; calcd for C30H29O4 453.2060) and trace trimers (m/z 679.3084 [3M − H]−; calcd for C45H43O6 679.3054) while that for treatment with 40 nM 2 showed negligible unreacted 1a and major peaks for the dimers and trimers (m/z 679.3084 [3M − H]−; calcd for C45H43O6 679.3054) with much smaller peaks for tetramers (m/z 905.4111 [4M − H]−; calcd for C60H57O8 905.4048) and pentamers (m/z 1131.4995 [5M − H]−; calcd for C75H71O10 1131.5042). Therefore, the extent of 1a oxidative polymerisation can be controlled by the concentration of 2 (ESI, Fig. S1 and S4‡).
The ability this chemistry provides to easily achieve a greater than >99% removal of 1a near neutral pH with the generation of a secondary insoluble waste stream which is entirely composed of polymerized BPA promises a very simple technique for removing BPA from near neutral pH waste streams provided other contaminants do not complicate the chemistry.
Degradation of BPA at pH 11
The pH 11 (0.01 M, phosphate) and 25 °C, 2/H2O2 oxidations of 1 were studied by HPLC, total organic carbon (TOC) analysis, 1H NMR spectroscopy, and ion chromatography. In the absence of H2O2, degradation of 1a over 1 h is negligible at pH 12. At pH 11 in the presence of 2 (1.5 × 10−7 M) and H2O2 (7.5 × 10−3 M), the concentration of 1a (1.5 × 10−4 M or 34244 μg L−1) decreased below the HPLC detection limit (1 × 10−7 M or 23 μg L−1) within 15 min. This represents a 99.9% removal without the generation of a secondary waste stream that requires additional treatment. No oligomeric degradation products were detected.
To further study the rapid and complete degradation of BPA by 2/H2O2 at pH 11, 1H NMR was employed. The spectrum of the parent BPA in water is simple allowing straightforward monitoring of the catalysed oxidation (Fig. 2). Although 2 is paramagnetic in the resting state,341 at 150 nM noticeable line broadening of the signals of 1a was not observed. The 1a aliphatic and aromatic resonances reduced quickly and were no longer visible by 7.25 min. Within 2.75 min, the oxidation produced two new singlets at δ 2.24 and 8.46 which were assigned to acetone and formate, respectively. These assignments were confirmed by spiking with authentic samples. Plots of integral intensity versus time (not shown) indicate that the rates of 1a decay and acetone formation are similar though the acetone product only accounts for 25% of the 1a aliphatic signals. After 2.75 min, an AA′BB′ pattern appeared (δ 6.65 and 7.15), as did three higher field (δ 1.62, 1.53, and 1.52) resonances. These signals were no longer distinguishable from the baseline at 15 min. At pH 11, the final 2/H2O2–1a solution was transparent.
As in other TAML degradation studies of phenols and systems with likely phenolic intermediates,309,318,325,326 ion chromatography (IC) of the final 1b–d reaction solutions confirms the deep oxidation of 1. When 1a (1.5 × 10−4 M) was subjected to 2/H2O2 (1.5 × 10−7 M/7 × 10−3 M) for 15 min at pH 11 (0.01 M carbonate), formate (1.7%) was the major observable product by IC. With the more electron-rich and more reactive 1b (see below for kinetic studies), formate was found at even lower yield (0.7%) under the same conditions. Corresponding 60 min degradations of 1c and 1d ([1] = 1.5 × 10−4 M, [2] = 4.5 × 10−7 M, [H2O2] = 7 × 10−3 M) liberated 62% of the chloride and 63% of the bromide, respectively.
Mechanistically relevant acid–base and redox properties of 1
The acid–base properties of 1 are pertinent to the interpretation of kinetic data for the processes described above. Therefore, we have studied the effects of pH on the speciation of 1a in the range of 8–12 by UV-vis spectroscopy. The data in Fig. 4 show the pH dependence of the UV-spectra. According to the literature, 1a has a pKa between 9.59 and 11.30.91 No well-defined isosbestic points were observed in our UV-vis study, suggesting that both phenolic hydroxides may undergo deprotonation in this pH range. The variation in the absorbance at 295 nm (A295) with pH shown in the inset to Fig. 4 could be fit to eqn (1), an analytical form for the dependence of the absorbance on [H+] which corresponds to the deprotonation sequence AH2 ⇄ AH− ⇄ A2− (AH2 is 1a) in which [1a]t is the total concentration of 1a, Ka1 and Ka2 are the first and the second dissociation constants for 1a, respectively, and εAH2, εAH, and εA are the extinction coefficients for the forms AH2, AH−, and A2−, respectively. The solid line in the inset is the calculated dependence of the A295 on the solution pH using the best-fit values of the parameters of eqn (1). The so-derived pKa1 and pKa2 of 9.4 ± 0.3 and 10.37 ± 0.07, respectively, indicate that at pH 8.5, the neutral form H2A is dominant while, at and above pH 11, the dianion A2− is the dominant form of 1a.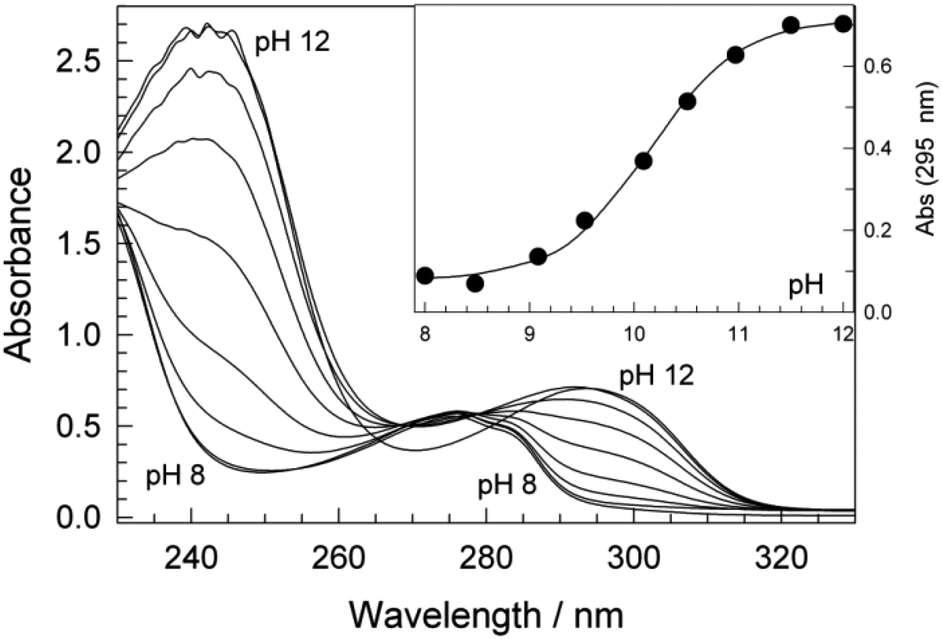 | ||
| Fig. 4 UV-vis variation with pH for 1a. Inset shows absorbance changes at 295 nm as a function of pH; the solid line is the calculated dependence using the best-fit values of eqn (1). Conditions: [1a] 1.5 × 10−5 M, 25 °C, 0.01 M phosphate. | ||
To characterize the relative tendency of substituted phenols 1a–d to undergo 1-electron oxidation, cyclic and differential pulse voltammetry methods have been applied. As was found previously,342,343 the 1a–d reduction potentials could not be determined in aqueous solutions. However, appropriate data could be obtained in acetonitrile via application of the differential pulse technique. Examples of the voltammograms are shown as the Inset to Fig. 6 and the corresponding reduction potentials of 1a–d are included in Table 1. As expected, the electron-donating methyl substituent lowers the reduction potential of 1b compared to that of 1a, and 1c is the most resistant 1 to oxidation.
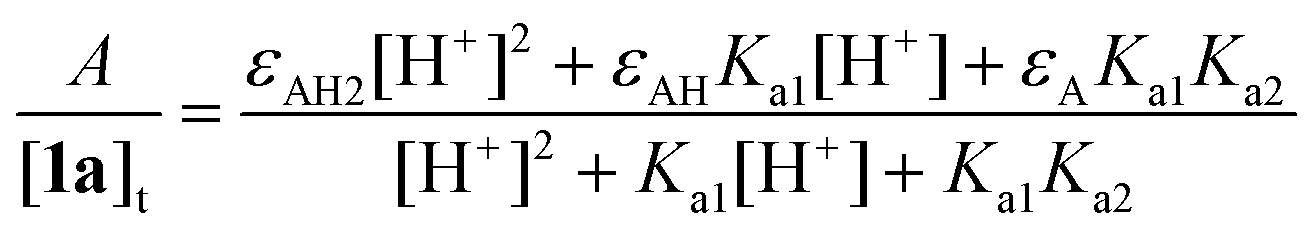 | (1) |
| 1 (R) | 10−4 × kII/M−1 s−1 | E′/V (vs. SCE) |
|---|---|---|
| 1b (Me) | 8 ± 3 | 0.181 ± 0.005 |
| 1a (H) | 1.0 ± 0.4 | 0.26 ± 0.01 |
| 1d (Br) | 0.72 ± 0.04 | 0.37 ± 0.01 |
| 1c (Cl) | 0.57 ± 0.08 | 0.41 ± 0.01 |
Mechanism of pH 8.5 TAML-catalysed 1a oxidation
Oligomers such as those reported are commonly produced in enzyme-catalysed polymerizations of phenols347–349 including 1a.350–354 The initial step is often proposed to be a one-electron oxidation of the AH2 or AH− forms of 1a to produce phenolate radicals which can then couple to give higher molecular weight products.350–354 Our study mirrors the enzymatic results. There is little doubt that the oxidation starts with 1-electron oxidation of either AH2 or AH− forms of 1a. In the former case, deprotonation of the primary radical-cation affords A and B (Scheme 2), precursors of the coupled C–C and C–O dimers 3 and 4, respectively (Fig. 3).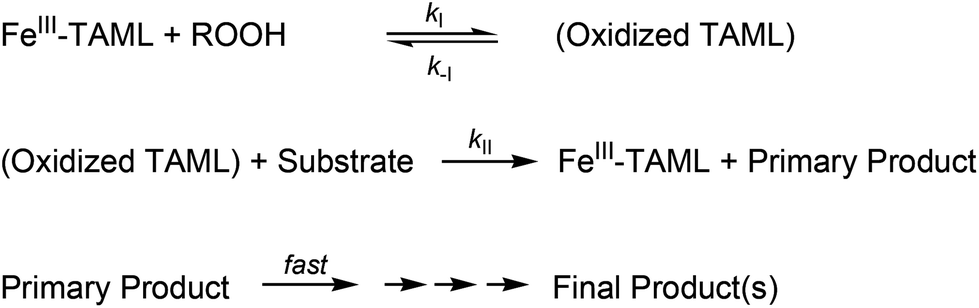 | ||
| Scheme 1 Stoichiometric mechanism of catalysis by TAML/peroxide. | ||
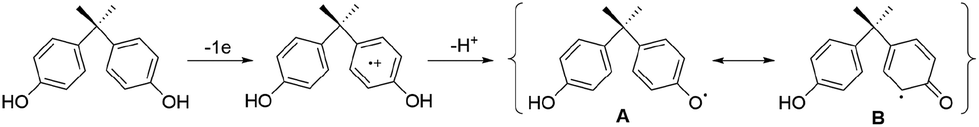 | ||
| Scheme 2 Initial steps of 2/H2O2 oxidative degradation of 1a at pH 8.5. | ||
Kinetic studies of pH 11, 2/H2O2 oxidation of 1a–d
TAML-catalysis of substrate (S) oxidations by peroxide usually follows the stoichiometric mechanism shown in Scheme 1. The initial rates of substrate oxidation (v) are well-modelled by eqn (2), the corresponding rate expression in which [Fe]t is the total initial concentration of all catalyst species.314 | (2) |
Obtaining the rate constants kII for oxidation of 1a–d by the initial rates method promised to deliver insight into the nature of the degradation process. As with many artificial peroxidase mimics,314 the step associated with kI is slow requiring that, when possible, the reaction conditions should be set to ensure that steady-state measurements allow for the determination of kII. For TAML 2 such conditions are favoured by high [H2O2] and basic pH around 11, where kI[H2O2] ≫ kII[S] usually applies and k−I can be assumed to be negligible.325,344 Under these conditions, eqn (2) simplifies to v = kII[S][Fe]t. Correspondingly, the initial rate is (i) independent of [H2O2] in the range of (0.35–1.40) × 10−2 M, (ii) proportional to the initial concentration of catalyst, [2], in the range of (0.375–1.50) × 10−7 M and, (iii) directly proportional to the concentration of substrate, [1a] (Fig. 5).
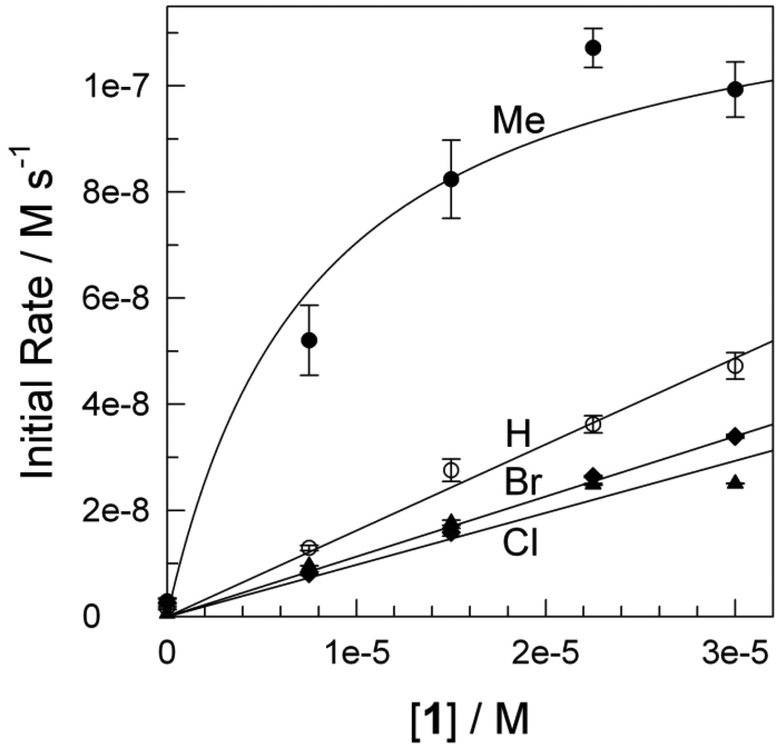 | ||
| Fig. 5 Dependence of the initial rate of oxidation of substituted Bisphenol A derivatives 1a–d (1: a = H, b = Me, c = Cl, d = Br) by H2O2 (7.5 × 10−3 M) catalysed by 2 (1.5 × 10−7 M). Conditions: 0.01 M phosphate (Na2HPO4), pH 11, 25 °C. | ||
Similar kinetic measurements were performed for 1c–d and the second-order rate constants kII were determined from the slopes of the linear plots for 1a, 1c and 1d (Fig. 5, Table 1). The oxidation of methyl-substituted 1b was considerably faster than all other cases and the initial rate showed saturation with increasing [1b], suggesting a contribution of the kI pathway to the overall rate. Therefore, the data were fitted to eqn (2) (assuming k−I ∼ 0) and the value of the thus calculated kII appears in Table 1.
We have developed a mathematical tool for modelling TAML catalysis345 which we have used to examine the comparative behaviour of 15 different catalysts in the oxidation of one substrate under one set of conditions327 and, more recently, the oxidation of 5 different substrates by 2 different catalysts and 2 different oxidants at two different pHs.321 We used the kII value for 2/H2O2 oxidation of 1a and the 2ki value of (7.7 ± 0.3) × 10−5 s−1 determined in 2/H2O2 oxidation of the azo dye Orange II at pH 11 and 25 °C,346 to estimate the percent removal of 1a by 150 nM 2 treatment. The predicted removal of ca. 100% agrees well with the observed removal of 99.9% providing further validation of the mathematical model.
The 1a–d phenols were characterized electrochemically to examine if rate-limiting electron transfer is the initial event upon encounter of these electron-rich substrates with the powerfully oxidizing TAML reactive intermediate. The negative slope found for the linear relationship between the values of logkII and the reduction potentials E′ (Fig. 6) supports this assignment of the initial step. This correlation and the observation of acetone as a final 1a degradation product are consistent with the proposed mechanism (Scheme 3).
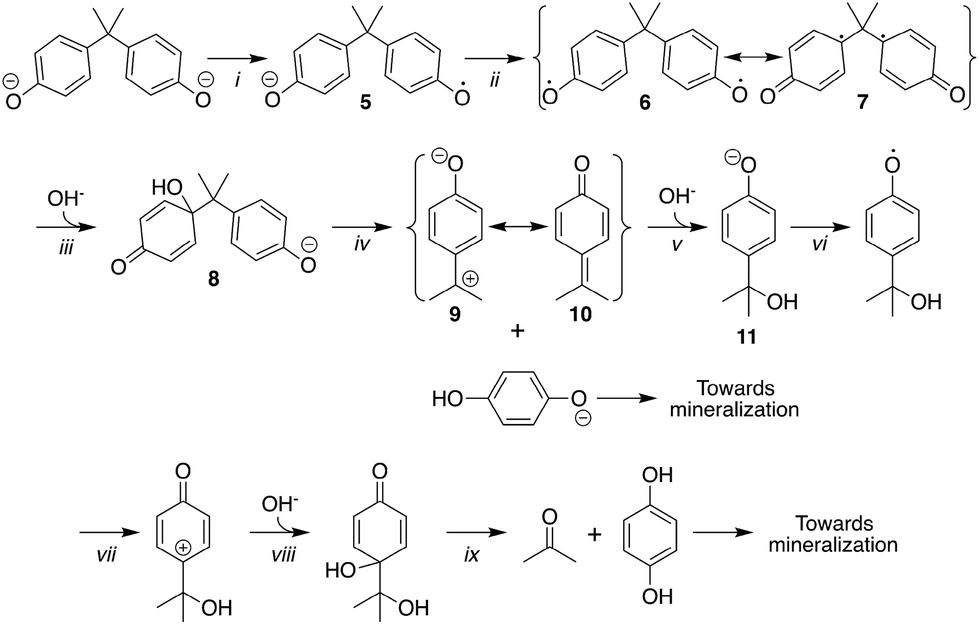 | ||
| Scheme 3 Tentative mechanism for 2/H2O2 oxidative degradation of 1a at pH 11 consistent with results and working assumptions derived in this work. | ||
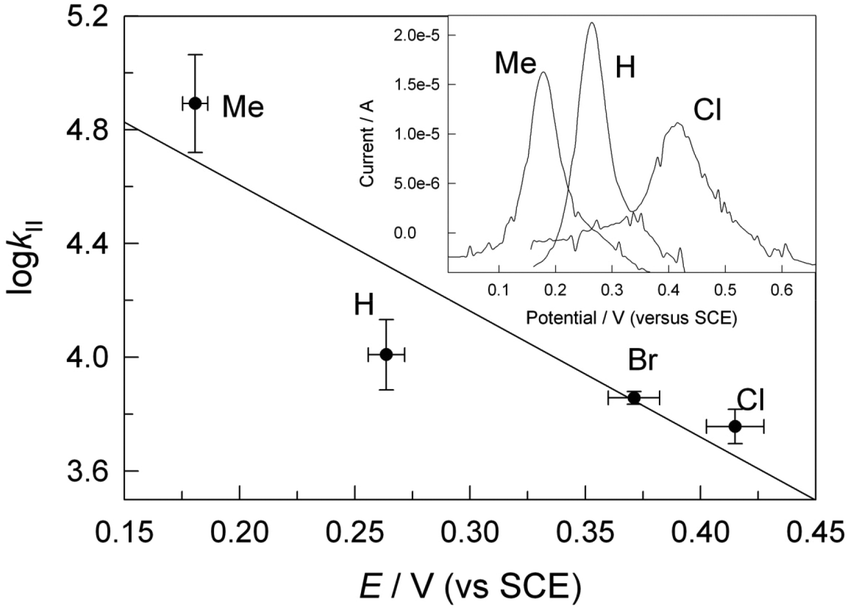 | ||
| Fig. 6 Relationship between logkII and experimentally determined reduction potentials of 1 obtained by differential pulse voltammetry in acetonitrile (Inset) of solutions of 1 ([1] ≈ 10−3 M). Conditions: 0.1 M [n-Bu4N]PF6 as supporting electrolyte, modulation time 0.05 s, interval time 0.1 s, step potential 0.002 V, modulation amplitude 0.005 V, scan rate 0.1 V s−1, glassy carbon electrode. | ||
Tentative mechanism of pH 11 TAML-catalysed 1 oxidation
Since the pKa2 of 1a (10.37 ± 0.07) indicates that the doubly deprotonated A2− form dominates the speciation of 1 at pH 11, and the kII values in Table 1 correlate with the reduction potentials (Fig. 6), and the observed removal agrees well with the removal predicted by the aforementioned mathematical tool, the initial step of pH 11, 2 catalysed 1 oxidation likely involves rate-limiting electron-transfer from A2− to the Oxidized TAML species (Scheme 1) to give the primary product 5 (Scheme 3, step i). Oxidation of the second phenolate would give a compound 6 having resonance form 7 (Scheme 3, step ii).355 Nucleophilic attack of hydroxide and intramolecular electron transfer would give 8 (step iii), an intermediate proposed in degradation of 1a by Sphingomonas sp. Strain TTNP3 via a type II ipso substitution mechanism thought to be enacted by a monooxygenase enzyme.356 Intermediate 8 can undergo heterolysis to give p-hydroquinone and the compound represented by resonance structures 9 and 10356 where the aromatization provides an important component of the driving force (step iv). The absence of p-hydroquinone in the 1H NMR spectra of the reaction mixture (Fig. 2) should not be interpreted as an indication that it is not generated. Molecules like p-hydroquinone are known to undergo fast oxidation by H2O2 under similar reaction conditions without involvement of TAMLs.325
Since, an intermediate having an AA′BB′ spin system was observed at δ 6.65 and 7.15 (Fig. 2, 7.25 min spectrum) and 4-(2-hydroxypropan-2-yl)phenolate (Scheme 3, 11), 4-(prop-1-en-2-yl)phenol (Scheme 4, 12), a tautomer of 10, and p-isopropylphenol, the reduction product of 12, have been observed in the product mixtures of bacterial,357 enzymatic6,75,335,350,358 thermal,276–279,359 and/or chemical degradations of 1a,360 including that of TTNP3,356 and the reaction was performed at basic pH, these compounds were investigated as candidates for the reaction intermediate. 1H NMR spectra of p-isopropylphenol and 12, which was generated from 1a as reported (Scheme 4),361,362 obtained under the reaction conditions showed AA′BB′ spin systems at δ 6.56 and 7.02 and δ 6.67 and 7.41, respectively, indicating that neither compound accumulated measurably during 2-catalysed oxidation of 1a. 1H NMR spectra of the crude products of the alkylation of p-hydroxyacetophenone with methylmagnesium bromide363 showed 11, the presence of which was further confirmed by ESI-MS. However, difficulties were encountered in the isolation of 11, as has been reported.363 As a result, we tentatively propose the intermediate to be 4-(2-hydroxypropan-2-yl)phenolate (Scheme 3, 11), the 1a analogue of a product observed in 1d oxidation by H2O2 catalysed by 2 immobilized in a layered double hydroxide composite.31011 can undergo two successive one electron oxidations, nucleophilic attack of hydroxide and heterolysis with proton transfer (Scheme 3, steps vi, vii, viii and ix, respectively) to give the observed acetone (Fig. 2) and p-hydroquinone which would undergo rapid catalysed or uncatalysed degradation.
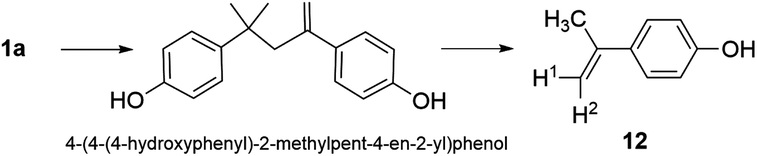 | ||
| Scheme 4 Synthesis of 12 from 1avia the intermediacy of 4-(4-(4-hydroxyphenyl)-2-methylpent-4-en-2-yl)phenol (see Experimental section). | ||
The material in the above Experimental section promises that, subject to successful real-world testing, a simple to deploy, technically effective, cheap to install and operate TAML/H2O2 process for removing 1 compounds from water is achievable. The literature covered in the first mini-review establishes ubiquitous 1a occurrences in products and water with broad exposures to humans and wildlife. In the following mini-review, we examine the literature describing the consequences of these exposures to underscore the importance of developing more effective treatment processes that, before deployment, are cleared of low-dose adverse effects.
Mini-review of BPA toxicity
Several derivatives of (4,4′)-dihydroxy-diphenyl methane, which differ in the alkyl substituents of the aliphatic carbon atom, including 1a, are oestrogens as was briefly communicated in 1936,23 more thoroughly reported in 1938,364 1940333 and 1944334 and reviewed in 1945.94 Given this history, it is not surprising that global 1a contamination can impact exposed organisms.1,74,75,128,218,219,365 In a 2008 report to the Canadian Government, authors concluded that, “Bisphenol A is acutely toxic to aquatic organisms and is a known endocrine disruptor”217 which, in 2010, led the Canadian Government to declare 1a to be toxic and add it to the List of Toxic Substances in Schedule 1 under section 64 of its Environmental Protection Act of 1999.421
In adult fish of many species, lab tests show that exposure to the levels of 1a found in surface waters (earlier mini-review) causes alteration of gene expression including stimulation of vitellogenin synthesis (a protein that is a precursor of egg yolk protein and a biomarker for exposure to oestrogens) in males, alteration of reproductive traits including reductions in male sperm quality, and delayed or no ovulation in females.74,366–372 In the embryonic and early life stages of fish development, such exposures have been observed to result in alteration of gene expression, reduction of heart rate, decreased eye pigmentation density, accelerated development, delayed hatching of embryos, testis-ova in males, hyperactivity in larva, and learning deficits in adult males.373–379
In addition to these impacts on the exposed fish, lab exposures at typical 1a surface water concentrations can cause effects in offspring not observed in the parental generation.365 These multigenerational effects can derive from both adult and developmental exposure to EDs and are measured by assessing the impacts of exposure on members of the initial population (F0) and each subsequent generation (Fn) versus unexposed controls (B0–Bn).77,96,380–382 One such study followed the effects of zebrafish exposure to 0.228 μg L−11a on the F0, F1, and F2 generations.365 In the continuously exposed F1 and F2 generations, decreased male/female sex ratios in the adult population were observed together with lowering of male sperm density and quality including decreased motility and ATP production as well as increased sperm lipid peroxidation. The majority of the F1 adverse effects on sperm quantity and quality were not found in F2 male offspring if, subsequent to the 1a exposure, the F1 generation was incubated for 150 days in water to which 1a was not added, highlighting that these effects may be reduced through reduction of 1a exposure—the decreased sperm ATP production persisted in F2 males. Increases in malformation and mortality at 8 days post fertilization in the F2 offspring of F1 males indicated male-mediated reproductive failures deriving from 1a exposure. In the gonads of F2 males, altered gene expression was observed leading to perturbations of signaling pathways including those regulating mitochondrial biogenesis and testis development. In the larvae of F2 parents, reduction in expression of DNA methyl transferases and the associated transcription factor was observed indicating that continuous 1a exposure can lead to alterations of the epigenome and may result in transgenerational effects such as the observed male specific reproductive failures.365
A US National Institute for Environmental Health and Safety panel has concluded that the available laboratory rodent studies provide sufficient evidence to be “confident” of human effects including effects on the male reproductive tract arising from adult exposures and effects on the organization of the reproductive tract of males, the brain and metabolism arising from developmental exposures.92 A US National Toxicology Program panel383 and the US Food and Drug Administration79 also concluded that there is “some concern” for effects on the brain, behavior, and prostate gland in foetuses, infants and children arising from exposure to 1a at levels currently observed in the human population (earlier mini-review). However, further studies accounting for contributions to human 1a exposure from dermal absorption and of the toxicokinetics following dermal absorption need to be performed.85 We proceed by highlighting laboratory studies that reflect the human effects of 1a exposure to the concentrations found in foetal, child and adult fluids and tissues (0.3–4.4 μg L−1 or 1.3–19 nM).120 Reviews of the epidemiological studies384–388 on the effects of human 1a exposure not discussed herein are available.68,389
Exposure to 1a at and slightly below the concentrations found in foetal, child and adult fluids and tissues can alter cellular development and produce mature cells which are improperly programmed. For example, exposure of HL-60 leukocytes to 1 nM 1a during neutrophilic differentiation induced by 1.25% dimethylsulfoxide and 25 ng mL−1 granulocyte colony-stimulating factor results in significant increases in PU.1 transcription factor activity and production of opsonized zymosan (OZ) receptor subunit CD18 and O2− stimulating NADPH-oxidase components p47phox and p67phox mRNA during differentiation via an oestrogen receptor independent mechanism, as well as differentiated cells with increased CD18 expression on the cell surface and OZ-stimulated O2− production suggesting that long-term 1a exposure may significantly affect human immunity.390
Exposure of day 7 human prostaspheres, prostate stem-progenitor cells, to 1a at human relevant concentrations resulted in rapid membrane-initiated oestrogen signalling with increased levels of p-Akt and p-Erk, downstream targets of membrane ERs, and Erk phosphorylation, which were sustained for at least 60 minutes and returned to baseline by 6 hours, a result similar to that obtained from exposure to the same concentration of E2, an endogenous hormone.391 Further insights into the effects of 1a exposure on prostate development have been gained through application of an in vivo renal graft model of chimeric human-rat prostate tissues. This model employs mice which express human prostate epithelial stem-progenitor cells that form normal human prostate epithelium, which produces prostate-specific antigen.392 Treatment of the host mice with testosterone and E2 for 1–4 months to model the elevated E2 levels of later life-staged men increases the transition of the human prostate cells from hyperplasia to prostatic intraepithelial neoplasia and adenocarcinoma demonstrating the role of E2 in cancer of the human prostate epithelium.391 Oral exposure of the host mice to 1a dosages giving free 1a serum levels of 0.39 and 1.35 μg L−1, comparable to the internal doses found in human umbilical cord blood and foetal and neonatal serum, for 2 weeks after renal grafting resulted in mice having reduced normal prostate incidences from the control 26% to 11 and 0%, respectively, increased benign lesion incidence from the control 74% to 89 and 100%, respectively, and increased malignant lesion incidence from the control 13% to 36 and 33%, respectively, upon initiation/promotion of hormonal carcinogenesis by administration of testosterone and E2 for 2–4 months.391 If, to model continuous exposure throughout development, prostaspheres were exposed to 1ain vitro prior to in vivo exposure, incidences of malignant lesions further increased to 45%. These results indicate that exposure of developing human prostate epithelium to doses of 1a relevant to those observed in human development increases its susceptibility to hormonal carcinogenesis.391
Exposure to 1a at and slightly below the average concentrations found in human blood also alters the functioning of developed cells. For example, exposure of developed human pancreatic β-cells to 1 nM 1a in the absence of glucose rapidly decreased the activity of KATP channels as effectively as exposure to 8 mM glucose, and in the presence of a stimulatory concentration of glucose (8 mM), exposure of human pancreatic islets of Langerhans to 1 nM 1a enhanced insulin secretion ca. 2 fold demonstrating that 1a exposure may be a risk for the development of type-2 diabetes.393 Exposure of human breast, subcutaneous and visceral adipose tissue explants and mature adipocytes to 1 and 10 nM 1a suppressed the release of adiponectin, an insulin sensitizer, a result similar to exposure to equimolar concentrations of E2.83,394 Exposure of human adipose explants to 10 nM 1a stimulated the release of inflammatory cytokines IL-6 and TNFα which promote insulin resistance. Taken together, the adipose tissue and adipocyte results indicate that exposure to 1a at population relevant levels may adversely effect metabolic homeostasis.83,394
Exposure to 1a at and slightly below the average concentrations found in human blood and serum also alters the behaviour of cancer cells in tumours. For example, exposure of human androgen-dependent prostatic adenocarcinoma cells, a model system for prostate tumours, to 1 nM 1a has been observed to activate AR-T887A leading to androgen independent cellular proliferation like that resulting from exposure to 0.1 nM dihydrotestosterone, an endogenous ligand, though 1a activation may occur indirectly through interaction with ERβ or other proteins.395 This result indicates that 1a exposure may ease the transition of prostatic adenocarcinomas to androgen independence,395 thereby challenging treatment.84 Exposure of both wild-type MCF7 breast cancer cells and an MCF7 subline, MCF7SH, which models the behaviour of long-term oestrogen deprived breast tumor cells, to 10 nM 1a has been observed to stimulate cellular growth, a response similar to, but weaker than that observed upon exposure to 10 nM E2, and this behaviour has been attributed to classical genomic activation of ERα.191
While the observation of weaker response to 1a than E2 would seem to reflect a general, lower potency of 1a, as would be anticipated to result from the known, weaker binding of 1a to classical oestrogen receptors, this is not always the case. For example, in vitro exposure of MCF-7 cells, which express both ERα and ERβ, MDA-MB-231 cells, which express ERβ only, and SKBR-3 cells, which express neither ERα nor ERβ to 0.1–100 nM 1a in the presence of 2 mM Ca2+ results in rapid Ca2+ influxes in all three cell types leading to increases in intracellular calcium concentrations ([Ca2+]i) comparable to those resulting from exposure to equimolar concentrations of E2.22 These results implicate 1a in non-genomic signalling pathways, such as membrane ERα (mERα) initiated signalling, and agree with those from in vitro studies employing rat pituitary tumor cell sublines GH3/B6/F10 (F10), which naturally expresses high levels of mERα, and GH3/B6/D9 (D9), which naturally expresses low levels of mERα. In vitro exposure of F10 cells to either 1 pM E221 or 1 pM 1a20 results in rapid and reversible influx of extracellular Ca2+ leading to nearly equal increases in [Ca2+]i, which results in prolactin secretion and can initiate signaling cascades that lead to changes in cellular protein phosphorylation that alter protein functioning. Exposure of D9 cells to either 1 pM E221 or 1 pM 1a20 does not alter the [Ca2+]i. Thus, 1a can be as potent as E2 in eliciting responses mediated by non-genomic pathways reinforcing that it is inappropriate to label 1a a weak oestrogen.20,22,393
Similar potency of BPA and EE2 has also been observed in vivo.396 This may result from the formation of a BPA metabolite that either syngergizes397 with BPA396 or is significantly more oestrogenic.17,396,398 One BPA metabolite produced by human liver S9 fractions in the presence of an NADPH-generating system is 4-methyl-2,4-bis(4-hydroxyphenyl)pent-1-ene (MBP, Fig. 1).17 MBP has several- to several thousand-fold increased oestrogenicity in cellular assays employing ERα and ERβ.17 One such assay showed MBP to be as oestrogenic as diethystilbestrol (DES, Fig. 1),17 prenatal exposure to which is known to increase the risk of breast42 and vaginal399 cancers.105 An in vivo uterotrophic assay using ovariectomized rats has shown MBP to be ca. 500 times more oestrogenic than BPA.400 The MBP oestrogenicity increase is thought to derive from the increased distance between the phenolic rings which resembles that of benzestrol (Fig. 1),401 a known oestrogen.28,402 Studies have also shown MBP to adopt a coplanar conformation like E2 with a similar inter-hydroxyl distance.17 Therefore, the effects of metabolites like MBP may contribute to the negative environmental403,404 and human health405 impacts of BPA, particularly if glucuronidation, the predominant, human, 1a metabolic pathway,14,15 which gives a monoglucuronide showing almost no oestrogenic activity,16 does not occur.17,18 Thus, BPA exposure of foetuses,68,120,406–410 which are largely incapable of glucuronidation,19,68,411–414 is particularly concerning and disruption of human development415 is a potential health impact of BPA and BPA metabolite exposure.398 This is especially troubling given the known effects of BPA on cellular programming which can increase susceptibility of some organs, including the prostate391,392,416 and mammary glands417,418 to the development of cancer.105,120 Since cytochrome p450 (CYP) operates in the liver fraction S9 metabolism that gives MBP17,18 and CYP isoforms are expressed in the human foetal liver,419in vivo foetal BPA exposure may also result in foetal MBP exposure.17 Unquestionably, the study of endocrine disruption phenomena associated with BPA metabolites should be a future research priority as there is enough already known to suggest that an under-recognized potent toxicity might be impacting human foetal development.
Along with numerous others, these studies of the effects of exposure to 1a on embryonic, early life stage and adult fish of many species and the zebrafish epigenome at environmentally relevant concentrations and of the effects of exposure to 1a on developing, developed, and cancer cells at concentrations relevant to those observed in the human population reinforce that 1a is an endocrine disruptor with negative environmental and health performances to challenge the on-going viability of many current applications.12,86,217,420
With these studies providing a useful background we now describe the toxicity assays used to examine the TAML/H2O21a treatments described in this work.
Toxicity of the treated 1a solutions
One-hour reaction mixtures (Table 2) were adjusted to neutral pH, quenched with catalase, and tested for acute toxicity using the Microtox assay.422 Samples obtained at pH 8, where solids form in the dominating oligomerization of this pH region, were filtered before the assay. Although the oligomers are insoluble, we emphasize that these species are important aspects of the environmental profile of pH 7 treatments. A thorough exploration of the endocrine activity of the water insoluble oligomers is beyond the scope of the current study—if precipitation occurs in real-world media this would be an excellent way to isolate waste 1a in a form having diminished potential for long range transport. The results (Table 2) show that samples treated with H2O2 in the presence of 2 were less toxic than those treated with H2O2 alone. An EC50 of >100% indicates that the undiluted sample was not toxic enough to affect 50% of the organisms. At pH 8, only 5 molar equivalents of H2O2 were necessary to effect this reduction in toxicity. At pH 12, the H2O2 requirement increased to 50 molar equivalents.| Reaction pH | H2O2 (equiv.) | EC50 (no 2)/% | EC50 (2-treated)/% |
|---|---|---|---|
| n/a | 0 | 21.2 ± 1.7 | 24.5 ± 1.3 |
| 8 | 5 | 20.7 ± 1.6 | >100 |
| 12 | 5 | 22.0 ± 0.8 | 38.0 ± 2.6 |
| 12 | 50 | 23.0 ± 0.3 | >100 |
Solution concentrations of 1a can be lowered to nearly zero by polymerizing treatment at pH 8.5 (Fig. 7a). It is prudent to show that these oxidised products, though effectively isolated, do not themselves have oestrogenic activity. This important point was illustrated by a recent study in which it was demonstrated that removal of EE2 from solution through partial oxidation produced soluble products that have similar oestrogenic activity as EE2 itself.330 Therefore, the oestrogenic activities of the 1a solutions were evaluated by the yeast oestrogen screen (YES)339 after oxidation with 2 at concentrations of 4, 16, 24 and 40 nM at time 0 and after 10 and 60 minute reaction times. The results are depicted in Fig. 7b. In all cases the oestrogenic activity was reduced, with the higher catalyst levels producing faster and more significant drops in activity. After 60 min, the solutions resulting from the reactions with 2 concentrations of 16, 24 and 40 nM showed almost no residual oestrogenic activity while the solutions resulting from reaction with 4 nM 2 showed approximately 75%. In all cases the residual oestrogenic activity correlated well with the amount of 1a remaining in solution. Therefore, it appears that the product solutions of oxidised 1a have no activity, and the 1a remaining can serve as an indication of the residual oestrogenicity of the treated solution.
 | ||
| Fig. 7 (a) Kinetic traces of 2 (concentrations shown) catalysed oxidation of 1a (43.8 μM) by H2O2 (4 mM) at pH 8.5 (0.01 M, carbonate) and 25 °C. Data points are each the mean of triplicate runs with estimated 3SD limits indicated. (b) Residual oestrogenic activity as a function of treatment time for some of the processes shown in (a). | ||
To further test for aquatic toxicity of 2/H2O2 treated 1a solutions, starting at 6 hours post fertilization, dechorionated zebrafish embryos were exposed to solutions containing 0.01 to 1% of the treated samples. This was done twice. In the first test, 50 μM 1a was treated with 2/H2O2 (20 nM/5 mM, respectively) at pH 7 (0.01 M, phosphate) and quenched with catalase at 12 h. From the unfiltered, agitated, treated solution, more dilute solutions (0.2–2 μM, based on the initial concentration of 1a) were prepared, and the embryos were exposed to the diluted solutions. In the second test, 80 μM 1a was treated with 2/H2O2 (200 nM/5 mM, respectively) at pH 7 (0.01 M, phosphate) and quenched with catalase at 12 h. From the unfiltered, agitated, treated solution, more dilute solutions (0.08–64 μM, based on the initial concentration of 1a) were prepared, and the embryos were exposed to the diluted solutions. With the treatment conditions chosen, no significant incidences of abnormality among any of the 22 endpoints were observed (ESI, Fig. S5‡). However, an insignificant increase in mortality was observed for the treated samples compared to the untreated samples.
Conclusion
In developing Green Chemistry, it is important that chemists come to understand the scope of the challenges posed by everyday-everywhere endocrine disruptors (EDs) to the sustainability of both the chemical enterprise and our complex global civilization. The most troubling such EDs, like BPA, invariably hold their protected positions in the economy because of seductive technical and cost performances that enable large, diverse, profitable markets. For sustainable chemicals, the health, environmental and fairness performances also have to be integral components of the value proposition. Understanding the negative performances of unsustainable chemicals helps in mapping the properties sustainable chemicals should not have. Key aspects of this understanding include the knowledge of which chemicals are and are not EDs and are and are not capable of eliciting low dose adverse effects by non-endocrine processes, the extent and routes by which the environment and people are exposed to commercial EDs, the environmental and human health consequences of ED exposures, the methods of assessment of endocrine activity423 including the TiPED,336 the mechanisms of the low dose adverse effects, the design approaches to attaining new and replacement chemicals free of such effects,307 and the stewardship methodologies that are currently deployed or might be deployed to better protect health and the environment from commercial EDs. This BPA case study traverses the appropriate multidisciplinary landscape with emphasis on the integration of chemistry and environmental health science in the development of endocrine disruption-free processes to aid the chemical enterprise and society in reducing BPA exposures. Importantly, the litany of unfortunate facts presented about BPA exposures and health and environmental performances is relieved to some extent by the possibility of reduced releases arising from the TAML/H2O2 technology mapped out in the empirical section.This experimental component demonstrates that TAML/H2O2 provides simple, effective water treatment methodologies, which depending on the pH, either decompose 1a or isolate it in low solubility oligomers. Both processes require only very low concentrations of 2 and H2O2 in further reflection of the remarkable efficiencies of the peroxidase enzymes that are faithfully mimicked by 2 and in marked contrast with the much higher relative iron- and peroxide-requiring Fenton processes. It remains to be established whether the current laboratory studies project to real world scenarios. These may include treatment of 1a-contaminated landfill leachates and paper plant processing solutions where the concentrations are similar to those employed in this study. In such scenarios, TAML/H2O2 would present an enzyme-mimicking method which in the case of 2 is comprised exclusively of biochemically common elements and has passed multiple TiPED assays that, in contrast with existing real world processes, avoids generation of 1a-contaminated sludges and associated subsequent releases to soil, that does not generate a 1a-contaminated adsorbent which must be replaced or regenerated at elevated temperature, that does not generate chlorinated forms of 1a, that does not generate a concentrated retentate, and that is remarkably simple to deploy using very low and cheap chemical inputs with all the positive potential consequences thereof for capital and operating expenses.
Finally, in order to avoid the habit or perception of greenwashing, a realistic perspective is essential to the integrity of green chemistry. We view the sustainability challenges posed by BPA as enormous—the experimental work presented could evolve into a solution for some of these problems but is, by no means, a general quick fix. BPA markets large and small are expanding rapidly, especially as the industry has learned how to produce even more effective replacements for glass and metal products. Huge new markets are developing such as those of plastic glass houses, and even houses, and automobile body parts that are comprised primarily of BPA. In this build-up, BPA's unfortunate health and environmental performances continue to be given short shrift. Continuation of the present BPA expansion trends without limits, technical corrections and more aggressive stewardship advances of multiple kinds will menace society with an ever increasing oestrogenization of the entire ecosphere.
Acknowledgements
Y. O. was a University of Auckland Doctoral Scholarship Recipient. E. S. B. was an NSF Predoctoral Fellow. A. D. R. was an Alexander von Humboldt Foundation Fellow during part of this project. M. R. M. was a CMU Steinbrenner Fellow. M. R. M. and S. K. were CMU Presidential Fellows. T. J. C. thanks the Heinz Endowments for funding and the Heinz Family Foundation for the Teresa Heinz Professorship in Green Chemistry. The authors thank Allan Carl Jackson III and Prof. Laura Vandenberg for valuable suggestions and the reviewers—especially the one who suggested splitting the review material into two mini-reviews, which greatly improved the logic and flow of the material presentation. The 1H NMR spectrometers of the Department of Chemistry's NMR Facility at Carnegie Mellon University were purchased in part with funds from the National Science Foundation (CHE-0130903).Notes and references
- R. K. Bhandari, S. L. Deem, D. K. Holliday, C. M. Jandegian, C. D. Kassotis, S. C. Nagel, D. E. Tillitt, F. S. vom Saal and C. S. Rosenfeld, Gen. Comp. Endocrinol., 2015, 214, 195–219 CrossRef CAS PubMed.
- R. K. Bhandari, F. S. vom Saal and D. E. Tillitt, Sci. Rep., 2015, 5, 9303 CrossRef CAS PubMed.
- R. T. Zoeller, T. R. Brown, L. L. Doan, A. C. Gore, N. E. Skakkebaek, A. M. Soto, T. J. Woodruff and F. S. Vom Saal, Endocrinology, 2012, 153, 4097–4110 CrossRef CAS PubMed.
- M. R. Mills, K. Arias-Salazar, A. Baynes, L. Q. Shen, J. Churchley, N. Beresford, C. Gayathri, R. R. Gil, R. Kanda, S. Jobling and T. J. Collins, Sci. Rep., 2015, 5, 10511 CrossRef CAS PubMed.
- C. A. Kinney, E. T. Furlong, S. D. Zaugg, M. R. Burkhardt, S. L. Werner, J. D. Cahill and G. R. Jorgensen, Environ. Sci. Technol., 2006, 40, 7207–7215 CrossRef CAS PubMed.
- Q. Huang and W. J. Weber Jr., Environ. Sci. Technol., 2005, 39, 6029–6036 CrossRef CAS PubMed.
- Z.-H. Liu, Y. Kanjo and S. Mizutani, Sci. Total Environ., 2009, 407, 731–748 CrossRef CAS PubMed.
- B. Borrell, Nature, 2010, 464, 1122–1124 CrossRef CAS PubMed.
- World BPA Production Grew by Over 372,000 Tonnes in 2012, Merchant Research & Consulting Ltd, 2013, https://mcgroup.co.uk/news/20131108/bpa-production-grew-372000-tonnes.html Search PubMed.
- Y. B. Wetherill, B. T. Akingbemi, J. Kanno, J. A. McLachlan, A. Nadal, C. Sonnenschein, C. S. Watson, R. T. Zoeller and S. M. Belcher, Reprod. Toxicol., 2007, 24, 178–198 CrossRef CAS PubMed.
- S. Flint, T. Markle, S. Thompson and E. Wallace, J. Environ. Manage., 2012, 104, 19–34 CrossRef CAS PubMed.
- S. C. Nagel, F. S. Vom Saal, K. A. Thayer, M. G. Dhar, M. Boechler and W. V. Welshons, Environ. Health Perspect., 1997, 105, 70–76 CrossRef CAS PubMed.
- W. V. Welshons, S. C. Nagel and F. S. vom Saal, Endocrinology, 2006, 147, s56–s69 CrossRef CAS PubMed.
- W. Völkel, T. Colnot, G. A. Csanády, J. G. Filser and W. Dekant, Chem. Res. Toxicol., 2002, 15, 1281–1287 CrossRef.
- J. J. Pritchett, R. K. Kuester and I. G. Sipes, Drug Metab. Dispos., 2002, 30, 1180–1185 CrossRef CAS.
- J. B. Matthews, K. Twomey and T. R. Zacharewski, Chem. Res. Toxicol., 2001, 14, 149–157 CrossRef CAS PubMed.
- S. i. Yoshihara, T. Mizutare, M. Makishima, N. Suzuki, N. Fujimoto, K. Igarashi and S. Ohta, Toxicol. Sci., 2004, 78, 50–59 CrossRef CAS PubMed.
- K. Okuda, T. Fukuuchi, M. Takiguchi and S. i. Yoshihara, Drug Metab. Dispos., 2011, 39, 1696–1703 CrossRef CAS PubMed.
- H. Yamada, I. Furuta, E. H. Kato, S. Kataoka, Y. Usuki, G. Kobashi, F. Sata, R. Kishi and S. Fujimoto, Reprod. Toxicol., 2002, 16, 735–739 CrossRef CAS PubMed.
- A. L. Wozniak, N. N. Bulayeva and C. S. Watson, Environ. Health Perspect., 2005, 113, 431–439 CrossRef CAS PubMed.
- N. N. Bulayeva, A. Wozniak, L. L. Lash and C. S. Watson, Am. J. Physiol.: Endocrinol. Metab., 2005, 288, E388–E397 CrossRef CAS PubMed.
- D. E. Walsh, P. Dockery and C. M. Doolan, Mol. Cell. Endocrinol., 2005, 230, 23–30 CrossRef CAS PubMed.
- E. C. Dodds and W. Lawson, Nature, 1936, 137, 996 CrossRef CAS.
- N. N. Bulayeva, B. Gametchu and C. S. Watson, Steroids, 2004, 69, 181–192 CrossRef CAS PubMed.
- G. Delbès, C. Levacher and R. Habert, Reproduction, 2006, 132, 527–538 CrossRef PubMed.
- G. S. Prins, L. Huang, L. Birch and Y. Pu, Ann. N. Y. Acad. Sci., 2006, 1089, 1–13 CrossRef CAS PubMed.
- J. Cross, Z. Werb and S. Fisher, Science, 1994, 266, 1508–1518 CAS.
- J. A. Katzenellenbogen, J. Med. Chem., 2011, 54, 5271–5282 CrossRef CAS PubMed.
- T. J. Collins, S. W. Gordon-Wylie and C. P. Horwitz, US Pat., 6054580, 2000 Search PubMed.
- B. L. Riggs and L. C. Hartmann, N. Engl. J. Med., 2003, 348, 618–629 CrossRef PubMed.
- M. L. Stefanick, Am. J. Med., 2005, 118, 64–73 CrossRef PubMed.
- B. J. A. Furr and V. C. Jordan, Pharmacol. Ther., 1984, 25, 127–205 CrossRef CAS PubMed.
- M. J. Meyers, J. Sun, K. E. Carlson, G. A. Marriner, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2001, 44, 4230–4251 CrossRef CAS PubMed.
- P. Christoforou, P. F. Christopoulos and M. Koutsilieris, Mol. Med., 2014, 20, 427–434 Search PubMed.
- S. Iwamuro, M. Yamada, M. Kato and S. Kikuyama, Life Sci., 2006, 79(23), 2165–2171 CrossRef CAS PubMed.
- K. Moriyama, T. Tagami, T. Akamizu, T. Usui, M. Saijo, N. Kanamoto, Y. Hataya, A. Shimatsu, H. Kuzuya and K. Nakao, J. Clin. Endocrinol. Metab., 2002, 87, 5185–5190 CrossRef CAS PubMed.
- C. Teng, B. Goodwin, K. Shockley, M. Xia, R. Huang, J. Norris, B. A. Merrick, A. M. Jetten, C. P. Austin and R. R. Tice, Chem.-Biol. Interact., 2013, 203, 556–564 CrossRef CAS PubMed.
- I. Writing Group for the Women's Health Initiative, J. Am. Med. Assoc., 2002, 288, 321–333 CrossRef.
- E. E. Hatch, A. L. Herbst, R. N. Hoover, K. L. Noller, E. Adam, R. H. Kaufman, J. R. Palmer, L. Titus-Ernstoff, M. Hyer, P. Hartge and S. J. Robboy, Cancer, Causes Control, 2001, 12, 837–845 CrossRef CAS PubMed.
- F. S. v. Saal, B. G. Timms, M. M. Montano, P. Palanza, K. A. Thayer, S. C. Nagel, M. D. Dhar, V. K. Ganjam, S. Parmigiani and W. V. Welshons, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2056–2061 CrossRef.
- J. R. Palmer, E. E. Hatch, R. S. Rao, R. H. Kaufman, A. L. Herbst, K. L. Noller, L. Titus-Ernstoff and R. N. Hoover, Am. J. Epidemiol., 2001, 154, 316–321 CrossRef CAS PubMed.
- J. R. Palmer, L. A. Wise, E. E. Hatch, R. Troisi, L. Titus-Ernstoff, W. Strohsnitter, R. Kaufman, A. L. Herbst, K. L. Noller, M. Hyer and R. N. Hoover, Cancer Epidemiol., Biomarkers Prev., 2006, 15, 1509–1514 CAS.
- A. Derfoul, F. J. Lin, E. M. Awumey, T. Kolodzeski, D. J. Hall and R. S. Tuan, J. Cell. Biochem., 2003, 89, 755–770 CrossRef CAS PubMed.
- R. R. Newbold, E. Padilla-Banks and W. N. Jefferson, Endocrinology, 2006, 147, s11–s17 CrossRef CAS PubMed.
- Chemical Bisphenol A (BPA), Classified As Toxic, http://www.env-health.org/news/latest-news/article/bpa-classified-as-toxic, (accessed June 29, 2017, 2017).
- EU finally recognises Bisphenol A as an endocrine disruptor for human health, http://www.env-health.org/resources/press-releases/article/eu-finally-recognises-bisphenol-a, (accessed June 16, 2017).
- Technavio, Global bis phenol-A market 2016–2020, 2016 Search PubMed.
- Y. Q. Huang, C. K. C. Wong, J. S. Zheng, H. Bouwman, R. Barra, B. Wahlström, L. Neretin and M. H. Wong, Environ. Int., 2012, 42, 91–99 CrossRef CAS PubMed.
- J. Oehlmann, M. Oetken and U. Schulte-Oehlmann, Environ. Res., 2008, 108, 140–149 CrossRef CAS PubMed.
- J. A. Brotons, M. F. Olea-Serrano, M. Villalobos, V. Pedraza and N. Olea, Environ. Health Perspect., 1995, 103, 608–612 CrossRef CAS PubMed.
- J. Rajasärkkä, M. Pernica, J. Kuta, J. Lašňák, Z. Šimek and L. Bláha, Water Res., 2016, 103, 133–140 CrossRef PubMed.
- Plastics Europe: Association of Plastics Manufacturers, Applications of Bisphenol A, http://www.bisphenol-a-europe.org/uploads/BPA applications.pdf.
- B. Bae, J. H. Jeong and S. J. Lee, Water Sci. Technol., 2002, 46, 381–387 CAS.
- T. Urase and K. Miyashita, J. Mater. Cycles Waste Manage., 2003, 5, 0077–0082 CrossRef CAS.
- O. Takahashi and S. Oishi, Environ. Health Perspect., 2000, 108, 931–935 CrossRef CAS PubMed.
- I. F. S. A. N. (INFOSAN), Bisphenol A (BPA) - Current state of knowledge and future actions by WHO and FAO, Report INFOSAN Information Note No. 5/2009 - Bisphenol A, World Health Organization, 2009.
- D.-C. Wang, C.-Y. Wang, W.-Y. Chiu and L.-W. Chen, J. Polym. Res., 1997, 4, 9–16 CrossRef CAS.
- Chemical Economics Handbook: Bisphenol A, IHS Markit, 2016 Search PubMed.
- R. P. Pohanish, Sittig's Handbook of Toxic and Hazardous Chemicals and Carcinogens, Elsevier Inc., Oxford, UK, Sixth edn., 2012 Search PubMed.
- T. Yamamoto and A. Yasuhara, Chemosphere, 1999, 38, 2569–2576 CrossRef CAS PubMed.
- H. Asakura, T. Matsuto and N. Tanaka, Waste Manage., 2004, 24, 613–622 CrossRef CAS PubMed.
- Y. Takao, H. C. Lee, S. Kohra and K. Arizono, J. Health Sci., 2002, 48, 331–334 CrossRef CAS.
- A. Goodson, W. Summerfield and I. Cooper, Food Addit. Contam., 2002, 19, 796–802 CrossRef CAS PubMed.
- M. A. Kamrin, Medscape Gen. Med., 2004, 6, 7 Search PubMed.
- F. S. vom Saal, S. Parmigiani, P. L. Palanza, L. G. Everett and R. Ragaini, Environ. Res., 2008, 108, 127–130 CrossRef CAS PubMed.
- M.-K. Yeo and M. Kang, Water Res., 2006, 40, 1906–1914 CrossRef CAS PubMed.
- J. Carlisle, D. Chan, M. Golub, S. Henkel, P. Painter and K. L. Wu, Toxicological Profile for Bisphenol A, Integrated Risk Assessment Branch Office of Environmental Health Hazard Assessment California Environmental Protection Agency, Sacramento, 2009, http://www.opc.ca.gov/webmaster/ftp/project_pages/MarineDebris_OEHHA_ToxProfiles/Bisphenol%20A%20Final.pdf Search PubMed.
- L. N. Vandenberg, R. Hauser, M. Marcus, N. Olea and W. V. Welshons, Reprod. Toxicol., 2007, 24, 139–177 CrossRef CAS PubMed.
- C. Nerín, C. Fernández, C. Domeño and J. Salafranca, J. Agric. Food Chem., 2003, 51, 5647–5653 CrossRef PubMed.
- T. Yamamoto and A. Yasuhara, Bunseki Kagaku, 2000, 49, 443–447 CrossRef CAS.
- J. Sajiki and J. Yonekubo, Chemosphere, 2003, 51, 55–62 CrossRef CAS PubMed.
- T. T. Schug, A. F. Johnson, L. S. Birnbaum, T. Colborn, L. J. G. Jr., D. P. Crews, T. Collins, A. M. Soto, F. S. v. Saal, J. A. McLachlan, C. Sonnenschein and J. J. Heindel, Mol. Endocrinol., 2016, 30, 833–847 CrossRef CAS PubMed.
- E. Diamanti-Kandarakis, J.-P. Bourguignon, L. C. Giudice, R. Hauser, G. S. Prins, A. M. Soto, R. T. Zoeller and A. C. Gore, Endocr. Rev., 2009, 30, 293–342 CrossRef CAS PubMed.
- D. A. Crain, M. Eriksen, T. Iguchi, S. Jobling, H. Laufer, G. A. LeBlanc and L. J. Guillette, Reprod. Toxicol., 2007, 24, 225–239 CrossRef CAS PubMed.
- H. Cabana, J. P. Jones and S. N. Agathos, Eng. Life Sci., 2007, 7, 429–456 CrossRef CAS.
- J. Corrales, L. A. Kristofco, W. B. Steele, B. S. Yates, C. S. Breed, E. S. Williams and B. W. Brooks, Dose-Response, 2015, 13, 1–29 CrossRef CAS PubMed.
- E. A. Miska and A. C. Ferguson-Smith, Science, 2016, 354, 59–63 CrossRef CAS PubMed.
- F. S. vom Saal and W. V. Welshons, Mol. Cell. Endocrinol., 2014, 398, 101–113 CrossRef CAS PubMed.
- N. Vandenberg Laura, A. Hunt Patricia, P. Myers John and S. vom Saal Frederick, Rev. Environ. Health, 2013, 28, 37 CAS.
- S. Eramo, G. Urbani, G. L. Sfasciotti, O. Brugnoletti, M. Bossù and A. Polimeni, Ann. Stomatol., 2010, 1, 14–21 Search PubMed.
- L. N. Vandenberg, I. Chahoud, J. J. Heindel, V. Padmanabhan, F. J. R. Paumgartten and G. Schoenfelder, Environ. Health Perspect., 2010, 118, 1055–1070 CrossRef CAS PubMed.
- T. Geens, D. Aerts, C. Berthot, J.-P. Bourguignon, L. Goeyens, P. Lecomte, G. Maghuin-Rogister, A.-M. Pironnet, L. Pussemier, M.-L. Scippo, J. Van Loco and A. Covaci, Food Chem. Toxicol., 2012, 50, 3725–3740 CrossRef CAS PubMed.
- N. Ben-Jonathan, E. R. Hugo and T. D. Brandebourg, Mol. Cell. Endocrinol., 2009, 304, 49–54 CrossRef CAS PubMed.
- S. S. Chang, Rev. Urol., 2007, 9, S13–S18 Search PubMed.
- K. Ćwiek-Ludwicka, Rocz. Pantsw. Zakl. Hig., 2015, 66, 299–307 Search PubMed.
- J. P. Myers, F. S. vom Saal, B. T. Akingbemi, K. Arizono, S. Belcher, T. Colborn, I. Chahoud, D. A. Crain, F. Farabollini, L. J. Guillette Jr., T. Hassold, S.-m. Ho, P. A. Hunt, T. Iguchi, S. Jobling, J. Kanno, H. Laufer, M. Marcus, J. A. McLachlan, A. Nadal, J. Oehlmann, N. Olea, P. Palanza, S. Parmigiani, B. S. Rubin, G. Schoenfelder, C. Sonnenschein, A. M. Soto, C. E. Talsness, J. A. Taylor, L. N. Vandenberg, J. G. Vandenbergh, S. Vogel, C. S. Watson, W. V. Welshons and R. T. Zoeller, Environ. Health Perspect., 2009, 117, 309–315 CrossRef CAS PubMed.
- Y. Luo, W. Guo, H. H. Ngo, L. D. Nghiem, F. I. Hai, J. Zhang, S. Liang and X. C. Wang, Sci. Total Environ., 2014, 473–474 Search PubMed , 619-641.
- B. L. Loeb, C. M. Thompson, J. Drago, H. Takahara and S. Baig, Ozone: Sci. Eng., 2012, 34, 64–77 CrossRef CAS.
- S. Renou, J. G. Givaudan, S. Poulain, F. Dirassouyan and P. Moulin, J. Hazard. Mater., 2008, 150, 468–493 CrossRef CAS PubMed.
- Y. Peng, Arabian J. Chem., 2017, 10, S2567–S2574 CrossRef CAS.
- C. A. Staples, P. B. Dorn, G. M. Klecka, S. T. O'Block and L. R. Harris, Chemosphere, 1998, 36, 2149–2173 CrossRef CAS PubMed.
- C. A. Richter, L. S. Birnbaum, F. Farabollini, R. R. Newbold, B. S. Rubin, C. E. Talsness, J. G. Vandenbergh, D. R. Walser-Kuntz and F. S. vom Saal, Reprod. Toxicol., 2007, 24, 199–224 CrossRef CAS PubMed.
- H. Inadera, Int. J. Med. Sci., 2015, 12, 926–936 CrossRef PubMed.
- U. V. Solmssen, Chem. Rev., 1945, 37, 481–598 CrossRef CAS PubMed.
- S. M. Rhind, Philos. Trans. R. Soc., B, 2009, 364, 3391–3401 CrossRef CAS PubMed.
- T. M. Edwards and J. P. Myers, Environ. Health Perspect., 2007, 115, 1264–1270 CrossRef CAS PubMed.
- E. Swedenborg, J. Rüegg, S. Mäkelä and I. Pongratz, J. Mol. Endocrinol., 2009, 43, 1–10 CrossRef CAS PubMed.
- B. O. Clarke and S. R. Smith, Environ. Int., 2011, 37, 226–247 CrossRef CAS PubMed.
- A. Kelessidis and A. S. Stasinakis, Waste Manage., 2012, 32, 1186–1195 CrossRef CAS PubMed.
- D. Fytili and A. Zabaniotou, Renewable Sustainable Energy Rev., 2008, 12, 116–140 CrossRef CAS.
- S. Kanuga, J. Am. Dent. Assoc., 2014, 145, 1272–1273 CrossRef PubMed.
- G. Gaustad, E. Olivetti and R. Kirchain, Resour., Conserv. Recycl., 2012, 58, 79–87 CrossRef.
- Aluminum Recycling and Processing for Energy Conservation and Sustainability, ed. J. A. S. Green, ASM International, Materials Park, Ohio, 2007 Search PubMed.
- K. Tuppurainen, I. Halonen, P. Ruokojärvi, J. Tarhanen and J. Ruuskanen, Chemosphere, 1998, 36, 1493–1511 CrossRef CAS.
- R. A. Keri, S.-M. Ho, P. A. Hunt, K. E. Knudsen, A. M. Soto and G. S. Prins, Reprod. Toxicol., 2007, 24, 240–252 CrossRef CAS PubMed.
- D. D. Seachrist, K. W. Bonk, S.-M. Ho, G. S. Prins, A. M. Soto and R. A. Keri, Reprod. Toxicol., 2016, 59, 167–182 CrossRef CAS PubMed.
- J.-H. Kang, Y. Katayama and F. Kondo, Toxicology, 2006, 217, 81–90 CrossRef CAS PubMed.
- J.-H. Kang, F. Kondo and Y. Katayama, Toxicology, 2006, 226, 79–89 CrossRef CAS PubMed.
- J.-H. Kang, D. Aasi and Y. Katayama, Crit. Rev. Toxicol., 2007, 37, 607–625 CrossRef CAS PubMed.
- M. Staniszewska, L. Falkowska, P. Grabowski, J. Kwaśniak, S. Mudrak-Cegiołka, A. R. Reindl, A. Sokołowski, E. Szumiło and A. Zgrundo, Arch. Environ. Contam. Toxicol., 2014, 67, 335–347 CrossRef CAS PubMed.
- C. Basheer, H. K. Lee and K. S. Tan, Mar. Pollut. Bull., 2004, 48, 1145–1167 CrossRef PubMed.
- A. Belfroid, M. van Velzen, B. van der Horst and D. Vethaak, Chemosphere, 2002, 49 CrossRef CAS.
- The New Plastics Economy: Rethinking the Future of Plastics, World Economic Forum, Ellen MacArthur Foundation and McKinsey & Company, 2016, http://www.ellenmacarthurfoundation.org/publications Search PubMed.
- J. Sajiki and J. Yonekubo, Chemosphere, 2004, 55, 861–867 CrossRef CAS PubMed.
- J.-H. Kang and F. Kondo, Chemosphere, 2005, 60, 1288–1292 CrossRef CAS PubMed.
- A. B. Hansen and P. Lassen, Screening of phenolic substances in the Nordic environments, Nordic Council of Ministers, Tema Nord, 2008 Search PubMed.
- G. Funakoshi and S. Kasuya, Chemosphere, 2009, 75, 491–497 CrossRef CAS PubMed.
- M. Ike, M. Y. Chen, E. Danzl, K. Sei and M. Fujita, Water Sci. Technol., 2006, 53, 153–159 CrossRef CAS PubMed.
- S. Schenker, M. Scheringer and K. Hungerbühler, Environ. Sci. Technol., 2014, 48, 5017–5024 CrossRef CAS PubMed.
- F. S. vom Saal, B. T. Akingbemi, S. M. Belcher, L. S. Birnbaum, D. A. Crain, M. Eriksen, F. Farabollini, L. J. Guillette, R. Hauser, J. J. Heindel, S.-M. Ho, P. A. Hunt, T. Iguchi, S. Jobling, J. Kanno, R. A. Keri, K. E. Knudsen, H. Laufer, G. A. LeBlanc, M. Marcus, J. A. McLachlan, J. P. Myers, A. Nadal, R. R. Newbold, N. Olea, G. S. Prins, C. A. Richter, B. S. Rubin, C. Sonnenschein, A. M. Soto, C. E. Talsness, J. G. Vandenbergh, L. N. Vandenberg, D. R. Walser-Kuntz, C. S. Watson, W. V. Welshons, Y. Wetherill and R. T. Zoeller, Reprod. Toxicol., 2007, 24, 131–138 CrossRef CAS PubMed.
- NHANES 2003–2004 Laboratory Data: Environmental Phenols, Centers for Disease Control and Prevention, 2003–2004, https://wwwn.cdc.gov/Nchs/Nhanes/2003-2004/L24EPH_C.htm Search PubMed.
- R. Kuruto-Niwa, Y. Tateoka, Y. Usuki and R. Nozawa, Chemosphere, 2007, 66, 1160–1164 CrossRef CAS PubMed.
- X. Ye, Z. Kuklenyik, L. L. Needham and A. M. Calafat, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 831, 110–115 CrossRef CAS PubMed.
- Y. Tateoka, J. Hum. Lact., 2014, 31, 474–478 CrossRef PubMed.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS PubMed.
- C. C. Montagner and W. F. Jardim, J. Braz. Chem. Soc., 2011, 22, 1452–1462 CrossRef CAS.
- C. Tang, J. Chen and Y. Zhang, Fresenius Environ. Bull., 2012, 21, 3911–3919 CAS.
- D. d. A. Azevedo, S. Lacorte, P. Viana and D. Barceló, J. Braz. Chem. Soc., 2001, 12, 532–537 CrossRef CAS.
- A. Colin, C. Bach, C. Rosin, J.-F. Munoz and X. Dauchy, Arch. Environ. Contam. Toxicol., 2014, 66, 86–99 CrossRef CAS PubMed.
- H. M. Kuch and K. Ballschmiter, Environ. Sci. Technol., 2001, 35 CrossRef CAS.
- V. A. Santhi, N. Sakai, E. D. Ahmad and A. M. Mustafa, Sci. Total Environ., 2012, 427, 332–338 CrossRef PubMed.
- N. Casajuana and S. Lacorte, Chromatographia, 2003, 57, 649–655 CAS.
- T. T. Makinwa and P. O. Uadia, World Environ., 2015, 5, 135–139 Search PubMed.
- J.-Y. Hu, T. Aizawa and S. Ookubo, Environ. Sci. Technol., 2002, 36, 1980–1987 CrossRef CAS PubMed.
- T. Yamamoto and A. Yasuhara, Chemosphere, 2002, 46, 1215–1223 CrossRef CAS PubMed.
- J.-H. Kang, K. Kito and F. Kondo, J. Food Prot., 2003, 66, 1444–1447 CrossRef CAS PubMed.
- A. Davis and J. H. Golden, J. Chromatogr., A, 1967, 26, 254–255 CrossRef CAS.
- S. R. Howe and L. Borodinsky, Food Addit. Contam., 1998, 15, 370–375 CrossRef CAS PubMed.
- E. M. Munguia-Lopez and H. Soto-Valdez, J. Agric. Food Chem., 2001, 49, 3666–3671 CrossRef CAS PubMed.
- C. Liao and K. Kannan, Environ. Sci. Technol., 2011, 45, 9372–9379 CrossRef CAS PubMed.
- J. López-Cervantes and P. Paseiro-Losada, Food Addit. Contam., 2003, 20, 596–606 CrossRef PubMed.
- N. Olea, R. Pulgar, P. Pérez, F. Olea-Serrano, A. Rivas, A. Novillo-Fertrell, V. Pedraza, A. M. Soto and C. Sonnenschein, Environ. Health Perspect., 1996, 104, 298–305 CrossRef CAS PubMed.
- D. Arenholt-Bindslev, V. Breinholt, A. Preiss and G. Schmalz, Clin. Oral Investig., 1999, 3, 120–125 CrossRef CAS PubMed.
- J. B. Lewis, F. A. Rueggeberg, C. A. Lapp, J. W. Ergle and G. S. Schuster, Clin. Oral Investig., 1999, 3, 107–113 CrossRef CAS PubMed.
- M. Noda, H. Komatsu and H. Sano, J. Biomed. Mater. Res., 1999, 47, 374–378 CrossRef CAS PubMed.
- G. Schmalz, A. Preiss and D. Arenholt-Bindslev, Clin. Oral Investig., 1999, 3, 114–119 CrossRef CAS PubMed.
- E. Y. K. Fung, N. O. Ewoldsen, H. A. J. St. Germain, D. B. Marx, C.-L. Miaw, C. Siew, H.-N. Chou, S. E. Gruninger and D. M. Meyer, J. Am. Dent. Assoc., 2000, 131, 51–58 CrossRef CAS PubMed.
- A. Zafra, M. del Olmo, R. Pulgar, A. Navalón and J. L. Vílchez, Chromatographia, 2002, 56, 213–218 CAS.
- A. S. Al-Hiyasat, H. Darmani and A. M. Elbetieha, Eur. J. Oral Sci., 2004, 112, 267–272 CrossRef CAS PubMed.
- N. Sasaki, K. Okuda, T. Kato, H. Kakishima, H. Okuma, K. Abe, H. Tachino, K. Tuchida and K. Kubono, J. Mater. Sci.: Mater. Med., 2005, 16, 297–300 CrossRef CAS PubMed.
- R. Joskow, D. B. Barr, J. R. Barr, A. M. Calafat, L. L. Needham and C. Rubin, J. Am. Dent. Assoc., 2006, 137, 353–362 CrossRef CAS PubMed.
- J. L. Carwile, H. T. Luu, L. S. Bassett, D. A. Driscoll, C. Yuan, J. Y. Chang, X. Ye, A. M. Calafat and K. B. Michels, Environ. Health Perspect., 2009, 117, 1368–1372 CrossRef CAS PubMed.
- E. M. Munguia-Lopez, E. Peralta, A. Gonzalez-Leon, C. Vargas-Requena and H. Soto-Valdez, J. Agric. Food Chem., 2002, 50, 7299–7302 CrossRef CAS PubMed.
- R. Braunrath, D. Podlipna, S. Padlesak and M. Cichna-Markl, J. Agric. Food Chem., 2005, 53, 8911–8917 CrossRef CAS PubMed.
- L. Grumetto, D. Montesano, S. Seccia, S. Albrizio and F. Barbato, J. Agric. Food Chem., 2008, 56, 10633–10637 CrossRef CAS PubMed.
- J. Yonekubo, K. Hayakawa and J. Sajiki, J. Agric. Food Chem., 2008, 56, 2041–2047 CrossRef CAS PubMed.
- J. E. Biles, T. P. McNeal, T. H. Begley and H. C. Hollifield, J. Agric. Food Chem., 1997, 45, 3541–3544 CrossRef CAS.
- G. D. Bittner, C. Z. Yang and M. A. Stoner, Environ. Health, 2014, 13, 41–41 CrossRef PubMed.
- Y. Sun, M. Wada, O. Al-Dirbashi, N. Kuroda, H. Nakazawa and K. Nakashima, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 749, 49–56 CrossRef CAS.
- A. D'Antuono, V. C. Dall'Orto, A. L. Balbo, S. Sobral and I. Rezzano, J. Agric. Food Chem., 2001, 49, 1098–1101 CrossRef.
- C. Brede, P. Fjeldal, I. Skjevrak and H. Herikstad, Food Addit. Contam., 2003, 20, 684–689 CrossRef CAS PubMed.
- Y. Takao, H. C. Lee, Y. Ishibashi, S. Kohra, N. Tominaga and K. Arizono, J. Health Sci., 1999, 45, P39–P39 CrossRef.
- C. Cox, Kicking the Can? Major Retailers Still Selling Canned Food with Toxic BPA, Center for Environmental Health, Oakland, CA, 2017 Search PubMed.
- J.-H. Kang and F. Kondo, Food Addit. Contam., 2002, 19, 886–890 CrossRef CAS PubMed.
- X.-L. Cao, J. Corriveau and S. Popovic, J. Agric. Food Chem., 2009, 57, 1307–1311 CrossRef CAS PubMed.
- A. K. Sakhi, I. T. L. Lillegaard, S. Voorspoels, M. H. Carlsen, E. B. Løken, A. L. Brantsæter, M. Haugen, H. M. Meltzer and C. Thomsen, Environ. Int., 2014, 73, 259–269 CrossRef CAS PubMed.
- J. E. Biles, T. P. McNeal and T. H. Begley, J. Agric. Food Chem., 1997, 45, 4697–4700 CrossRef CAS.
- L. K. Ackerman, G. O. Noonan, W. M. Heiserman, J. A. Roach, W. Limm and T. H. Begley, J. Agric. Food Chem., 2010, 58, 2307–2313 CrossRef CAS PubMed.
- X.-L. Cao, G. Dufresne, S. Belisle, G. Clement, M. Falicki, F. Beraldin and A. Rulibikiye, J. Agric. Food Chem., 2008, 56, 7919–7924 CrossRef CAS PubMed.
- H.-W. Kuo and W.-H. Ding, J. Chromatogr., A, 2004, 1027, 67–74 CrossRef CAS PubMed.
- B. M. Thomson and P. R. Grounds, Food Addit. Contam., 2005, 22, 65–72 CrossRef CAS PubMed.
- N. Rastkari, M. Yunesian and R. Ahmadkhaniha, Iran. J. Environ. Health. Sci. Eng., 2011, 8, 95–100 CAS.
- N. C. Maragou, E. N. Lampi, N. S. Thomaidis and M. A. Koupparis, J. Chromatogr., A, 2006, 1129, 165–173 CrossRef CAS PubMed.
- T. Yoshida, M. Horie, Y. Hoshino, H. Nakazawa, M. Horie and H. Nakazawa, Food Addit. Contam., 2001, 18, 69–75 CrossRef CAS PubMed.
- B. S. Shin, C. H. Kim, Y. S. Jun, D. H. Kim, B. M. Lee, C. H. Yoon, E. H. Park, K. C. Lee, S.-Y. Han, K. L. Park, H. S. Kim and S. D. Yoo, J. Toxicol. Environ. Health, Part A, 2004, 67, 1971–1985 CrossRef CAS PubMed.
- D. Zalko, C. Jacques, H. Duplan, S. Bruel and E. Perdu, Chemosphere, 2011, 82, 424–430 CrossRef CAS PubMed.
- S. Biedermann, P. Tschudin and K. Grob, Anal. Bioanal. Chem., 2010, 398, 571–576 CrossRef CAS PubMed.
- S.-Y. Lu, W.-J. Chang, S. O. Sojinu and H.-G. Ni, Chemosphere, 2013, 92, 1190–1194 CrossRef CAS PubMed.
- A. M. Hormann, F. S. vom Saal, S. C. Nagel, R. W. Stahlhut, C. L. Moyer, M. R. Ellersieck, W. V. Welshons, P.-L. Toutain and J. A. Taylor, PLoS One, 2014, 9, e110509 Search PubMed.
- T. Mendum, E. Stoler, H. VanBenschoten and J. C. Warner, Green Chem. Lett. Rev., 2011, 4, 81–86 CrossRef CAS.
- T. Geens, L. Goeyens, K. Kannan, H. Neels and A. Covaci, Sci. Total Environ., 2012, 435–436, 30–33 CrossRef CAS PubMed.
- M. R. Bernier and L. N. Vandenberg, PLoS One, 2017, 12, e0178449 Search PubMed.
- N. K. Wilson, J. C. Chuang, C. Lyu, R. Menton and M. K. Morgan, J. Exposure Anal. Environ. Epidemiol.., 2003, 13, 187–202 CrossRef CAS PubMed.
- N. K. Wilson, J. C. Chuang, M. K. Morgan, R. A. Lordo and L. S. Sheldon, Environ. Res., 2007, 103, 9–20 CrossRef CAS PubMed.
- N. K. Wilson, J. C. Chuang and C. Lyu, J. Exposure Anal. Environ. Epidemiol., 2001, 11, 449–458 CrossRef CAS PubMed.
- P. Fu and K. Kawamura, Environ. Pollut., 2010, 158, 3138–3143 CrossRef CAS PubMed.
- C. Liao, F. Liu, Y. Guo, H.-B. Moon, H. Nakata, Q. Wu and K. Kannan, Environ. Sci. Technol., 2012, 46, 9138–9145 CrossRef CAS PubMed.
- R. A. Rudel, J. G. Brody, J. D. Spengler, J. Vallarino, P. W. Geno, G. Sun and A. Yau, J. Air Waste Manage. Assoc., 2001, 51, 499–513 CAS.
- R. A. Rudel, D. E. Camann, J. D. Spengler, L. R. Korn and J. G. Brody, Environ. Sci. Technol., 2003, 37, 4543–4553 CrossRef CAS PubMed.
- T. Oka and Y. Oshige, Anal. Sci., 2000, 16, 1071–1076 CrossRef.
- A. Vivacqua, A. G. Recchia, G. Fasanella, S. Gabriele, A. Carpino, V. Rago, M. L. Di Gioia, A. Leggio, D. Bonofiglio, A. Liguori and M. Maggiolini, Endocrine, 2003, 22, 275–284 CrossRef CAS PubMed.
- K. Inoue, S. Murayama, K. Takeba, Y. Yoshimura and H. Nakazawa, J. Food Compos. Anal., 2003, 16, 497–506 CrossRef CAS.
- X.-L. Cao, J. Corriveau, S. Popovic, G. Clement, F. Beraldin and G. Dufresne, J. Agric. Food Chem., 2009, 57, 5345–5351 CrossRef CAS PubMed.
- A. G. Asimakopoulos, M. Elangovan and K. Kannan, Environ. Sci. Technol., 2016, 50, 13539–13547 CrossRef CAS PubMed.
- J. Sajiki, R. Yanagibori and Y. Kobayashi, Nippon Eiseigaku Zasshi, 2010, 65, 467–470 CrossRef CAS PubMed.
- P. Viñas, I. López-García, N. Campillo, R. E. Rivas and M. Hernández-Córdoba, Anal. Bioanal. Chem., 2012, 404, 671–678 CrossRef PubMed.
- E. S. Kwak, A. Just, R. Whyatt and R. L. Miller, Open Allergy J., 2009, 2, 45–50 CrossRef CAS PubMed.
- R. E. Dodson, M. Nishioka, L. J. Standley, L. J. Perovich, J. G. Brody and R. A. Rudel, Environ. Health Perspect., 2012, 120, 935–943 CrossRef CAS PubMed.
- A. Myridakis, E. Fthenou, E. Balaska, M. Vakinti, M. Kogevinas and E. G. Stephanou, Environ. Int., 2015, 83, 1–10 CrossRef CAS PubMed.
- C. Liao and K. Kannan, Environ. Sci. Technol., 2011, 45, 6761–6768 CrossRef CAS PubMed.
- W. J. Jackson, Jr. and W. R. Darnell, US Pat., 4507256, 1983 Search PubMed.
- M. Gehring, L. Tennhardt, d. Vogel, D. Weltin and B. Bilitewski, in Waste Management and the Environment II. WIT Transactions on Ecology and the Environment, ed. C. A. Brebbia, S. Kungulos, V. Popov and H. Itoh, WIT Press, Southampton, Boston, 2004, vol. 78, ch. 294, pp. 294–300 Search PubMed.
- H. Fukazawa, K. Hoshino, T. Shiozawa, H. Matsushita and Y. Terao, Chemosphere, 2001, 44, 973–979 CrossRef CAS PubMed.
- A. Ozaki, Y. Yamaguchi, T. Fujita, K. Kuroda and G. Endo, Food Chem. Toxicol., 2004, 42, 1323–1337 CrossRef CAS PubMed.
- A. M. Vinggaard, W. Körner, K. H. Lund, U. Bolz and J. H. Petersen, Chem. Res. Toxicol., 2000, 13, 1214–1222 CrossRef CAS PubMed.
- M. J. Lopez-Espinosa, A. Granada, P. Araque, J. M. Molina-Molina, M. C. Puertollano, A. Rivas, M. Fernández, I. Cerrillo, M. F. Olea-Serrano, C. López and N. Olea, Food Addit. Contam., 2007, 24, 95–102 CrossRef CAS PubMed.
- K. Peltonen and J. Pukkila, J. Chromatogr., A, 1988, 439, 375–380 CrossRef CAS PubMed.
- K. Peltonen, J. Anal. Appl. Pyrolysis, 1986, 10, 51–57 CrossRef CAS.
- T. Zhang, J. Xue, C.-z. Gao, R.-l. Qiu, Y.-x. Li, X. Li, M.-z. Huang and K. Kannan, Environ. Sci. Technol., 2016, 50, 4045–4053 CrossRef CAS PubMed.
- X. Bi, B. R. T. Simoneit, Z. Wang, X. Wang, G. Sheng and J. Fu, Atmos. Environ., 2010, 44, 4440–4445 CrossRef CAS.
- T. Yamamoto, A. Yasuhara, H. Shiraishi and O. Nakasugi, Chemosphere, 2001, 42, 415–418 CrossRef CAS PubMed.
- H. Yin, Y. Zhou, J. Xu, S. Ai, L. Cui and L. Zhu, Anal. Chim. Acta, 2010, 659, 144–150 CrossRef CAS PubMed.
- H. Yin, L. Cui, S. Ai, H. Fan and L. Zhu, Electrochim. Acta, 2010, 55, 603–610 CrossRef CAS.
- Tox Town: Polyvinyl Chloride (PVC), U.S. National Library of Medicine Division of Specialized Information Services, Bethesda, 2017, https://toxtown.nlm.nih.gov/text_version/chemicals.php?id=84 Search PubMed.
- H. P. H. Arp, N. A. O. Morin, S. E. Hale, G. Okkenhaug, K. Breivik and M. Sparrevik, Waste Manage., 2017, 60, 775–785 CrossRef CAS PubMed.
- S. Lee, C. Liao, G.-J. Song, K. Ra, K. Kannan and H.-B. Moon, Chemosphere, 2015, 119, 1000–1006 CrossRef CAS PubMed.
- Screening Assessment for the Challenge: Phenol, 4,4′-(1-methylethylidene)bis-(Bisphenol A), Environment Canada and Health Canada, Ottowa, 2008, http://www.ec.gc.ca/ese-ees/default.asp?lang=En&n=3C756383-1 Search PubMed.
- Consultation Document: Phenol, 4,4′-(1-methylethylidene)bis-(Bisphenol A), Environment Canada, Gatineau, Quebec, 2009, https://www.ec.gc.ca/lcpe-cepa/79A93C66-1EE0-4F72-89C1-114DFD7B7292/pba-eng.pdf Search PubMed.
- M. Wright-Walters, C. Volz, E. Talbott and D. Davis, Sci. Total Environ., 2011, 409, 676–685 CrossRef CAS PubMed.
- USEPA, Advancing sustainable Materials Management: Facts and Figures, United States Environmental Protection Agency Office of Land and Emergency Management, 2014, https://www.epa.gov/sites/production/files/2016-11/documents/<?pdb_no 2014?>2014<?pdb END?>_smmfactsheet_508.pdf Search PubMed.
- B. Bilitewski, G. Härdtle and K. Marek, Waste Management, Springer, New York, 1997 Search PubMed.
- D. Vogel, B. Bilitewski and L. D. Nghiem, Int. J. Environ. Waste Manage., 2007, 1, 338–350 CrossRef CAS.
- J. Schwarzbauer, S. Heim, S. Brinker and R. Littke, Water Res., 2002, 36, 2275–2287 CrossRef CAS PubMed.
- T. Wintgens, M. Gallenkemper and T. Melin, Water Sci. Technol., 2003, 48, 127–134 CAS.
- K. Yamada, T. Urase, T. Matsuo and N. Suzuki, Mizu Kankyo Gakkaishi, 1999, 22, 40–45 CrossRef CAS.
- N. Morin, H. P. H. Arp and S. E. Hale, Environ. Sci. Technol., 2015, 49, 7675–7683 CrossRef CAS PubMed.
- N. Paxéus, Water Sci. Technol., 2000, 42, 323–333 Search PubMed.
- A. Rigol, A. Latorre, S. Lacorte and D. Barceló, Environ. Toxicol. Chem., 2004, 23, 339–347 CrossRef CAS PubMed.
- M. Fürhacker, S. Scharf and H. Weber, Chemosphere, 2000, 41, 751–756 CrossRef.
- H. Fukazawa, M. Watanabe, F. Shiraishi, H. Shiraishi, T. Shiozawa, H. Matsushita and Y. Terao, J. Health Sci., 2002, 48, 242–249 CrossRef CAS.
- J. Jackson and R. Sutton, Sci. Total Environ., 2008, 405, 153–160 CrossRef CAS PubMed.
- Bisphenol A Alternatives in Thermal Paper, U.S. Environmental Protection Agency, 2015, https://www.epa.gov/sites/production/files/2015-09/documents/bpa_ch5.pdf Search PubMed.
- H.-B. Lee, T. E. Peart, J. Chan and G. Gris, Water Qual. Res. J. Can., 2004, 39, 57–63 CAS.
- C. Höhne and W. Püttmann, Environ. Sci. Pollut. Res., 2008, 15, 405–416 CrossRef PubMed.
- V. G. Samaras, A. S. Stasinakis, D. Mamais, N. S. Thomaidis and T. D. Lekkas, J. Hazard. Mater., 2013, 244–245, 259–267 CrossRef CAS PubMed.
- H. Zhou, X. Huang, X. Wang, X. Zhi, C. Yang, X. Wen, Q. Wang, H. Tsuno and H. Tanaka, Environ. Monit. Assess., 2010, 161, 107–121 CrossRef CAS PubMed.
- J. Zhao, Y. Li, C. Zhang, Q. Zeng and Q. Zhou, J. Hazard. Mater., 2008, 155, 305–311 CrossRef CAS PubMed.
- Z. Zhang, M. Le Velly, S. M. Rhind, C. E. Kyle, R. L. Hough, E. I. Duff and C. McKenzie, Sci. Total Environ., 2015, 515–516, 1–11 CAS.
- K. A. Langdon, M. S. J. Warne, R. J. Smernik, A. Shareef and R. S. Kookana, Sci. Total Environ., 2011, 409, 1075–1081 CrossRef CAS PubMed.
- T. A. Ternes, H. Andersen, D. Gilberg and M. Bonerz, Anal. Chem., 2002, 74, 3498–3504 CrossRef CAS.
- O. Braga, G. A. Smythe, A. I. Schäfer and A. J. Feitz, Environ. Sci. Technol., 2005, 39, 3351–3358 CrossRef CAS PubMed.
- S. Chu and C. D. Metcalfe, J. Chromatogr., A, 2007, 1164, 212–218 CrossRef CAS PubMed.
- S. Song, M. Song, L. Zeng, T. Wang, R. Liu, T. Ruan and G. Jiang, Environ. Pollut., 2014, 186, 14–19 CrossRef CAS PubMed.
- K. A. Hicks, M. L. M. Fuzzen, E. K. McCann, M. J. Arlos, L. M. Bragg, S. Kleywegt, G. R. Tetreault, M. E. McMaster and M. R. Servos, Environ. Sci. Technol., 2017, 51, 1811–1819 CrossRef CAS PubMed.
- H. Fromme, T. Küchler, T. Otto, K. Pilz, J. Müller and A. Wenzel, Water Res., 2002, 36, 1429–1438 CrossRef CAS PubMed.
- C. A. Kinney, E. T. Furlong, D. W. Kolpin, M. R. Burkhardt, S. D. Zaugg, S. L. Werner, J. P. Bossio and M. J. Benotti, Environ. Sci. Technol., 2008, 42, 1863–1870 CrossRef CAS PubMed.
- E. Z. Harrison, S. R. Oakes, M. Hysell and A. Hay, Sci. Total Environ., 2006, 367, 481–497 CrossRef CAS PubMed.
- D. P. Mohapatra, S. K. Brar, R. D. Tyagi and R. Y. Surampalli, J. Xenobiot., 2011, 1, 3 CrossRef.
- J. Hall, in Workshop on Problems Around Sludge: Proceedings, ed. H. Langenkamp and L. Mamo, European Commission Joint Research Centre, 1999, ch. Session 3: Technology and innovative options related to sludge management, pp. 155–172 Search PubMed.
- NeBRA , A National Biosolids Regulation, Quality, End Use and Disposal Survey (Final Report), North East Biosolids and Residuals Association (NEBRA), Tamworth, NH, 2007.
- S. M. Rhind, C. E. Kyle, H. Ruffie, E. Calmettes, M. Osprey, Z. L. Zhang, D. Hamilton and C. McKenzie, Environ. Pollut., 2013, 181, 262–270 CrossRef CAS PubMed.
- R. E. Alcock, S. P. McGrath and K. C. Jones, Environ. Toxicol. Chem., 1995, 14, 553–560 CrossRef CAS.
- E. Eljarrat, G. Marsh, A. Labandeira and D. Barceló, Chemosphere, 2008, 71, 1079–1086 CrossRef CAS PubMed.
- M.-J. Wang, S. P. McGrath and K. C. Jones, Environ. Sci. Technol., 1995, 29, 356–362 CrossRef CAS PubMed.
- K. A. Langdon, M. S. J. Warne, R. J. Smernik, A. Shareef and R. S. Kookana, Chemosphere, 2012, 86, 1050–1058 CrossRef CAS PubMed.
- K. A. Langdon, M. S. Warne, R. J. Smernik, A. Shareef and R. S. Kookana, Sci. Total Environ., 2013, 447, 56–63 CrossRef CAS PubMed.
- C. Paul, S. M. Rhind, C. E. Kyle, H. Scott, C. McKinnell and R. M. Sharpe, Environ. Health Perspect., 2005, 113, 1580–1587 CrossRef PubMed.
- H. W. Erhard and S. M. Rhind, Sci. Total Environ., 2004, 332, 101–108 CrossRef CAS PubMed.
- P. A. Fowler, N. J. Dorà, H. McFerran, M. R. Amezaga, D. W. Miller, R. G. Lea, P. Cash, A. S. McNeilly, N. P. Evans, C. Cotinot, R. M. Sharpe and S. M. Rhind, Mol. Hum. Reprod., 2008, 14, 269–280 CrossRef CAS PubMed.
- P. M. Lind, M. Gustafsson, S. A. B. Hermsen, S. Larsson, C. E. Kyle, J. Örberg and S. M. Rhind, Sci. Total Environ., 2009, 407, 2200–2208 CrossRef CAS PubMed.
- M. Bellingham, P. A. Fowler, M. R. Amezaga, S. M. Rhind, C. Cotinot, B. Mandon-Pepin, R. M. Sharpe and N. P. Evans, Environ. Health Perspect., 2009, 117, 1556–1562 CrossRef CAS PubMed.
- M. Bellingham, P. A. Fowler, M. R. Amezaga, C. M. Whitelaw, S. M. Rhind, C. Cotinot, B. Mandon-Pepin, R. M. Sharpe and N. P. Evans, J. Neuroendocrinol., 2010, 22, 527–533 CrossRef CAS PubMed.
- P. M. Lind, D. Öberg, S. Larsson, C. E. Kyle, J. Örberg and S. M. Rhind, Sci. Total Environ., 2010, 408, 2340–2346 CrossRef CAS PubMed.
- M. Bellingham, M. R. Amezaga, B. Mandon-Pepin, C. J. B. Speers, C. E. Kyle, N. P. Evans, R. M. Sharpe, C. Cotinot, S. M. Rhind and P. A. Fowler, Mol. Cell. Endocrinol., 2013, 376, 156–172 CrossRef CAS PubMed.
- R. G. Lea, M. R. Amezaga, B. Loup, B. Mandon-Pépin, A. Stefansdottir, P. Filis, C. Kyle, Z. Zhang, C. Allen, L. Purdie, L. Jouneau, C. Cotinot, S. M. Rhind, K. D. Sinclair and P. A. Fowler, Sci. Rep., 2016, 6, 22279 CrossRef CAS PubMed.
- S. Hombach-Klonisch, A. Danescu, F. Begum, M. R. Amezaga, S. M. Rhind, R. M. Sharpe, N. P. Evans, M. Bellingham, C. Cotinot, B. Mandon-Pepin, P. A. Fowler and T. Klonisch, Mol. Cell. Endocrinol., 2013, 367, 98–108 CrossRef CAS PubMed.
- R. L. Hough, P. Booth, L. M. Avery, S. Rhind, C. Crews, J. Bacon, C. D. Campbell and D. Tompkins, Waste Manage., 2012, 32, 117–130 CrossRef CAS PubMed.
- L. K. Dodgen, J. Li, D. Parker and J. J. Gan, Environ. Pollut., 2013, 182, 150–156 CrossRef CAS PubMed.
- S. Sidhu, B. Gullett, R. Striebich, J. Klosterman, J. Contreras and M. DeVito, Atmos. Environ., 2005, 39, 801–811 CrossRef CAS.
- M. Šala, Y. Kitahara, S. Takahashi and T. Fujii, Chemosphere, 2010, 78, 42–45 CrossRef PubMed.
- Y. Kitahara, S. Takahashi, M. Tsukagoshi and T. Fujii, Chemosphere, 2010, 80, 1281–1284 CrossRef CAS PubMed.
- L.-H. Lee, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2859–2873 CrossRef.
- S. Tsuge, T. Okumoto, Y. Sugimura and T. Takeuchi, J. Chromatogr. Sci., 1969, 7, 253–256 CrossRef CAS.
- Y. Ito, H. Ogasawara, Y. Ishida, H. Ohtani and S. Tsuge, Polym. J., 1996, 28, 1090–1095 CrossRef CAS.
- C. Puglisi, L. Sturiale and G. Montaudo, Macromolecules, 1999, 32, 2194–2203 CrossRef CAS.
- M. Blazsó, Polymer, 1991, 32, 590–596 CrossRef.
- H. Nakagawa, S. Wakatsuka, S. Tsuge and T. Koyama, Polym. J., 1988, 20, 9–16 CrossRef CAS.
- N. Grassie, M. I. Guy and N. H. Tennent, Polym. Degrad. Stab., 1986, 14, 125–137 CrossRef CAS.
- H. Nakagawa, S. Tsuge and T. Koyama, J. Anal. Appl. Pyrolysis, 1987, 12, 97–113 CrossRef CAS.
- R. E. Miller, US Pat., 4147531, 1979 Search PubMed.
- N. R. C. U. C. o. H. E. o. W. Incineration, in Waste Incineration & Public Health, National Academies Press, Washington, DC, 2000, vol. 3 Search PubMed.
- T. A. Association , Aluminum: The Element of Sustainability: A North American Aluminum Industry Sustainability Report, 2011.
- J. P. Aittola, J. Paasivirta and A. Vattulainen, Chemosphere, 1993, 27, 65–72 CrossRef CAS.
- European Union Risk Assessment Report, 4,4′-Isopropylidenediphenol (Bisphenol-A), European Commission-Joint Research Centre, Institute for Health and Consumer Protection, Luxembourg, 2008.
- L. B. Barber, G. K. Brown and S. D. Zaugg, in Analysis of Environmental Endocrine Disruptors, American Chemical Society, 1999, vol. 747, ch. 7, pp. 97–123 Search PubMed.
- USEPA, Wastewater Technology Fact Sheet: Chlorine Disinfection, United States Environmental Protection Agency Office of Water, 1999, https://www3.epa.gov/npdes/pubs/chlo.pdf Search PubMed.
- R. Bolger, T. E. Wiese, K. Ervin, S. Nestich and W. Checovich, Environ. Health Perspect., 1998, 106, 551–557 CrossRef CAS PubMed.
- R. Kuruto-Niwa, Y. Terao and R. Nozawa, Environ. Toxicol. Pharmacol., 2002, 12, 27–35 CrossRef CAS PubMed.
- Y. Yang, L. Lu, J. Zhang, Y. Yang, Y. Wu and B. Shao, J. Chromatogr., A, 2014, 1328, 26–34 CrossRef CAS PubMed.
- S. Chu, G. D. Haffner and R. J. Letcher, J. Chromatogr., A, 2005, 1097, 25–32 CrossRef CAS PubMed.
- G. Mascolo, V. Locaputo and G. Mininni, J. Chromatogr., A, 2010, 1217, 4601–4611 CrossRef CAS PubMed.
- Micropollutants in municipal wastewater: Processes for advanced removal in wastewater treatment plants, The Swiss Federal Office for the Environment, Bern, 2012, https://www.bafu.admin.ch/bafu/en/home/topics/water/water--publications/publications-water/micropollutants-municipal-wastewater-summary.html Search PubMed.
- J. Margot, C. Kienle, A. Magnet, M. Weil, L. Rossi, L. F. de Alencastro, C. Abegglen, D. Thonney, N. Chèvre, M. Schärer and D. A. Barry, Sci. Total Environ., 2013, 461–462, 480–498 CrossRef CAS PubMed.
- Y. Yoon, P. Westerhoff, S. A. Snyder and M. Esparza, Water Res., 2003, 37, 3530–3537 CrossRef CAS PubMed.
- I. Gültekin, V. Mavrov and N. H. Ince, J. Adv. Oxid. Technol., 2009, 12, 242–248 Search PubMed.
- S. Baig, G. Hansmann and B. Paolini, Water Sci. Technol., 2008, 58, 451–458 CrossRef CAS PubMed.
- M. Deborde, S. Rabouan, P. Mazellier, J.-P. Duguet and B. Legube, Water Res., 2008, 42, 4299–4308 CrossRef CAS PubMed.
- T. Kamiya, T. Yamauchi, J. Hirotsuji and M. Fujita, Ozone: Sci. Eng., 2005, 27, 389–395 CrossRef CAS.
- M. Deborde, S. Rabouan, J.-P. Duguet and B. Legube, Environ. Sci. Technol., 2005, 39, 6086–6092 CrossRef CAS PubMed.
- P.-O. Nordström and G. O'Kelly, Tighter recycled fiber markets: Softwood strikes back!, McKinsey&Company, 2013 Search PubMed.
- K. Pivnenko, D. Laner and T. F. Astrup, Environ. Sci. Technol., 2016, 50, 12302–12311 CrossRef CAS PubMed.
- J. Morris, Int. J. Life Cycle Assess., 2005, 10, 273–284 CrossRef.
- K. R. Bruce, L. O. Beach and B. K. Gullett, Waste Manage., 1991, 11, 97–102 CrossRef CAS.
- B. K. Gullett, K. R. Bruce, L. O. Beach and A. M. Drago, Chemosphere, 1992, 25, 1387–1392 CrossRef CAS.
- E. P. Management, pH Measurement in Deinking Mills (Secondary Fiber), Pulp and Paper, 2013 Search PubMed.
- M. Suhr, G. Klein, I. Kourti, M. R. Gonzalo, G. G. Santonja, S. Roudier and L. D. Sancho, Best Available Technologies (BAT) Reference Document for the Production of Pulp, Paper and Board, European Commission Joint Research Centre Institute for Prospective Technological Studies, Luxembourg, 2015 Search PubMed.
- L. Truong, M. A. DeNardo, S. Kundu, T. J. Collins and R. L. Tanguay, Green Chem., 2013, 15, 2339–2343 RSC.
- W. C. Ellis, C. T. Tran, R. Roy, M. Rusten, A. Fischer, A. D. Ryabov, B. Blumberg and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 9774–9781 CrossRef CAS PubMed.
- S. S. Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K.-W. Schramm and T. J. Collins, Science, 2002, 296, 326–328 CrossRef PubMed.
- C. Wang, J. Gao and C. Gu, Environ. Sci. Technol., 2017, 51, 488–496 CrossRef CAS PubMed.
- T. J. Collins, Acc. Chem. Res., 2002, 35, 782–790 CrossRef CAS PubMed.
- A. Chanda, D.-L. Popescu, F. T. de Oliveira, E. L. Bominaar, A. D. Ryabov, E. Münck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606–619 CrossRef CAS PubMed.
- D. Banerjee, F. M. Apollo, A. D. Ryabov and T. J. Collins, Chem. – Eur. J., 2009, 15, 10199–10209 CrossRef CAS PubMed.
- A. D. Ryabov and T. J. Collins, Adv. Inorg. Chem., 2009, 61, 471–521 CrossRef CAS.
- T. J. Collins, S. K. Khetan and A. D. Ryabov, in Handbook of Green Chemistry, ed. P. T. Anastas and R. H. Crabtree, WILEY-VCH Verlag GmbH & KgaA, Weinheim, 2009, ch. 3, pp. 39–77 Search PubMed.
- S. Kundu, A. Chanda, J. V. K. Thompson, G. Diabes, S. K. Khetan, A. D. Ryabov and T. J. Collins, Catal. Sci. Technol., 2015, 5, 1775–1782 CAS.
- L. Q. Shen, E. S. Beach, Y. Xiang, D. J. Tshudy, N. Khanina, C. P. Horwitz, M. E. Bier and T. J. Collins, Environ. Sci. Technol., 2011, 45, 7882–7887 CrossRef CAS PubMed.
- A. Chanda, S. K. Khetan, D. Banerjee, A. Ghosh and T. J. Collins, J. Am. Chem. Soc., 2006, 128, 12058–12059 CrossRef CAS PubMed.
- S. Kundu, A. Chanda, L. Espinosa-Marvan, S. K. Khetan and T. J. Collins, Catal. Sci. Technol., 2012, 2, 1165–1172 CAS.
- L. L. Tang, M. A. DeNardo, C. Gayathri, R. R. Gil, R. Kanda and T. J. Collins, Environ. Sci. Technol., 2016, 50, 5261–5268 CrossRef CAS PubMed.
- L. L. Tang, M. A. DeNardo, C. J. Schuler, M. R. Mills, C. Gayathri, R. R. Gil, R. Kanda and T. J. Collins, J. Am. Chem. Soc., 2017, 139, 879–887 CrossRef CAS PubMed.
- S. Kundu, A. Chanda, S. K. Khetan, A. D. Ryabov and T. J. Collins, Environ. Sci. Technol., 2013, 47, 5319–5326 CrossRef CAS PubMed.
- D. Banerjee, A. L. Markley, T. Yano, A. Ghosh, P. B. Berget, E. G. Minkley, S. K. Khetan and T. J. Collins, Angew. Chem., Int. Ed., 2006, 45, 3974–3977 CrossRef CAS PubMed.
- C. P. Horwitz, D. R. Fooksman, L. D. Vuocolo, S. W. Gordon-Wylie, N. J. Cox and T. J. Collins, J. Am. Chem. Soc., 1998, 120, 4867–4868 CrossRef CAS.
- N. Chahbane, D.-L. Popescu, D. A. Mitchell, A. Chanda, D. Lenoir, A. D. Ryabov, K.-W. Schramm and T. J. Collins, Green Chem., 2007, 9, 49–57 RSC.
- D. A. Mitchell, A. D. Ryabov, S. Kundu, A. Chanda and T. J. Collins, J. Coord. Chem., 2010, 63, 2605–2618 CrossRef CAS.
- M. A. DeNardo, M. R. Mills, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2016, 138, 2933–2936 CrossRef CAS PubMed.
- TAML® Catalytic Oxidant Activators in the Pulp and Paper Industry, ed. T. J. Collins, C. P. Horwitz, A. D. Ryabov, L. D. Vuocolo, S. Sen Gupta, A. Ghosh, N. L. Fattaleh, Y. Hangun, B. Steinhoff, C. A. Noser, E. Beach, D. Prasuhn Jr., T. Stuthridge, K. G. Wingate, J. Hall, L. J. Wright, I. Suckling and R. W. Allison, American Chemical Society, Washington, DC, 2002 Search PubMed.
- N. W. Shappell, M. A. Vrabel, P. J. Madsen, G. Harrington, L. O. Billey, H. Hakk, G. L. Larsen, E. S. Beach, C. P. Horwitz, K. Ro, P. G. Hunt and T. J. Collins, Environ. Sci. Technol., 2008, 42, 1296–1300 CrossRef CAS PubMed.
- J. L. Chen, S. Ravindran, S. Swift, L. J. Wright and N. Singhal, Water Res., 2012, 46, 6309–6318 CrossRef CAS PubMed.
- A. D. Ryabov, Adv. Inorg. Chem., 2013, 65, 118–163 CrossRef.
- R. T. Malecky, E. S. Beach, C. P. Horwitz and T. J. Collins, Presented in part at the 232nd ACS National Meeting, San Francisco, CA, 2006.
- N. R. Campbell, Proc. R. Soc. London, Ser. B, 1940, 129, 528–538 CrossRef CAS.
- E. E. Reid and E. Wilson, J. Am. Chem. Soc., 1944, 66, 967–969 CrossRef CAS.
- T. Hirano, Y. Honda, T. Watanabe and M. Kuwahara, Biosci. Biotechnol., Biochem., 2000, 64, 1958–1962 CrossRef CAS PubMed.
- T. T. Schug, R. Abagyan, B. Blumberg, T. J. Collins, D. Crews, P. L. DeFur, S. M. Dickerson, T. M. Edwards, A. C. Gore, L. J. Guillette, T. Hayes, J. J. Heindel, A. Moores, H. B. Patisaul, T. L. Tal, K. A. Thayer, L. N. Vandenberg, J. Warner, C. S. Watson, F. S. v. Saal, R. T. Zoeller, K. P. O'Brien and J. P. Myers, Green Chem., 2013, 15, 181–198 RSC.
- P. George, Biochem. J., 1953, 55, 220–230 CrossRef CAS PubMed.
- H. A. Balsiger, R. de la Torre, W.-Y. Lee and M. B. Cox, Sci. Total Environ., 2010, 408, 1422–1429 CrossRef CAS PubMed.
- E. J. Routledge and J. P. Sumpter, Environ. Toxicol. Chem., 1996, 15, 241–248 CrossRef CAS.
- L. Truong, D. M. Reif, L. St. Mary, M. C. Geier, H. D. Truong and R. L. Tanguay, Toxicol. Sci., 2014, 137, 212–233 CrossRef CAS PubMed.
- A. Ghosh, A. D. Ryabov, S. M. Mayer, D. C. Horner, D. E. Prasuhn, S. Sen Gupta, L. Vuocolo, C. Culver, M. P. Hendrich, C. E. F. Rickard, R. E. Norman, C. P. Horwitz and T. J. Collins, J. Am. Chem. Soc., 2003, 125, 12378–12379 CrossRef CAS PubMed.
- M. A. Mendez, M. F. Suarez and M. T. Cortes, J. Electroanal. Chem., 2006, 590, 181–189 CrossRef CAS.
- I. L. Kogan and K. Yakushi, Electrochim. Acta, 1998, 43, 2053–2060 CrossRef CAS.
- D.-L. Popescu, M. Vrabel, A. Brausam, P. Madsen, G. Lente, I. Fabian, A. D. Ryabov, R. van Eldik and T. J. Collins, Inorg. Chem., 2010, 49, 11439–11448 CrossRef CAS PubMed.
- M. Emelianenko, D. Torrejon, M. DeNardo, A. Socolofsky, A. Ryabov and T. Collins, J. Math. Chem., 2014, 52, 1460–1476 CrossRef CAS.
- M. A. DeNardo, Ph.D. Dissertation, Carnegie Mellon, March 26, 2016 Search PubMed.
- J. S. Dordick, M. A. Marletta and A. M. Klibanov, Biotechnol. Bioeng., 1987, 30, 31–36 CrossRef CAS PubMed.
- M. Reihmann and H. Ritter, Adv. Polym. Sci., 2006, 194, 1–49 CrossRef CAS.
- S. Kobayashi, H. Kurioka and H. Uyama, Macromol. Rapid Commun., 1996, 17, 503–508 CrossRef CAS.
- H. Sakuyama, Y. Endo, K. Fujimoto and Y. Hatano, J. Biosci. Bioeng., 2003, 96, 227–231 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama, T. Ushiwata, T. Uchiyama, J. Sugihara and H. Kurioka, Macromol. Chem. Phys., 1998, 199, 777–782 CrossRef CAS.
- H. Uyama, N. Maruichi, H. Tonami and S. Kobayashi, Biomacromolecules, 2002, 3, 187–193 CrossRef CAS PubMed.
- J. Kulys, R. Vidziunaite, A. Ziemys and I. Bratkovskaja, Biologija, 2007, 53, 40–44 CAS.
- Y. H. Kim, E. S. An, S. Y. Park, J.-O. Lee, J. H. Kim and B. K. Song, J. Mol. Catal. B: Enzym., 2007, 44, 149–154 CrossRef CAS.
- S. Arulmozhiraja, M. L. Coote, Y. Kitahara, M. Juhász and T. Fujii, J. Phys. Chem. A, 2011, 115, 4874–4881 CrossRef CAS PubMed.
- B. Kolvenbach, N. Schlaich, Z. Raoui, J. Prell, S. Zühlke, A. Schäffer, F. P. Guengerich and P. F. X. Corvini, Appl. Environ. Microbiol., 2007, 73, 4776–4784 CrossRef CAS PubMed.
- W. Zhang, K. Yin and L. Chen, Appl. Microbiol. Biotechnol., 2013, 97, 5681–5689 CrossRef CAS PubMed.
- T. Fukuda, H. Uchida, Y. Takashima, T. Uwajima, T. Kawabata and M. Suzuki, Biochem. Biophys. Res. Commun., 2001, 284, 704–706 CrossRef CAS PubMed.
- N. Grassie, M. I. Guy and N. H. Tennent, Polym. Degrad. Stab., 1985, 12, 65–91 CrossRef CAS.
- J.-H. Kim, P.-K. Park, C.-H. Lee, H.-H. Kwon and S. Lee, J. Membr. Sci., 2008, 312, 66–75 CrossRef CAS.
- S. H. Dai, C. Y. Lin, D. V. Rao, F. A. Stuber, P. S. Carleton and H. Ulrich, J. Org. Chem., 1985, 50, 1722–1725 CrossRef CAS.
- W.-F. Chen, H.-Y. Lin and S. A. Dai, Org. Lett., 2004, 6, 2341–2343 CrossRef CAS PubMed.
- R. W. Murray and H. Gu, J. Org. Chem., 1995, 60, 5673–5677 CrossRef CAS.
- E. C. Dodds and W. Lawson, Proc. R. Soc. London, Ser. B, 1938, 125, 222–232 CrossRef CAS.
- J. Chen, Y. Xiao, Z. Gai, R. Li, Z. Zhu, C. Bai, R. L. Tanguay, X. Xu, C. Huang and Q. Dong, Aquat. Toxicol., 2015, 169, 204–214 CrossRef CAS PubMed.
- P. Sohoni, C. R. Tyler, K. Hurd, J. Caunter, M. Hetheridge, T. Williams, C. Woods, M. Evans, R. Toy, M. Gargas and J. P. Sumpter, Environ. Sci. Technol., 2001, 35, 2917–2925 CrossRef CAS PubMed.
- F. Lahnsteiner, B. Berger, M. Kletzl and T. Weismann, Aquat. Toxicol., 2005, 75, 213–224 CrossRef CAS PubMed.
- A. Hatef, S. M. H. Alavi, A. Abdulfatah, P. Fontaine, M. Rodina and O. Linhart, Ecotoxicol. Environ. Saf., 2012, 76, 56–62 CrossRef CAS PubMed.
- H. Hayashi, A. Nishimoto, N. Oshima and S. Iwamuro, J. Toxicol. Sci., 2007, 32, 91–96 CrossRef CAS PubMed.
- D. L. Villeneuve, N. Garcia-Reyero, B. L. Escalon, K. M. Jensen, J. E. Cavallin, E. A. Makynen, E. J. Durhan, M. D. Kahl, L. M. Thomas, E. J. Perkins and G. T. Ankley, Environ. Sci. Technol., 2012, 46, 51–59 CrossRef CAS PubMed.
- S. Liu, F. Qin, H. Wang, T. Wu, Y. Zhang, Y. Zheng, M. Li and Z. Wang, Aquat. Toxicol., 2012, 122–123, 19–27 CrossRef CAS PubMed.
- F. Qin, L. Wang, X. Wang, S. Liu, P. Xu, H. Wang, T. Wu, Y. Zhang, Y. Zheng, M. Li, X. Zhang, C. Yuan, G. Hu and Z. Wang, Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol., 2013, 157, 192–202 CrossRef CAS PubMed.
- C. D. Metcalfe, T. L. Metcalfe, Y. Kiparissis, B. G. Koenig, C. Khan, R. J. Hughes, T. R. Croley, R. E. March and T. Potter, Environ. Toxicol. Chem., 2001, 20, 297–308 CrossRef CAS PubMed.
- K. S. Saili, M. M. Corvi, D. N. Weber, A. U. Patel, S. R. Das, J. Przybyla, K. A. Anderson and R. L. Tanguay, Toxicology, 2012, 291, 83–92 CrossRef CAS PubMed.
- W. Lee, C.-W. Kang, C.-K. Su, K. Okubo and Y. Nagahama, Chemosphere, 2012, 88, 945–952 CrossRef CAS PubMed.
- J. Wang, X. Liu, H. Wang, T. Wu, X. Hu, F. Qin and Z. Wang, Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol., 2010, 152, 313–320 CrossRef PubMed.
- E. Chung, M. C. Genco, L. Megrelis and J. V. Ruderman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17732–17737 CrossRef CAS PubMed.
- S. H. Lam, M. M. Hlaing, X. Zhang, C. Yan, Z. Duan, L. Zhu, C. Y. Ung, S. Mathavan, C. N. Ong and Z. Gong, PLoS One, 2011, 6, e28273 CAS.
- Y. Gibert, S. Sassi-Messai, J.-B. Fini, L. Bernard, D. Zalko, J.-P. Cravedi, P. Balaguer, M. Andersson-Lendahl, B. Demeneix and V. Laudet, BMC Dev. Biol., 2011, 11, 1–18 CrossRef PubMed.
- T. R. Baker, R. E. Peterson and W. Heideman, Toxicol. Sci., 2013, 135, 241–250 CrossRef CAS PubMed.
- M. D. Anway and M. K. Skinner, Endocrinology, 2006, 147, s43–s49 CrossRef CAS PubMed.
- D. Crews, A. C. Gore, T. S. Hsu, N. L. Dangleben, M. Spinetta, T. Schallert, M. D. Anway and M. K. Skinner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5942–5946 CrossRef CAS PubMed.
- M. Shelby , NTP CERHR MON, 2008, 22, v, vii–ix, 1–64 passim.
- D. Melzer, N. E. Rice, C. Lewis, W. E. Henley and T. S. Galloway, PLoS One, 2010, 5, e8673 Search PubMed.
- D. Li, Z. Zhou, D. Qing, Y. He, T. Wu, M. Miao, J. Wang, X. Weng, J. R. Ferber, L. J. Herrinton, Q. Zhu, E. Gao, H. Checkoway and W. Yuan, Hum. Reprod., 2010, 25, 519–527 CrossRef CAS PubMed.
- D.-K. Li, Z. Zhou, M. Miao, Y. He, J. Wang, J. Ferber, L. J. Herrinton, E. Gao and W. Yuan, Fertil. Steril., 2011, 95(2), 625–630.e4 CrossRef CAS PubMed.
- D.-K. Li, Z. Zhou, M. Miao, Y. He, D. Qing, T. Wu, J. Wang, X. Weng, J. Ferber, L. J. Herrinton, Q. Zhu, E. Gao and W. Yuan, J. Androl., 2010, 31, 500–506 CrossRef CAS PubMed.
- D.-K. Li, M. Miao, Z. Zhou, C. Wu, H. Shi, X. Liu, S. Wang and W. Yuan, PLoS One, 2013, 8, e65399 CAS.
- J. R. Rochester, Reprod. Toxicol., 2013, 42, 132–155 CrossRef CAS PubMed.
- H. Watanabe, R. Adachi, K. Kusui, A. Hirayama, T. Kasahara and K. Suzuki, Int. Immunopharmacol., 2003, 3, 1601–1608 CrossRef CAS PubMed.
- G. S. Prins, W.-Y. Hu, G.-B. Shi, D.-P. Hu, S. Majumdar, G. Li, K. Huang, J. L. Nelles, S.-M. Ho, C. L. Walker, A. Kajdacsy-Balla and R. B. van Breemen, Endocrinology, 2014, 155, 805–817 CrossRef PubMed.
- W.-Y. Hu, G.-B. Shi, H.-M. Lam, D.-P. Hu, S.-M. Ho, I. C. Madueke, A. Kajdacsy-Balla and G. S. Prins, Endocrinology, 2011, 152, 2150–2163 CrossRef CAS PubMed.
- S. Soriano, P. Alonso-Magdalena, M. García-Arévalo, A. Novials, S. J. Muhammed, A. Salehi, J.-A. Gustafsson, I. Quesada and A. Nadal, PLoS One, 2012, 7, e31109 CAS.
- E. Hugo, T. Brandebourg, J. Woo, J. Loftus, J. Alexander and N. Ben-Jonathan, Environ. Health Perspect., 2008, 116, 1642–1647 CrossRef CAS PubMed.
- Y. B. Wetherill, C. E. Petre, K. R. Monk, A. Puga and K. E. Knudsen, Mol. Cancer Ther., 2002, 1, 515–524 CAS.
- R. Steinmetz, N. G. Brown, D. L. Allen, R. M. Bigsby and N. Ben-Jonathan, Endocrinology, 1997, 138, 1780–1786 CrossRef CAS PubMed.
- S. F. Arnold, D. M. Klotz, B. M. Collins, P. M. Vonier, L. J. Guillette and J. A. McLachlan, Science, 1996, 272, 1489–1492 CAS.
- S. i. Yoshihara, M. Makishima, N. Suzuki and S. Ohta, Toxicol. Sci., 2001, 62, 221–227 CrossRef CAS PubMed.
- A. L. Herbst, H. Ulfelder and D. C. Poskanzer, N. Engl. J. Med., 1971, 284, 878–881 CrossRef CAS PubMed.
- K. Okuda, M. Takiguchi and S. i. Yoshihara, Toxicol. Lett., 2010, 197, 7–11 CrossRef CAS PubMed.
- M. E. Baker and C. Chandsawangbhuwana, PLoS One, 2012, 7, e46078 CAS.
- R. B. Stebbins and E. W. Blanchard, Endocrinology, 1945, 36, 305–312 CrossRef CAS.
- H. Ishibashi, N. Watanabe, N. Matsumura, M. Hirano, Y. Nagao, H. Shiratsuchi, S. Kohra, S.-i. Yoshihara and K. Arizono, Life Sci., 2005, 77, 2643–2655 CrossRef CAS PubMed.
- A. Yamaguchi, H. Ishibashi, S. Kohra, K. Arizono and N. Tominaga, Aquat. Toxicol., 2005, 72, 239–249 CrossRef CAS PubMed.
- S.-H. Liu, C.-C. Su, K.-I. Lee and Y.-W. Chen, Sci. Rep., 2016, 6, 39254 CrossRef CAS PubMed.
- H. Miyakoda, M. Tabata, S. Onodera and K. Tkeda, J. Health Sci., 1999, 45, 318–323 CrossRef CAS.
- Y. Ikezuki, O. Tsutsumi, Y. Takai, Y. Kamei and Y. Taketani, Hum. Reprod., 2002, 17(11), 2839–2841 CrossRef CAS PubMed.
- G. Schönfelder, W. Wittfoht, H. Hopp, C. E. Talsness, M. Paul and I. Chahoud, Environ. Health Perspect., 2002, 110, A703–A707 CrossRef.
- Y. J. Lee, H.-Y. Ryu, H.-K. Kim, C. S. Min, J. H. Lee, E. Kim, B. H. Nam, J. H. Park, J. Y. Jung, D. D. Jang, E. Y. Park, K.-H. Lee, J.-Y. Ma, H.-S. Won, M.-W. Im, J.-H. Leem, Y.-C. Hong and H.-S. Yoon, Reprod. Toxicol., 2008, 25, 413–419 CrossRef CAS PubMed.
- S. M. Engel, B. Levy, Z. Liu, D. Kaplan and M. S. Wolff, Reprod. Toxicol., 2006, 21, 110–112 CrossRef CAS PubMed.
- J. Matsumoto, H. Yokota and A. Yuasa, Environ. Health Perspect., 2002, 110, 193–196 CrossRef CAS PubMed.
- C. P. Strassburg, A. Strassburg, S. Kneip, A. Barut, R. H. Tukey, B. Rodeck and M. P. Manns, Gut, 2002, 50, 259–265 CrossRef CAS PubMed.
- M. W. Coughtrie, B. Burchell, J. E. Leakey and R. Hume, Mol. Pharmacol., 1988, 34, 729–735 CAS.
- G. M. Pacifici, M. Kubrich, L. Giuliani, M. de Vries and A. Rane, Eur. J. Clin. Pharmacol., 1993, 44, 259–264 CrossRef CAS PubMed.
- O. Tsutsumi, J. Steroid Biochem. Mol. Biol., 2005, 93, 325–330 CrossRef CAS PubMed.
- S.-M. Ho, W.-Y. Tang, J. Belmonte de Frausto and G. S. Prins, Cancer Res., 2006, 66, 5624–5632 CrossRef CAS PubMed.
- L. N. Vandenberg, M. V. Maffini, C. M. Schaeberle, A. A. Ucci, C. Sonnenschein, B. S. Rubin and A. M. Soto, Reprod. Toxicol., 2008, 26, 210–219 CrossRef CAS PubMed.
- L. Speroni, M. Voutilainen, M. L. Mikkola, S. A. Klager, C. M. Schaeberle, C. Sonnenschein and A. M. Soto, Sci. Rep., 2017, 7, 40806 CrossRef CAS PubMed.
- M. Kitada, T. Kamataki, K. Itahashi, T. Rikihisa and Y. Kanakubo, J. Biol. Chem., 1987, 262, 13534–13537 CAS.
- A. Schecter, T. T. Schug, S. A. Vogel, L. N. Vandenberg, J. Braun, R. Hauser, J. A. Taylor, F. S. v. Saal and J. J. Heindel, Dioxins and Health: Including Other Persistent Organic Pollutants and Endocrine Disruptors, Wiley & Sons, New York, 2012 Search PubMed.
- Proposed Notice Requiring the Preparation and Implementation of Pollution Prevention Plans with Respect to Bisphenol A In Industrial Effluents, Canada Gazette, Public Works and Government Services Canada, Ottawa, Canada, 2010, http://publications.gc.ca/gazette/archives/p1/2010/2010-10-16/pdf/g1-14442.pdf Search PubMed.
- F. G. Doherty, Water Qual. Res. J. Can., 2001, 36, 475–518 CAS.
- F. Manservisi, C. B. Marquillas, A. Buscaroli, J. Huff, M. Lauriola, D. Mandrioli, M. Manservigi, S. Panzacchi, E. K. Silbergeld and F. Belpoggi, Environ. Health Perspect., 2017, 125, 289–295 Search PubMed.
Footnotes |
| † We dedicate this publication, which has occupied us for 15 years, to the memory of our great friend and extraordinary visionary into endocrine disruption, Dr Theodora Emily Decker Colborn (b.1928–d.2014), to our brilliant colleague who coined the term ‘Endocrine Disruption’, Dr Pete Myers, and to the great father of BPA low dose toxicity research, Prof. Frederick S. vom Saal—their work (and that of other ED researchers) can arm all chemists with a better understanding of the relationships between everyday-everywhere chemicals and the future good to inspire proactive advances in sustainable chemistry. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc01415e |
| This journal is © The Royal Society of Chemistry 2017 |