
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalytic access to nonracemic γ-oxo esters via stereoselective reduction using ene-reductases†
Nikolaus G.
Turrini
a,
Răzvan C.
Cioc
b,
Daan J. H.
van der Niet
b,
Eelco
Ruijter
 b,
Romano V. A.
Orru
b,
Mélanie
Hall
a and
Kurt
Faber
*a
b,
Romano V. A.
Orru
b,
Mélanie
Hall
a and
Kurt
Faber
*a
aDepartment of Chemistry, University of Graz, 8010 Graz, Austria. E-mail: kurt.faber@uni-graz.at
bDepartment of Chemistry and Pharmaceutical Sciences, Vrije Universiteit Amsterdam, 1081 HZ Amsterdam, The Netherlands
First published on 21st October 2016
Abstract
The asymmetric bioreduction of α,β-unsaturated γ-keto esters using ene-reductases from the Old Yellow Enzyme family proceeds with excellent stereoselectivity and high conversion levels, covering a broad range of acyclic and cyclic derivatives. Various strategies were employed to provide access to both enantiomers, which are versatile precursors of bioactive molecules. The regioselectivity of hydride addition on di-activated alkenes was elucidated by isotopic labeling experiments and showed strong preference for the keto moiety as activating/binding group as opposed to the ester. Finally, chemoenzymatic synthesis of (R)-2-(2-oxocyclohexyl)acetic acid was achieved in high ee on a preparative scale combining enzymatic reduction followed by ester hydrogenolysis.
Introduction
γ-Oxo esters are versatile molecules owing to the presence of two reactive functional groups that allow their straightforward incorporation into more complex scaffolds. So far, these compounds have been widely employed as precursors of (bi)cyclic compounds, including bicyclic (spiro)lactams and γ-butyrolactone derivatives. Typical examples are five- and six-membered 2-(3-oxocycloalkyl) or 2-(2-oxocycloalkyl)acetic acid esters or 4-oxopentanoic acid derivatives. Chiral γ-oxo esters, in particular, find broad applications in the synthesis of biologically active molecules, such as therapeutic drugs and natural products (Scheme 1).1 Several synthetic protocols have been developed, often based on kinetic resolution of racemic γ-oxo esters or corresponding carboxylic acids, which suffers from loss of material (50% maximum theoretical yield). Hence, asymmetric strategies are preferred, as they contribute to enhanced atom economy, but must display exquisite stereoselectivity to comply with regulations on the marketing of single enantiomeric drugs.2 In this context, biocatalysis offers an attractive approach to enantiopure γ-oxo esters by contributing to low environmental impact combined with high stereoselectivity usually encountered with enzymes. For instance, Pietruszka et al. developed a stereoselective bienzymatic approach to trisubstituted γ-butyrolactone with up to 90% overall yield, where ene-reductases were employed for the asymmetric reduction of ethyl 2,3-dimethyl-4-oxo-pent-2-enoate).3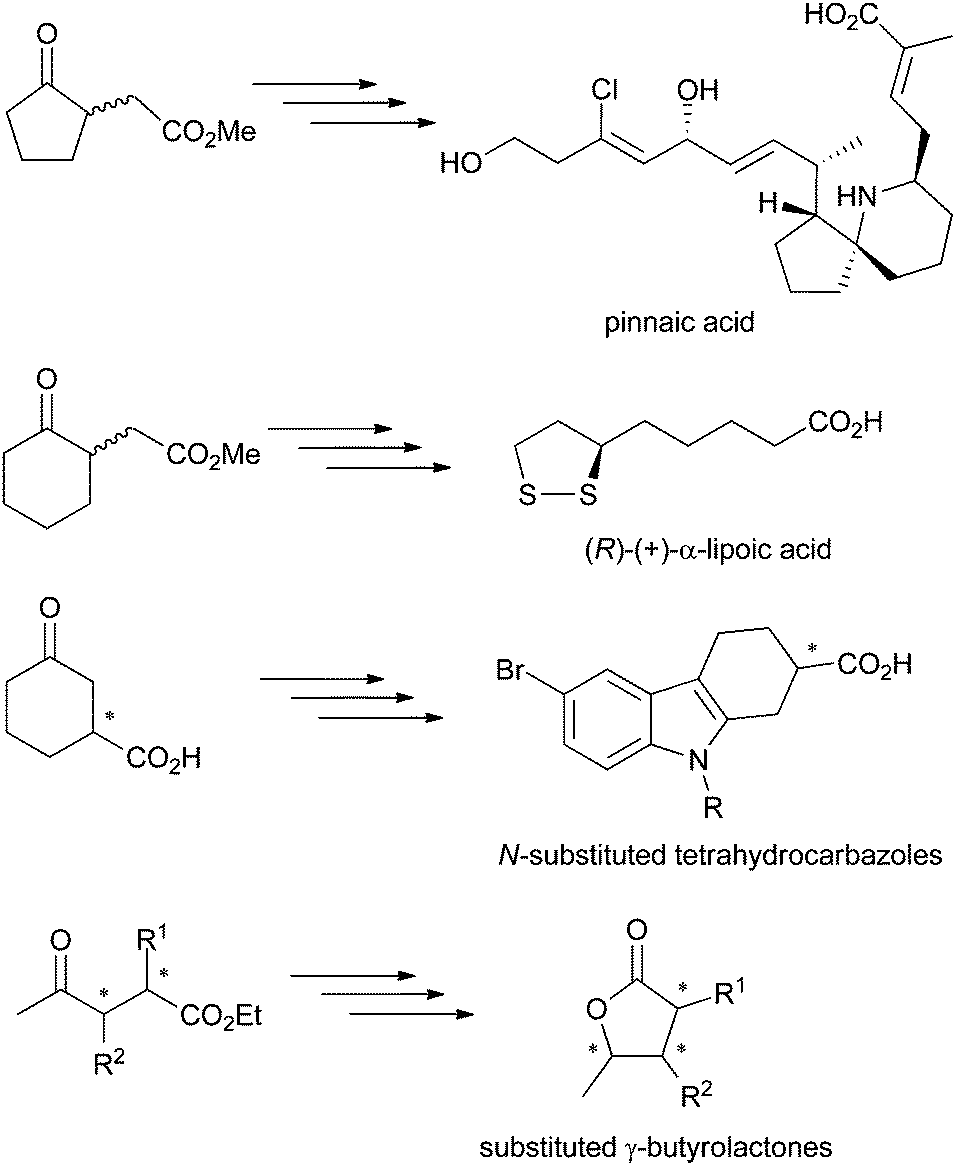 | ||
| Scheme 1 γ-Oxo acids and esters employed as precursors of biologically active molecules.1 | ||
Ene-reductases are flavoproteins of the Old Yellow Enzyme (OYE) family that show broad substrate tolerance in the asymmetric reduction of activated alkenes. An electron-withdrawing group (EWG) attached to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is mandatory to (i) anchor the substrate in the enzyme's active site via H-bonding, which (ii) enhances the polarization of the CC bond.
C bond is mandatory to (i) anchor the substrate in the enzyme's active site via H-bonding, which (ii) enhances the polarization of the CC bond.
Nicotinamide cofactor is required as external hydride source and is recycled using cheap reducing equivalents, such as glucose or formate. Mechanistic investigations have shown that a hydride is delivered from N5 of reduced flavin onto Cβ, while protonation occurs at Cα in a trans-fashion through a tyrosine residue. The EWG is typically a nitro, ketone, aldehyde, ester or nitrile group (Scheme 2). Several di-activated alkenes have been successfully reduced by ene-reductases with the intention to reduce the electron density of the CC bond and/or to generate di-functionalized molecules. In this context, the concept of ‘supported substrate activation’ was proposed, according to which the presence of an additional α-electron-withdrawing group enhances the polarization of the CC bond, leading to enhanced conversion levels.4 Although not participating in substrate binding, particularly α-halogens increased substrate reactivity, in line with increased CC bond polarization. For substrates bearing two possible anchoring groups, such as fumaric/maleic diesters, γ-oxo and β-cyano unsaturated esters, the actual activating group could be identified via deuterium labeling experiments. However, so far, no general rule could be delineated to predict the favored binding mode.3,5
 | ||
| Scheme 2 Bioreduction of activated alkenes 1–14a by ene-reductases at the expense of nicotinamide cofactor. | ||
Along these considerations, several members of the OYE family were selected for the preparation of enantiopure γ-oxo esters as precursors of bioactive molecules in an environmentally friendly fashion featuring mild reaction conditions in aqueous systems. In addition, mechanistic investigations were conducted to elucidate the preferred activating/binding groups. The data obtained facilitate the design of substrates for the asymmetric bioreduction of substituted γ-oxoesters.
Results and discussion
Screening of type I–V substrates
A range of unsaturated γ-oxo esters was prepared with focus on diversity around the central CC bond (types I–V, Fig. 1): substrates were straight-chain, or 5- and 6-membered cyclic derivatives, and the position of the CC bond was varied from endo- to exo-cyclic; the position of the ester group (non-/conjugated to the CC bond) as well as the type of alkoxy moiety was altered. These variations within type I–V substrates provide multiple handles to control activity and stereoselectivity of OYEs via substrate-based stereocontrol.6
 | ||
| Fig. 1 Unsaturated γ-oxo esters investigated as substrate library for the bioreduction catalyzed by ene-reductases. | ||
Acyclic 3-methyl-4-oxopent-2-enoic acid esters 1a and 2a (type I) were highly reactive and were reduced by all enzymes in up to full conversion, with 2a appearing more reactive than 1a (>99% conv. for both E/Z-isomers in all cases, entries 3- and 4, Table 1). All enzymes converted (E)-1a and (E)-2a to (S)-1b and (S)-2b, respectively, with typically high stereoselectivity (up to 96% ee); only OPR1, OPR3 and EBP1 displayed lower stereopreference. In contrast, the corresponding (Z)-isomers of 1a and 2a were converted to the opposite (R)-enantiomers of 1b and 2b, in line with previous observations on (E/Z)-isomeric pairs of substituted alkenes. The switch in stereopreference is obvious for (E/Z)-isomers of α-mono-β-di-substituted mono-activated substrates with conserved binding mode due to the orientation of β-substituents on opposite sides (Scheme 3a). However, in case of α-di-β-mono-substituted monoactivated substrates, the substrate must flip within the enzyme active site (‘right/left’, Scheme 3b); in addition, docking studies (corroborated by experimental results) showed that rotamers of methyl (E/Z)-2-chloropentenoate further contributed to increasing the complexity of binding modes.6b With di-activated alkenes, the picture is even more complex, since two groups can act as binding motif, which increases the number of possible binding modes, especially when carbonyl groups are involved. The lower ee values observed for (Z)-2a can be attributed to spontaneous (E/Z)-isomerisation that occurred after substrate synthesis, yielding (E)-2a, which furnished (S)-2b upon bioreduction (data not shown).
 | ||
| Scheme 3 Binding modes of (E/Z)-isomeric mono-activated substrates via His–His/Asn resulting in opposite enantiomeric products. (a) Conserved binding of α-mono-β-di-substituted substrates; (b) ‘right/left’ flip α-di-β-mono-substituted substrates. | ||
| Entry | Substrate | OYE1 | OYE2 | OYE3 | NCR | YqjM | OPR1 | OPR3 | XenA | EBP1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: Substrate (10 mM) added from a stock solution in DMSO or EtOH in Tris-HCl buffer (50 mM, pH 7.5) containing NADH (15 mM) and the biocatalyst (final protein concentration 100 μg mL−1, ∼2.5 μM). Reaction mixture shaken for 24 h at 30 °C and 120 rpm. | |||||||||||
| 1 | (E)-1a | Conversion/% | >99 | >99 | >99 | >99 | 80 | >99 | 90 | 54 | 74 |
| ee/% | 92(S) | 94(S) | 96(S) | 94(S) | 96(S) | 72(S) | 80(S) | 96(S) | 32(S) | ||
| 2 | (Z)-1a | Conversion/% | >99 | >99 | >99 | >99 | 32 | >99 | >99 | 34 | 44 |
| ee/% | 76(R) | 76(R) | 78(R) | 90(R) | 38(R) | 48(R) | 56(R) | 54(R) | 42(R) | ||
| 3 | (E)-2a | Conversion/% | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 |
| ee/% | 94(S) | 96(S) | 96(S) | 94(S) | 94(S) | 76(S) | 82(S) | 94(S) | 56(S) | ||
| 4 | (Z)-2a | Conversion/% | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 |
| ee/% | 71(R) | 42(R) | 15(R) | 73(R) | 60(R) | 30(R) | 34(R) | 35(R) | 64(R) | ||
Activities and stereoselectivities on cyclic substrates showed great variations depending on the type of substrate and enzyme (Table 2). Overall, type II compounds comprising 3a, 4a, 11a and 12a showed broadest acceptance by the panel of enzymes, showing up to full conversion and perfect stereoselectivity (Table 2, entries 1, 2, 11 and 12). Although cyclic enones with β-alkyl substitution generally tend to be poorly reactive, derivatives with β-carboxylic ester groups are highly reactive.7 These data indicate that an additional electron-withdrawing group efficiently enhances substrate activation. Most enzymes, in particular YqjM and OPR1, were slightly more efficient on ethyl esters yielding the (R)-enantiomers of 4b and 12b in >99% conversion and >99% ee. While OYE1 showed high (R)-stereoselectivity, OYE3 was only modestly selective and OYE2 consistently produced the opposite (S)-enantiomer, highlighting that predictions based on sequence similarity are elusive, bearing in mind that OYE1 and OYE2 share 92% identity and astonishing 95% similarity, while OYE2 and OYE3 show 82% identity and 89% similarity.8
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | Substrate | OYE1 | OYE2 | OYE3 | NCR | YqjM | OPR1 | OPR3 | XenA | Control | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: Substrate (10 mM) added from a stock solution in DMSO or EtOH in Tris-HCl buffer (50 mM, pH 7.5) containing NADH (15 mM) and the biocatalyst (final protein concentration 100 μg mL−1, ∼2.5 μM). Reaction mixture shaken for 24 h at 30 °C and 120 rpm; control experiments were performed in absence of enzyme. Conversion values (%) were obtained from GC analysis (see Experimental section). b Side product detected on GC-MS (MWproduct + 16; likely the epoxide product10), amount (based on GC) indicated in brackets. n.a. not applicable; n.d. not determined. | |||||||||||
| 1 | 3a | Conversion/% | >99 | >99 | >99 | >99 | >99 | >99 | >99 | >99 | <1 |
| ee/% | 68(R) | 80(S) | 73(R) | 75(R) | 94(R) | 91(R) | 92(R) | >99(R) | n.a. | ||
| 2 | 4a | Conversion/% | >99 | >99 | >99 | >99 | >99 | >99 | 83 | 68 | <1 |
| ee/% | 88(R) | 66(S) | rac | 90(R) | >99(R) | >99(R) | 78(R) | >99(R) | n.a. | ||
| 3 | 5a | Conversion/% | 69 | <1 | <1 | 99 | 68 | 65 | 77 | 32 | <1 |
| ee/% | 94(R) | n.a. | n.a. | 46(R) | 34(R) | 50(R) | 38(R) | 40(R) | n.a. | ||
| 4 | endo-6a | Conversion/% | 90 | 4 | 3 | >99 | 49 (15)b | 73 | 87 | 23 | <1 |
| ee/% | 48(R) | rac | rac | 30(R) | 32(R) | 34(R) | 26(R) | 50(R) | n.a. | ||
| 5 | exo-6a | Conversion/% | 36 | 23 | 77 | 22 | 18 | 14 | 15 | 22 | 22 |
| ee/% | rac | rac | rac | rac | rac | rac | rac | rac | rac | ||
| 6 | endo-8a | Conversion/% | <1 | <1 | <1 | 3 | 13 (3)b | 11 | 8 (3)b | 4 (1)b | <1 |
| ee/% | n.a. | n.a. | n.a. | 90(R) | 76(R) | 72(R) | 78(R) | 86(R) | n.a. | ||
| 7 | exo-8a | Conversion/% | 43 | 18 | 95 | 17 | 13 | 9 | 4 | 10 | 12 |
| ee/% | 20(S) | rac | rac | rac | rac | 74(R) | rac | rac | rac | ||
| 8 | endo-9a | Conversion/% | 3 | <1 | 1 | 7 | 42 (6)b | 20 (3)b | 20 (7)b | 35 | <1 |
| ee/% | 61(R) | n.a. | n.d. | 88(R) | 78(R) | 68(R) | 80(R) | 84(R) | n.a. | ||
| 9 | exo-9a | Conversion/% | 19 | 14 | 81 | 15 | 9 | 11 | 11 | 19 | 15 |
| ee/% | rac | rac | rac | rac | rac | rac | rac | rac | rac | ||
| 10 | endo-10a | Conversion/% | 19 | <1 | <1 | 75 | 9 | 3 | 5 | 13 | <1 |
| ee/% | 86(R) | n.a. | n.a. | >97(R) | 20(R) | n.d. | n.d. | 48(R) | n.a. | ||
| 11 | 11a | Conversion/% | 70 | 3 | 34 | 98 | >99 | >99 | 62 (16)b | 18 | <1 |
| ee/% | 86(R) | 46(S) | rac | 88(R) | 94(R) | >99(R) | 80(R) | >99(R) | n.a. | ||
| 12 | 12a | Conversion/% | 65 | 4 | 30 | 99 | >99 | >99 | 60 (29)b | 76 | <1 |
| ee/% | >99(R) | 76(S) | 22(R) | 98(R) | >99(R) | >99(R) | 90(R) | >99(R) | n.a. | ||
Upon changing substitution position (β- to α-), and conjugation (–CO2R to –CH2CO2R), both conversion levels and ee values dropped significantly with type III substrates (5a, endo-6a, endo-8a, endo-9a and endo-10a), e.g.5avs.3a or endo-9avs.12a (entries 1/3 and 8/12, Table 2), most prominently with six-membered ring substrates. OYE2 and OYE3 were most dramatically affected (max. 4% conversion with endo-6a in type III series, entry 4, Table 2). This is caused by loss of activation due to insertion of CH2 between the CC bond and the second EWG yielding monoactivated substrates. Based on previous experience on the influence of the size of substituents on ene-reductase stereoselectivity,5a,9 a bulkier benzyl ester was introduced (endo-10a). This approach proved successful with NCR, as not only was the conversion boosted (from max. 7% with endo-9a to 75%) but also the stereoselectivity was perfect [>97% (R), entry 10, Table 2], which even surpassed the values obtained with five-membered ring derivatives of type II (max. 46% ee with 5a). The labile character of free acid 7a in aqueous medium prevented analysis of the reaction samples (data not shown).
Alternatively, products obtained by bioreduction of type III compounds can also be obtained from type IV substrates. However, the latter approach did not prove effective. In most cases, the reactivity of the substrate was so high that NADH itself could reduce the CC bond non-stereoselectively. Enzymes on average did not provide substantial increase in conversion levels compared to controls in the absence of enzymes using excess of NADH (with the exception of OYE3), and products were racemic (entries 5, 7 and 9, Table 2). Although the use of co-solvents could suppress the NADH-mediated background reaction, enzyme activity was also strongly diminished (data not shown). In situ recycling of NADH (via GDH/glucose/cat. NAD+) did not influence the outcome of the reaction (data not shown). The enhanced reactivity towards NADH but decreased influence on OYEs is intriguing, since type IV substrates are di-activated alkenes and resemble type II compounds, with the only difference in the position of the alkyl chain (ring) with respect to the activating groups. This suggests an overall poor fit of type IV compounds in the enzyme active site. This was corroborated by the non-reactivity of type V substrates (13a–14a), which display even more steric hindrance and a similar backbone (data not shown). Bulky exo-10a was converted only by OYE3 with max. 3% conversion (data not shown).
Scale up
In order to demonstrate the preparative-scale applicability of this method, the bioreduction of endo-10a was optimized. Use of a FDH- or GDH-based recycling system was not advantageous with regard to the conversion (data not shown). Various co-solvents were assessed to enhance the solubility of the lipophilic substrate (EtOH, DMSO and tert-butyl methyl ether). 10% v/v TBME provided a slight increase in conversion (+4%) while maintaining a high ee value for 10b. Scale-up of the reaction was performed using 400 mg of endo-10a. New aliquots of enzyme and fresh NADH were added after 8 h, 24 h, 32 h and 48 h. Prolonged reaction times and use of fresh catalyst and cofactor led to 81% conversion and >97% ee. Isolation and purification of (R)-10b proved challenging and resulted in 27% isolated yield (108 mg, >97% ee). Finally, deprotection of (R)-10bvia hydrogenation on Pd/C was successful and furnished the corresponding acid (R)-7b in 70% yield (49 mg, Scheme 4 and ESI†). These results demonstrate that variation of the ester moiety is a powerful strategy to enhance substrate acceptance, since 7b could not be obtained directly from 7a. | ||
| Scheme 4 Access to (R)-2-(2-oxocyclohexyl)acetic acid [(R)-7b] via chemoenzymatic bioreduction-deprotection using NCR. | ||
Isotopic labeling studies
The question which of the EWGs in di-activated alkenes serves as the anchor in the active site can be studied via incorporation of deuterium in the final product based on the known mechanism of OYE homologues (hydride addition at Cβ to the binding group and trans-protonation at Cα, Scheme 2).3,5b,c,11 In an attempt to rationalize the complex stereodivergent pattern obtained on type I–IV substrates, the following strategy was adopted (exemplified for 3a in Scheme 5): incorporation of D-labeled hydride at Cβ (via GDH-based NADD recycling using glucose-d1) or D-labeled proton at Cα (using buffered D2O) was implemented in the bioreduction of (E)- and (Z)-2a with NCR, of 3a with OYE2 and XenA, of 5a with NCR and of exo-8a with OYE3 (Table 3). The reactions were run on larger scale (8–16 mL) to allow purification and NMR analysis of deuterated products. To explain the stereochemical outcome of bioreductions (Tables 1 and 2), two types of flip of the substrate in the active site can be envisaged: (i) a ‘bottom-top’ flip switching the activating/binding group or (ii) a 180° ‘right/left’ flip of the substrate around the CC bond axis (Scheme 3b), which maintains the binding group but results in a hydride attack from opposite faces.5a In addition, s-cis and trans-rotamers cannot be excluded with 2a and 3a.6b Analysis of NMR spectra (ESI†) clearly indicated that for 2a, 3a and 5a, the ketone carbonyl group is the dominating activating/anchoring group (going in line with its superior electron-withdrawing property relative to the ester), which allows only a single binding mode for 3a and 5a. With exo-8a, the labeling results are challenging to interpret, suggesting that racemic product results from mixed binding modes (data not shown).
 | ||
| Scheme 5 Elucidation of the anchor group of di-activated substrates (exemplified for 3a) in ene-reductases via incorporation of deuterium. | ||
| Entry | Substrate | Method | Yieldc/% | ee/% | Anchor |
|---|---|---|---|---|---|
| a Reaction conditions: Substrate (10 mM) in Tris-HCl buffer (50 mM, pH 7.5) containing 1 mM of NAD+, 10 U of GDH and 20 mM glucose-d1 and the biocatalyst (final protein concentration 100 μg mL−1, ∼2.5 μM). Reaction mixture shaken for 24 h (second aliquot of enzyme added after 8 h) at 30 °C and 120 rpm and extracted with CDCl3. b Reaction conditions: Substrate (10 mM), cofactor NADH (15 mM) and the biocatalyst (final protein concentration 100 μg mL−1, ∼2.5 μM) in a buffer prepared using D2O and DCl solution for adjustment of the pD. Reaction mixture shaken for 24 h (second aliquot of enzyme added after 8 h) at 30 °C and 120 rpm and extracted with CDCl3. c Isolated yield (conversion indicated in brackets); n.d. not determined. | |||||
| 1 | (E)-2a | NCR/NADDa | 53 (>99) | >99(S) | Ketone |
| 2 | (Z)-2a | NCR/D2Ob | 40 (>99) | 72(R) | Ketone |
| 3 | 3a | OYE2/NADDa | 62 (n.d.) | 76(S) | Ketone |
| 4 | 3a | XenA/NADDa | 54 (n.d.) | >99(R) | Ketone |
| 5 | 5a | NCR/D2Ob | 38 (97) | 62(R) | Ketone |
| 6 | exo-8a | OYE3/D2Ob | 63 (95) | rac | Mixed |
Determination of absolute configuration
Absolute configurations were assigned by co-injection with authentic reference materials, by comparison with literature data (retention times and elution order on GC or HPLC under identical experimental conditions, or optical rotation). Lipase-catalyzed transesterification of 1b to 2b using immobilized CAL-B proceeds without change of absolute configuration and allowed determination of the absolute configuration of 1b. The absolute configuration of 2b was determined based on known (S)-stereoselectivity of YqjM in the reduction of substrate (E)-2a.3 Similarly, the absolute configuration of 3b and 11b was assigned based on the known (R)-stereoselectivity of YqjM on 3a and 11a.12 CAL-B catalyzed transesterification to corresponding ethyl esters allowed determination of the absolute configuration of 4b and 12b. The absolute configuration of 5b and 6b was determined by comparison of retention times on GC from literature using identical temperature program and GC column.13 The absolute configuration of 8b was determined via bioreduction of rac-8b (synthesis described in ESI†) with baker's yeast, which proceeds via kinetic resolution and converts (R)-8b to (1R,2S)-8c (with minor amounts of (1S,2S)-8c), leaving (S)-8b unreacted (Scheme 6).14 Transesterification of (S)-8b to ethyl ester (S)-9b allowed assignment of the absolute configuration of 9b. Finally, the absolute configuration of 10b was determined by deprotection of the benzyl ester to yield the corresponding acid 7b, followed by esterification to methyl ester 8b (procedure in ESI†). Comparison of retention times on GC allowed assignment of the absolute configuration.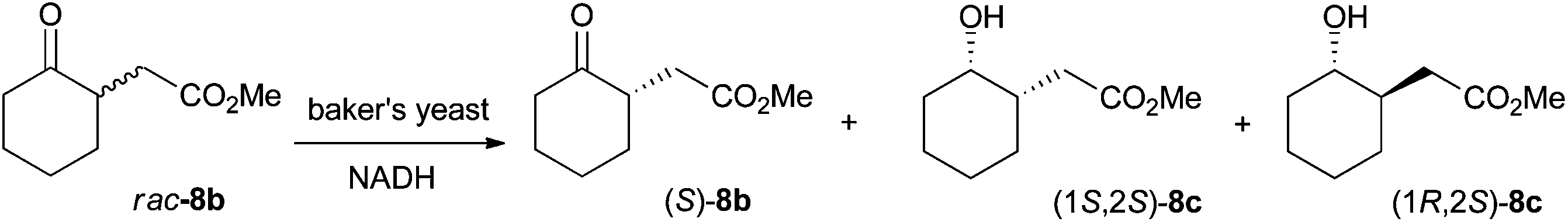 | ||
| Scheme 6 Baker's yeast catalyzed bioreduction of rac-8b for determination of absolute configuration.14 | ||
Conclusions
With few exceptions, ene-reductases from the OYE family have been so far applied to the reduction of comparably simple molecules bearing only a single activating group.15 Although a single strong electron-withdrawing group, e.g. –CHO, –CRO, –NO2, sufficiently activates the CC bond and ensures good reactivity with ene-reductases, the corresponding products are limited in view of follow-up chemistry. Here, we focused on the preparation of a broad range of nonracemic α- and β-substituted γ-oxo esters, which offer two reactive ‘handles’ – carbonyl and ester functionality – for further transformations. To date, the catalytic behaviour of ene-reductases cannot be predicted based on amino acid sequence or protein structure, even for highly homologous proteins.16 Consequently, access to a particular stereoisomeric product requires rigorous screening of substrates and proteins, which leads to identification of matching enzyme–substrate pairs. Access to both enantiomeric products may be achieved via enzyme- or substrate-based stereocontrol.6a In summary, (i) for type I substrates, OYE3 is the catalyst of choice; (ii) with type II substrates, YqjM, OPR1 and NCR are excellent candidates for both 5- and 6-membered rings; (iii) overall, ethyl esters are slightly superior within type I and II substrates; (iv) with type III substrates, the use of a larger ester group in combination with NCR is necessary to achieve high conversion and ee, which was demonstrated on preparative scale (>100 mg final ester product in optically pure form); (v) type IV/V compounds are not suitable as substrates (Fig. 1). In general, the ketone carbonyl group is preferred over the ester moiety for binding of the substrate in the enzyme active site, in line with its superior electron-withdrawing properties.
Experimental
General
GC-MS analyses were performed on a HP Agilent Technologies 6890 Series GC system equipped with a 5973 mass selective detector, a 7683 Series injector and an Agilent HP-5 MSI column (30 m, 0.25 mm ID, 0.25 μm film thickness). GC-FID measurements were carried out on a HP Agilent Technologies 7890A GC system equipped with a FID detector and a 7693 Autosampler or a 7683B Injector in combination with a 7683 Series Autosampler. For HPLC analysis, a Shimadzu LC-20AD HPLC system with a DGU-20A5 degasser, a SIL-20AC autosampler, SPD-M20A diode array detector and a CTO-20AC column oven, equipped with a Chiralcel OD-H column (25 × 0.46 cm) was used. All enzymes were overexpressed and purified as reported (see ESI† for details on cloning and purification procedures)17 and used as purified protein (>90% purity): OYE1 (Old Yellow Enzyme 1 from Saccharomyces pastorianus), OYE2 (Old Yellow Enzyme 2 from Saccharomyces cerevisiae), OYE3 (Old Yellow Enzyme 3 from Saccharomyces cerevisiae), NCR (NAD(P)H-dependent 2-cyclohexen-1-one reductase from Zymomonas mobilis), YqjM (from Bacillus subtilis), OPR1 (12-Oxophytodienoate reductase 1 from Lycopersicon esculentum), OPR3 (12-Oxophytodienoate reductase 3 from Lycopersicon esculentum), XenA (Xenobiotic reductase A from Pseudomonas putida II-B), EBP1 (Estrogen binding protein from Candida albicans).Standard procedure for bioreduction reactions
To a solution of Tris-HCl buffer (800 μL, 50 mM, pH 7.5) containing the substrate (10 mM) and the cofactor NADH (15 mM) was added an aliquot of the respective enzyme (final protein concentration 100 μg mL−1, ∼2.5 μM). When a GDH recycling system was employed, the buffer contained 100 μM NAD+, 20 mM glucose and 10 U GDH. Substrates were added from a stock solution in DMSO or EtOH to overcome solubility problems (final concentration of DMSO or EtOH 1% v/v). Control experiments in the absence of enzyme were included in every screening and all reactions were run in duplicates. The reaction mixture was shaken for 24 h at 30 °C and 120 rpm. Afterwards, the reaction was extracted with EtOAc (2 × 500 μL), the combined organic layers were dried over Na2SO4 and transferred into GC-vials. Products were identified by comparison with authentic reference material (commercially available or synthesized according to published methods, for details see ESI†).Scale-up of reaction with endo-10a and NCR
The reaction was performed according to the standard procedure adapted to 400 mg endo-10a (1.6 mmol, 400 mg). Samples were placed in the shaker at 30 °C and 120 rpm. Additional aliquots of NCR and NADH were added after 8 h, 24 h, 32 h and 48 h. After 62 h, the samples were transferred into a separatory funnel, extracted with tert-butyl methyl ether (3 × 30 mL), ethyl acetate (3 × 30 mL) and CH2Cl2 (3 × 30 mL). The combined organic layers were dried over Na2SO4 and the solvent was evaporated under reduced pressure, yielding product 10b with remaining endo-10a (81% conversion). Purification via flash column chromatography (hexane/ethyl acetate 3:1) afforded the product 10b in 27% isolated yield (108 mg). Low isolated yield was due to losses during extraction and incomplete elution from flash column chromatography. For spectra see ESI.†
General procedure for isotopic labeling using deuterated buffer
The buffer was prepared using D2O and DCl-solution for adjustment of the pD. To a solution of Tris-DCl buffer (800 μL, 50 mM, pH 7.5) containing the substrate (10 mM) and NADH (15 mM), an aliquot of the respective enzyme (final protein concentration 100 μg mL−1, ∼2.5 μM) was added. Substrates were added from a stock solution in DMSO or EtOH to overcome solubility problems (final concentration of DMSO or EtOH 1% v/v). The experiments were run in 10–20 Eppendorf tubes (800 μL each). The mixture was shaken 30 °C and 120 rpm and another aliquot of enzyme was added after 8 h to ensure high conversion. After 24 h, the samples were transferred into a separatory funnel, extracted with CDCl3 (3 × 6 mL), and the combined organic layers were dried over Na2SO4. Evaporation of the solvent under reduced pressure yielded the desired product. The samples were taken up in CDCl3 and analyzed by NMR and GC (for conversion, yield and ee see Table 3).General procedure for isotopic labeling using NADD
To a solution of Tris-HCl buffer (800 μL, 50 mM, pH 7.5) containing the substrate (10 mM), 1 mM of NAD+, 10 U of GDH and 20 mM glucose-d1 was added an aliquot of the respective enzyme (final protein concentration 100 μg mL−1, ∼2.5 μM). Substrates were added from a stock solution in DMSO or EtOH to overcome solubility problems (final concentration of DMSO or EtOH 1% v/v). The reaction was performed in 10–20 Eppendorf tubes (800 μL each). The mixture was shaken at 30 °C and 120 rpm; another aliquot of enzyme was added after 8 h to ensure high conversion. After 24 h, the samples were transferred into a separatory funnel, extracted with CDCl3 (3 × 6 mL), and the combined organic layers were dried over Na2SO4. Evaporation of the solvent under reduced pressure yielded the desired product. The samples were taken up in CDCl3 and analyzed by NMR and GC (for conversion, yield and ee see Table 3).Procedure for CAL-B catalyzed transesterification
To a solution of methyl ester (10 mM of 1b, 3b or 11b obtained in >99% ee by bioreduction) in 1 mL EtOAc, 10 mg of Novozym 435 lipase were added and the reaction was run at 30 °C and 120 rpm until full conversion was reached as analyzed by GC-MS. The lipase was removed by filtration and the corresponding ethyl ester (2b, 4b or 12b) was analyzed by GC-FID.Bioreduction of rac-8b using baker's yeast14
20 mg baker's yeast (Spar Natur Pur® Bio Hefe) were rehydrated in 1 mL KPi buffer (50 mM, pH 7.5) for 30 min at 30 °C and 120 rpm. Afterwards, rac-8b (20 mM) was added from a EtOH stock (200 mM). After 14 h, the yeast was removed by filtration over celite and washed with EtOAc (3 × 2 mL), the aqueous and organic phases were evaporated under reduced pressure (Speedvac), the residue was taken up in EtOAc, ultrasonicated for 30 s, dried over Na2SO4 and transferred into GC-vials for measurement.Analytical methods
The enantiomeric excess of 10b was determined via HPLC using a Daicel Chiralcel OD-H column (250 mm × 4.6 mm, 5 μm), n-heptane/i-PrOH (95:5 v/v, isocratic) at 30 °C and 1 mL min−1. Retention times: endo-10a: 14.2 min, exo-10a: 7.9 min, (S)-10b: 9.1 min, (R)-10b: 11.9 min.
Acknowledgements
The research leading to these results has received funding from the Innovative Medicines Initiative Joint Undertaking project CHEM21 (http://www.chem21.eu) under grant agreement no. 115360, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in kind contribution. Barbara Grischek, Tamara Reiter, Somayyeh Gandomkar and Olivia Laggner (University of Graz) are thanked for their excellent technical assistance. Klaus Zangger and Bernd Werner (University of Graz) are acknowledged for their assistance in recording NMR spectra.Notes and references
- (a) H. Wu, H. Zhang and G. Zhao, Tetrahedron, 2007, 63, 6454 CrossRef CAS; (b) D. Enders, O. Niemeier and T. Balensiefer, Angew. Chem., Int. Ed., 2006, 45, 1463 CrossRef CAS PubMed; (c) M. Ganaha, S. Yamauchi and Y. Kinoshita, Biosci. Biotechnol. Bioeng., 1999, 63, 2025 CrossRef CAS PubMed; (d) S.-I. Hashimoto, T. Shinoda, Y. Shimada, T. Honda and S. Ikegami, Tetrahedron Lett., 1987, 28, 637 CrossRef CAS; (e) Z. Yin, L. R. Whittell, Y. Wang, S. Jergic, C. Ma, P. J. Lewis, N. E. Dixon, J. L. Beck, M. J. Kelso and A. J. Oakley, J. Med. Chem., 2015, 58, 4693 CrossRef CAS PubMed.
- A. G. Draffan, G. R. Evans and J. A. Henshilwood, Chirality and Biological Activity, in Burger’s Medicinal Chemistry and Drug Discovery, Wiley, 2003, vol. 1, pp. 781–826 Search PubMed.
- T. Classen, M. Korpark, M. Schölzel and J. Pietruszka, ACS Catal., 2014, 4, 1321 CrossRef CAS.
- G. Tasnadi, C. K. Winkler, D. Clay, N. Sultana, W. M. F. Fabian, M. Hall, K. Ditrich and K. Faber, Chem. – Eur. J., 2012, 18, 10362 CrossRef CAS PubMed.
- (a) C. Stueckler, C. K. Winkler, M. Hall, B. Hauer, M. Bonnekessel, K. Zangger and K. Faber, Adv. Synth. Catal., 2011, 353, 1169 CrossRef CAS; (b) E. Brenna, F. G. Gatti, A. Manfredi, D. Monti and F. Parmeggiani, Adv. Synth. Catal., 2012, 354, 2859 CrossRef CAS; (c) E. Brenna, M. Crotti, F. G. Gatti, A. Manfredi, D. Monti, F. Parmeggiani, A. Pugliese and D. Zampieri, J. Mol. Catal. B: Enzym., 2014, 101, 67 CrossRef CAS; (d) C. K. Winkler, D. Clay, N. G. Turrini, H. Lechner, W. Kroutil, S. Davies, S. Debarge, P. O'Neill, J. Steflik, M. Karmilowicz, J. W. Wong and K. Faber, Adv. Synth. Catal., 2014, 356, 1878 CrossRef CAS PubMed.
- (a) C. Stueckler, M. Hall, H. Ehammer, E. Pointner, W. Kroutil, P. Macheroux and K. Faber, Org. Lett., 2007, 9, 5409 CrossRef CAS PubMed; (b) G. Oberdorfer, K. Gruber, K. Faber and M. Hall, Synlett, 2012, 1857 CAS.
- (a) B. Kosjek, F. J. Fleitz, P. G. Dormer, J. T. Kuethe and P. N. Devine, Tetrahedron: Asymmetry, 2008, 19, 1403 CrossRef CAS; (b) M. Hall, C. Stueckler, B. Hauer, R. Stuermer, T. Friedrich, M. Breuer, W. Kroutil and K. Faber, Eur. J. Org. Chem., 2008, 1511 CrossRef CAS; (c) R. Agudo and M. T. Reetz, Chem. Commun., 2013, 49, 10914 RSC.
- Y. S. Niino, S. Chakraborty, B. J. Brown and V. Massey, J. Biol. Chem., 1995, 270, 1983 CrossRef CAS PubMed.
- C. K. Winkler, C. Stueckler, N. J. Mueller, D. Pressnitz and K. Faber, Eur. J. Org. Chem., 2010, 6354 CrossRef CAS.
- N. J. Mueller, C. Stueckler, M. Hall, P. Macheroux and K. Faber, Org. Biomol. Chem., 2009, 7, 1115 CAS.
- K. Durchschein, S. Wallner, P. Macheroux, K. Zangger, W. M. F. Fabian and K. Faber, ChemBioChem, 2012, 13, 2346 CrossRef CAS PubMed.
- D. J. Bougioukou, S. Kille, A. Taglieber and M. T. Reetz, Adv. Synth. Catal., 2009, 351, 3287 CrossRef CAS.
- J. R. de Alaniz, M. S. Kerr, J. L. Moore and T. Rovis, J. Org. Chem., 2008, 73, 2033 CrossRef PubMed.
- M. Ganaha, Y. Funabiki, M. Motoki, S. Yamauchi and Y. Kinoshita, Biosci., Biotechnol., Biochem., 1998, 62, 181 CrossRef CAS PubMed.
- K. Faber and M. Hall, in Science of Synthesis: Biocatalysis in Organic Synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Georg Thieme Verlag, Stuttgart, 2015, vol. 2, p. 213 Search PubMed.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892 CrossRef CAS.
- C. K. Winkler, D. Clay, E. van Heerden and K. Faber, Biotechnol. Bioeng., 2013, 110, 3085 CrossRef CAS PubMed.
- J. B. Tuttle, S. G. Ouellet and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 12662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and NMR data. See DOI: 10.1039/c6gc02493a |
| This journal is © The Royal Society of Chemistry 2017 |