
Electrochemical synthesis of ammonia directly from N2 and water over iron-based catalysts supported on activated carbon†
Baochen
Cui
ab,
Jianhua
Zhang
ab,
Shuzhi
Liu
*ab,
Xianjun
Liu
ab,
Wei
Xiang
ab,
Longfei
Liu
ab,
Hongyu
Xin
ab,
Matthew J.
Lefler
c and
Stuart
Licht
*c
aCollege of Chemistry and Chemical Engineering, Northeast Petroleum University, Daqing 163318, China. E-mail: cuibaochen2005@163.com; Tel: +86 459 6504758
bProvince Key Laboratory of Oil and Natural Gas Chemical Industry, Northeast Petroleum University, Daqing 163318, China
cDepartment of Chemistry, George Washington University, Washington DC 20052, USA. E-mail: slicht@gwu.edu; Tel: +1 202 994 6121
First published on 23rd November 2016
Abstract
A new green methodology for the CO2-free synthesis of ammonia from air and water is presented. The conventional production of H2 utilizes fossil fuels and causes a massive greenhouse gas release, making ammonia production one of the most energy intensive and highest CO2 emitting manufacturing processes. In 2014 we introduced an alternative method for efficient ammonia synthesis that utilizes water (along with N2) instead of H2 based on electrolysis of nano-structured catalyst suspensions of Fe2O3 in low temperature aqueous or higher temperature molten hydroxide electrolytes. Here, this is replaced with a solid Fe2O3 catalyst confined to activated charcoal opening pathways to improve the rate and efficiency of ammonia production. Cyclovoltammetric studies show that Fe2O3/AC catalysts can inhibit competing hydrogen reduction and enhance reduction of iron. This iron-based catalyst supported on activated carbon (Fe2O3/AC) was prepared for use as an electrocatalyst for the electrochemical synthesis of ammonia in molten hydroxide (NaOH–KOH) directly from wet N2 at atmospheric pressure. XRD analysis shows that the catalyst exhibits a Fe2O3 structure. At 250 °C, a voltage of 1.55 V with a current density of 49 mA cm−2 yielded the highest rate of ammonia formation, 8.27 × 10−9 mol (s cm2)−1. The highest coulombic efficiency for the 3e− per ammonia formation, 13.7%, was achieved at 1.15 V with a lower average current density of 11 mA cm−2. This is a promising simple technology for the sustainable synthesis of ammonia in the future.
1. Introduction
Ammonia (NH3) is the second most commonly produced chemical in the world.1 The global industrial production of ammonia was ∼145 million metric tons in 2014.2 Ammonia is not only a major and important raw material for industry and agriculture, but is also a significant energy storage intermediate and a vital source of hydrogen (the hydrogen content in liquid ammonia is 17.6 wt% compared to methanol's 12.5 wt% hydrogen content).3–7 The primary industrial method for ammonia synthesis is the Haber–Bosch process, created by Fritz Haber in 1905 and developed for the industry by Carl Bosch in 1910, in which nitrogen gas and hydrogen gas react (N2 + 3H2 → 2NH3) over an enriched iron or cobalt catalyst at ∼450 °C under high pressure ranging from 15–30 MPa.8A green, low carbon footprint alternative is needed to replace the old CO2-extensive Haber–Bosch process to mitigate associated climate effects. In the energy-intensive Haber–Bosch process, almost all of the hydrogen gas is produced by the steam reformation of natural gas (CH4 + 2H2O → 4H2 + CO2), which consumes 3–5% of the global natural gas supply and annually emits 200 million tonnes of CO2 into the atmosphere.9,10 As a green alternative, water, rather than methane, is an ideal hydrogen source due to its natural abundance, cleanliness, and uniformity of distribution over the world. Therefore, it is of industrial interest to develop an electrochemical process that uses renewable electricity to convert water (as an alternative hydrogen source) and either N2 or air into ammonia under more reasonable conditions, such as atmospheric pressure and lower temperatures. This novel process directly utilizes water in place of high-purity hydrogen that is currently required for the existing Haber–Bosch-based production of ammonia, and will provide a green ammonia synthesis pathway without carbon dioxide emission.
In 2009, Skodra and Stoukides reported that NH3 was electrochemically produced from steam and N2 in electrolytic cells using SrCe0.95Yb0.05O3−δ as a solid electrolyte at 450–700 °C.11 Ammonia was produced with a maximum formation rate of 4.0 × 10−13 mol (s cm2)−1 at 650 °C using a precious metal (Ru/MgO) catalyst as the cathode. The low activity is attributed primarily to the poor conductivity of the working electrode. In addition, the Ru/MgO catalyst is expensive due to the use of the precious metal ruthenium.
Non-noble, low-cost catalysts and molten salt electrolytes have both garnered attention as opening alternative pathways to more practical ammonia syntheses and to enhance the conductivity of the synthesis electrolyte. Amar et al. reported that ammonia was successfully synthesized from H2O and N2 using CoFe2O4–Ce0.8Gd0.18Ca0.02O2−δ and Sm0.5Sr0.5CoO3−δ–Ce0.8Gd0.18Ca0.02O2−δ composites as the cathode and anode, respectively, and a composite of Ce0.8Gd0.18Ca0.02O2−δ–(Li,Na,K)2CO3 as the conducting electrolyte.12 A maximum ammonia formation rate of 6.5 × 10−11 mol (s cm2)−1 and coulombic efficiency (comparing each 3 mol or Faraday of electron consumed to each mole of ammonia synthesized) of 0.095% were observed at 400 °C and 1.6 V. One of the low-cost catalysts, perovskite oxides, have drawn considerable interest owing to their ease of synthesis, good catalytic activity, low cost, and thermal stability.13 When using the perovskite oxide La0.6Sr0.4Fe0.8Cu0.2O3−δ as the electrocatalyst in place of CoFe2O4 for the electrochemical synthesis of ammonia directly from wet nitrogen, based on the same electrolyte and anode, the maximum ammonia formation rate was found to be 5 × 10−11 mol (s cm2)−1 at 400 °C when a voltage of 1.4 V was applied.14 It has also been reported by Lan et al. that using the perovskite oxide La0.8Cs0.2Fe0.8Ni0.2O3−δ as the electrocatalyst and a composite electrolyte of Ce0.8Gd0.2O2−δ and (Li,Na,K)2CO3 for the electrochemical synthesis of ammonia directly from wet air (3 mol% H2O), the ammonia formation rate and coulombic efficiency reached 9.21 × 10−11 mol (s cm2)−1 and 0.06% at 400 °C and 1.4 V.15 These values increased to 1.23 × 10−10 mol (s cm2)−1 (at 1.4 V) and 0.52% (at 1.5 V) at 400 °C when wet N2 (3 mol% H2O) was fed into the reactor instead of wet air. Lan et al. also further reported that by replacing the La0.8Cs0.2Fe0.8Ni0.2O3−δ electrocatalyst with a perovskite oxide Pr0.6Ba0.4Fe0.8Cu0.2O3−δ, ammonia formation rates of 1.07 × 10−10 mol (s cm2)−1 and 1.83 × 10−10 mol (s cm2)−1 were obtained at 400 °C and a voltage of 1.4 V with wet N2.16 When wet air was used, the highest coulombic efficiency was 0.77% at 1.5 V, and in the case of wet N2, the highest coulombic efficiency was 5.33% at 1.7 V.
In 2005, Murakami et al. reported that using the eutectic mixture of LiCl–KCl–CsCl–Li3N as the electrolyte, a glassy carbon rod as the working electrode, and a nitrogen gas electrode consisting of porous nickel as the counter electrode for electrolytic synthesis of ammonia from water and N2, the average ammonia synthesis rate was 2 × 10−8 mol (s cm2)−1 and the current efficiency reached 23% at 300 °C and 2.9 V.17 However, when glassy carbon was used as the anode material, the electrode was consumed, emitting carbon dioxide instead of oxygen. The BDD electrode can work as a non-consumable anode for ammonia synthesis without emitting carbon dioxide in the melt containing nitride ions.18
Ammonia tends to decompose at high temperatures, therefore, a low temperature synthesis is necessary to avoid this degradation.19 In 2000, Kordali et al. reported that using a Nafion separator in aqueous 2 M KOH electrolyte with a Ru/C cathode enabled ammonia synthesis from water and nitrogen at a rate and maximum (coulombic) efficiency of 2.78 × 10−12 mol (s cm2)−1 and 0.9% at 20 °C, and at 90 °C, a maximum rate of 2.1 × 10−11 mol (s cm2)−1 at 0.2% efficiency.20 In 2013, it was reported that ammonia was formed directly from air and H2O by applying 1.6 V at room temperature and one atmosphere, resulting in an ammonia synthesis rate of 1.14 × 10−9 mol (s cm2)−1 and about 0.52% efficiency by using Pt/C on a gas diffusion layer at both electrodes and a Nafion 211 membrane as the electrolyte.17 Ammonia is a weak base and readily reacts with an acidic membrane, reducing the proton conductivity. The authors also reported that the highest ammonia formation rate (9.37 × 10−10 mol (s cm2)−1) and efficiency (0.83%) were obtained by using a Li+/H+/NH4+ mixed conducting electrolyte (a Nafion 211 membrane treated with 0.1 M Li2SO4 aqueous solution before use) and Pt/C at both electrodes at 80 °C when a voltage of 1.2 V was applied.21
In 2014, we presented a carbon dioxide (CO2)-free electrochemical pathway in which ammonia (NH3) is produced by electrolysis of N2 and steam at 200 °C in a molten hydroxide suspension of nano-Fe2O3 at nickel and Monel electrodes.22 The coulombic efficiency of the reaction is high, ∼35%, when the current density is low (2 mA cm−2), but decreases to 7% at higher current densities (25 mA cm−2), producing NH3 at maximum rates of 2.4 × 10−9 and 6.7 × 10−9 mol (s cm2)−1, respectively. Constant current electrolysis is driven at 1.2 V for a 2 mA cm−2 reaction, or at 1.4 V for a 25 mA cm−2 reaction.22 We found the relationship between coulombic efficiency and current density to be inversely proportional. At 200 mA cm−2, over 90% of the applied current drives H2, rather than NH3, formation.23 Many studies also observed that N2 reduction will necessarily compete with the hydrogen gas evolution in the electrosynthesis of ammonia using water or steam as a reactant in lieu of hydrogen.14,18,22 We also found that the simple dispersion of nano-iron oxides in the electrolyte was not conducive to long-term stability of the cell, as electrostatics tend to coagulate the nanoparticles over time.22
In this work, for the first time, we demonstrated that ammonia can be synthesized directly from N2 and H2O over an iron-based catalyst supported on activated carbon with supplied electricity, which can be obtained from renewable resources, such as solar or wind.
2. Experimental
Sodium hydroxide (AR, >96% NaOH) and potassium hydroxide (AR, >82% KOH) are combined to form the eutectic molten hydroxide electrolyte. Iron nitrate (AR, ≥98.5% Fe(NO3)3·9H2O, Tianjin Guangfu Fine Chemical Research Institute, China) and activated carbon (Tianjin Nankai Chemical Factory, China) are used for the preparation of Fe2O3-loaded activated carbon powder. Ferric oxide (Fe2O3, AR, 69.8–70.1% Fe, Tianjin Damao Chemical Reagent Factory, China) is used as a catalyst for control experiment. 2 mm pure Ni wire (99.5%), pure Ni shim (99.95%), and 304 stainless steel mesh (200 mesh) were used to make electrodes.Fe(NO3)3·9H2O was used as the iron source, and activated carbon was used as a support for the preparation of Fe2O3-loaded activated carbon powder (Fe2O3/AC). Prior to loading, activated carbon was ground and screened with 200 mesh screens. Then, a calculated amount of Fe(NO3)3·9H2O (the weight ratio of iron to activated carbon is 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) was dissolved in deionized water. Fe(NO3)3·9H2O was loaded on activated carbon by an initial impregnation for 24 hours via an aqueous solution soak, and the mixture was dried for 3 h at 105 °C, followed by calcinations for 3 h at 200, 300 and 400 °C in air.
:50) was dissolved in deionized water. Fe(NO3)3·9H2O was loaded on activated carbon by an initial impregnation for 24 hours via an aqueous solution soak, and the mixture was dried for 3 h at 105 °C, followed by calcinations for 3 h at 200, 300 and 400 °C in air.
Transmission electron microscopy (TEM) was performed with a JEM-2100 apparatus (JEOL Ltd) at an acceleration voltage of 200 kV. The Fe compound obtained on the carbon was identified to be Fe2O3 by X-ray diffraction (XRD) with a Cu Kα source (Rigaku D/MAX-2200). Each sample was scanned from 2θ 10 to 80°. Cyclic voltammetry (CV) measurements were carried out in a two-electrode cell on an electrochemical workstation (CS350, Wuhan CorrTest instruments Co. Ltd, China). The two-electrode cell consisted of a working electrode of 304 stainless steel mesh (200 mesh, 2 × 2.5 cm), a counter electrode of nickel shim (2 × 2.5 cm, also used as a reference electrode), and the electrolyte of molten 50/50 mol% NaOH/KOH eutectic.
The ammonia electrosynthesis chamber contained a 304 stainless steel mesh (200 mesh, 2 × 2.5 cm) cathode and a nickel shim (2 × 2.5 cm) anode with 7 g of Fe2O3/AC suspended in 70 g of molten hydroxide electrolyte. The crucible, made of alumina (99.7%, OD 40 mm, height 170 mm, China), is sealed to allow gas inlet at the cathode and gas outlet from the head space. External connections for the electrodes are spot welded to 2 mm Ni wire (enclosed in an alumina tube with an inner diameter of 3 mm). The reactants, N2 (99.999%, 250 ml min−1) and water vapor, are bubbled through the mesh over the cathode at atmospheric pressure. The combined gas products exit through an exit tube in the chamber head space. Exit gas is bubbled through an ammonia water trap consisting of a 500 mL of 0.001 M H2SO4 solution, changed regularly for ammonia analysis. Ammonia concentration was determined by absorption at 697 nm on a visible spectrophotometer (722E, Shanghai Spectrum Instruments Co. Ltd, China) in a conventional 1 cm path length cuvette by the salicylic method.22
The cell is placed in a 1800 W band heater and insulated by rigid thermal shock resistant ceramic insulation without a binder. Temperature is monitored by thermocouple and controlled by a temperature controller (708P, YUDIAN automation technology Co. Ltd, China). Constant voltage electrolysis was performed and controlled by a LANHE battery testing system (CT2001A, Wuhan LAND electronics Co. Ltd, China), and the current–time curve was recorded at the same time.
3. Results and discussion
In order to consider the potential for electrochemical synthesis of ammonia, a thermodynamic evaluation of the synthesis of ammonia from H2O and N2 was carried out.24–26 We described a mechanism of the ammonia synthesis from H2O and N2 by electrolysis, consistent with our previous experimental measurements in which iron oxide is electrochemically reduced to iron (eqn (1)) and chemically reacts with N2 and water (eqn (2)).23| Fe2O3 → 2Fe + 3/2O2 | (1) |
| 3H2O + N2 + 2Fe → 2NH3 + Fe2O3. | (2) |
Combining eqn (1) with eqn (2) provides the electrolytic synthesis of ammonia from N2 and water in eqn (3):
| N2 + 3H2O → 2NH3 + 3/2O2 | (3) |
Whereas the chemical combination of nitrogen and hydrogen to form ammonia is thermodynamically favored, it is kinetically disfavored, requiring high pressure, high temperature, and catalysts, and also requiring pre-formation of the hydrogen reactant. The chemical reaction of nitrogen, water, and iron is also thermodynamically favored, but requires neither high temperature, high pressure, nor hydrogen. We calculate the Gibbs free energies of eqn (1)–(3), as summarized in Fig. 1. Whereas these energy calculations show the standard Gibbs free energy change of eqn (1) and (3) to be positive, thus rendering these reactions non-spontaneous, the reaction listed as eqn (2) has a negative ΔG at temperatures below 350 °C, therefore the reaction occurs at these temperatures without energy input from external sources (but consumes iron by oxidation, unless the iron is electrochemically regenerated in accordance with eqn (1)).
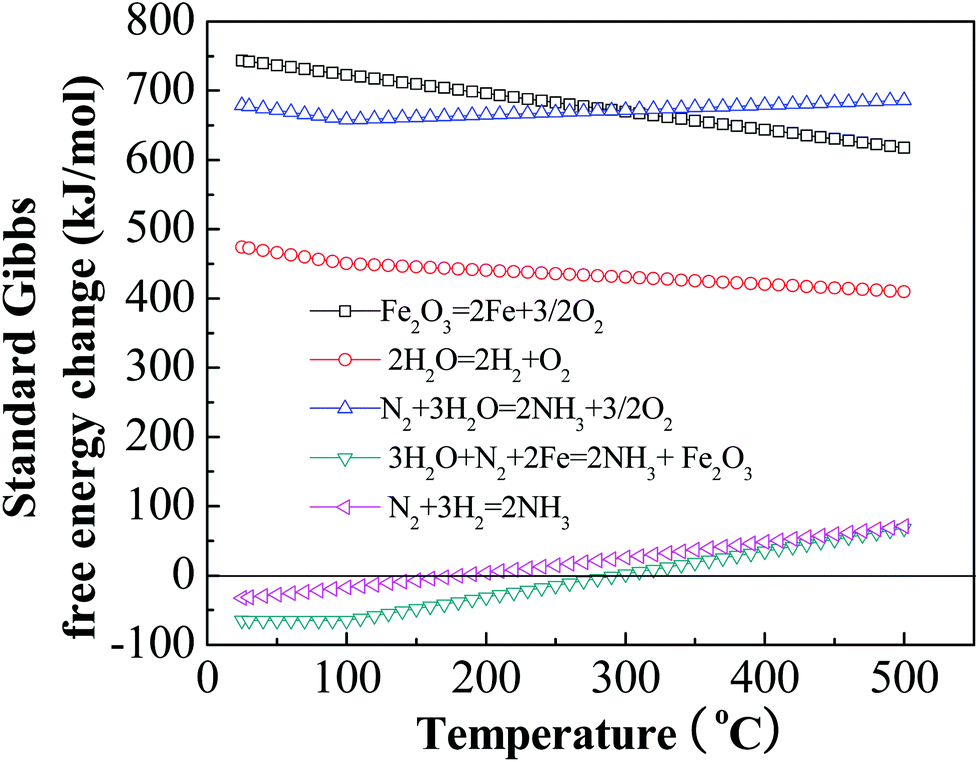 | ||
| Fig. 1 The Gibbs free energy change for electrochemical synthesis of ammonia from N2 and H2O in a molten hydroxide suspension of Fe2O3-based catalysts. Calculations of Gibbs free energy changes are based on the temperature variation of the individual species’ thermochemical data. | ||
The thermodynamic electrolysis potential (standard reduction potential) for each reaction at a given temperature, E°(T), can be derived with the equation E°(T) = ΔG°(T)/nF, where ΔG°(T) is the Gibbs free energy change at a temperature (T), n is the electrons transferred, and F is Faraday's constant. For example, according to Fig. 2, at 250 °C, 1.18 V is required for the reduction of iron oxide, 1.15 V for ammonia formation, and 1.13 V is necessary for water splitting. It is expected that ammonia would be produced as long as the applied voltage is higher than the electrolysis potential for the reduction of Fe2O3 to Fe (Fig. 2). Additionally, it is thermodynamically feasible (as shown in Fig. 2) that along with the reduction of Fe2O3 to Fe, the electrolysis of water into hydrogen and oxygen eqn (4) should occur simultaneously in the molten hydroxide chamber due to the electrolysis potential for water splitting being below that of Fe2O3 reduction.
| 2H2O → 2H2 + O2 | (4) |
 | ||
| Fig. 2 Thermodynamic electrolysis potentials for electrochemical synthesis of ammonia from N2 and H2O in a molten hydroxide suspension of Fe2O3-based catalysts. | ||
Cyclic voltammetry studies were carried out with a two-electrode cell assembly that had the 304 stainless steel mesh electrode as the working electrode and nickel shim as the counter electrode and the reference electrode. The electrolyte used was a molten hydroxide (a NaOH–KOH eutectic electrolyte). Cyclic voltammetry measurements were recorded at a sweep rate of 100 mV s−1 and in the range of −1.25 to 0 V. The voltammograms of the 304 stainless steel mesh electrode during the initial three cycles are shown in Fig. 3a. One cathodic peak, Ic1, was observed, at around −0.05 V, which corresponds to the hydrogen evolution peak in a NaOH–KOH eutectic electrolyte without adding catalysts. Evolution of hydrogen gas bubbles was readily observed visually on the cathode. After the cyclic voltammetry measurement, the Fe2O3 powder was added to the electrolyte, and then cyclic voltammetry studies were conducted again. From Fig. 3b, it can be seen that the hydrogen evolution peak, Ic1, is present at around −0.45 V. The current formed another cathodic peak, Ic2, at around −0.82 V, which corresponds to the reduction peak of iron deposition. The reduction peak of iron deposition was separated from the hydrogen evolution peak. Upon the sweep reversal in the anodic direction, the current formed one small anodic peak, Ia1, at around −0.23 V, which corresponds to the oxidation of deposited iron, and then diminished. Finally, we added Fe2O3/AC to the electrolyte, and then cyclic voltammetry studies were carried out again. As shown in Fig. 3c, both the hydrogen evolution peak, Ic1, and the reduction peak of iron deposition, Ic2, moved to a less negative potential, and the current under the hydrogen evolution peak decreased while current under the reduction peak of iron deposition increased. This could be attributed to the absorption of Fe2O3/AC on the cathode, which would inhibit hydrogen reduction and enhance reduction of iron. In contrast, as seen in Fig. 3d, the cyclic voltammetry studies show that the two cathodic peaks (Ic1 and Ic2) and anodic peak (Ia1) do not appear when the AC powder was added to the electrolyte, again verifying inhibition of hydrogen evolution by the addition of AC powder.
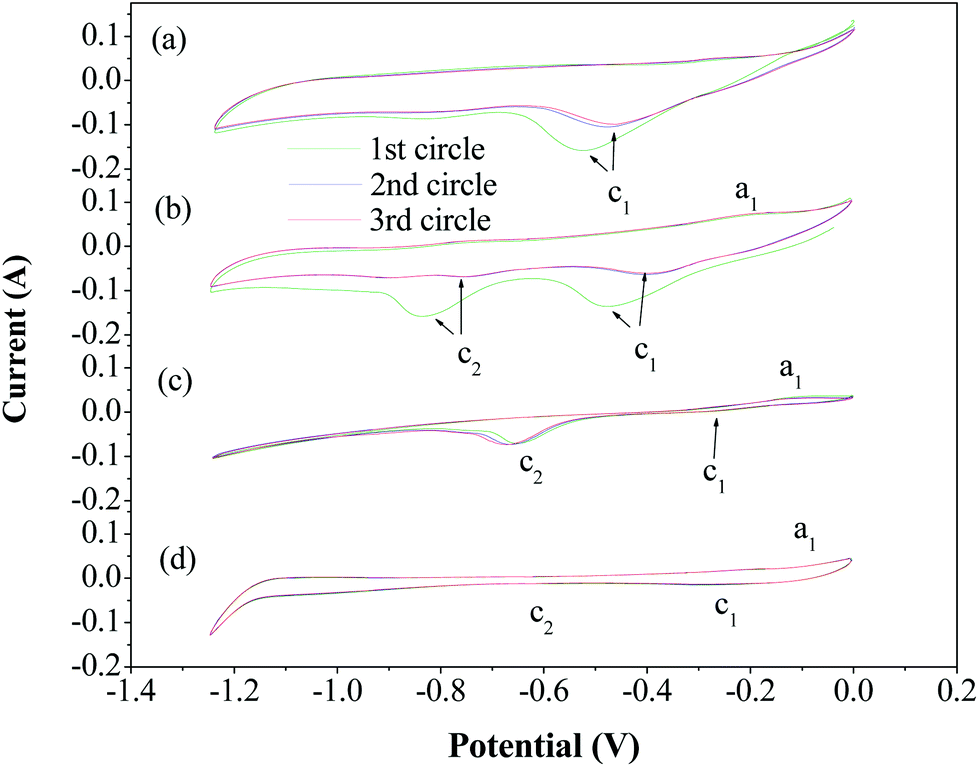 | ||
| Fig. 3 Cyclic voltammograms for: (a) a molten NaOH–KOH eutectic electrolyte, (b) a molten NaOH–KOH eutectic electrolyte with added Fe2O3 powder, (c) a molten NaOH–KOH eutectic electrolyte with added Fe2O3 powder and the Fe2O3/AC catalyst, and (d) a molten NaOH–KOH eutectic electrolyte with added AC powder. Potential scan rates of 100 mV s−1. | ||
The synthesis of ammonia from wet N2 was successfully improved using the Fe2O3/AC catalyst at 250 °C and atmospheric pressure. Fig. S1† presents the coulombic efficiency and the ammonia formation rate at 250 °C with different catalysts or without catalysts in wet N2 when a voltage of 1.15 V is applied. As can be seen, no ammonia was synthesized without catalysts or with AC. Coulombic efficiency remains low for ammonia formation over Fe2O3 powder while larger efficiencies are observed when using a mix of Fe2O3 powder and Fe2O3/AC as the catalyst. The highest coulombic efficiencies result from using only Fe2O3/AC. This can be attributed to the higher viscosity of the mix of Fe2O3 powder and Fe2O3/AC. A similar trend was observed for the ammonia formation rates; despite Fe2O3 powder presenting a high ammonia formation rate at the beginning of the reaction, the rate drops off sharply with reaction time.
Fig. S2† shows a photograph of the electrolyte containing Fe2O3/AC catalysts, cathode, and anode subsequent to electrolysis. The system has settled without bubbling N2 for 2 hours at electrolysis temperature of 250 °C. The crucible is removed after cooling to room temperature. Here we observe that Fe2O3/AC catalysts have neither settled at the bottom of the crucible nor floated to the top of the electrolyte, but have dispersed uniformly throughout the electrolyte. This result differs from the nano-iron oxides used in our previous research.22,23 It is well known that activated carbon can have a high degree of microporosity, given that just one gram of activated carbon has an extremely large surface area. The solid or skeletal density of activated carbon will typically range between 2.0 and 2.1 g cm−3,27 which is close to the density of molten NaOH–KOH eutectic electrolyte, resulting in extremely effective dispersion of activated carbon powder into molten NaOH–KOH eutectic electrolyte. Fig. S3† probes the stability of the ammonia formation rate and efficiency for the NH3 electrolysis over 21 h. The ammonia formation rate and efficiency decrease over the first 7 h and are nearly stable at 1.3–1.68 × 10−9 mol (s cm2)−1, and 6.4–7.2% over the next 14 h of electrolysis, respectively. Fig. S4† shows the morphology images of the Fe2O3/AC catalyst before use, after 1 h electrolysis, 3 h electrolysis and 21 h electrolysis. For the fresh Fe2O3/AC catalyst, a large number of Fe2O3 particles were observed to be supported on AC, furthermore, the Fe2O3 crystallites exhibited a spherical shape with good dispersion. The diameter of Fe2O3 particles was approximately within 10–30 nm, as shown in Fig. S4a.† In contrast, as electrolysis went on, the Fe2O3 crystallite size became bigger, as shown in Fig. S4b–d.†
Fig. 4 uses powder X-ray diffraction to illustrate the temperature dependence of the Fe2O3 structure. Similar to activated carbon, the catalyst calcined at 200 °C did not show the peaks of Fe2O3 due to the low calcination temperature. The catalyst calcined at 300 °C gave sharp 2-theta peaks, which is consistent with the library XRD of Fe2O3 (MDI Jade 5.0, PDF 33-0664). The crystallite size of the Fe2O3 particles was calculated from the half width of the diffraction peak (2θ = 35.64) using the Scherrer equation, D = 0.9λ/βcosθ, where D is the particle size, λ is the X-ray wavelength (Cu Kα radiation, λ = 0.154056 nm), θ is the diffraction angle, and β is the line width. The results showed that the crystallite size of the Fe2O3 particles was about 33.9 nm, which was consistent with that observed by TEM.
 | ||
| Fig. 4 XRD analysis of the Fe2O3/AC catalysts calcined at different temperatures. Blue vertical lines: XRD library magnetite (Fe3O4) spectra, PDF: 75-0449. Red vertical lines: XRD library hematite (Fe2O3) spectra, PDF: 33-0664. | ||
The catalyst calcined at 400 °C presented 2-theta peaks at 30.28, 35.76, 43.47, 53.94, 57.59 and 63.17°, which is consistent with the library XRD of Fe3O4 (MDI Jade 5.0, PDF 75-0499), indicating that Fe3O4 was produced. However, the 2-theta peaks at 35.76, 43.47, 53.94 and 57.59° corresponded to Fe3O4 as well as Fe2O3, that is, those peaks are the sum peaks of Fe3O4 and Fe2O3. Contrary to the catalyst calcined at 300 °C, the peaks at 56.15, 57.43 and 71.94° (corresponding to Fe2O3) disappeared for the catalyst calcined at 400 °C. All these seem to indicate that Fe2O3 crystals decreased while Fe3O4 was produced, and therefore, we suggested that Fe2O3 was partly reduced to Fe3O4 (3Fe2O3 + 1/2C = 2Fe3O4 + 1/2CO2) when calcined at 400 °C. In addition, weight loss of activated carbon with 400 °C calcinations is higher than that with 300 °C calcinations. Fig. S5† shows that the catalyst calcined at 300 °C obtained the highest ammonia formation rate and coulombic efficiency.
Fig. S6† presents the ammonia formation rate and the coulombic efficiency at different weight ratios of iron to activated carbon. As can be seen at low Fe/AC ratios, both the ammonia formation rate and the coulombic efficiency increase. With increasing Fe/AC ratio, the coulombic efficiency approaches a stable maximum, and the ammonia formation rate exhibits a maximum at a Fe/AC ratio of 7/50.
All electrode reactions were driven by electrode potentials in agreement with the previous thermodynamic evaluations on the synthesis of ammonia. A control of the electrode potential is important to ensure the occurrence of desired reactions, high process efficiency, and satisfactory product quality. The comparison of the rates between different electrode reduction reactions needs to be made based on the difference of the applied potential, E, and the thermodynamic electrolysis potential, E°. The difference is defined as the overpotential, η, where η = |E° − E|. Fig. 5a shows the recorded current density change versus the time of electrolysis during the ammonia synthesis process for different potentials at 250 °C over a period of 1 h. The applied potential of 1.15 V (η = 0 V) is about equal to the thermodynamic electrolysis potential required for ammonia formation (see Fig. 2), and the maximum applied voltage is 1.55 V (η = 0.40 V). As can be seen in Fig. 5a, at lower applied voltages, (≤1.35 V) the current density tends to decrease in time, while it is stable at higher applied potentials. The electrolytic cell demonstrated good stability under applied voltage. Current density increased directly with increased applied voltage, which ranged from 1.15 to 1.55 V with 0.1 V intervals. An especially sizeable jump in current density occurred between 1.45 V and 1.55 V.
 | ||
| Fig. 5 (a) Current density under different applied voltages and times. Cathode was supplied with N2 and water. (b) Ammonia formation rate and coulombic efficiency. | ||
We also investigated the effects of applied voltage at 250 °C on the ammonia formation rate. As shown in Fig. 5b, there is a slight increase in the ammonia formation rate when the applied voltage was increased from 1.15 to 1.45 V. However, the rate increased significantly when the applied voltage was further increased to 1.55 V, at which point the ammonia formation rate reached its maximum value (8.27 × 10−9 mol (s cm2)−1). This higher rate results in the formation of more products and unwanted by-products over the cathode surface, which could potentially be the cause of the increase in current density seen in Fig. 5a. Compared to the ammonia formation rate, coulombic efficiency for ammonia formation decreased with increasing applied voltage, from 13.7% at 1.15 V to 4.91% at 1.55 V. The increase of the ammonia formation rate with the corresponding decrease of coulombic efficiencies indicates that there is more than one process occurring on the cathode surface, and that the competitive hydrogen evolution reaction (HER) is dominant at a higher applied voltage.14
4. Conclusions
The electrolysis of water into hydrogen and oxygen competes with the electrosynthesis of ammonia from N2 and water or steam in molten hydroxide with a suspension of Fe2O3-based catalysts. The cyclic voltammetric studies show that Fe2O3/AC catalysts can inhibit hydrogen reduction and enhance reduction of iron. XRD analysis revealed that Fe2O3 structures appear on the iron-based catalysts supported on activated carbon at a calcination temperature of 300 °C. For the first time, electrochemical synthesis of ammonia directly from wet N2 has been achieved at 250 °C in a molten hydroxide electrolyte with a suspension of Fe2O3/AC catalysts at atmospheric pressure. A maximum ammonia production rate of 8.27 × 10−9 mol (s cm2)−1 has been achieved with an applied voltage of 1.55 V and an average current density of 49 mA cm−2, while the highest coulombic efficiency for ammonia formation is 13.7% at 1.15 V and an average current density of 11 mA cm−2.Acknowledgements
This work was supported by the Natural Science Foundation of Heilongjiang Province, China (Grant No. B2015011). S. Z. Liu is grateful to the Foundation of Northeast Petroleum University, China for partial support of this study. S. Licht is grateful to the United States National Science Foundation grant 1505830 for partial support of this study.Notes and references
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14579 CrossRef CAS.
- U.S. Dept. of Interior, Mineral Commodity Summaries 2016, 119. http://minerals.usgs.gov/minerals/pubs/mcs/2016/mcs2016.pdf (Aug. 25, 2016).
- C. Zamfirescu and I. Dincer, J. Power Sources, 2008, 185, 459–465 CrossRef CAS.
- W. H. Avery, Int. J. Hydrogen Energy, 1988, 13, 761–773 CrossRef CAS.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- R. Liu and G. Xu, Chin. J. Chem., 2010, 28, 139–142 CrossRef CAS.
- G. Xu and R. Liu, Chin. J. Chem., 2009, 27, 677–680 CrossRef CAS.
- J. Pool, E. Lobkovsky and P. Chirik, Nature, 2004, 427, 527–530 CrossRef CAS PubMed.
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press, Cambridge, MA, 2004 Search PubMed.
- R. Strait and M. Nagvekar, Carbon dioxide capture and storage in the nitrogen and syngas industries, Nitrogen + Syngas, 2010, vol. 303, pp. 1–3. (Jan–Feb) at http://www.kbr.com/Newsroom/Publications/Articles/Carbon-Dioxide-Capture-and-Storage-in-the-Nitrogen-Syngas-Industries.pdf Search PubMed.
- A. Skodra and M. Stoukides, Solid State Ionics, 2009, 180, 1332–1336 CrossRef CAS.
- I. A. Amar, C. T. G. Petit, G. Mann, R. Lan, P. J. Skabara and S. W. Tao, Int. J. Hydrogen Energy, 2014, 39, 4322–4330 CrossRef CAS.
- G. Pecchi1, M. G. Jiliberto1, E. J. Delgado, L. E. Cadús and J. L. G. Fierro, J. Chem. Technol. Biotechnol., 2011, 86, 1067–1073 CrossRef.
- I. A. Amar, R. Lan and S. W. Tao, J. Electrochem. Soc., 2014, 6, H350–H354 CrossRef.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. W. Tao, Electrochim. Acta, 2014, 123, 582–587 CrossRef CAS.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. W. Tao, Appl. Catal., B, 2014, 152–153, 212–217 CrossRef CAS.
- T. Murakami, T. Nohira, T. Goto, Y. H. Ogata and Y. Ito, Electrochim. Acta, 2005, 50, 5423–5426 CrossRef CAS.
- T. Murakami, T. Nohira, Y. Araki, T. Goto, R. Hagiwara and Y. H. Ogata, Electrochem. Solid-State Lett., 2007, 10, E4–E6 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Sci. Rep., 2013, 3, 1145, DOI:10.1038/srep01145.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674 RSC.
- R. Lan and S. Tao, RSC Adv., 2013, 3, 18016–18021 RSC.
- S. Licht, B. Cui, B. Wang, F. F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- F.-F. Li and S. Licht, Inorg. Chem., 2014, 53, 10042–10044 CrossRef CAS PubMed.
- S. Licht, J. Phys. Chem. C, 2009, 113, 16283–16292 CAS.
- Thermochemical data are available online at U.S. National Institute of Standards and Technology ChemWeb: http://webbook.nist.gov/chemistry/form-ser.html.
- Glenn Research Center, National Aeronautics and Space Administration (NASA), ThermoBuild access to NASA Glenn thermodynamic CEA database, 2006, data available at http://www.grc.nasa.gov/WWW/CEAWeb/ceaThermoBuild.htm.
- https://en.wikipedia.org/wiki/Activatedcarbon (access 10 June, 2016).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02386j |
| This journal is © The Royal Society of Chemistry 2017 |