
A promising low pressure methanol synthesis route from CO2 hydrogenation over Pd@Zn core–shell catalysts†
Fenglin
Liao
a,
Xin-Ping
Wu
 b,
Jianwei
Zheng
c,
Molly Meng-Jung
Li
a,
Anna
Kroner
d,
Ziyan
Zeng
e,
Xinlin
Hong
e,
Youzhu
Yuan
c,
Xue-Qing
Gong
*b and
Shik Chi Edman
Tsang
*a
b,
Jianwei
Zheng
c,
Molly Meng-Jung
Li
a,
Anna
Kroner
d,
Ziyan
Zeng
e,
Xinlin
Hong
e,
Youzhu
Yuan
c,
Xue-Qing
Gong
*b and
Shik Chi Edman
Tsang
*a
aWolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford, OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
bKey Laboratory for Advanced Materials, Centre for Computational Chemistry and Research Institute of Industrial Catalysis, College of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, P.R. China. E-mail: xgong@ecust.edu.cn
cState Key Laboratory of Physical Chemistry of Solid Surfaces, National Engineering Laboratory for Green Chemical Production of Alcohols-Ethers-Esters, iChEM, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
dDiamond Light Source Ltd, Harwell Science and Innovation, Chilton, Didcot, Oxfordshire OX11 0DE, UK
eDepartment of Chemistry, Wuhan University, Wuhan 430072, P.R. China
First published on 27th October 2016
Abstract
At present, there is no low pressure methanol synthesis from CO2/H2 with high yield despite the presence of an upstream process of aqueous phase reforming (APR) of biomass derivatives on an industrial scale for CO2/H2 production at ca. 2 MPa. This is due to the intrinsic thermodynamics of the system which leads to particularly high CO levels at low pressure through reversed water gas shift reaction (RWGS) for most studied catalysts. Here we report a new Pd@Zn core–shell catalyst that offers a significantly higher kinetic barrier to CO/H2O formation in CO2 hydrogenation to reduce the CO levels but facilitates CH3OH formation at or below 2 MPa with CH3OH selectivity maintained at ca. 70% compared to ca. 10% over industrial Cu catalysts. The corresponding methanol yield at 2 MPa reaches 6.1 gmethanol gactive metal−1 h−1, which is comparable to the best reported value among a wide variety of catalysts under 5 MPa. It is thus believed that this active Pd based catalyst opens up a promising possibility for low pressure and temperature methanol production using a renewable biomass resource for fossil-fuel-starved countries.
Introduction
Catalytic methanol production is one of the most important reactions in industry due to the wide applications of methanol in chemical synthesis.1,2 Currently, methanol is produced via two steps: a high temperature process (at >850 °C, 2 MPa) that breaks down the resilient methane molecule (natural gas) to synthesis gas (CO/H2 syngas) by steam reforming over an Ni based catalyst, followed by syngas rearrangement to methanol over a Cu/ZnO catalyst at 250 °C, 5–10 MPa.3,4 Thus, this two-step process is non-renewable and energy inefficient, and also the conditions for the two steps are incompatible with each other.5,6 In particular, the transformation of syngas to methanol is done under high pressure, requiring specialized equipment and incurring increasing costs. Owing to the exhaustion of fossil fuels and the accompanying emission of CO2 as the primary greenhouse gas,7,8 it would be attractive to use renewable methanol as a chemical platform for future fuels and chemical production. This so-called methanol economy is carbon neutral.1 As one of the primary renewable energy resources, biomass is regarded as a promising alternative to fossil fuels.9 The low temperature aqueous phase reforming (APR) of biomass derivatives on an industrial scale for CO2/H2 production at 2 MPa was recently demonstrated.10 Thus, a downstream catalytic CO2 hydrogenation to methanol (CO2 + 3H2 → CH3OH + H2O) under similar conditions to couple with the above CO2/H2 processes would be highly desirable. Much of current research focuses on Cu catalysts for this reaction. As reviewed by Saito,11 Cu/ZnO based catalysts not only appear to be active for CO/H2 conversion but they also exhibit superior activity in CO2 hydrogenation to methanol among a wide variety of catalysts. Unfortunately, CO can be favourably produced through the reversed water gas shift (RWGS) reaction route (CO2 + H2 → CO + H2O) during methanol synthesis, especially under low pressure conditions. For Cu-based catalysts, methanol selectivity above 50% commonly requires a pressure over 5–10 MPa.11 For pressures at or below 2 MPa, the rate of RWGS is generally 1–3 orders of magnitude higher than that of methanol formation over the CuZn surface,12 leading to extremely low methanol selectivity. To couple with the CO2/H2 production from low pressure biomass reforming, we are therefore interested in exploring non-Cu based catalysts for methanol synthesis at or below 2 MPa. It was recently demonstrated that In2O3/ZrO2 is highly selective for methanol synthesis from CO2/H2 but the methanol yield is only 3 gmethanol gactive metal−1 h−1 even under 5 MPa with a high ratio of H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 (4–8), which is much lower than the best reported value of 6.4 gmethanol gactive metal−1 h−1 over Pd/Ga2O3.11,13 Pd commonly shows similar catalytic properties to Cu upon modification but is more active and less susceptible to sintering and poisoning. However, typical Pd-based catalysts (Pd/ZnO, Pd/Ga2O3etc.) are not selective for methanol synthesis (see results later), due to parallel RWGS, particularly at low pressure. In this communication, we report a series of highly active Pd@Zn core–shell nano-structures with variable compositions that show a progressively enhanced ability to suppress the RWGS selectively with increasing Zn content. As a result, a methanol selectivity of 70% with a high yield of 6.1 gmethanol gactive metal−1 h−1 over this novel Pd@Zn catalyst was achieved at a low pressure of 2 MPa which is far beyond the value of industrial Cu catalysts (10%). This high selectivity and yield hold promise for using this catalyst to couple methanol production downstream to biomass reforming based on similar reaction conditions (Scheme 1).
:CO2 (4–8), which is much lower than the best reported value of 6.4 gmethanol gactive metal−1 h−1 over Pd/Ga2O3.11,13 Pd commonly shows similar catalytic properties to Cu upon modification but is more active and less susceptible to sintering and poisoning. However, typical Pd-based catalysts (Pd/ZnO, Pd/Ga2O3etc.) are not selective for methanol synthesis (see results later), due to parallel RWGS, particularly at low pressure. In this communication, we report a series of highly active Pd@Zn core–shell nano-structures with variable compositions that show a progressively enhanced ability to suppress the RWGS selectively with increasing Zn content. As a result, a methanol selectivity of 70% with a high yield of 6.1 gmethanol gactive metal−1 h−1 over this novel Pd@Zn catalyst was achieved at a low pressure of 2 MPa which is far beyond the value of industrial Cu catalysts (10%). This high selectivity and yield hold promise for using this catalyst to couple methanol production downstream to biomass reforming based on similar reaction conditions (Scheme 1).
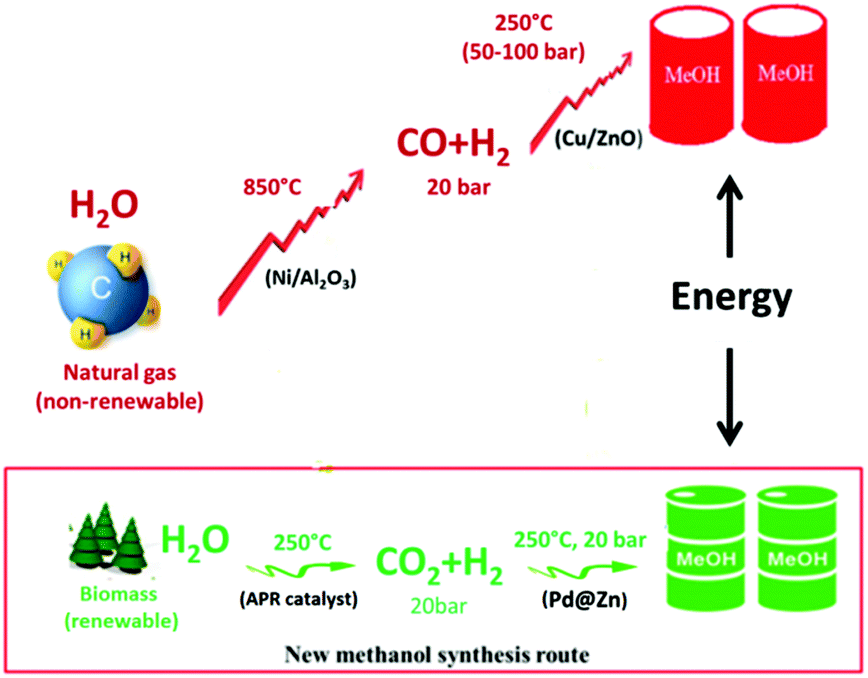 | ||
| Scheme 1 A renewable-based low temperature, low pressure methanol production process via catalytic hydrogenation of CO2 over Pd@Zn in comparison with the conventional syngas route over Cu/ZnO. | ||
Experimental
The design and synthesis of the catalyst
To investigate alternative catalysts to conventional Cu catalysts, we chose supported Pd catalysts but modified with Zn atoms. It has been reported that Pd nanoparticles on ZnO supports display high activity for both catalytic methanol synthesis from CO2/H214 and methanol decomposition to CO2/H2 (reverse reaction of methanol synthesis).15 Previous research also indicated that a small quantity of Zn can be reduced from the ZnO support, which decorated the Pd, accounting for its high catalytic performance.16 Thus, our supported PdZn/ZnO was synthesized through the reduction of PdO on the rod-like ZnO. The reduction of PdO to Pd(0) was completed readily at room temperature after which the produced Pd particles catalyzed a small degree of reduction of ZnO to Zn(0) at an elevated temperature. However, it was found that these reduced PdZn/ZnO catalysts were not selective for methanol synthesis particularly at low pressure (see Fig. 7), presumably due to the small quantity of Zn(0) on the Pd. A higher reduction temperature for the synthesis of dispersed PdZn with a higher Zn(0) content was also not successful due to extensive sintering. We have reported earlier that the addition of foreign semi-conductive material to a semiconductor support, forming a type II heterojunction, can facilitate a deeper reduction of the support to decorate the overlying noble metal nanoparticle at a mild temperature. Hence, this maintains the small primary metal particle size.17 Thus, CdSe was added, which introduced staggered energy levels to the ZnO to form a typical type II hetero-junction.
The ZnO–CdSe support was first synthesized by a sequential growth of CdSe and ZnO. CdSe particles were obtained through the reaction of Na2SeSO3 and Cd(NO3)2. The Na2SeSO3 aqueous solution was prepared by refluxing Se powder in an aqueous Na2SO3 solution at 80 °C overnight. 0.64 g Cd(NO3)2 and 14.70 g sodium citrate were dissolved into 100 mL water to form a solution which was then mixed with freshly prepared 0.1 M Na2SeSO3 (100 mL) into a flask and heated in a water bath at 60 °C for 15 minutes. The red precipitate was collected by centrifugation at 5000 rpm for 10 minutes and extensively washed, after which the supernatant was decanted and discarded. The growth of ZnO rods was carried out by Zeng's method.18 1.487 g zinc nitrate [Zn(NO3)2·6H2O] and 6.000 g NaOH were dissolved in 10 mL deionized water (the molar ratio of Zn2+ to OH− was 1:30). Some CdSe particles produced previously were dispersed into 100 mL ethanol which was added to the solution containing a Zn precursor. (To control the ratio of ZnO:CdSe in the support, various amounts of CdSe were added for samples 1–4. The accurate compositions of the samples were analysed by inductive coupled plasma-atomic emission spectrometry (ICP-AES).) 5 mL ethylenediamine (EDA) was also added to the mixture, which was then transferred to a 250 mL covered plastic container. This was stirred at room temperature until the red mixture turned white. The white crystalline product was then collected by centrifugation and was washed with deionized water and pure ethanol. The final product was dried in an oven at 60 °C for 12 h. The loading of 5 wt% Pd onto the above synthesized support was achieved by the impregnation method; the ZnO–CdSe support was immersed into a Pd(NO3)2 ethanol solution and the mixture was stirred at 50 °C until the ethanol solvent evaporated. The collected powder was calcined in air at 450–500 °C for 2 h.
Catalytic test of a series of Pd@Zn samples in CO2 hydrogenation
Catalyst tests for hydrogenation of CO2 were carried out in a tubular fixed bed reactor (12.7 mm outside diameter) by using a catalyst weight of 0.1 g. A CO2/H2 reaction mixture with a molar ratio of 1:2.8 was fed at a rate of 30 stp mL min−1 (stp = standard temperature and pressure; P = 101.3 kPa, T = 298 K) through the catalyst bed. Before each test, the catalyst was pre-reduced at 280 °C for 2 h under the H2 flow (20 stp mL min−1). The products were analysed by a gas chromatograph equipped with a thermal conductivity detector (TCD). Each sample was tested at least twice to confirm the reproducibility of the data. A comparison of the catalytic performance of fresh and used samples under identical conditions can be found in the ESI.†
Catalyst characterization
530 eV) were performed on beamline B18 at Diamond Light Source (Diamond, UK) to obtain information about the local structure of a noble metal (the nearest-neighbour interatomic distances and coordination number). Also, it was used to estimate the composition of the metal particles. The Diamond installation comprises a 3 GeV electron storage ring with typical currents of 200 mA. The B18 is a bending magnet beamline which has been designed to deliver monochromatic X-rays in the energy range of 2 to 35 keV. A Si (311) double crystal monochromator was used for energy selection with a resolution of 1 eV. X-ray absorption spectroscopy data were collected at ambient temperature in transmission mode using optimized ionization chambers as detectors. The fluorescence spectra were acquired using I0 and a high count rate fluorescence 9-element Ge detector. The EXAFS data analysis was performed using IFEFFIT 1 with Horae packages 2 (Athena and Artemis). All spectra were calibrated with Pd foil as a reference to avoid small energy shifts of the nanocatalyst. The amplitude parameter was obtained from EXAFS data analysis of the Pd foil with a known coordination number (equals to 12), which was used as a fixed input parameter in all fits to allow coordination number (CN) refinement.
In this work, we have only performed a first shell data analysis under the assumption of a single scattering. The curve-fitting analyses were done in k3 space with a range of 2–12. The best fit was selected according to the lowest R value throughout EXAFS analysis.
:CO = 1. The samples were measured on a Micromeritics ASAP 2020 (M+C) analyzer. The samples with variable concentration of CdSe (samples 1–4) were pre-reduced at 523 K and evacuated for 1 h at 523 K to ensure a high surface cleanness. An initial isotherm was measured in an adsorbate pressure range of 100–600 Torr at a precisely controlled temperature of 308 K. Subsequently evacuation at 308 K for 30 min was conducted to remove the weak reversibly adsorbed CO molecules before measuring another repetitive isotherm under the same conditions as the initial one. The difference between the repetitive and the initial isotherm is denoted as the uptake of irreversibly chemisorbed CO on catalysts.
The powders of sample 1 (5 wt% Pd, 0.0 wt% CdSe) and sample 4 (5 wt% Pd, 26.4 wt% CdSe) were pressed into pellets and loaded onto the sample holder. The sample was then flushed with 5% H2/Ar (20 mL min−1) for 10 min and then reduced for 1 h at 250 °C. After the pre-reduction, vacuum conditions were applied to the cell until the pressure was lower than 1 × 10−6 bar and then backgrounds were recorded at 50 °C, 100 °C, 150 °C, 200 °C, and 250 °C.
After collecting the backgrounds, a mixture of CO2/H2 (3:100) was passed through the reduced sample pellet and then the in situ FTIR spectra were collected at 50 °C, 100 °C, 150 °C, 200 °C, and 250 °C, maintaining the sample at each temperature for 10 min.
Computational method
The PBE functional was used to do the spin-polarized DFT calculations by using the Vienna ab initio Simulation Package (VASP).19 The project-augmented wave (PAW)20 method was used to describe the interaction between atomic cores and electrons. Wave functions were expanded in plane waves with an energy cutoff of 400 eV. For integrations over the Brillouin zone, we used a 5 × 5 × 1 Monkhorst-Pack grid for all calculations. The structure optimizations were converged until the Hellman–Feynman force on each ion was less than 0.02 eV Å−1. The calculated lattice parameter of the bulk Pd was 3.95 Å, which is in good agreement with the values reported from previous studies.21,22The Pd(111)-based surfaces were modeled by 4-layer slabs repeated in a 4 × 4 surface unit cell with the bottom layer being fixed to the bulk parameters, while the other layers were allowed to fully relax. To avoid interactions between slabs, all slabs were separated by a vacuum gap greater than 10 Å. Previous experimental studies23,24 suggested that the Zn atoms are contained entirely and distributed uniformly in the topmost layer of the Pd(111) substrate after annealing when the Zn coverage is under 0.5 monolayer (ML). For coverage around 0.5 ML, the p(2 × 1) PdZn (1:1) surface alloy is formed. Further increasing the Zn coverage will not break the (2 × 1) phase. Instead, Zn will infiltrate into the sub-surface layer first and then to the deeper layers. Accordingly, we constructed a p(2 × 1) 2 ML–PdZn (1:1) surface alloy deposited Pd(111) (case D in Scheme 2) representing a Pd surface with 1 ML Zn decoration for comparison with pure Pd(111) (case A in Scheme 2).
 | ||
| Scheme 2 The demonstration of cases A and D, A: Pd(111), D: 2 ML Pd1Zn1/Pd(111), respectively. The bigger and smaller balls represent the first and second layer atoms, respectively. | ||
Transition states were located by using the climbing-image nudged elastic band (CI-NEB) method.25–27
Results and discussion
The samples were prepared by adding CdSe quantum dots into the Pd/ZnO system to modify the reduction behaviour of the ZnO support during H2 pre-treatment. The existence of CdSe in the ZnO support was confirmed by X-ray diffraction. As shown in Fig. 1, the intense peaks labelled as △ are indexed into wurtzite ZnO while the diffraction peaks at 24.5°, 27.0° and 28.5° in samples 2–4 are assigned to the facets of (100), (002) and (101) of CdSe which indicate the presence of a CdSe phase in the supports. The concentrations of CdSe in the supports were determined by ICP-AES and the results are labelled in all the figures. The reduction process of ZnO–CdSe is described in Scheme 3: the staggered energy levels of ZnO and CdSe allow thermally excited electrons (e) to reside in the conduction band of ZnO whilst holes (+) reside in the valence band of CdSe. The accumulated holes (electron depleted Se) in CdSe can be relaxed by reacting them with the spill-over H from neighbouring Pd to produce H2Se. The excited electrons in the ZnO conduction band (composed of Zn 4s) will preferentially reduce Zn(2+) to Zn(0) to maintain electronic neutrality. As a result, the concentration of Se (1.1 wt%) in the reduced sample 4 determined by energy-dispersive X-ray spectroscopy (EDX) is much lower than that of Cd (8.8 wt%) as shown in Table 1 since Se species escape from the system in the form of H2Se gas. Consequently various numbers of Zn atoms were derived from the refractory support to decorate on the surface of the Pd particles.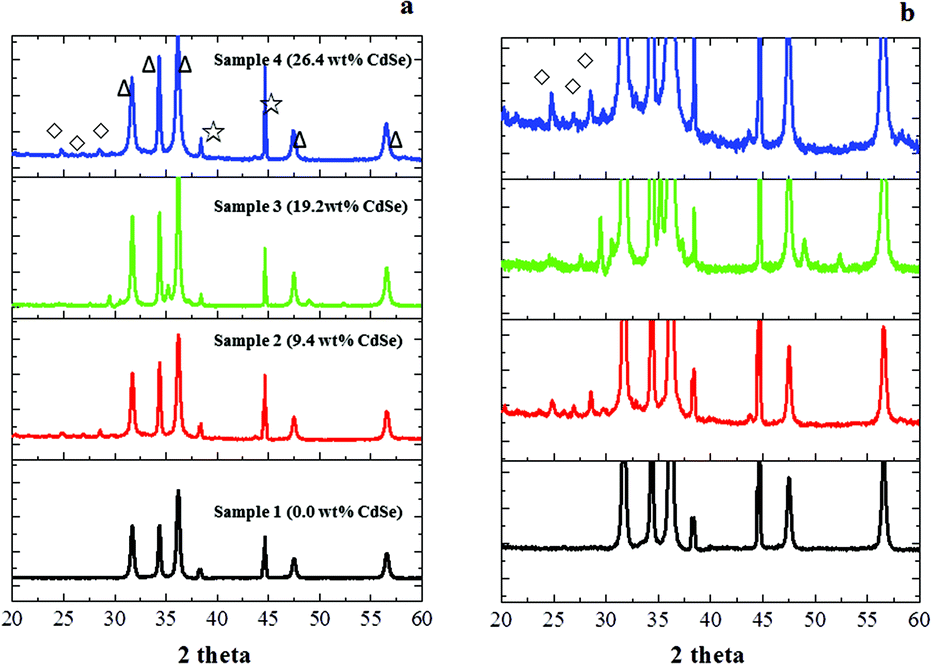 | ||
| Fig. 1 (a) The XRD patterns of samples 1–4 with various contents of CdSe; (b) the corresponding enlarged patterns showing CdSe peaks. △, ☆ and ◊ indicate the diffraction peaks of ZnO, sample holder and CdSe. | ||
 | ||
| Scheme 3 The proposed reduction process of ZnO promoted by CdSe. | ||
| Element | Pd | Zn | Cd | Se |
|---|---|---|---|---|
| Weight % | 8.0 | 82.1 | 8.8 | 1.1 |
After reduction, the Zn X-ray photoelectron spectra (XPS) of a series of reduced Pd/ZnO–CdSe samples shown in Fig. 2a clearly indicate a downshift of binding energy from 1021.3 eV to 1020.1 eV while no obvious change is observed in the corresponding Pd XPS spectra. This implies that the surface concentration of Zn(0) (1020.1 eV) increases at the expense of Zn(2+) (1021.3 eV) upon reduction by increasing the CdSe dopant concentration in Pd/ZnO.
 | ||
| Fig. 2 (a) The XPS curves of Zn in a series of reduced Pd/ZnO–CdSe (5 wt% Pd) samples (dotted lines indicate the binding energy of Zn(2+) (1021.3 eV) and Zn(0) (1020.1 eV) with reference to the added boron nitride (BN), N 398.2 eV); (b) the corresponding XPS curves of Pd in the samples 1–4. | ||
The extended X-ray absorption fine structure (EXAFS) results at the Pd K-edge (24350 eV) are shown in Fig. 3, 4 and Table 2. The direct observation of a shorter scattering path of 2.57 Å compared with a typical 2.70 Å Pd–Pd scattering path28 is assigned to the first shell scattering pair of Pd–Zn. This indicates a strong atomic interaction between the Pd and Zn atoms. Meanwhile, the number of neighbouring Zn (NZn) around each absorbing Pd atom rises at the expense of neighbouring Pds (NPd) with increasing CdSe content.
 | ||
| Fig. 3 The number of neighbouring Pd (NPd) and Zn (NZn) around each Pd absorbing atom as derived from the EXAFS. | ||
 | ||
| Fig. 4 k 3 Space EXAFS curves of Pd for a series of reduced Pd/ZnO–CdSe samples: (a) sample 1 (0.0 wt% CdSe), (b) sample 2 (9.4 wt% CdSe), (c) sample 3 (19.2 wt% CdSe), (d) sample 4 (26.4 wt% CdSe). (Blue: experiment data; red: fitting data.) | ||
| Sample no. | Total CN | ΔE (eV) | Pd–Pd distance (Å) | D–W factor (Å2) | Pd–Zn distance (Å) | D–W factor (Å2) | R-Factor |
|---|---|---|---|---|---|---|---|
| “CN” is coordination number; “ΔE” is the energy difference between the experimental absorption energy and the calculated value in curve fitting; “D–W” is Debye Waller; “R factor” is the indication of the quality of curve fitting. | |||||||
| 1 | 7.8(8) | −5.0 | 2.67(1) | 0.015(2) | 2.56(1) | 0.004(1) | 2.1% |
| 2 | 7.7(7) | −4.5 | 2.71(1) | 0.014(2) | 2.56(1) | 0.008(1) | 0.8% |
| 3 | 8.0(5) | −0.8 | 2.72(1) | 0.014(2) | 2.57(1) | 0.009(1) | 0.9% |
| 4 | 8.3(6) | −1.7 | 2.73(1). | 0.007(1) | 2.59(1) | 0.014(1) | 1.2% |
The transmission electron microscopy (TEM) images of reduced samples 4 and 1 are shown in Fig. 5 and 6, respectively. Sample 4 (5 wt% Pd, 26.4 wt% CdSe) with the highest concentration of CdSe depicts an imperfect core (dark)–shell (light) structure of 4–5 nm on ZnO rod support (Fig. 5e), corresponding to a mixed phase with an irregular atom arrangement. Lattice fringes of ca. 0.23 and 0.26 nm are observed in the core and shell regions, respectively. The former value corresponds to the (111) plane of pure Pd29 while the latter corresponds to the Zn metal (002) facet or Zn rich ZnPd alloy with similar lattice parameters.30 This implies that the Pd surface is heavily doped with Zn atoms through the formation of a Pd@Zn bimetallic phase at the interface with a high Zn content. In contrast, no core–shell structure is seen for sample 1 (5 wt% Pd, 0.0 wt% CdSe) with a slightly smaller nanoparticle size (3–4 nm) due to a lower Zn(0) content, which is consistent with the XPS results.
 | ||
| Fig. 5 (a, b) TEM and (d, e, f) high resolution TEM (HRTEM) images for reduced sample 4 (5 wt% Pd, 26.4 wt% CdSe) showing core–shell Pd containing particles; (c) the particle size distribution of decorated Pd particles. 0.32 nm corresponds to the lattice spacing of ZnO support. | ||
 | ||
| Fig. 6 (a, b) TEM and (d, e, f) HRTEM images for reduced sample 1 (5 wt% Pd, 0.0 wt% CdSe); (c) the particle size distribution of Pd particles. | ||
The numbers of exposed Pd active sites of the reduced Pd/ZnO–CdSe samples (5 wt% Pd) were determined by the chemisorption uptake of CO with a Pd:CO = 1. The results are displayed in Table 3. Clearly, the number of exposed Pd atoms decreases with the Zn decoration due to the formation of a zinc rich surface but there is still a significant exposure of Pd atoms with no total coverage of the Pd surface by Zn atoms.
| Sample no. | 1 (0.0 wt% CdSe, NZn = 2.5) | 2 (9.4 wt% CdSe, NZn = 3.5) | 3 (19.2 wt% CdSe, NZn = 4.2) | 4 (26.4 wt% CdSe, NZn = 5.0) |
|---|---|---|---|---|
| Number of exposed Pd atoms (μmol gcat−1) | 55.3 | 35.7 | 25.8 | 13.5 |
The number of exposed Cu sites with commercial Cu catalysts was measured by N2O uptake and the value is 520 μmol gcat−1.
From the above results, the supported Pd@Zn bimetallic particles show the intimate contact of these two elements which may modify the electronic properties of Pd. The catalytic performances of the as-prepared Pd@Zn samples in CO2 hydrogenation to methanol under a low pressure of 2 MPa were studied (a mixture of CO2:H2 = 1:2.8 was flowed through the loaded catalyst (0.1 g) in a fixed-bed reactor at a flow rate of 30 mL min−1). The plots of the catalytic results of a series of Pd@Zn (5 wt% Pd) samples versus the value of NZn around Pd are summarized in Fig. 7. The NZn value was derived from EXAFS indicating the Zn content in the Pd@Zn nanoparticles. A commercial Cu/ZnO based catalyst (HiFUEL-R120 ca. 50 wt% Cu) was employed as a reference. With the surface areas of Pd and Cu taken into account (active sites per gram catalyst as shown in Table 3), the methanol TOFs were calculated over the Pd and Cu catalysts (Fig. 7d). From samples 1 to 4, the TOF values for Pd increase dramatically with respect to the increasing Zn content. The TOF reaches 1.9 × 10−1 s−1 for sample 4, which is about 350 times greater than the commercial Cu catalyst under the same reaction conditions. As far as we are aware, the TOF value of sample 4 represents the highest TOF among all the reported values using a H2:CO2 ratio of 3:1. The activity of sample 4 in terms of weight time yield has reached 6.1 gmethanol gactive metal−1 h−1 at 270 °C, 2.0 MPa (Fig. 7b), which as far as we are aware, is the highest reported value for CO2 hydrogenation under 2.0 MPa. This value is much higher than the best reported value for a Cu-based catalyst (1.48 gmethanol gactive metal−1 h−1) over Cu/ZnO/Ga2O3 (50 wt% Cu) at 5 MPa and is comparable to the best reported results over Pd in Pd/Ga2O3 (6.4 gmethanol gactive metal−1 h−1) but significantly decreases the applied pressure from 5 MPa to 2 MPa (shown in Fig. 8a).11 Compared to the commercial Cu catalyst of 0.1 gmethanol gactive metal−1 h−1, our sample has 60 times higher activity under the same reaction conditions. The detailed comparison of our sample 4 with the other reported catalysts in the literature is shown in Fig. 8a and Table S1 in the ESI.†
 | ||
| Fig. 7 (a) The plots of methanol selectivities; (b) the methanol weight time yields; for a series of Pd@Zn samples in CO2 hydrogenation at 2.0 MPa with WHSV = 18000 mL h−1 g−1versus number of Zn atoms (NZn) around Pd derived from EXAFS at 250 °C (red), 270 °C (black). (c) The pressure effects on methanol selectivity for Pd@Zn (sample 4) and the commercial Cu catalyst at 250 °C in the range of 2–4.5 MPa. (d) The TOF values of Pd in a series of Pd@Zn samples (5 wt% Pd) in methanol synthesis from CO2 hydrogenation at 2.0 MPa, with WHSV = 18000 mL h−1 g−1, with reference to the commercial Cu catalyst. The dotted line represents the calculated thermodynamic values (taking both methanol synthesis and RWGS equilibria into account). | ||
 | ||
| Fig. 8 (a) The comparison of catalytic performance of sample 4 with those of the reported catalysts in the literature.11 The detailed data are shown in Table S1 in the ESI.† (b) The calculated methanol yield, (c) CO yield, and (d) TOF of CO formation at 2 MPa in a temperature range of 210–270 °C for samples 1 and 4 and the commercial Cu catalyst. The dotted line represents thermodynamic values (taking both methanol synthesis and RWGS equilibria into account). | ||
Besides the activity (TOF), the methanol selectivity also dramatically increases according to the number of neighbouring Zn around Pd in Pd@Zn catalysts (Fig. 7a). The nearly 80% methanol selectivity of sample 4 at 250 °C, 4.5 MPa is far beyond that of commercial Cu catalysts (30%) (Fig. 7c). Surprisingly, sample 4 with the highest content of Zn(0) (NZn = 5.0) maintains a high methanol selectivity of 70% at 2 MPa with weight hourly space velocity (WHSV) = 18000 mL h−1 g−1 (Fig. 7a and c). The clear difference between the commercial Cu catalyst and the Pd@Zn surface shows the significance of heavy decoration of Pd with Zn in relation to methanol synthesis. By decreasing the operating pressure from 4.5 MPa to 2.0 MPa (Fig. 7c), the methanol selectivity for the commercial Cu catalyst sharply decreases to below 10%, which is governed by thermodynamics. In contrast, the methanol selectivities over the sample 4 (Pd@Zn) catalyst especially at low pressures are surprisingly well beyond the thermodynamic prediction (indicated by the dotted line in Fig. 7c), indicating that the reaction rate of RWGS is significantly suppressed on the Zn rich Pd@Zn surface.
To derive the apparent activation energies for RWGS, the catalytic data over various catalysts were collected under variable temperature at 2 MPa as shown Fig. 8(b–d). Through the Arrhenius equation (ln(K) = ln(A) − ΔEa/RT where K is the reaction rate constant, A is the Arrhenius factor, ΔEa is the activation energy, R = 8.314 J mol−1 K−1, T is the temperature), the apparent activation energies for RWGS on sample 4, conventional Pd/ZnO (sample 1) and the commercial Cu catalyst were indeed found to be 98 kJ mol−1, 71 kJ mol−1 and 69 kJ mol−1, respectively (the Arrhenius plots for RWGS are shown in Fig. 9). The higher activation barrier of this Zn rich Pd@Zn catalyst promoted by CdSe (sample 4) for RWGS significantly reduces the CO production and raises methanol selectivity compared with the conventional Pd/ZnO and Cu/ZnO. Consequently, the methanol yield on sample 4 appears to have overcome the restriction of thermodynamics calculated based on the equilibrium for the elementary steps of RWGS and methanol synthesis, reaching a higher value with the elevated temperature as shown in Fig. 8b. These results clearly indicate that Pd@Zn with a high content of Zn are promising low-pressure catalysts for methanol synthesis from CO2 hydrogenation, offering an unusually high kinetic barrier for RWGS.
 | ||
| Fig. 9 The Arrhenius plots for RWGS on (a) the commercial Cu catalyst from 170 °C to 230 °C (the data in 250–270 °C is approaching the equilibrium), (b) sample 1 (5 wt% Pd, 0 wt% CdSe) and (c) sample 4 (5 wt% Pd, 26.4 wt% CdSe) from 210 °C to 270 °C; (d) the values of calculated activation energy for the samples. | ||
The characterization after testing (Fig. S1 and S2†) and the catalytic performance of the used sample 4 (recovered from the first testing) (Table S2†) are shown for comparison in the ESI.† It is evident that the typical sample 4 shows reproducible catalytic performance in the repeated testing despite a degree of metal sintering after the testing.
In situ Fourier transform infrared (FTIR) spectroscopy shows some key surface intermediates and confirms the drastic attenuation in CO production on the Pd@Zn surface (Fig. 10). For sample 1 (shown in Fig. 10a and b), a series of vibration bands were detected corresponding to adsorbed CO (linear mode, 2000 cm−1 and bridge mode, 1860 cm−1),31 formate (1595 cm−1, 1395 cm−1),32 dioxymethylene (1230 cm−1),33 and formaldehyde (1502 cm−1, 1702 cm−1)34 species similar to those previously reported over the Cu surface. The formate formation was observed at 50 °C and with rising temperature, the vibration bands for CO, dioxymethylene and formaldehyde become more intense (Fig. 10b). For sample 4 (shown in Fig. 10c), almost no CO signal was detected even at 250 °C but the vibration intensities of dioxymethylene and formaldehyde clearly increased at a higher temperature. It is generally believed that the first step of CO2/H2 activation generates two different surface intermediates of adsorbed HCOO and COOH, which leads to different reaction routes, as shown in Fig. 10e. (Note that the IR vibration bands of the adsorbed COOH cannot be differentiated from that of HCOO due to their similar structures.)35 The hydrogenation of HCOO produces methanol via dioxymethylene and formaldehyde, whereas the decomposition of COOH yields the CO product. The growing signals of adsorbed CO with those of dioxymethylene and formaldehyde at elevated temperature imply parallel pathways for CO and methanol synthesis on sample 1. The absence of a CO signal in sample 4 means that the RWGS is suppressed on the Pd@Zn surface, which may also reflect the selective blockage of CO2 hydrogenation to COOH. (Given that CO is formed from the decomposition of surface COOH.)35
 | ||
| Fig. 10 In situ FTIR spectra of the adsorbed species on the reduced surface. (a) Sample 1 (5 wt% Pd, 0.0 wt% CdSe). (c) Sample 4 (5 wt% Pd, 26.4 wt% CdSe) in CO2 hydrogenation (a flow gas of 3 mol% CO2 in H2 was passed through the selected catalyst pellet of 20 mg) at various temperatures. (b) A combined sample 1 FTIR spectra. (d) The assignments of selected vibration bands in FTIR. Both samples 1 and 4 were pre-reduced in H2 at 250 °C for 1 h. (e) The reported possible pathways in CO2 hydrogenation.35,36 | ||
From the selected potential energy surfaces obtained from our density functional theory (DFT) calculations (see calculation details in the Experimental section) as shown in Fig. 11, the occurrence of surface COOH is preferred over HCOO at pure Pd(111) (case A, Scheme 2). In contrast, with 2 layers of Pd1Zn1 ((case D, 1 ML (monolayer) Zn coverage, Scheme 2)) deposition, HCOO formation is much more favoured. Therefore, on sample 4, which has the highest Zn content, the selective formation of HCOO over COOH as the intermediate leads to enhanced methanol selectivity through further multi-step hydrogenation. It is also noted that the formation barrier of COOH on the PdZn surface (1.43 eV) is higher than that on the Pd(111) surface (1.21 eV), which implies a higher kinetic barrier for the RWGS reaction as COOH is the precursor of CO. This is consistent with our experimental results (Fig. 9).
 | ||
| Fig. 11 (a) The adsorption modes of COOH and HCOO on the Pd surfaces of cases A (pure Pd(111)) and D (2 layers Pd1Zn1 on Pd(111)), respectively. (Top: top view; bottom: side view) Those adsorption energies for cases B (0.25 ML (monolayer) Zn coverage on Pd denoted as Pd3Zn1/Pd(111)) and C (0.5 ML Zn coverage on Pd denoted as Pd1Zn1/Pd(111)) were also calculated. They showed intermediate values between cases A and D, hence cases A and D were used for comparison. The structure of case D was optimized from an entire monolayer of Zn at Pd(111) that was relatively less stable with respect to two mixed layers of Pd1Zn1 at Pd(111). This is indicative of the preferential formation of a saturated surface alloy. Pd and Zn atoms are in blue and orange, C, O and H atoms are in grey, red and white, respectively. (b) The calculated energy profiles for CO2 hydrogenation to adsorbed COOH and HCOO species at Pd cases A and D. (The numbers labelled in the figure are in units of eV representing the corresponding reaction activation barriers. * indicates the adsorbed state.) | ||
The calculation results of the further transformations of trans-COOH over Pd cases are illustrated in Fig. 12; the decomposition to CO is the most favourable reaction route with the lowest activation barrier among the three possible pathways, which confirms the rapid CO production from trans-COOH. But once the formation of trans-COOH is suppressed, CO production could be inhibited.
 | ||
| Fig. 12 The calculated reaction pathways of CO2 hydrogenation at cases A (pure Pd(111)) and D (2 ML–Pd1Zn1/Pd(111)). The numbers labeled in the figure are in units of eV representing the corresponding reaction barriers. (Note that there is an equilibrium trans-COOH/cis-COOH isomer pair on the catalyst surface and the COOH mentioned in the text represents trans-COOH; the corresponding structures of the intermediates are shown in Fig. S3.†) | ||
DFT calculation was also employed to investigate the catalytic properties of the CuZn surface. The results of pure Cu(111) (case A) and Cu(111) with 1 ML Zn decoration (case D, case E) are selected for comparison with Pd surfaces. Case E with 1 ML Zn atoms deposited on Cu(111) is the most stable configuration of a given composition according to our stability analyses (Fig. 13), and is 0.33 eV more stable in total energy than case D (2 ML–Cu1Zn1 surface alloy deposited on Cu(111)).
 | ||
| Fig. 13 The calculated structures of Cu-based catalysts; (a) Cu(111), (b–e) various configurations of 1 ML Zn decoration on Cu(111). Cu and Zn atoms are in brown and purple, respectively. The bigger and smaller balls represent the first and second layer atoms, respectively. Cases D and E are labelled and selected to compare with PdZn surfaces due to the lower energies than other ones. The yellow lines indicate the surface Zn atoms distribution. | ||
This clearly indicates that the Cu(111) surface can be totally covered by 1 ML Zn atoms (case E) but in the case of the Pd(111) surface two layers of Pd1Zn1 would be created instead.
Given the fact that case D of Cu has geometric structures similar to the corresponding Pd case, this was also taken into consideration in this work.
Interestingly, it is observed that HCOO is also formed preferentially over COOH on the Cu based surfaces (note the negative potential energy difference values of adsorbed HCOO and COOH species shown in Table 4) as well as on the PdZn surfaces. Malte Behrens et al. have recently reported a similar stabilization of adsorbed HCOO when Zn atoms are introduced on Cu step sites.37 However, the Cu-based surfaces exhibit much lower methanol selectivity than PdZn, under low pressure in particular. To further investigate the difference in the catalytic properties of Pd and Cu, we calculated the H adsorption energies at the various Pd and Cu surfaces. The calculation results in Table 4 clearly show that the H adsorption energies on Cu(111) decorated by Zn are low (0.16, 0.06 and −0.50 eV for cases A, D and E, respectively), which leads to low activity for H2 dissociation on Cu-based surfaces. At low operating pressures, this could lead to a low surface H coverage that slows down the further hydrogenation of HCOO into methanol. In addition, the decomposition of COOH to adsorbed CO and OH is less dependent on the concentration of surface H, which rapidly consumes the produced COOH in the first step and shifts the equilibrium between HCOO/COOH. In contrast, the Pd@Zn based surfaces show superior ability in activating H2 as indicated by the higher H adsorption energies (0.57 and 0.31 eV for cases A and D in Table 4). In principle, this can further help to produce methanol readily from the hydrogenation of preferentially formed HCOO under pressures as low as 2 MPa. Consequently, an enhanced methanol selectivity with a high yield can be observed over the Pd@Zn catalyst.
| Surface | Case | H adsorption energy (eV) | E HCOO*–ECOOH* (eV) |
|---|---|---|---|
| Cu-Based | A (Cu(111)) | 0.16 | −0.79 |
| D (2 ML Cu1Zn1/Cu(111)) | 0.06 | −0.80 | |
| E (1 ML Zn/Cu(111)) | −0.50 | −1.11 | |
| Pd-Based | A (Pd(111)) | 0.57 | +0.18 |
| D (2 ML Pd1Zn1/Pd(111)) | 0.31 | −0.70 |
Conclusions
In conclusion, in methanol synthesis from the CO2 hydrogenation reaction, the Zn enriched Pd@Zn core–shell bimetallic catalysts prepared via CdSe can stabilize surface HCOO species over COOH hence suppressing CO production from the RWGS reaction. This novel Pd surface with heavy Zn decoration offers a superior capability for H2 adsorption and activation compared to the Cu surface, leading to a superior performance for methanol production from CO2/H2. A methanol yield of 6.1 gmethanol gactive metal−1 h−1 with a high selectivity over 70% is obtained even at a pressure as low as 2.0 MPa, which is comparable to the best reported yield under 5 MPa in the literature. With the increasing demand for greener methanol synthesis from renewable resources, the superior catalytic performance of this novel Pd-based surface under low pressure provides a perfect means to couple it with the upstream aqueous reforming CO2/H2 production processes, as depicted in Scheme 1. It is believed that a new, low-pressured and integrated methanol synthesis process at or below 2 MPa can be developed based on the above findings.Acknowledgements
The authors wish to thank EPSRC, UK (Oxford) and NSFC-21421004, 21373153, 21322307, China for the financial support for this collaborative work and are grateful to the Chinese Scholarship Council (CSC) of China for granting a PhD scholarship to FL to work at Oxford. The ECUST group acknowledges the computing time at the National Super Computing Center in Jinan, China. The authors thank Diamond Light Source to allow a rapid access to the EXAFS facilities on B18.Notes and references
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- L. K. Rihko-Struckmann, A. Peschel, R. Hanke-Rauschenbach and K. Sundmacher, Ind. Eng. Chem. Res., 2010, 49, 11073–11078 CrossRef CAS.
- J. Wu, M. Saito, M. Takeuchi and T. Watanabe, Appl. Catal., A, 2001, 218, 235–240 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Appl. Catal., 1988, 36, 1–65 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 1993, 18, 305–324 CrossRef.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- L. M. Romeo, et al. , Energy Convers. Manage., 2008, 49, 2809–2814 CrossRef CAS.
- G. Bolye, Renewable Energy–Power for a Sustainable Future, Oxford University Press, Oxford, UK, 1996 Search PubMed.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS.
- M. Saito, Catal. Surv. Jpn., 1998, 2, 175–184 CrossRef CAS.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- O. Martin, A. J. Martin, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferre and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- N. Iwasa, H. Suzuki, M. Terashita, M. Arai and N. Takezawa, Catal. Lett., 2004, 96, 75–78 CrossRef CAS.
- Y. H. Chin, R. Dagle, J. Hu, A. C. Dohnalkova and Y. Wang, Catal. Today, 2002, 77, 79–88 CrossRef CAS.
- H. Zhang, et al. , ACS Catal., 2014, 4, 2379–2386 CrossRef CAS.
- F. Liao, et al. , ChemCatChem, 2015, 7, 230–235 CrossRef CAS.
- B. Liu and H. C. Zeng, Langmuir, 2004, 20, 4196–4204 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251–14269 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- A. A. B. Padama and H. Kasai, J. Chem. Phys., 2014, 140, 244707 CrossRef PubMed.
- R. S. Johnson, et al. , Phys. Chem. Chem. Phys., 2013, 15, 7768–7776 RSC.
- J. M. MacLeod, J. A. Lipton-Duffin, A. Baraldi, R. Rosei and F. Rosei, Phys. Chem. Chem. Phys., 2013, 15, 12488–12494 RSC.
- J. A. Lipton-Duffin, et al. , Phys. Chem. Chem. Phys., 2014, 16, 4764–4770 RSC.
- G. Mills, H. Jónsson and G. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Takenaka, Y. Shigeta, E. Tanabe and K. Otsuka, J. Phys. Chem. B, 2004, 108, 7656–7664 CrossRef CAS.
- J. Zhang, Y. Xu and B. Zhang, Chem. Commun., 2014, 50, 13451–13453 RSC.
- D. Pradhan, S. Sindhwani and K. T. Leung, J. Phys. Chem. C, 2009, 113, 15788–15791 CAS.
- A. Karelovic, ACS Catal., 2013, 3, 2799–2812 CrossRef CAS.
- J. Araña, et al. , Appl. Surf. Sci., 2004, 239, 60–71 CrossRef.
- T. Chen, et al. , J. Phys. Chem. C, 2007, 111, 8005–8014 CAS.
- G. Y. Popova, T. V. Andrushkevich, Y. A. Chesalov and E. S. Stoyanov, Kinet. Catal., 2000, 41, 885–891 Search PubMed.
- Y.-F. Zhao, et al. , J. Catal., 2011, 281, 199–211 CrossRef CAS.
- M. Y. Clarence, et al. , Catal. Lett., 2009, 128, 221–226 CrossRef.
- M. Behrens, et al. , Science, 2012, 336, 893–897 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02366e |
| This journal is © The Royal Society of Chemistry 2017 |