
Hyperbranched polyesters by polycondensation of fatty acid-based ABn-type monomers†
Blandine
Testud
ab,
Didier
Pintori
c,
Etienne
Grau
ab,
Daniel
Taton
*ab and
Henri
Cramail
*ab
aUniversité de Bordeaux, Laboratoire de Chimie des Polymères Organiques, UMR 5629, Bordeaux INP/ENSCBP, 16 avenue Pey-Berland, F-33607 Pessac Cedex, France. E-mail: taton@enscbp.fr; cramail@enscbp.fr
bCentre National de la Recherche Scientifique, Laboratoire de Chimie des Polymères Organiques, UMR 5629, F-33607 Pessac Cedex, France
cITERG, 11 rue Gaspard Monge, Parc Industriel Bersol 2, F-33600 Pessac Cedex, France
First published on 27th October 2016
Abstract
Widely available vegetable oils were readily derivatized into chemically pure ABn-type monomers (n = 2 or 3). Their polymerization led to unprecedented hyperbranched polyesters. Four different AB2/AB3-type monomers bearing one A-type methyl ester and two or three B-type alcohol functions were purposely synthesized via two elementary steps, i.e. epoxidation of the internal double bond of the vegetable oil precursors followed by ring-opening of the epoxy groups under acidic conditions. The polycondensation of these bio-sourced monomers was performed in bulk, in the presence of an appropriate catalyst, giving access to modular hyperbranched polyesters with tunable properties. Among the catalysts tested, zinc acetate, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and sodium methoxide proved the most effective, allowing the achievement of molar masses in the range 3000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and dispersities varying from 2 to 15, depending on the initial conditions. The degree of branching, DB, as determined by 1H NMR spectroscopy, was found to be between 0.07 and 0.45. The as-devised hyperbranched polyesters displayed either amorphous or semi-crystalline properties, as a function of the selected AB2/AB3-type initial monomers, with a glass transition temperature, Tg, ranging from −33 to 9 °C and a decomposition temperature at 5 wt% of the sample, Td5%, varying from 204 to 340 °C.
000 g mol−1 and dispersities varying from 2 to 15, depending on the initial conditions. The degree of branching, DB, as determined by 1H NMR spectroscopy, was found to be between 0.07 and 0.45. The as-devised hyperbranched polyesters displayed either amorphous or semi-crystalline properties, as a function of the selected AB2/AB3-type initial monomers, with a glass transition temperature, Tg, ranging from −33 to 9 °C and a decomposition temperature at 5 wt% of the sample, Td5%, varying from 204 to 340 °C.
Introduction
The last three decades have witnessed the rapid development of hyperbranched polymers (HBPs).1 The first intentional synthesis of HBPs dates back to 1987 with the polyphenylenes of Kim and Webster,2 long after Flory had predicted in the 1950s that ABn-type monomers should afford soluble and highly branched materials of a globular shape, via a self-condensation polymerization process.3 The field is still expanding, reaching more than 500 publications in recent years.4 HBPs are a special class of dendritic materials and possess as common features with dendrimers a high branching density as well as a compact and globular architecture. This unusual structure endows them with unique properties as compared to their linear counterparts. Of particular interest are their lower viscosity both in solution and in the molten state, an improved solubility and a high functionality. Indeed, their large number of terminal functional groups offers the possibility for further modifications, providing an additional method for the variation of HBPs’ properties and broadening the scope of their potential applications. In contrast to regular dendrimers,5 HBPs are prepared in one pot, hence they exhibit an overall structure that is not flawless. In particular, HBPs are characterized by the presence of linear units coexisting with dendritic and terminal units, which is mirrored by their degree of branching (DB) that is most often lower than unity. In addition, related one-pot synthetic methods to obtain HBPs are usually more straightforward and more economically viable, compared to dendrimer synthesis.Tremendous synthetic efforts have been made in the past few decades to access a wide range of HBPs, from the polycondensation of ABn-type monomers to less conventional polymerization methods.4,6–11 In this context, hyperbranched polyesters have been the most investigated. This is due to the wide availability of various suitable monomers and the relative ease of synthesis of this subclass of HBPs. The success of hyperbranched polyesters (HBPEs) has resulted in the marketing of miscellaneous Boltorn™ products based on 2,2-bis(methylol)propionic acid (bis-MPA).12,13 As a matter of fact, HBPEs have been nearly exclusively designed starting from fossil resources.14–18 In a context of price volatility and uncertain supply of oil and gas, combined with environmental concerns (global warming, waste production, etc.), there is yet an urgent need to release the chemical industry from its dependence on petroleum resources.19 In recent years, particular attention has been paid to biomass as a sustainable source of carbon.20,21 Among available renewable resources, vegetable oils and their derivatives, i.e. fatty acid methyl esters (FAMEs), represent a promising feedstock for the polymer industry owing to their abundant availability, relatively low cost and inherent degradability.22–28 Interest in FAMEs is also motivated by the presence of both ester and double bonds enabling countless derivatizations and designs of a wide variety of functional building blocks and related materials.
The synthesis of vegetable oil-based linear polyesters has been extensively studied in the past decade,29–31 including by our group.32 In contrast, only a handful of studies have been dedicated to HBPEs derived from plant oils. Related examples have involved the polycondensation of ABn-type monomer precursors (n ≥ 2). For this purpose, selective modifications/derivatizations of hydroxy-containing fatty acid derivatives have been implemented. For instance, Meier et al. and Li et al. have resorted to the thiol–ene addition of 1-thioglycerol onto methyl-10-undecenoate, as a means to access an AB2-type monomer, the polycondensation of which, in the presence of organic or metallic catalysts, has led to bio-based HBPEs.33,34 Meier et al.33 have also reported on the polycondensation of the same AB2-type monomer using glycerol as a core molecule, and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a catalyst at 120 °C, resulting in HBPEs with moderate molar masses (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 3500–4400 g mol−1, Đ = 1.87–2.85). Li et al.34 have achieved higher molar masses (n = 11400–60400 g mol−1) though with broader dispersities (Đ = 5.2–25.3), upon metallic catalysis based on Ti(OBu)4, Sb2O3 or Zn(OAc)2 operating at 160–170 °C. DB values determined by quantitative 13C NMR have been found in the range 0.38–0.54, in agreement with the value of 0.5 predicted by theory.35 As for Petrović et al., they have developed two synthetic pathways to HBPEs from hydroxylated FAMEs obtained either by (i) hydrogenation36 of epoxidized soybean oil or by (ii) hydroformylation.37,38 High molar masses (up to 42000 g mol−1) have been obtained only in the latter case, with a weight average hydroxyl functionality of the polymer up to 23.2. Note, however, that no DB value has been reported. Two other reports have dealt with HBPE synthesis via the A2 + B3 approach, through the copolymerization of various α,ω-diacids with glycerol.39,40
n = 3500–4400 g mol−1, Đ = 1.87–2.85). Li et al.34 have achieved higher molar masses (n = 11400–60400 g mol−1) though with broader dispersities (Đ = 5.2–25.3), upon metallic catalysis based on Ti(OBu)4, Sb2O3 or Zn(OAc)2 operating at 160–170 °C. DB values determined by quantitative 13C NMR have been found in the range 0.38–0.54, in agreement with the value of 0.5 predicted by theory.35 As for Petrović et al., they have developed two synthetic pathways to HBPEs from hydroxylated FAMEs obtained either by (i) hydrogenation36 of epoxidized soybean oil or by (ii) hydroformylation.37,38 High molar masses (up to 42000 g mol−1) have been obtained only in the latter case, with a weight average hydroxyl functionality of the polymer up to 23.2. Note, however, that no DB value has been reported. Two other reports have dealt with HBPE synthesis via the A2 + B3 approach, through the copolymerization of various α,ω-diacids with glycerol.39,40
In this contribution, we describe a platform of new ABn-type building blocks based on FAMEs and the synthesis of related vegetable oil-based HBPEs with tunable properties. For this purpose, sunflower, castor and rapeseed oils were used as raw materials for the synthesis of chemically pure monomers of ABn-type (n = 2 or 3), featuring an ester (A) and two or three alcohol (B) moieties. The polytransesterification of these monomers was optimized, by varying the initial experimental conditions. Insights into their fine structure are provided both by 1H NMR spectroscopy and MALDI ToF mass spectrometry. Their crystallization ability and thermal stability are also assessed by DSC and TGA analyses.
Experimental
Materials
1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), meta-chloroperbenzoic acid (m-cpba, <77%), anhydrous zinc acetate (Zn(OAc)2, 99.99% trace metals basis), sodium methoxide (powder, 95%), glycerol (99%), anhydrous tert-butanol (ACS reagent, ≥99.0%), phosphoric acid (85%) and hydrogen peroxide (30%) were obtained from Sigma Aldrich. Methyl-10-undecenoate (>96.0%) was supplied by TCI Europe. All products and solvents were used as received except otherwise mentioned. The solvents were of reagent grade quality and were purified when necessary according to the methods reported in the literature. Methyl-9,10-dihydroxystearate (M2HS), methyl-9,10,12-trihydroxystearate (M3HS) and methyl-13,14-dihydroxybehenate (M2HB) were synthesized by ITERG (Pessac, France), and their synthesis is described further.Methods
Size exclusion chromatography (SEC) analyses were performed in THF as the eluent (1 mL min−1) at 40 °C, on a PL-GPC 50 Plus Integrated GPC from Polymer Laboratories-Varian with a series of four columns from TOSOH [TSKgel TOSOH: HXL-L (guard column 6.0 mm ID × 4.0 cm L); G4000HXL (7.8 mm ID × 30.0 cm L); G3000HXL (7.8 mm ID × 30.0 cm L) and G2000HXL (7.8 mm ID × 30.0 cm L)]. The elution times of the filtered samples were monitored using RI detectors with a calibration curve based on low dispersity polystyrene standards (PS). Trichlorobenzene was added as a flow marker.
Differential scanning calorimetry (DSC) measurements were carried out on a DSC Q100 apparatus from TA Instruments. For each sample, two cycles from −100 to 150 °C (except otherwise mentioned) were performed at 10 °C min−1. Glass transition and melting temperatures were calculated based on the second heating run.
Thermogravimetric analyses (TGA) were performed on two different apparatus from TA Instruments depending on the availability: TGA Q50 and Q500 at a heating rate of 10 °C min−1 under a nitrogen atmosphere from room temperature to 700 °C.
MALDI ToF mass spectrometry (MS) analyses were performed by the Centre d'Etude Structurale et d'Analyse des Molécules Organiques (CESAMO, Bordeaux, France) on a Voyager mass spectrometer (Applied Biosystems). The instrument is equipped with a pulsed N2 laser (337 nm) and a time-delayed extracted ion source. Spectra were recorded in the positive-ion mode using a reflectron and with an accelerating voltage of 20 kV. Samples were dissolved in THF at 10 mg ml−1. The IAA matrix (trans-3-indoleacrylic acid) was prepared by dissolving 10 mg in 1 ml of THF. A methanol solution of a cationization agent (NaI, 10 mg ml−1) was also prepared. The solutions were combined in a 10:1:1 volume ratio of matrix to sample to the cationization agent. One to two microliters of the obtained solution were deposited onto the sample target and vacuum-dried.
Synthetic procedures
Epoxidation step. Methyl esters of high oleic sunflower oil (40 kg, 133.3 mol) and formic acid (1.7 kg, 37.8 mol) were added in a reactor equipped with a mechanical stirrer, a dropping funnel and a condenser. The resulting mixture was heated at 40 °C for 1 hour under stirring. Hydrogen peroxide (35%, 24.4 kg, 251.6 mol) was added dropwise to the reactor at 40 °C, using a dropping funnel to maintain the temperature in the reactor close to 70–75 °C. As the reaction is exothermic, a cooling system was used to cool down the reactor. The reaction was monitored by gas chromatography and epoxide titration. The reaction mixture was then cooled down to room temperature and the aqueous phase was discarded. The organic layer was washed with an aqueous solution of sodium hydroxide (0.1 N) until the pH became neutral. The organic phase was then dried under vacuum at 60 °C to afford a clear and slightly yellow liquid.
Hydroxylation step. Epoxidized fatty acid methyl esters (10 kg) were placed along with an aqueous solution of phosphoric acid (12% w/w, 5 kg) and tert-butanol (3 kg) as the solvent in a reactor equipped with a condenser and a mechanical stirrer. The resulting mixture was heated at 90 °C under vigorous stirring. The reaction was monitored by gas chromatography. When the reaction was completed, the aqueous phase was discarded at 50 °C. tert-Butanol was eliminated under vacuum distillation. The organic phase was then washed with hot water until the pH reached 6–7 and was dried under vacuum to afford a white solid. M2HS was then recrystallized in cyclohexane (3 times, 40–60 g L−1) and dried under vacuum to afford a white solid powder. The product was then dissolved in a minimum of dichloromethane (DCM) and injected in a flash chromatography apparatus from Grace. The constituents were separated on a silica column, using a dichloromethane–methanol gradient and an Evaporative Light Scattering Detector (ELSD). Two fractions were collected, corresponding to M2HS and its acid form, respectively. The M2HS purity was determined by GC (97.8%).
The procedure followed to prepare methyl-13,14-dihydroxybehenate (M2HB) and methyl-9,10,12-trihydroxystearate (M3HS) was nearly identical to that described for M2HS; details are provided hereafter.
:methanol gradient (95:5). The AB3-type monomer was obtained as a white powder in a rather low yield (25%). Purity: 98.1%.
Epoxidation step. Methyl undecenoate (15 g, 0.076 mol) and m-cpba (39.2 g, 0.227 mol) were stirred at room temperature in DCM (20 mL g−1 of product) overnight. 3-Chlorobenzoic acid formed as a side product precipitated in DCM. The reaction mixture was first filtered to remove 3-chlorobenzoic acid, and washed with aqueous sodium sulfite Na2SO3 (3 × 50 mL), aqueous sodium bicarbonate NaHCO3 (4 × 50 mL) and brine (2 × 50 mL) until the pH became neutral. The organic phase was then dried over anhydrous magnesium sulfate, filtered and DCM was removed on a rotary evaporator to afford methyl-10-epoxyundecenoate. Yield: 92%.
Hydroxylation step. Methyl-10-epoxyundecenoate (2.5 g) was charged in a round-bottom flask equipped with a mechanical stirrer, an oil bath and a condenser. The epoxidized intermediate was dissolved in 50 mL of a 1
:1 (v/v) mixture of water and tert-butanol under stirring. After the addition of phosphoric acid (85% w/w, 3 wt%), the reaction flask was heated under reflux at 90 °C. 4 hours later, the aqueous phase was discarded at 50 °C and tert-butanol was removed under vacuum distillation. After the addition of 50 mL DCM, the organic phase was washed twice with water (2 × 50 mL) and brine (1 × 50 mL), dried over anhydrous magnesium sulfate and filtered and then the solvent was removed on a rotary evaporator. M2HU was obtained as a white powder (purity: 99%). Yield: 57%.
Results and discussion
Synthesis of bio-based ABn-type monomers
Unsaturated FAMEs serving as raw plant oil materials were first chemically modified, so as to achieve different precursors of AB2/AB3-type consisting of methyl ester (A) and alcohol (B) reactive functions. The conversion of A-type FAMEs into ABn-type monomers (n = 2 or 3) was accomplished following a two-step straightforward procedure, involving (i) epoxidation of the double bond and (ii) subsequent ring-opening of the as-formed epoxide under acidic conditions. Derivatization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds into 1,2-diols through epoxidation has been widely applied to vegetable oil derivatives.41 Upon using methyl oleate, it leads to the formation of methyl-9,10-dihydroxystearate (M2HS), i.e. an AB2-type monomer (Scheme 1). Other substrates of interest were also considered, on the basis of the same strategy. Thus, methyl euricate, a C22-carbon atom-containing precursor (instead of C18 for methyl oleate), allowed accessing methyl-13,14-dihydroxybehenate (M2HB). The latter monomer was expected to provide HBPEs with a higher alkyl chain length between branching points compared to M2HS (11 CH2vs. 7 CH2). Similar modification of methyl ricinoleate, where a hydroxyl function is already present, yielded the AB3-type monomer precursor denoted as M3HS. Lastly, methyl undecenoate (C11:1) was selected to prepare methyl-10,11-dihydroxyundecanoate (M2HU), an AB2-type monomer, the polycondensation of which should lead to HBPEs free of dangling chains.
C double bonds into 1,2-diols through epoxidation has been widely applied to vegetable oil derivatives.41 Upon using methyl oleate, it leads to the formation of methyl-9,10-dihydroxystearate (M2HS), i.e. an AB2-type monomer (Scheme 1). Other substrates of interest were also considered, on the basis of the same strategy. Thus, methyl euricate, a C22-carbon atom-containing precursor (instead of C18 for methyl oleate), allowed accessing methyl-13,14-dihydroxybehenate (M2HB). The latter monomer was expected to provide HBPEs with a higher alkyl chain length between branching points compared to M2HS (11 CH2vs. 7 CH2). Similar modification of methyl ricinoleate, where a hydroxyl function is already present, yielded the AB3-type monomer precursor denoted as M3HS. Lastly, methyl undecenoate (C11:1) was selected to prepare methyl-10,11-dihydroxyundecanoate (M2HU), an AB2-type monomer, the polycondensation of which should lead to HBPEs free of dangling chains.
 | ||
| Scheme 1 Synthetic route to novel ABn-type fatty acid-based monomers and related hyperbranched polyesters. | ||
All monomer precursors were obtained as white powders in yields higher than 60%, except for M3HS (≈25%). The low yield in the latter case could be explained by the hydroxylation of methyl ricinoleate which resulted in a mixture of four diastereoisomers, only one of them being able to crystallize.42–45 The structure of the so-formed AB2- or AB3-type monomers was assessed by 1H NMR spectroscopy (see ESI Fig. S1†). GC analyses revealed a chemical purity higher than 94% in all cases.
Synthesis of hyperbranched polyesters by self-polycondensation of AB2- and AB3-type monomers
All polymerizations were carried out in bulk, in line with many of the Principles of Green Chemistry,46 following a two-step-one-pot methodology illustrated in Scheme 1. After a two-hour oligomerization stage at 120 °C, the temperature was raised to 160 °C, and dynamic vacuum was applied. Polycondensations were thus performed in the melt, and crude polymers were analyzed without further purification (Table 1).| Entry | Catalyst | x (%) |
na (g mol−1) |
Đ |
|---|---|---|---|---|
| a Determined by SEC in THF; calibration with PS standards. Procedure: 2 hours at 120 °C under N2 followed by 13 hours at 160 °C under dynamic vacuum. | ||||
| 1 | Ti(OBu)4 | 44 | 1100 | 1.09 |
| 2 | Ti(OiPr)4 | 48 | 1200 | 1.08 |
| 3 | Zn(OAc)2 | 98 | 3500 | 2.71 |
| 4 | DBTO | 39 | 1100 | 1.09 |
| 5 | Sb2O3 | 45 | 1200 | 1.08 |
| 6 | TBD | 100 | 4100 | 2.46 |
| 7 | m-TBD | 21 | 1000 | 1.10 |
| 8 | DABCO | No polymerization | ||
| 9 | DBU | |||
| 10 | NaOMe | 100 | 6100 | 3.08 |
Various commercially available transesterification catalysts were screened with a loading of 1.5 wt% relative to the monomer. A more systematic investigation was undertaken with M2HS (Table 1). Because transition metal catalysts currently dominate condensation polymerization processes at the industrial scale, a selection of zinc, tin, titanium and antimony-based catalysts was considered first. Organic catalysts were also investigated in line with the development of environmentally friendly metal-free processes.47,48 Guanidines including 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), its N-methyl derivative, namely, 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (m-TBD), the amidine base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and the tertiary amine 1,4-diazabicyclo[2.2.2]octane (DABCO) were thus examined. Finally, sodium methoxide (NaOMe) was also tested as it is widely used in oleochemistry as transesterification catalyst of crude vegetable oils.49
Most of the organometallic catalysts tested, including titanium(IV) butoxide and isopropoxide, Ti(OBu)4 and Ti(OiPr)4, respectively, dibutyltin(IV) oxide (DBTO) and antimony trioxide (Sb2O3), along with the organic base m-TBD showed a poor efficiency toward the polycondensation of M2HS. DABCO and DBU did not show any activity at all. Much better results were achieved with zinc acetate, Zn(OAc)2, TBD and NaOMe. Different reaction outcomes were thus noted from one catalyst to another, which might be correlated to the reaction mechanism in each case. Regarding the difference between m-TBD and TBD used as organocatalysts, one can hypothesize that TBD acts as an efficient dual catalyst due to both its H-donor and H-acceptor capability.47,48 In contrast, MTBD is known to behave as a Brønsted base that appears poorly active under the conditions tested here.
Based on this catalyst screening, Zn(OAc)2, TBD and NaOMe were selected for further polymerization/optimization utilizing the other ABn-type bio-sourced monomers.
As summarized in Table 2, M2HS, M3HS and M2HB were efficiently polymerized at 160 °C, monomer conversion over 95% being reached within 15 hours. SEC analyses indicated the formation of HBPEs with molar masses (Mn) in between 3000 and 6100 g mol−1, corresponding to an average number degree of polymerization  ranging from 10 to 12. However, since these molar masses were determined using linear PS standards, they are only apparent values and should be taken with caution, real values being likely much higher. HBPEs are indeed expected to display much smaller hydrodynamic radii than that of linear homologues or of PS standards of the same molar mass. Dispersity values higher than 2 were obtained, as expected for hyperbranched materials prepared by the polycondensation of ABn-type monomers.50 Higher molar masses, Mn = 7600 g mol−1, were reached by increasing either the reaction time, from 15 to 24 hours, and/or the reaction temperature up to 170 °C, as highlighted with M2HS and M2HB (Table 2, P1 vs. P2, P3 vs. P4 and P5, P7 vs. P8). The impact was even more important on dispersities, significantly broader values being obtained (>10) in agreement with the theory. However, higher polymerization temperatures (≥200 °C) and longer reaction times (over 40 hours) were found to favor the formation of insoluble gel-like materials.
ranging from 10 to 12. However, since these molar masses were determined using linear PS standards, they are only apparent values and should be taken with caution, real values being likely much higher. HBPEs are indeed expected to display much smaller hydrodynamic radii than that of linear homologues or of PS standards of the same molar mass. Dispersity values higher than 2 were obtained, as expected for hyperbranched materials prepared by the polycondensation of ABn-type monomers.50 Higher molar masses, Mn = 7600 g mol−1, were reached by increasing either the reaction time, from 15 to 24 hours, and/or the reaction temperature up to 170 °C, as highlighted with M2HS and M2HB (Table 2, P1 vs. P2, P3 vs. P4 and P5, P7 vs. P8). The impact was even more important on dispersities, significantly broader values being obtained (>10) in agreement with the theory. However, higher polymerization temperatures (≥200 °C) and longer reaction times (over 40 hours) were found to favor the formation of insoluble gel-like materials.
| Entry | Monomer | Catalyst | T (°C) |
M
nb (g mol−1) |
Đ | DBFreyc |
T
gd (°C) |
T
md (°C) |
T
cd (°C) |
T
de 5 wt% (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Temperature of polymerization. b SEC in THF – calibration PS standards. c 1H NMR. d DSC – 10 °C min−1. e Determined by TGA – 10 °C min−1 under N2. f Duration of polymerization increased from 15 to 24 hours. nd. = not determined. | ||||||||||
| P1 | M2HS | Zn(OAc)2 | 160 | 3500 | 2.71 | 0.07 | −27 | — | — | 246 |
| P2 | Zn(OAc)2 | 170 | 6300 | >15 | 0.26 | −30 | — | — | 252 | |
| P3 | TBD | 160 | 4100 | 2.46 | 0.35 | −24 | — | — | 320 | |
| P4 | TBD | 170 | 5300 | 3 | 0.38 | −24 | — | — | 324 | |
| P5 | TBD6 | 170 | 7600 | >12 | 0.41 | −22 | — | — | 338 | |
| P6 | NaOMe | 160 | 6100 | 3.08 | 0.29 | −20 | — | — | 340 | |
| P7 | M2HB | Zn(OAc)2 | 160 | 3000 | 1.93 | 0.09 | — | nd. | nd. | nd. |
| P8 | Zn(OAc)2f |
160 | 5600 | >11 | 0.18 | — | 20 | 14 | 260 | |
| P9 | TBD | 160 | 5600 | 3.05 | 0.33 | — | 22 | 12 | 298 | |
| P10 | NaOMe | 160 | 9200 | 3.27 | 0.30 | — | 23 | 12 | 332 | |
| P11 | M2HU | Zn(OAc)2 | 140 | 2400 | 2.33 | nd. | — | 61 | 44 | 204 |
| P12 | TBD | 140 | 2400 | 2.67 | 0.25 | — | 54 | 36 | 230 | |
| P13 | M3HS | Zn(OAc)2 | 160 | 3800 | 2.58 | nd. | −3 | — | — | 254 |
| P14 | TBD | 160 | 5600 | 4.05 | nd. | −1 | — | — | 305 | |
| P15 | NaOMe | 160 | 4600 | 2.83 | nd. | 3 | — | — | 299 | |
In contrast to the other bio-based monomers, M2HU was preferably polymerized at 140 °C. Due to the higher reactivity of its primary alcohols, it was indeed noted that gelation occurred more rapidly at 160 °C, i.e. in less than 10 hours. Yet, a further decrease of the polymerization temperature to 120 °C yielded oligomers only (Mn < 900 g mol−1). Within 8 hours at 140 °C, full conversions were achieved using both zinc acetate and TBD as catalysts, leading to a molar mass around 2500 g mol−1.
All these experiments revealed the importance of suitable experimental conditions to achieve soluble HBPEs. According to Flory's theory, the statistical polycondensation of ABn-type monomers yields HBPs without any risk of gelation provided that (i) A reacts exclusively with B and (ii) the reactions do not involve internal cyclization.3 However, in practice, side reactions can take place during HBP synthesis and can also be responsible for the formation of insoluble materials due to cross-linking. Syntheses involving M2HS were further scrutinized by analyzing M2HS-based HBPEs by MALDI-ToF mass spectroscopy (Fig. 1).
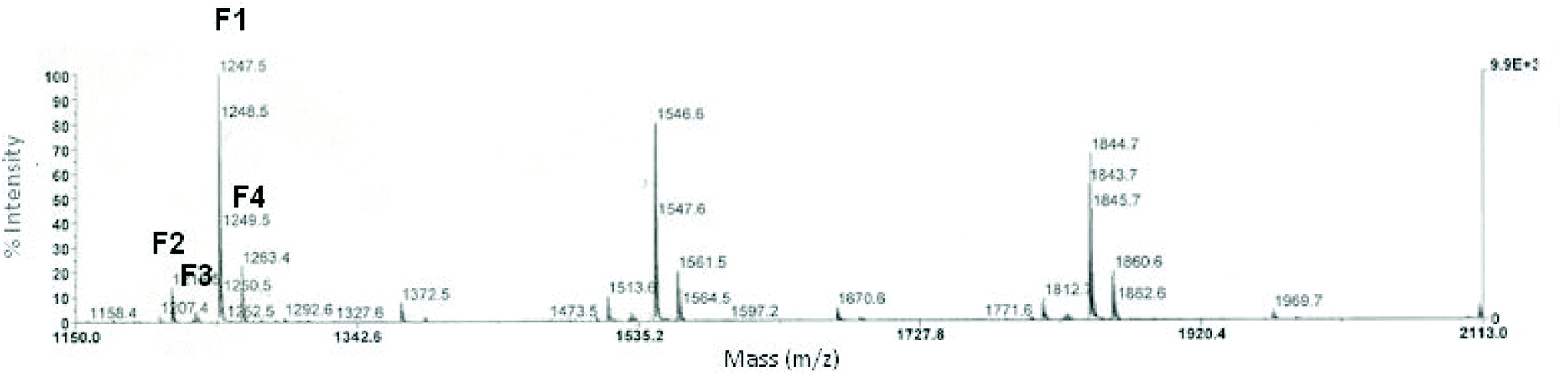 | ||
| Fig. 1 MALDI-ToF MS analysis of M2HS-based HBPE (matrix: trans-3-indoleacrylic acid, entry P3, Table 2). | ||
Four distinct families of populations were eventually detected with an expected peak-to-peak mass increment of 298.1 g mol−1 matching the repeating unit. Population F1 corresponds to the targeted HBPE chains derived from M2HS, i.e. n × (Munit) + MMeOH + MNa where Munit = MM2HS − MMeOH = 298.1 g mol−1. Population F2 shows a mass difference of −32 g mol−1, compared to the former regular HBPE structure. This was ascribed to the presence of lactone units (n × (Munit) + MNa), as the result of an esterification reaction occurring by an intramolecular pathway. The latter side reaction could either involve the carbonyl group of the focal point and a hydroxyl function of the same HBPE molecule (F2, Scheme 2), or could take place via a hydroxyl-ester interchange reaction between groups of the same branch (F′2, Scheme 2). As for population F3, it was characterized by m/z −18 g mol−1 with regard to F1 (Scheme 2), and it could be explained by the loss of a water molecule. In other words, this population reflected the occurrence of intramolecular etherification between two hydroxyl groups: n × (Munit) + MMeOH + MNa − MH2O. It is noteworthy that, if intramolecular cyclizations through ester (F2) or ether (F3) bond formation reduced achievable molar masses of these bio-based HBPEs, then they did not yield cross-linked materials. Gelation observed experimentally would thus be due to extensive etherification side reactions taking place intermolecularly, and thus leading to more than one focal group per macromolecule, i.e. at n × (Munit) + 2 × MMeOH + MNa − MH2O. Accordingly, a series of peaks (= population F4) was observed, at m/z +14 g mol−1 (= MMeOH − MH2O), and assigned to ether bridges between polymeric chains (Scheme 3).
 | ||
| Scheme 2 Proposed side reactions during bio-based HBPE synthesis and related populations: cyclization by intramolecular esterification (F2), by intramolecular hydroxyl-ester interchange between groups of the same branch (F′2) and by intramolecular etherification (F3). | ||
 | ||
| Scheme 3 Intermolecular etherification accounting both for the presence of population F4 and for gel formation. | ||
Although these side reactions could not be quantified, they appeared to occur whatever the polymerization catalyst employed (Zn(OAc)2 and TBD), and thus competed with the transesterification reaction during the whole polymerization process. These observations were supported by SEC analyses, as illustrated in Fig. 2, comparing SEC-RI traces of aliquots of the TBD-catalyzed polycondensation of M2HS. In the early stages of reaction, indeed, chromatograms displayed the pattern of a typical step-growth polymerization of ABn-type monomers. The observation of multimodal distributions was consistent with the formation of intermediate oligomers of increasing degrees of polymerization  . The disappearance of the peak at 27.9 minutes, assigned to M2HS, evidenced a complete conversion within the first 4 hours of polymerization. After 15 hours of reaction, signals attributed to dimers and trimers at 26.4 and 25.3 minutes, respectively, were split into two discrete peaks, as the result of lactone formation by cyclization, as discussed above. In the absence of a focal point constituted of a methyl ester function, these cyclic oligomers cannot react further during polymerization and remain as such until the last stage of the reaction, thereby contributing to the broadening of the molar mass distribution. Beyond 20 hours of polymerization, a shoulder was observed in the higher molar mass region, likely as the consequence of both intermolecular transesterification and etherification side reactions. This intermolecular etherification reaction can create a second focal point in the polymer chain and ultimately lead to crosslinking.
. The disappearance of the peak at 27.9 minutes, assigned to M2HS, evidenced a complete conversion within the first 4 hours of polymerization. After 15 hours of reaction, signals attributed to dimers and trimers at 26.4 and 25.3 minutes, respectively, were split into two discrete peaks, as the result of lactone formation by cyclization, as discussed above. In the absence of a focal point constituted of a methyl ester function, these cyclic oligomers cannot react further during polymerization and remain as such until the last stage of the reaction, thereby contributing to the broadening of the molar mass distribution. Beyond 20 hours of polymerization, a shoulder was observed in the higher molar mass region, likely as the consequence of both intermolecular transesterification and etherification side reactions. This intermolecular etherification reaction can create a second focal point in the polymer chain and ultimately lead to crosslinking.
 | ||
| Fig. 2 SEC-RI traces of aliquots withdrawn at different reaction times, during the TBD-catalyzed polycondensation of M2HS. | ||
Characterization of HBPEs
The chemical structure of the as-formed HBPEs was first confirmed by FT-IR analysis (see ESI Fig. S3†). In all spectra, absorption bands characteristic of both the ester and the hydroxyl functions were observed at 1734 and 3400 cm−1, respectively. To gain a better insight into their fine structure, and in particular to determine the DB value of these HBPEs, analysis by 1H NMR spectroscopy was realized. Fréchet et al.51 first expressed the DB as follows: | (1) |
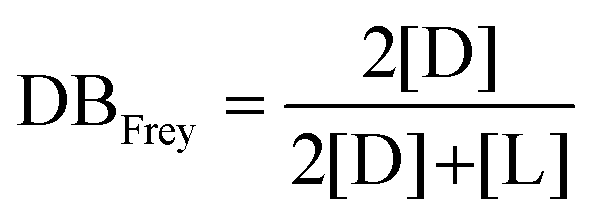 | (2) |
Both definitions lead to similar results at high conversions. In the present study, eqn (2) was used for the determination of DB.
Importantly, well-separated signals assigned to the three different subunits, T, L and D, could be clearly distinguished by 1H NMR spectroscopy, allowing the DB value to be accurately determined. As an illustration, Fig. 3 (see also Fig. S4 in the ESI†) depicts the 1H NMR spectrum of P3 of Table 2. The steep decrease in the intensity of the peak assigned to the methoxy group at 3.66 ppm confirmed the polyester synthesis. Consistently with the 1H NMR spectrum of M2HS, the peak at 3.38 ppm was attributed to the terminal unit (T), i.e. to the protons adjacent to the vicinal diol on α-carbon atoms (Ha). After the first substitution, protons Hb and Hc, which are characteristic of the linear unit (L), are no longer equivalent and thus display down-fielded signals at 3.56 and 4.81 ppm, respectively. It should be noted that chemical shifts are equivalent after esterifying either the 9 or the 10-position. Finally, the peak at 4.99 ppm was unambiguously assigned to the protons Hd of the dendritic unit (D).
 | ||
| Fig. 3 1H NMR spectrum of P3 prepared by the polycondensation of M2HS in CDCl3 (Table 2). | ||
Similarly, HBPEs derived from M2HB and M2HU were successfully characterized by 1H NMR spectroscopy (see ESI, Fig. S5, S6 and Table S1†). In the case of the AB3-type monomer, however, as-formed HBPEs are not characterized by three but by four different units, including the terminal (T), the linear (L), the semi-dendritic (sD) and the perfectly dendritic (D) units. Since the three hydroxyl groups of M3HS are not chemically equivalent, the number of accessible configurations is increased to 8, leading to a complex NMR spectrum (see ESI, Fig. S7†) that did not allow us to determine the DB value in this case.
The DB was found to dramatically depend on the type of catalyst employed with both AB2-type monomers investigated in this work. While zinc acetate appeared to favor the formation of weakly branched structures (DB < 0.26), both TBD and NaOMe gave HBPEs with a DB up to 0.45 (see ESI, Fig. S8†).
We also attempted to determine the DB value as a function of the conversion, on the basis of previous studies by Hölter and Frey35 and by Yan, Müller et al.,52 who expressed the conversion dependence of the DB as follows:
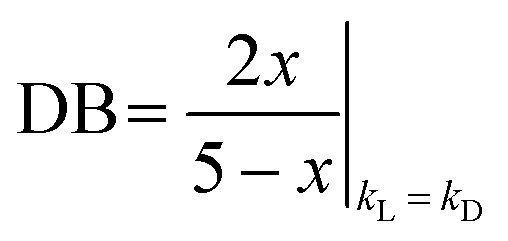 | (3) |
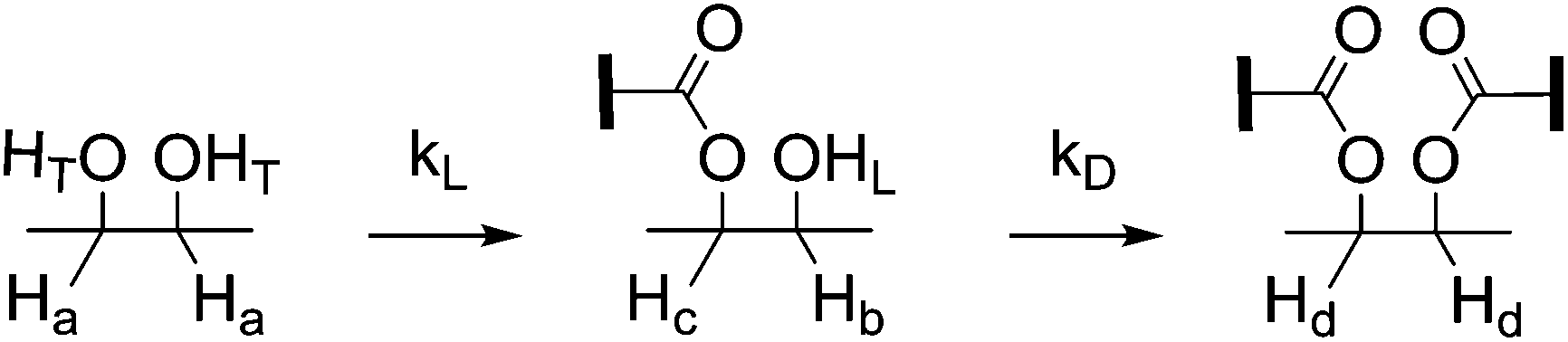 | ||
| Scheme 4 Rate constants of the formation of linear and dendritic units: kL and kD. | ||
Here, both the Zn(OAc)2 and the TBD-catalyzed polytransesterifications of M2HS were monitored by 1H NMR spectroscopy. Theoretical and experimental data are compared in Fig. 4. The discontinuity of the curves showing the evolution of DB with the ester group conversion (x) is explained by the fact that the first three data were collected during the oligomerization stage at 120 °C (see above), while subsequent data were obtained when the polymerization took place at 160 °C under dynamic vacuum. This curve clearly indicates that the two hydroxyl functions of M2HS do not exhibit the same reactivity, irrespective of the catalyst, experimental points always remaining below the theoretical line. The maximum DB value of 0.4 with TBD and of 0.2 with Zn(OAc)2 denotes a reactivity ratio r (r = kL/kD) equal to 0.5 and to 0.1, respectively.53
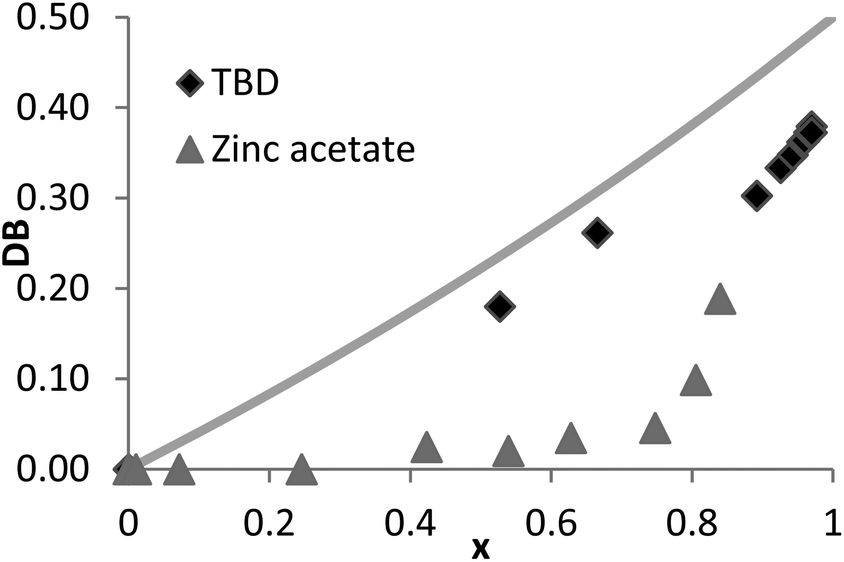 | ||
| Fig. 4 Conversion dependence of the degree of branching (DB) for the polycondensation of M2HS using TBD or zinc acetate as the catalyst (solid lines: theoretical data, symbols: NMR data). | ||
Thermal properties
The thermal properties of our bio-based HBPEs were assessed by DSC analysis. Glass transition and melting temperatures were thus determined from the second heating scan at 10 °C min−1 and crystallization temperatures from the first cooling scan at the same rate (Table 2). As expected, all HBPEs derived from M2HS and M3HS proved amorphous. Compounds obtained from M3HS showed higher glass transition temperatures, i.e. in between −10 and 9 °C, compared to M2HS-based polyesters (−32.5 ≤ Tg ≤ −20 °C), which was obviously explained by the higher functionality of the AB3-type monomer (M3HS vs. M2HS). Unlike linear polymers (see ESI Fig. S9†), the Tg value of HBPs is greatly affected both by the nature and the content of end-groups (T units).11 With the number of polar end-groups increasing with the monomer functionality, more interactions likely develop within M3HS-based HBPEs, compared to M2HS-based homologues of similar molar masses. Note however that the branching density of these renewable HBPEs did not seem to impact Tg values.Interestingly, both HBPEs derived from M2HB and M2HU displayed semi-crystalline properties. DSC thermograms are given in the ESI (Fig. S10†). The higher flexibility of M2HB-derived HBPEs, which consist of a higher chain-length between branching points compared to M2HS (11 carbon atoms vs. 7), allowed the development of crystalline zones. The semi-crystalline character of M2HB-based HBPEs was confirmed by wide-angle X-ray diffraction (WAXS) measurements (see ESI; Fig. S11†). The absence of pendant alkyl chains acting as plasticizers also appeared to endow the HBPEs derived from M2HU with semi-crystalline properties. In this case, complex melting and crystallization patterns were observed, probably due to the unsymmetrical nature of the initial diol that could affect the chain mobility and prevent the formation of stable crystals.
Lastly, thermal stabilities of these HBPEs were investigated by TGA under non-oxidative conditions, and at a heating rate of 10 °C min−1 (Table 2). Typical degradation profiles are given in the ESI (Fig. S12 and S13†). HBPEs thus showed typical thermal stabilities for oil-derived polymers,32 with a 5% weight loss (Td5%) up to 332 °C. HBPEs derived from M2HU were found to exhibit the lowest heat resistance of this series, likely due to their lower molar masses (n ≤ 2500 g mol−1). Overall, HBPEs prepared using zinc acetate (P1, P2, P7, P8, P11 and P13) appeared less stable than their homologues obtained with TBD or NaOMe. The thermal degradation of the former compounds was found to be accelerated at high temperatures, likely due to residual traces of Zn(OAc)2 catalyzing their depolymerization (see ESI Fig. S14†), as already reported for more conventional polyesters, such as poly(ethyleneterephthalate)54 or poly(tetramethylene succinate).55
Conclusion
Sunflower, castor and rapeseed oils can serve as raw materials to be readily derivatized into AB2- or AB3-type monomer substrates featuring methyl ester and alcohol functions, by means of epoxidation followed by the ring-opening of epoxy-rings. The subsequent polycondensation of these bio-based monomers can be performed in bulk, giving access to modular hyperbranched polyesters through repeated transesterification reactions. Zinc acetate, TBD and sodium methoxide-three catalytic systems operating by a specific activation mechanism, are the most effective, achieving molar masses in the range 3000–10000 g mol−1 and dispersities varying from 2 to 15. Depending on the initial conditions and on the monomer considered, either amorphous or semi-crystalline hyperbranched polyesters, with a Tg value in the range −33 °C to 9 °C, a thermal stability above 300 °C, and with a degree of branching that could be varied from 0.07 to 0.45, can thus be synthesized. Due to the presence of numerous hydroxyl groups both in linear and terminal units, such hyperbranched polyesters are likely amenable to facile post-polymerization modification, allowing for further tuning of the properties of these renewable branched materials.
Funding sources
This work was performed, in partnership with the SAS PIVERT, within the framework of the French Institute for the Energy Transition (Institut pour la Transition Energétique (ITE) P. I. V. E. R. T. (http://www.institut-pivert.com)) selected as an Investment for the Future (“Investissements d'Avenir”). This work was supported, as part of the Investments for the Future, by the French Government under the reference ANR-001-01.Acknowledgements
The authors thank the University of Bordeaux, Bordeaux INP, CNRS, Aquitaine Regional Council, the SAS PIVERT and ITERG for the support of this research. Besides, the writers acknowledge P. Castel and C. Absalon from CESAMO for MALDI-ToF MS experiments and A.Bentaleb from CRPP for the WAXS measurements.Notes and references
- D. Yan, C. Gao and H. Frey, Hyperbranched polymers: Synthesis, Properties, and Applications, John Wiley & Sons, Inc., 2011 Search PubMed.
- Y. H. Kim and O. W. Webster, Macromolecules, 1992, 25, 5561–5572 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718–2723 CrossRef CAS.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- D. A. Tomalia, J. B. Christensen and U. Boas, Dendrimers, Dendrons and Dendritic polymers: Discovery, Applications and the Future, Cambridge University Press, 2012 Search PubMed.
- Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685–1698 CrossRef CAS.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453–571 CrossRef CAS.
- M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233–1285 CrossRef CAS.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- Perstorp, Boltorn, advancing performance & confort, https://www.perstorp.com/-/media/files/perstorp/pb/boltorn.pdf (accessed September 2016).
- E. Žagar and M. Žigon, Prog. Polym. Sci., 2011, 36, 53–88 CrossRef.
- A. Hult, M. Johansson and E. Malmström, in Branched Polymers II, ed. J. Roovers, Springer, Berlin, Heidelberg, 1999, vol. 143, pp. 1–34 Search PubMed.
- M. G. McKee, S. Unal, G. L. Wilkes and T. E. Long, Prog. Polym. Sci., 2005, 30, 507–539 CrossRef CAS.
- X. Zhang, Prog. Org. Coat., 2010, 69, 295–309 CrossRef CAS.
- H. Kricheldorf, in Polycondensation: History and results, Springer, Berlin, Heidelberg, 2014, pp. 147–159 Search PubMed.
- A. Ghosh, S. Banerjee and B. I. Voit, Adv. Polym. Sci., 2015, 266, 27–124 CrossRef CAS.
- S. Sorrell, J. Speirs, R. Bentley, A. Brandt and R. Miller, Energy Policy, 2010, 38, 5290–5295 CrossRef.
- C. Okkerse and H. van Bekkum, Green Chem., 1999, 1, 107–114 RSC.
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- J. C. Ronda, G. Lligadas, M. Galià and V. Cádiz, Eur. J. Lipid Sci. Technol., 2011, 113, 46–58 CrossRef CAS.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Biomacromolecules, 2010, 11, 2825–2835 CrossRef CAS PubMed.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef CAS.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- O. Türünç and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2013, 115, 41–54 CrossRef.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- O. Türünç and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 1822–1826 CrossRef PubMed.
- Y. Bao, J. He and Y. Li, Polym. Int., 2013, 62, 1457–1464 CrossRef CAS.
- D. Hölter, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef.
- Z. S. Petrović and I. Cvetković, Contemp. Mater., 2012, III, 63–71 Search PubMed.
- Z. S. Petrović, I. Cvetković, J. Milić, D. Hong and I. Javni, J. Appl. Polym. Sci., 2012, 125, 2920–2928 CrossRef.
- J. Milic, I. Teraoka and Z. S. Petrovic, J. Appl. Polym. Sci., 2012, 125, 586–594 CrossRef.
- Y. Zhang, Y. Yang, J. Cai, W. Lv, W. Xie, Y. Wang and R. A. Gross, in Biobased Monomers, Polymers, and Materials, ed. P. B. Smith and R. A. Gross, ACS Symposium series, Washington, 2012, p. 111 Search PubMed.
- E. Wagner, F. Bruchmann, D. Haering, P. Keller and T. Pouhe, US7081509, 2006 Search PubMed.
- A. Köckritz and A. Martin, Eur. J. Lipid Sci. Technol., 2008, 110, 812–824 CrossRef.
- T. W. Findley, D. Swern and J. T. Scanlan, J. Am. Chem. Soc., 1945, 67, 412–414 CrossRef CAS.
- R. Subra Rao and K. T. Achaya, J. Sci. Ind. Res., 1960, 19B, 482–484 Search PubMed.
- A. F. McKay and A. R. Bader, J. Org. Chem., 1948, 13, 75–85 CrossRef CAS PubMed.
- M. A. Khuddus, Y. Usui and D. Swern, J. Am. Oil Chem. Soc., 1973, 50, 524–528 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. Fèvre, J. Vignolle, Y. Gnanou and D. Taton, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, p. 67 Search PubMed.
- A. Demirbas, Energy Convers. Manage., 2008, 49, 125–130 CrossRef CAS.
- D. Yan and Z. Zhou, Macromolecules, 1999, 32, 819–824 CrossRef CAS.
- C. Hawker, R. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 4588, 4583–4588 CrossRef.
- D. Yan, A. H. E. Müller and K. Matyjaszewski, Macromolecules, 1997, 30, 7024–7033 CrossRef CAS.
- H. Galina, J. B. Lechowicz and M. Walczak, Macromolecules, 2002, 35, 3261–3265 CrossRef CAS.
- T. Shah, J. Bhatty, G. Gamlen and D. Dollimore, Polymer, 1984, 25, 1333–1336 CrossRef CAS.
- J. Yang, S. Zhang, X. Liu and A. Cao, Polym. Degrad. Stab., 2003, 81, 1–7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of monomers in DMSO-d6/FT-IR spectra of HBPEs/2D NMR spectra of M2HS-derived HBPE in CDCl3/1H NMR spectra of M2HB, M2HU, M3HS-derived HBPEs and LPE in CDCl3 or DMSO-d6/catalyst dependence of DB values for M2HS and M2HB-based HBPEs/molar mass dependence of DB values for HBPEs prepared by the TBD-catalyzed polycondensation of M2HS/DSC curves of HBPEs and LPE/WAXS patterns of M2HB-derived HBPEs/TGA and TGA derivative curves of HBPEs. See DOI: 10.1039/c6gc02294d |
| This journal is © The Royal Society of Chemistry 2017 |