
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Yogurt inhibits intestinal barrier dysfunction in Caco-2 cells by increasing tight junctions†
Kelley K.
Putt
a,
Ruisong
Pei
a,
Heather M.
White
 b and
Bradley W.
Bolling
*a
b and
Bradley W.
Bolling
*a
aDepartment of Food Science, University of Wisconsin-Madison, 1605 Linden Dr., Madison, WI 53706, USA. E-mail: bwbolling@wisc.edu; Fax: +1-608-262-6872; Tel: +1-608-890-0212
bDepartment of Animal Science, University of Wisconsin-Madison, 1675 Observatory Drive, Madison, WI 53706, USA
First published on 9th January 2017
Abstract
Chronic inflammation disrupts intestinal barrier function and may contribute to the pathology of obesity and other diseases. The goal of this study was to determine the mechanism by which yogurt improves intestinal barrier function. Caco-2 cells were differentiated on Transwell inserts and used as a model of intestinal barrier permeability. Transepithelial electrical resistance (TEER) and flux of 4 kDa fluorescein isothiocyanate-dextran (FD) and lucifer yellow (LY) were used as indicators of monolayer integrity and paracellular permeability. Immunofluorescence microscopy and real time quantitative polymerase chain were used to assess the localization and expression of tight junction proteins known to regulate intestinal permeability. Differentiated cells were treated with a vehicle control (C), inflammatory stimulus (I) (interleukin-1β, tumor necrosis factor-α, interferon-γ, and lipopolysaccharide), or I and 0.03 g mL−1 yogurt (IY). After 48 h, I reduced Caco-2 TEER by 46%, while IY reduced TEER by only 27% (P < 0.0001). FD and LY flux reflected TEER measurements, with IY having significantly lower permeability than I (P < 0.05). Yogurt also improved localization of occludin and zona occludens protein 1 (ZO-1) at tight junctions of differentiated Caco-2 cells. IY increased Caco-2 claudin-1, ZO-1, and occludin mRNA relative to I (P < 0.05). In a simulated digestion, the barrier-improving bioactivity of yogurt was maintained through the gastric phase, but was reduced to the level of I after intestinal digestion (P < 0.05). Therefore, yogurt improved inflammation-disrupted intestinal barrier function in a Caco-2 model by increasing tight junctions, but the beneficial effect on barrier function was reduced at latter stages of digestion.
1 Introduction
Intestinal barrier permeability contributes to the systemic inflammation observed with obesity, inflammatory bowel disease, and other chronic disease.1,2 Increased intestinal barrier permeability allows greater translocation of endotoxin and other pro-inflammatory components present in the lumen, perpetuating chronic inflammation.3 These inflammatory stimuli and other endogenous cytokines can directly affect the intestinal barrier by reducing tight junction protein localization and expression.4Dietary components such as probiotics, polyphenols, lipids, and proteins can improve intestinal barrier function by inhibiting inflammation or by directly affecting barrier function.5–8 Yogurt and other low-fat dairy products may modulate intestinal barrier function. Low-fat dairy consumption reduced biomarkers of inflammation in overweight and obese individuals compared to a low-dairy control diet.9,10 In elderly individuals, low-fat yogurt consumption decreased plasma endotoxin and lipopolysaccharide binding protein, suggesting improved intestinal barrier function.11 In mice, low-fat yogurt prevented trinitrobenzene sulfonic acid-induced intestinal inflammation and reduced Toll-like receptor 4 positive intestinal T-cells.12 Given these studies, it is unclear if yogurt can directly affect intestinal barrier function in the presence of low-grade inflammatory stimuli.
Differentiated human Caco-2 cells are a useful model of the intestinal barrier with functional tight junction complexes.13,14 Thus, we hypothesized that yogurt will improve intestinal barrier function in Caco-2 cells subjected to chronic inflammatory stimuli. Therefore, the aim of this study was to determine the effects of yogurt treatment on barrier integrity and the localization and expression of tight junctions in a Caco-2 model of inflammation-perturbed barrier function.
2 Materials and methods
2.1 Chemicals and supplies
Low-fat yogurt (strawberry flavor) was from Yoplait (Minneapolis, MN) purchased through a local distributor. Dulbecco's Modified Eagle Medium (DMEM, high glucose), qualified fetal bovine serum (FBS), MEM non-essential amino acid solution (NEAA), Hank's balanced salt solution (HBSS), penicillin/streptomycin, and TRIzol reagent were obtained from Life Technologies (Grand Island, NY). Mouse anti-occludin, Mouse anti-ZO1, Alexa Fluor 488 Goat anti-mouse, and rhodamine phalloidin were purchased from ThermoFisher (Waltham, MA). RNeasy mini kits and RNase-free DNase were purchased from Qiagen (Valencia, CA), and RNase-free water was from Dot Scientific (Burton, MI). The iScript™ cDNA synthesis kit, iTaq™ Universal SYBR® Green Supermix, and Bio-Rad CFX96 system and software were obtained from Bio-Rad (Hercules, CA). HPLC-grade chloroform, isopropanol, and ethanol were obtained from Fisher Scientific (Pittsburgh, PA). Interleukin 1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), lipopolysaccharide (LPS), porcine pancreatin, porcine bile extract, porcine pepsin, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), fluorescein isothiocyanate-dextran (FD, 4 kDa), lucifer yellow (LY, 457 Da), Triton-X-100, bovine serum albumin (BSA), 4′,6-diamidino-2-phenylindole (DAPI) and all other reagents were purchased from Sigma Aldrich (St Louis, MO).2.2 Caco-2 cell culture maintenance
Caco-2 cells were obtained from ATCC (ATCC® HTB-37™), and were between passages 20–45 for all experiments (Manassas, VA). Cells were cultured in high glucose DMEM supplemented with 10% (v/v) FBS, 1% (v/v) MEM NEAA, and 1% (v/v) penicillin/streptomycin. Cells were incubated at 37 °C and 5% CO2, and were subcultured at 80–90% confluence every 3–4 days. Near confluence, cells were detached with trypsin, counted, and seeded at a density of 2 × 105 cells per mL onto polycarbonate membrane Transwell inserts with 0.4 μm pore size (Corning, Inc; Lowell, MA). Cells were cultured for 21 days to reach differentiation, and growth media was refreshed was refreshed every 2–3 days.2.3 Induction of barrier dysfunction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 1:100 (w/v) in regular supplemented growth medium or with the inflammatory cocktail before application to cell monolayers and further incubation. Routinely, experiments were conducted at 1:30 (w/v) dilution of yogurt powder in culture media, corresponding to a 1:8 (w/v) dilution of yogurt on a wet weight basis.
:10 to 1:100 (w/v) in regular supplemented growth medium or with the inflammatory cocktail before application to cell monolayers and further incubation. Routinely, experiments were conducted at 1:30 (w/v) dilution of yogurt powder in culture media, corresponding to a 1:8 (w/v) dilution of yogurt on a wet weight basis.
| Nutrient | Content per 100 g |
|---|---|
| Ingredients: Cultured pasteurized grade A low fat milk, sugar, strawberries, modified corn starch, nonfat milk, Kosher gelatin, citric acid, tricalcium phosphate, colored with carmine, natural flavor, pectin, vitamin A acetate, vitamin D3. Contained disclaimer that product meets National Yogurt Association criteria for live and active culture yogurt. (Contains Lactobacillus bulgaricus and Streptococcus thermophilus with at least 107 cultures per g at manufacture.) | |
| Total fat (g) | 1 |
| Carbohydrate (g) | 20 |
| Protein (g) | 3 |
| Cholesterol (mg) | 4 |
| Sodium (mg) | 50 |
| Calcium (mg) | 130 |
| Vitamin A (IU) | 440 |
| Vitamin D (μg) | 2.0 |
2.3.2.1 Simulated digestion of yogurt. Yogurt was subjected to in vitro digestion as described in ESI Fig. 1,† based on the method previously described by Minekus et al.19 Samples were collected after the stages of gastric digestion and full digestion (gastric and intestinal phase) and then snap frozen in liquid nitrogen. Digested samples were then resuspended in regular supplemented growth medium at the same mass ratio as undigested samples (1
:30, w/v) and applied to cell monolayers. Cells were treated with gastric digesta diluted with inflammatory cocktail, intestinal digesta diluted with inflammatory cocktail, or a digesta vehicle control diluted with inflammatory cocktail.
2.4 Transepithelial electrical resistance (TEER)
TEER was used as a measure of cell monolayer integrity and was assessed before and after all treatments. TEER was measured based on previously described approaches.20,21 Growth media was decanted from culture plates and inserts, and cells were then washed with HBSS containing 5 mM HEPES. HBSS/HEPES was added to the basolateral and apical compartments, in 1 and 0.2 mL volumes, respectively, and the plates were incubated at 37 °C and 5% CO2 for 30 min. Plates were then transferred to a hot plate set at 37 °C and TEER was measured using an epithelial volt-ohm meter with a chopstick electrode (Millicell ERS-2, EMD Millipore, Billerica, MA). The electrode was immersed at a 90° angle with one tip in the basolateral chamber and the other in the apical chamber. Care was taken to avoid electrode contact with the monolayer and triplicate measurements were recorded for each monolayer. An insert without cells was used as a blank and its mean resistance was subtracted from all samples. Unit area resistance was then calculated by dividing resistance values by the effective membrane area (0.33 cm2). For untreated, fully differentiated monolayers, TEER values were routinely 500–700 ohm cm2 (n = 22).2.5 Determination of paracellular permeability
Paracellular permeability was determined by the flux of FD and LY through differentiated Caco-2 monolayers as previously described.6,22 FD and LY were dissolved in 37 °C HBSS/HEPES at 1 mg mL−1 and 0.5 mg mL−1, respectively. The FD/LY/HBSS mixture (0.2 mL) was then added to the apical compartment of each insert, while 1.0 mL of HBSS was added to the basolateral well. The plate was covered with foil to prevent light inactivation of the fluorescent markers and placed in a shaker incubator set at 37 °C at 150 rpm. Every 4 h for 16 h, 0.3 mL aliquots were taken from the basolateral chamber and placed into a black, clear bottom 96 well plate (Fisher Scientific, Toronto, ON). After sampling, the basolateral compartment was decanted and replenished with a new 1.0 mL aliquot of 37 °C HBSS, and further incubated until the next sampling. The fluorescence intensity of the basolateral HBSS aliquots was measured immediately after sampling using excitation/emission wavelengths of 485/530 nm and 428/540 nm, for FD and LY respectively using a VarioSkan Flash Reader (Thermo Scientific, Waltham, MA).Flux (μg h−1) and the apparent permeability coefficient, Papp (cm s−1) were determined for each well. Cumulative flux values were determined as the sum of marker (μg mL−1) from each 4 h interval. The 16 h flux was reported as μg h−1. The Papp was determined according to eqn (1), where Papp is the apparent permeability coefficient (cm s−1), dQ is the amount of fluorescent marker on the basolateral side (mol mL−1), dt is the function of time per second (1 s−1), A is the surface area of the membrane (cm2), and C0 is the initial concentration on the apical side (mol mL−1).
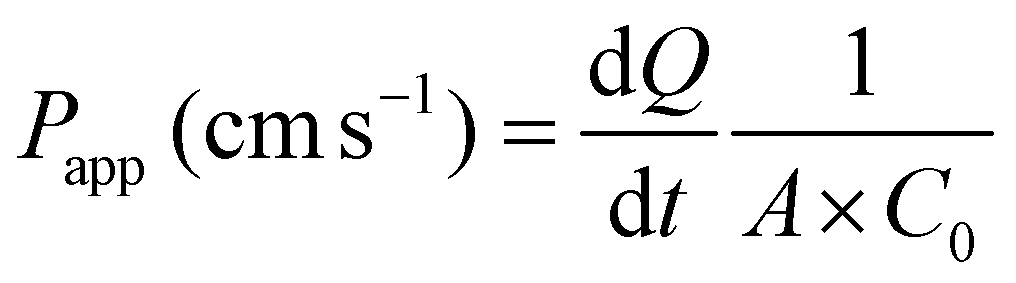 | (1) |
2.6 Immunofluorescence microscopy localization of tight junction proteins
Localization of occludin and ZO-1 to the tight junction was analyzed with immunofluorescence microscopy, as adapted from previously described methods.6 Fully differentiated Caco-2 cell monolayers were cultured and treated with the vehicle control, inflammatory cocktail, or inflammatory cocktail with yogurt for 48 h as described above. Following these treatments, Caco-2 cell monolayers were washed with PBS. For occludin staining, cells were fixed with 75% (v/v) ethanol in PBS for 30 min and then permeabilized with 75% (v/v) acetone in PBS for 3 min. For ZO-1 staining, cells were fixed with 2% (v/v) formaldehyde in PBS for 30 min, and incubated with 1% (v/v) Triton-X-100 in PBS 3 times for 5 min each to permeabilize cells. For both protein stains, monolayers were then washed and blocked for 30 min with 1% (w/v) bovine serum albumin (BSA) in PBS. Cells were then incubated with either mouse anti-occludin (1:150) or mouse anti-ZO-1 (1:50). Primary antibodies were incubated overnight at 4 °C for occludin, and 4 h at 37 °C for ZO-1 as described by others.6 Cells were then washed again with PBS and incubated with the secondary antibody, Alexa Fluor 488 goat anti-mouse (1:200), and the actin stain rhodamine-phalloidin (4 U mL−1) for 1 h at 23 °C. Cells were then washed again with PBS, and excised from the Transwell insert (∼0.5 cm × 0.5 cm squares) using a scalpel. The permeable support membrane was then mounted cell side up between a slide and coverslip with DAPI-containing mounting medium.
Microscopy of the mounted membranes was performed on a Zeiss LSM 510 Meta Confocal Laser Scanning Microscope (Carl Zeiss AG, Oberkochen, Germany). Scan areas were chosen based on intact cell structures and even staining, avoiding the edges of the square cutouts. Microscope settings are further described in ESI Tables 1 and 2.† Images were processed and analyzed using ZEN 2010 software (Carl Zeiss AG, Oberkochen, Germany). Intensity was quantified using grayscale analysis in ImageJ software (NIH, Bethesda, MD). In order to quantify protein localization to the tight junction, relative grayscale intensities were measured for each image, consistent with previously published protocols.6,23 Because the amount of tight junction space is highly dependent on the number of cells, the grayscale density intensity was divided by the number of cells shown in each image. Data was expressed as density intensity per cell, relative to the control.
2.7 Gene expression analysis of tight junction proteins
Cells were harvested from permeable inserts using 0.25% Trypsin and counted by a Scepter Cell Counter (EMD Millipore, Darmstadt, Germany). A target of 5 × 106 cells was transferred to a centrifuge tube and pelleted at 120g for 10 min at room temperature. RNA was extracted from Caco-2 cells using the TRIzol reagent following the manufacturer's protocol and then suspended in RNase-free water. An RNeasy mini kit was then used to purify the extracted RNA, according to the manufacturer's protocol. Qubit (ThermoFisher Scientific, Waltham, MA) was used to determine RNA concentration through fluorometric quantitiation. A volume containing 1 μg of RNA was then reverse transcribed to cDNA using an iScript™ cDNA synthesis kit, at a 20 μL reaction volume.Real time quantitative polymerase chain reaction (RT-qPCR) was performed using iTaq™ Universal SYBR® Green Supermix on a Bio-Rad CFX96 system according to the manufacturer's protocol. Primers for ZO-1 (TJP1), claudin-1 (CLDN1), occludin (OCLN), ribosomal protein large P0 (RPLP0), and 18s RNA (RNA18S5) were designed and selected using the NCBI primer design tool and previous publications.24,25 Primers were manufactured at 25 nmol scale by Integrated DNA Technologies (Coralville, IA). Primer sequences are listed in ESI Table 3.†RPLP0 and RNA18S5 were selected as reference genes after verification of maintained expression across treatments. Gene expression was normalized to the arithmetic mean of RPLP0 and RNA18S5, as previously validated in Caco-2 cells.26,27 The RT-qPCR reaction volume was 10 μL, with 2 μL 20-times diluted cDNA and 8 μL of mastermix (SYBR® Green Supermix, 2.5 μM forward and reverse primers). A standard curve was prepared, using a 6 point, 4-fold serial dilution of a cDNA sample pool (3 μL taken from each sample and combined).28 Each run also included a no cDNA sample and no reverse transcriptase sample as controls. Unknown sample concentrations were determined using the relative standard curve method with the Bio-Rad CFX Manager 3.1 software (Bio-Rad, Hercules, CA).
2.8 Statistical analysis
Data were reported as mean ± SD or SEM as indicated in tables and figures. Experiments were routinely performed in at least triplicate, as indicated in tables and figures. Normality of distribution and equality of variance was determined by visual inspection of data followed by the Brown–Forsythe test in GraphPad Prism 6.05 software (GraphPad Software, La Jolla, CA). Statistical significance was determined using one-way ANOVA followed by Tukey HSD test using JMP Pro 11.0.0 software (SAS, Cary, NC). Two-way repeated measures ANOVA followed by Tukey's multiple comparison test was performed using GraphPad Prism 6.05 software. Differences were considered significant when P < 0.05.3 Results and discussion
We investigated the extent low-fat yogurt affected the barrier function of differentiated Caco-2 cells exposed to an inflammatory stimulus (Fig. 1). The inflammatory cocktail reduced Caco-2 TEER by 38% from the initial value at 24 h after the inflammatory stimulus, continuing to 72 h after application (Fig. 1A). The 48 h treatment time was selected for future experiments because it produced a reproducible reduction in TEER and also allowed sufficient exposure to the test treatment. Yogurt did not prevent TEER reduction by the inflammatory cocktail at 24 h, but significantly improved TEER by 48 h after stimulation compared to the inflammatory cocktail alone (Fig. 1A), to a similar extent as the 100 μM genistein control (Fig. 1B). Cell viability was not affected by treatments (data not shown). Thus, yogurt did not prevent the initial inflammation-induced barrier disruption, but helped hasten recovery after the inflammatory stimulus. The ability of yogurt to improve Caco-2 barrier function was independent of inflammation, as the 1:30 dilution treatment increased TEER by 45% in cells not exposed to the inflammatory cocktail (ESI Fig. 2†).
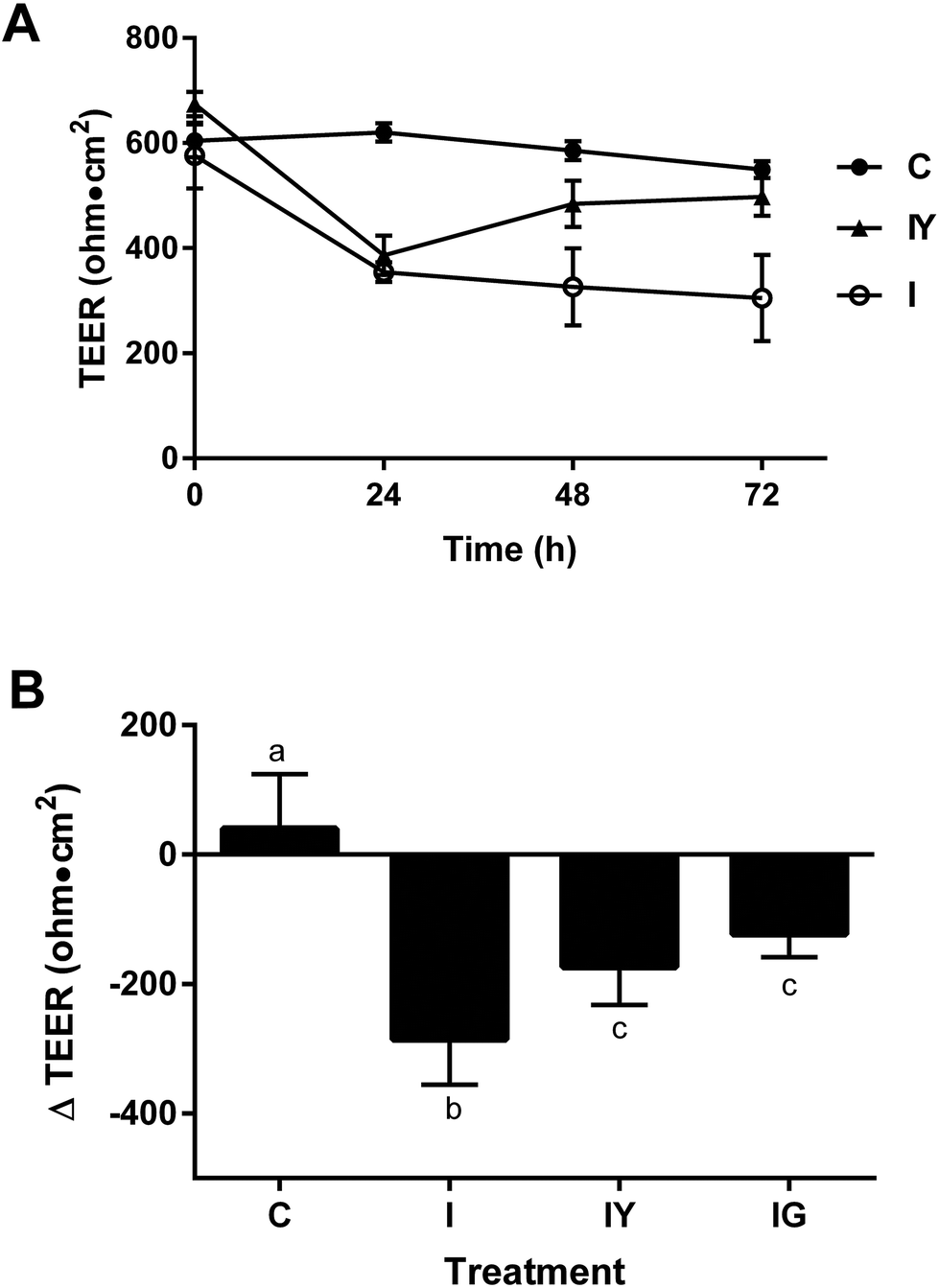 | ||
| Fig. 1 Yogurt increases TEER of differentiated Caco-2 cell monolayers subjected to an inflammatory stimulus. A. Caco-2 cells were treated with a vehicle control (growth media) (C), inflammatory stimulus (I) consisting of 25 ng mL−1 IL-1β, 50 ng mL−1 TNF-α, 50 ng mL−1 IFN-γ, and 1 μg ml−1 LPS, or I and yogurt (1:30, w/v, IY) from 0 to 72 h. B. Data are means ± SD, n = 6, P < 0.0001 by treatment, time, and their interaction by two-way ANOVA. B. Caco-2 cells treated with C, I, IY, or I with 100 μM genistein for 48 h. Data are means ± SD, n = 3 for IG, n = 15–22 for others. Means with different letters were significantly different as determined by one-way ANOVA followed by Tukey-HSD test (P < 0.05). | ||
The dose relevance of this study is an important consideration. A 1:30 dilution lyophilized yogurt powder represents a 1:8 dilution of fresh yogurt. Gastric dilutions depend on the amount of yogurt consumed, consumption of fluids or other foods, and gastric secretions. If 8 oz. (226 g) yogurt is consumed with a 1000 g meal and 100 mL liquid, it would approximate the expected dilution of yogurt in the upper gastrointestinal system. It should be considered that regular yogurt consumption would also result in a cyclical exposure to the diluted yogurt due to peristalsis.
TEER is a non-specific marker of Caco-2 barrier function, so we sought to confirm that TEER differences in this system were related to paracellular permeability. After 48 h, the inflammatory cocktail increased flux of both LY and FD through the Caco-2 barrier (Fig. 2). Yogurt with the inflammatory cocktail inhibited the flux of FD and LY 12 and 16 h after addition to the apical compartment, to levels similar to the vehicle control. Papp reflected TEER data, but were not statistically significant for the 16 h course (ESI Table 4†).
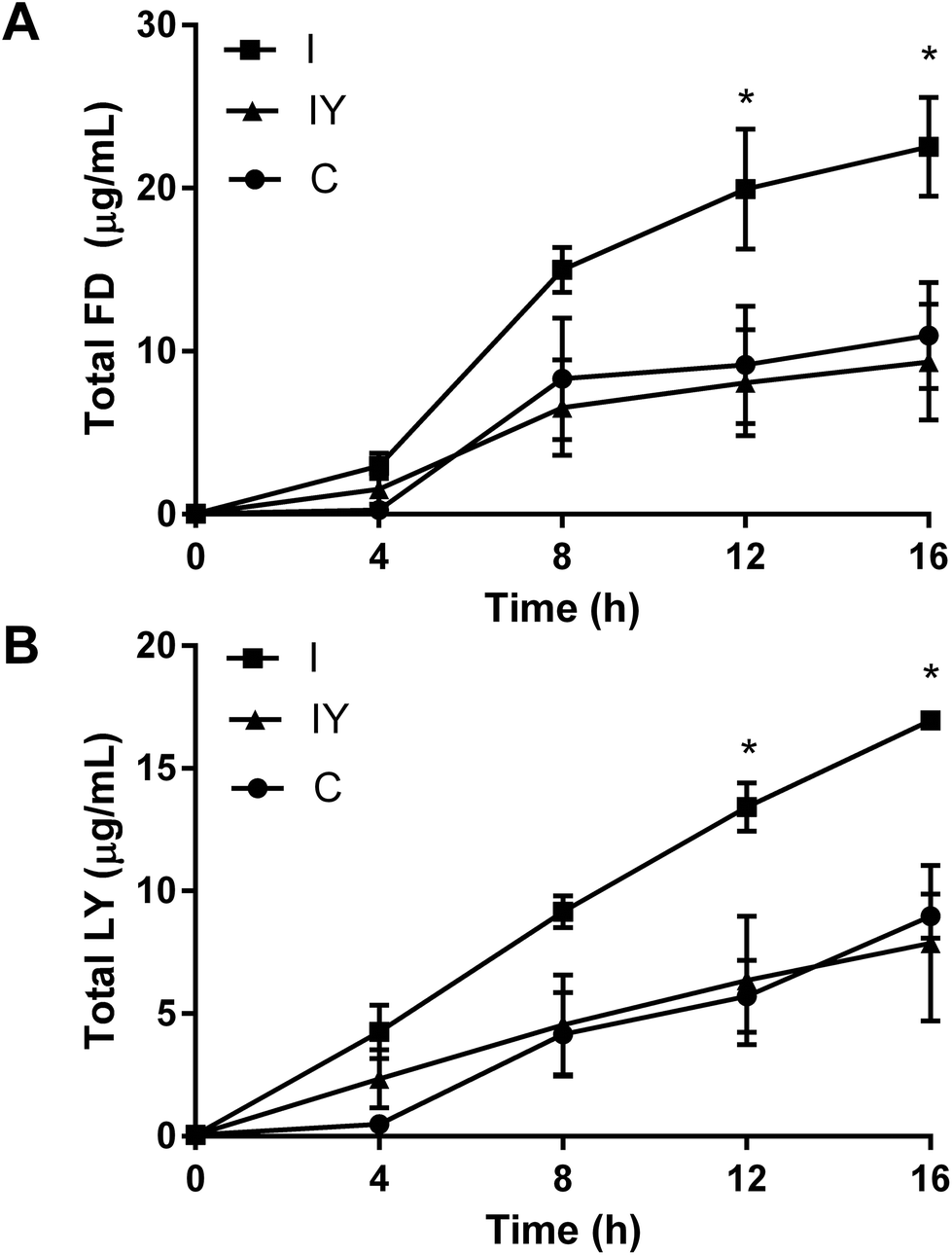 | ||
| Fig. 2 Flux of A. fluorescein isothiocyanate-dextran (FD), 4 kDA and B. lucifer yellow (LY) in differentiated Caco-2 cells exposed to control (C), inflammatory stimulus (I), or inflammatory stimulus and yogurt (1:30, w/v, IY) for 48 h. Values are means ± SEM, n = 2–3, where two-way ANOVA was P < 0.0001 for time, P < 0.1468 for treatment, and <0.0001 for their interaction for FD, and P < 0.0001 for time, P < 0.1582 for treatment, and <0.0001 for their interaction for LY. *P < 0.05 for I compared to C and IY by Tukey's multiple comparison test. | ||
Inverse and non-linear correlations of TEER and paracelluar permeability have been observed in previous studies with Caco-2 cells.29,30 Increased paracellular permeability may increase the localized inflammatory response, and is associated with obesity and chronic intestinal inflammation in rodent models of disease.31,32 Thus, the improved barrier function and reduced paracellular permeability observed in the Caco-2 model may be an anti-inflammatory mechanism of yogurt.
Immunofluorescence microcopy was performed to test the hypothesis that yogurt inhibits Caco-2 intestinal barrier disruption by the inflammatory cocktail through increased localization of occludin and ZO-1 to the tight junction. Fluorescent images confirmed Caco-2 cell monolayer formation and the presence of fully functional tight junctions, actin networks, and nuclei (Fig. 3). Occludin and ZO-1 formed a continuous mesh-like pattern between adjacent cells indicating localization between cells. DAPI and actin staining confirmed that Caco-2 cells were intact and not merged. Cross-sectional 3D stacked renderings of cell monolayers confirmed the localization of occludin and ZO-1, actin, and nuclei (ESI Fig. 3–5†). As expected, occludin was localized to the apical end of the lateral membrane, and ZO-1 was more distributed vertically throughout the cytoplasm. The inflammatory stimulus reduced occludin fluorescence intensity to 74% of the control (Fig. 3). Yogurt with the inflammatory stimulus increased occludin intensity to 123% of the control. ZO-1 exhibited a similar, but non-significant (P = 0.3726), trend as occludin. ZO-1 fluorescence intensity of the inflammatory stimulus was 77% of the control, while yogurt with the inflammatory stimulus was 94% of the control.
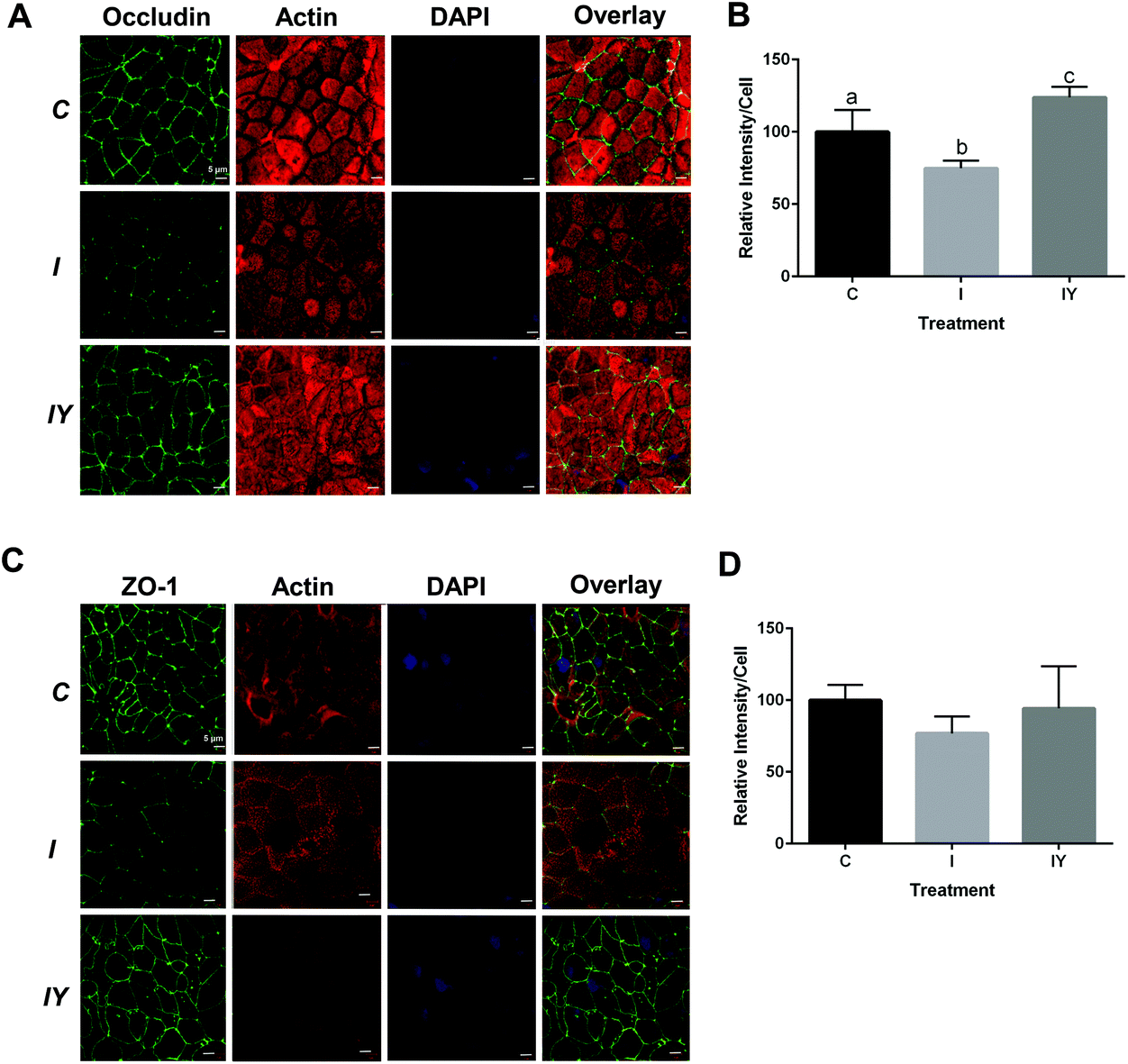 | ||
| Fig. 3 Yogurt treatment modulates occludin and ZO-1 in differentiated Caco-2 cells exposed to an inflammatory stimulus. Caco-2 cells were differentiated and treated for 48 h with control media, media with an inflammatory stimulus as described in Fig. 1, or the inflammatory stimulus with yogurt (1:30, w/v). A. Representative immunofluorescence images of occludin; B. immunofluorescence analysis of occludin; C. representative immunofluorescence images of ZO-1; D. immunofluorescence analysis of ZO-1. Data is presented as means ± SD, n = 3, bars bearing different letters were significantly different by one-way ANOVA followed by Tukey-HSD test if P < 0.05. | ||
Modulation of tight junction proteins by inflammation and dietary treatments has been previously described. In previous reports, an inflammatory stimuli reduced staining of tight junction proteins occludin and ZO-1 in Caco-2 cells.6,33 Caco-2 cells treated with Lactobacillus plantarum MB452 had more occludin and ZO-1 compared to an untreated control.8 In T84 cells, pretreatment with Bifidobacterium infantis conditioned medium prevented TNF-α and IFN-γ induced disruption of occludin and claudin-1 at the tight junction.34
Intestinal barrier disruption can be a result of redistribution of tight junction proteins to the interior of cells, changing occludin and ZO-1 distribution from continuous to punctate.35 In a prior report, intracellular pools of Caco-2 occludin and claudin-1 were increased upon inflammatory exposure, suggesting internalization of the tight junction proteins.33 However, in the present study, Caco-2 cells did not exhibit occludin or ZO-1 internalization or punctate formation. Thus, redistribution of the tight junction proteins does not appear to be a mechanism of improved barrier function by yogurt in the present model.
Because yogurt treatment resulted in increased TEER and reduced paracellular permeability without tight junction redistribution, we hypothesized that changes in mRNA expression of tight junction proteins were responsible for the improved barrier function by the addition of yogurt to the inflammatory cocktail. RT-qPCR was used to determine if yogurt increased gene expression of occludin (OCLN), ZO-1 (TJP1), and claudin-1 (CLDN1) in Caco-2 monolayers because of their importance in regulating paracellular permeability. In cells exposed to the inflammatory stimulus, OCLN was 0.62 of the control, while OCLN of yogurt and the inflammatory stimulus was 1.38 of the control (Fig. 4). The inflammatory cocktail TJP1 was 0.49 of the control, but yogurt with the inflammatory cocktail TJP1 was 1.07, significantly higher than the inflammatory stimulus. The increased TJP1 and OCLN in yogurt treated cells relative to the inflammatory stimulus were consistent with the trends of ZO-1 and occludin observed by immunofluorescence microscopy (Fig. 4). Furthermore, the inflammatory stimulus CLDN1 was 0.90 of the control cells, but 2.383 after treatment with yogurt and the inflammatory stimuls, significantly higher than both the control and the inflammatory stimulus. Thus, other tight junction proteins may also have a role in improvement in Caco-2 cells. The effect of yogurt with the inflammatory stimulus on Caco-2 TEER, Papp, and tight junction proteins are consistent. Together, these results appear to indicate that yogurt improves intestinal barrier function through increasing the expression of tight junction proteins.
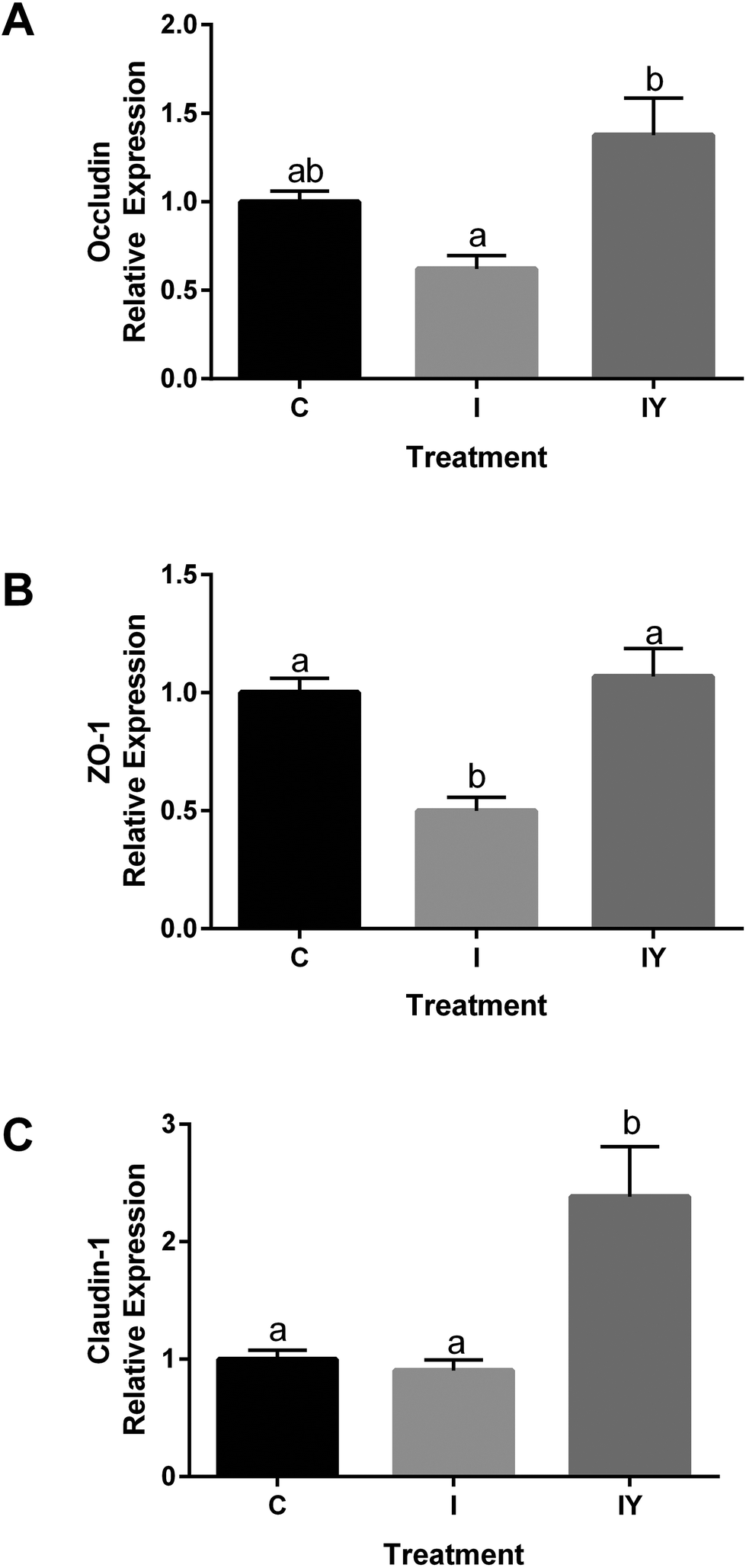 | ||
| Fig. 4 Relative mRNA expression of A. occludin (OCLN); B. ZO-1 (TJP1); and C. claudin-1 (CLDN1) in differentiated Caco-2 cells exposed to control (C), inflammatory stimulus (I), or inflammatory stimulus and yogurt (1:30, w/v, IY). mRNA expression is presented as relative to control. Values are means ± SD, n = 6, bars bearing different letters were significantly different by one-way ANOVA followed by Tukey-HSD test (P < 0.05). | ||
A prior study linked increased mRNA expression of Caco-2 tight junction proteins to improved intestinal barrier function after exposure to Lactobacillus plantarum MB452.8 Caco-2 cells treated with the casein peptide NPWDQ had increased occludin expression, as well as increased TEER.36 Glutamine also increases expression of occludin and ZO-1 in Caco-2 cells.37,38
To better model the function of yogurt through the digestive system, yogurt was subjected to in vitro simulated digestion. Digestion stages had differential effects on improvement of Caco-2 barrier disruption by the inflammatory stimulus (Fig. 5). The gastric digested samples improved inflammatory disruption of TEER, having ΔTEER values similar to undigested samples. Fully digested yogurt, representing complete gastric and intestinal digestion did not restore inflammation-induced Caco-2 barrier dysfunction. Control digestive fluids had no significant effect on TEER function. Thus, the progression of intestinal digestion resulted in a loss of yogurt bioactivity toward the intestinal barrier.
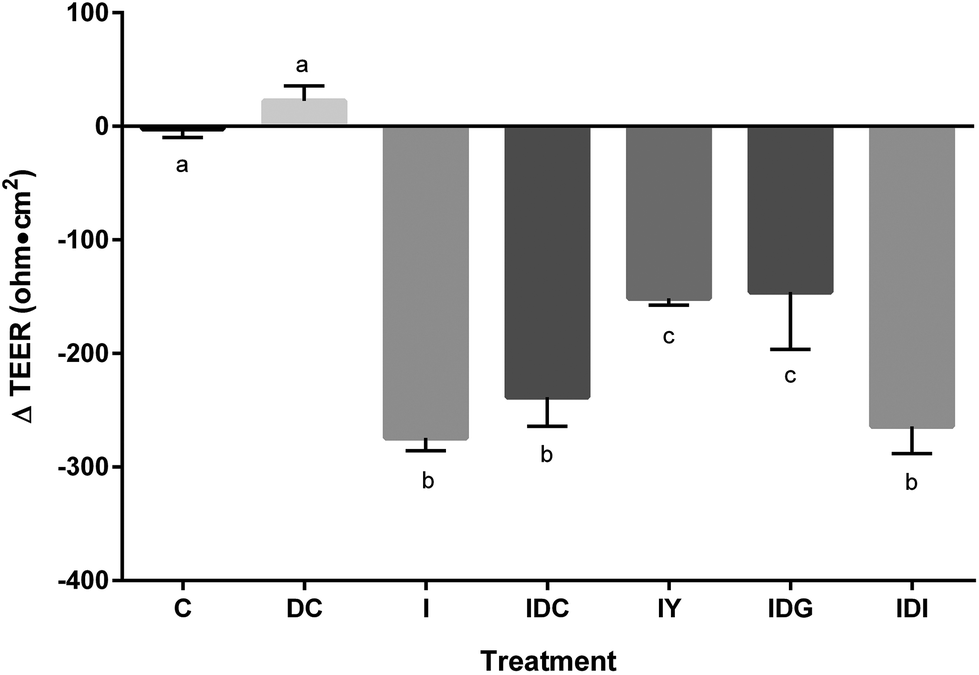 | ||
| Fig. 5 In vitro simulated digestion of yogurt differentially affects inflammation-induced Caco-2 barrier dysfunction. Differentiated Caco-2 cells were treated with a vehicle control (C), digest control (DC) (all fluids used in digestion), inflammatory stimulus (I) (25 ng mL−1 IL-1β, 50 ng mL−1 TNF-α, 50 ng mL−1 IFN-γ, and 1 μg mL−1 LPS), I and DC (IDC), I and 0.03 g mL−1 undigested yogurt (IY), I and 0.03 g mL−1 gastric phase digested yogurt (IDG), or I and 0.03 g mL−1 intestinal phase digested yogurt (IDI) for 48 h. Values are means ± SD, with n = 6 for each treatment. Means with different letters were significantly different as determined by one-way ANOVA followed by Tukey-HSD test (P < 0.05). | ||
Although we did not determine the component(s) responsible for this effect, others have suggested calcium, microorganisms, and proteins modulate intestinal barrier function. Calcium, magnesium, and lactose applied at levels reported in milk increased Caco-2 TEER relative to a glucose control.39 Extracellular calcium increases tight junction function through direct interaction with claudin.40 Probiotic bacteria and bacteria-conditioned media also increase TEER by modulating tight junction proteins.8,34 β-Lactoglobulin increased TEER through modulation of the cytoskeletal structure in Caco-2 cells.41 Casein-derived peptides and amino acids have also improved Caco-2 TEER, primarily through the increased expression of the tight junction proteins.36–38 For example, NPWDQ increased Caco-2 TEER and also up-regulated occludin expression.36
The loss of bioactivity of digested yogurt may help explain the components responsible for its effect on intestinal barrier function. Minerals such as calcium are not affected by in vitro digestion. Lipids and probiotics are unlikely to have significant bioactivity, as the yogurt did not have probiotics and was low-fat. The active component was likely proteins and/or peptides lost upon treatment with pancreatin in the model digestion. For example, yogurt is rich in β-lactoglobulin,42 and its application to Caco-2 cells increased TEER and reduced paracellular flux of FD.43 In contrast, the peptide NPWDQ does not appear to be a functional component, as it is resistant to degradation by digestive enzymes.36
There are several limitations to this study that should be considered. Yogurt is typically consumed fresh, and the extent freeze-drying alters the bioactive component or bacterial viability was not determined. The impact of freeze-drying on yogurt culture viability depends on the bacterial strain, food matrix, and process conditions.44 Yogurt cultures do not appear to maintain significant viability or colonize the intestine, and the extent that viability may contribute to their potential to modulate barrier function is unknown.45 Also, we did not extensively investigate the dose–response effect beyond TEER, and barrier permeability and tight-junction expression may have different dose responses than TEER. Furthermore, the Caco-2 model as used in the present study does not recapitulate the complexity of the digestive system and bacterial–host interactions. Other agents may also affect intestinal barrier function, including other inflammatory cytokines, immunocytes, and microbiota–host interactions.
4 Conclusions
Intestinal barrier dysfunction contributes to the pathology of obesity, inflammatory bowel disease, and colon cancer. Yogurt restored inflammation-induced intestinal barrier dysfunction of Caco-2 cells. In the presence of inflammatory stimulus, yogurt increased Caco-2 occludin, ZO-1, and claudin-1 mRNA expression and increased occludin at the tight junctions. Simulated digestion of the yogurt indicated the functional components are not stable throughout the entire gastrointestinal tract. Taken together, these results suggest that the protective effect of yogurt against intestinal barrier permeability induced by inflammation would occur prior to proteolysis in the small intestine. Stabilizing the bioactive component through digestion would be necessary to provide this functional benefit in the lower gastrointestinal tract.Conflict of interest
The authors report no conflicts of interest.Acknowledgements
All imaging was performed at the Newcomb Imaging Center, Department of Botany, UW-Madison.Notes and references
- P. C. Calder, N. Ahluwalia, F. Brouns, T. Buetler, K. Clement, K. Cunningham, K. Esposito, L. S. Jonsson, H. Kolb and M. Lansik, Br. J. Nutr., 2011, 106, S5–S78 CrossRef CAS PubMed.
- J. M. Moreno-Navarrete, M. Sabater, F. Ortega, W. Ricart and J. M. Fernandez-Real, PLoS One, 2012, 7, e37160 CAS.
- P. D. Cani, J. Amar, M. A. Iglesias, M. Poggi, C. Knauf, D. Bastelica, A. M. Neyrinck, F. Fava, K. M. Tuohy, C. Chabo, A. Waget, E. Delmée, B. Cousin, T. Sulpice, B. Chamontin, J. Ferrières, J.-F. Tanti, G. R. Gibson, L. Casteilla, N. M. Delzenne, M. C. Alessi and R. Burcelin, Diabetes, 2007, 56, 1761–1772 CrossRef CAS PubMed.
- W. Cui, L. X. Li, C. M. Sun, Y. Wen, Y. Zhou, Y. L. Dong and P. Liu, Braz. J. Med. Biol. Res., 2010, 43, 330–337 CrossRef CAS PubMed.
- C. Carrasco-Pozo, P. Morales and M. Gotteland, J. Agric. Food Chem., 2013, 61, 5291–5297 CrossRef CAS PubMed.
- P. Beguin, A. Errachid, Y. Larondelle and Y. J. Schneider, Food Funct., 2013, 4, 923–931 CAS.
- S. Drago, R. El Asmar, M. Di Pierro, M. Grazia Clemente, A. Tripathi, A. Sapone, M. Thakar, G. Iacono, A. Carroccio, C. D'Agate, T. Not, L. Zampini, C. Catassi and A. Fasano, Scand. J. Gastroenterol., 2006, 41, 408–419 CrossRef CAS PubMed.
- R. C. Anderson, A. L. Cookson, W. C. McNabb, Z. Park, M. J. McCann, W. J. Kelly and N. C. Roy, BMC Microbiol., 2010, 10, 316 CrossRef CAS PubMed.
- M. B. Zemel and X. Sun, J. Nutr., 2008, 138, 1047–1052 CAS.
- M. B. Zemel, X. Sun, T. Sobhani and B. Wilson, Am. J. Clin. Nutr., 2010, 91, 16–22 CrossRef CAS PubMed.
- E. J. Schiffrin, A. Parlesak, C. Bode, J. C. Bode, M. A. van't Hof, D. Grathwohl and Y. Guigoz, Br. J. Nutr., 2009, 101, 961–966 CrossRef CAS PubMed.
- S. Chaves, G. Perdigon and A. de Moreno de LeBlanc, J. Food Prot., 2011, 74, 801–811 CrossRef CAS PubMed.
- I. J. Hidalgo, T. J. Raub and R. T. Borchardt, Gastroenterol., 1989, 98, 736–749 CrossRef.
- Y. Sambuy, I. De Angelis, G. Ranaldi, M. L. Scarino, A. Stammati and F. Zucco, Cell Biol. Toxicol., 2005, 21, 1–26 CrossRef CAS PubMed.
- J. Van De Walle, A. Hendrickx, B. Romier, Y. Larondelle and Y. J. Schneider, Toxicol. In Vitro, 2010, 24, 1441–1449 CrossRef CAS PubMed.
- R. B. Sartor, Gastroenterol., 1994, 106, 533–539 CrossRef CAS.
- S. Noda, S. Tanabe and T. Suzuki, J. Agric. Food Chem., 2012, 60, 4628–4633 CrossRef CAS PubMed.
- S. Piegholdt, K. Pallauf, T. Esatbeyoglu, N. Speck, K. Reiss, L. Ruddigkeit, A. Stocker, P. Huebbe and G. Rimbach, Free Radical Biol. Med., 2014, 70, 255–264 CrossRef CAS PubMed.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carriere, R. Boutrou, M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Menard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. Wickham, W. Weitschies and A. Brodkorb, Food Funct., 2014, 5, 1113–1124 CAS.
- M. D. L.-F. Blume, F. Gieseler and T. Kunze, Pharmazie, 2010, 65, 19–24 Search PubMed.
- B. Srinivasan, A. R. Kolli, M. B. Esch, H. E. Abaci, M. L. Shuler and J. J. Hickman, J. Lab. Autom., 2015, 20, 107–126 CrossRef CAS PubMed.
- L. M. Feighery, S. W. Cochrane, T. Quinn, A. W. Baird, D. O'Toole, S. E. Owens, D. O'Donoghue, R. J. Mrsny and D. J. Brayden, Pharm. Res., 2008, 25, 1377–1386 CrossRef CAS PubMed.
- C. Terryn, M. Sellami, C. Fichel, M. D. Diebold, S. Gangloff, R. Le Naour, M. Polette and J. M. Zahm, Cytometry, Part A, 2013, 83, 235–241 CrossRef PubMed.
- A. Orlando, M. Linsalata, M. Notarnicola, V. Tutino and F. Russo, BMC Microbiol., 2014, 14, 19 CrossRef PubMed.
- A. Sapone, L. de Magistris, M. Pietzak, M. G. Clemente, A. Tripathi, F. Cucca, R. Lampis, D. Kryszak, M. Carteni, M. Generoso, D. Iafusco, F. Prisco, F. Laghi, G. Riegler, R. Carratu, D. Counts and A. Fasano, Diabetes, 2006, 55, 1443–1449 CrossRef CAS PubMed.
- R. A. Vreeburg, S. Bastiaan-Net and J. J. Mes, Food Funct., 2011, 2, 124–129 CAS.
- N. Maubon, M. Le Vee, L. Fossati, M. Audry, E. Le Ferrec, S. Bolze and O. Fardel, Fundam. Clin. Pharmacol., 2007, 21, 659–663 CrossRef CAS PubMed.
- T. M. Grala, J. R. Roche, J. K. Kay, A. G. Rius, H. M. White, S. S. Donkin, M. D. Littlejohn, R. G. Snell and C. V. Phyn, J. Dairy Sci., 2014, 97, 838–850 CrossRef CAS PubMed.
- G. Ranaldi, R. Consalvo, Y. Sambuy and M. L. Scarino, Toxicol. Vitro, 2003, 17, 761–767 CrossRef CAS.
- G. Ranaldi, K. Islam and Y. Sambuy, Antimicrob. Agents Chemother., 1994, 38, 1239–1245 CrossRef CAS PubMed.
- P. D. Cani, S. Possemiers, T. Van de Wiele, Y. Guiot, A. Everard, O. Rottier, L. Geurts, D. Naslain, A. Neyrinck, D. M. Lambert, G. G. Muccioli and N. M. Delzenne, Gut, 2009, 58, 1091–1103 CrossRef CAS PubMed.
- M. A. A. Schepens, A. J. Schonewille, C. Vink, E. M. van Schothorst, E. Kramer, T. Hendriks, R.-J. Brummer, J. Keijer, R. van der Meer and I. M. J. Bovee-Oudenhoven, J. Nutr., 2009, 139, 1525–1533 CrossRef CAS PubMed.
- F. Wang, W. V. Graham, Y. Wang, E. D. Witkowski, B. T. Schwarz and J. R. Turner, Am. J. Pathol., 2005, 166, 409–419 CrossRef CAS PubMed.
- J. B. Ewaschuk, H. Diaz, L. Meddings, B. Diederichs, A. Dmytrash, J. Backer, M. Looijer-van Langen and K. L. Madsen, Am. J. Physiol.: Gastrointest. Liver Physiol., 2008, 295, G1025–G1034 CrossRef CAS PubMed.
- P. Farshori and B. Kachar, J. Membr. Biol., 1999, 170, 147–156 CrossRef CAS PubMed.
- H. Yasumatsu and S. Tanabe, Br. J. Nutr., 2010, 104, 951–956 CrossRef CAS PubMed.
- N. Li, P. Lewis, D. Samuelson, K. Liboni and J. Neu, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 287, G726–G733 CrossRef CAS PubMed.
- S. Beutheu, I. Ghouzali, L. Galas, P. Dechelotte and M. Coeffier, Clin. Nutr., 2013, 32, 863–869 CrossRef CAS PubMed.
- K. A. Staples, R. J. Durham and M. R. Jones, Aust. J. Dairy Technol., 2005, 60, 127–131 CAS.
- A. S. Yu, M. H. Cheng and R. D. Coalson, J. Biol. Chem., 2010, 285, 37060–37069 CrossRef CAS PubMed.
- K. Hashimoto, T. Nakayama and M. Shimizu, Biosci., Biotechnol., Biochem., 1998, 62, 1819–1821 CrossRef CAS PubMed.
- N. Hassan, M. Takasugi, K. Yamada and M. Sugano, Food Sci. Technol. Int., 1997, 3, 56–60 CrossRef CAS.
- K. Hashimoto, K. Takeda, T. Nakayama and M. Shimizu, Biosci., Biotechnol., Biochem., 1995, 59, 1951–1952 CrossRef CAS PubMed.
- Y. Shao, S. Gao, H. Guo and H. Zhang, J. Dairy Sci., 2014, 97, 1270–1280 CrossRef CAS PubMed.
- R. Pei, D. A. Martin, D. M. DiMarco and B. W. Bolling, Crit. Rev. Food Sci. Nutr., 2017 DOI:10.1080/10408398.2014.883356.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental Fig. 1–5 and Supplemental Tables 1–4. See DOI: 10.1039/c6fo01592a |
| This journal is © The Royal Society of Chemistry 2017 |