 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: origins, current status and discussion
Lee
Brammer

Department of Chemistry, University of Sheffield, Brook Hill S3 7HF, UK. E-mail: lee.brammer@sheffield.ac.uk; Web: http://https://www.sheffield.ac.uk/chemistry/staff/profiles/lee_brammer
First published on 5th October 2017
Abstract
The role of the closing lecture in a Faraday Discussion is to summarise the contributions made to the Discussion over the course of the meeting and in so doing capture the main themes that have arisen. This article is based upon my Closing Remarks Lecture at the 203rd Faraday Discussion meeting on Halogen Bonding in Supramolecular and Solid State Chemistry, held in Ottawa, Canada, on 10–12th July, 2017. The Discussion included papers on fundamentals and applications of halogen bonding in the solid state and solution phase. Analogous interactions involving main group elements outside group 17 were also examined. In the closing lecture and in this article these contributions have been grouped into the four themes: (a) fundamentals, (b) beyond the halogen bond, (c) characterisation, and (d) applications. The lecture and paper also include a short reflection on past work that has a bearing on the Discussion.
1. Introduction: history and foundations
Halogen bonding in its modern incarnation has been the subject of study for approximately 20 years, but is underpinned by many important earlier contributions, sometimes noted in historic introductions to the field and sometimes overlooked. Early papers from the 19th century have more recently been recognised as being observations of halogen bonding, the earliest among these being the association of iodine (I2) with ammonia.1,2 Some of this work pre-dates the development of ideas that led to early definitions of the hydrogen bond,3–5 the intermolecular interaction to which halogen bonding is most often compared. Studies by Hassel in the 1950s/60s,6–8 which are the subject of his Nobel Prize lecture,9 provided a clear characterisation of halogen bonds, not yet named as such, by crystallographic means and notably established their linear geometry. The role of crystallography and solid-state chemistry continues to be important in the study of halogen bonds and their applications, as reflected in the title of this Discussion and many of the papers contributed. These include those by Riley and Tran (DOI: 10.1039/c7fd000106a), Alavi et al. (DOI: 10.1039/c7fd00064b), Kennepohl et al. (DOI: 10.1039/c7fd0076f), Edwards et al. (DOI: 10.1039/c7fd00072c), Politzer et al. (DOI: 10.1039/c7fd00062f), Southern et al. (DOI: 10.1039/c7fd00087a), Vargas-Baca et al. (DOI: 10.1039/c7fd00075h), Guru Row et al. (DOI: 10.1039/c7fd00084g), Mosquera et al. (DOI: 10.1039/c7fd00079k), Aakeröy et al. (DOI: 10.1039/c7fd00080d), Fourmigué et al. (DOI: 10.1039/c7fd00067g), Lloyd et al. (DOI: 10.1039/c7fd000108h), and Friščić et al. (DOI: 10.1039/c7fd00114b).The formation and geometries of interactions analogous to halogen bonds, but involving in the Lewis acidic role main group elements from other groups in the periodic table, notably groups 14–16 and 18, was well established in the 1960s and 70s. Reviews by Bent on donor–acceptor interactions,10 and by Alcock on secondary bonding,11 describe these interactions in some detail and cover many of the points being discussed in the recent rediscovery and resurgence of interest in what are now being termed tetrel bonds (group 14), pnictogen bonds (group 15), chalcogen bonds (group 16) and aerogen bonds (group 18). Such interactions are now most commonly described in terms of electrostatic interactions between a lone pair of electrons (or other electron-rich site) on one molecule and an electropositive site referred to as the σ-hole and situated opposite to a σ-bond formed by the main-group element.12,13 Bent described these interactions in the framework of electron donor–acceptor complexes, requiring an orbital interaction model. The debate regarding which is the more appropriate bonding description, and whether both are models are should be invoked, was an important theme of the present Discussion, such as that of session 1 (DOI: 10.1039/c7fd90062g). Alcock introduced the term secondary bond in 1971 to describe the linear intermolecular interaction in which a main group element serves in a Lewis acidic role.11 The first paragraph of Alcock’s review, entitled “Secondary bonding to nonmetallic elements”, is reproduced here (Chart 1), and sets out the description of secondary bonding, which should be familiar to all who are currently studying these interactions. The more recent subdivision and renaming of secondary bonding in terms of the group names for the elements in the periodic table (see Section 3) is currently the subject of an IUPAC project to provide definitions associated with these names. There is certainly a discussion to be had in this regard, notably in how far classes of analogous interactions should be subdivided and separately named. In this context one reasonably might ask, for example, whether it is appropriate to subdivide halogen bonds into a series of named interactions as iodine bonds, bromine bonds, chlorine bonds and (possibly) fluorine bonds.
 | ||
| Chart 1 Definition of the term secondary bond by Alcock. Reproduced from ref. 11 with permission from Elsevier. | ||
In the 1980s, Parthasarathy and Murray-Rust described the angular dependence of electrophilic and nucleophilic interactions with terminal halogens based on observations from crystal sructures.14 Desiraju later described type I and type II halogen–halogen interactions, which have different geometries, the latter of which is consistent with halogen bonding involving the interaction of an electrophilic halogen with a nucleophilic halogen.15,16 Our own work later emphasised the importance of the anisotropic charge distribution and resulting electrostatic potential around terminal halogens in developing an understanding of electrophilic interactions, predominantly hydrogen bonding, with terminal halogens,17 but ultimately led us to become involved in studying halogen bonding.18–20
It is often difficult to single out individual papers that have made a difference in advancing a field of study, but here I will note two papers that appeared almost 20 years ago, both of which contributed significantly to the renaissance of interest in what is now termed halogen bonding by providing a point of comparison with hydrogen bonding, an interaction that was well-established in the minds of chemists and those working in the molecular sciences. The review in 1999 by Legon21 on structural characterisation of gas-phase complexes (Fig. 1) demonstrated the similarities between hydrogen bonding and halogen bonding, which had been systematically developed in experimental spectroscopic studies over a number of years, but was perhaps not well known to those with interests in self-assembly in the condensed phases. The complexes studied provide experimental rigour to underpin the fundamental understanding of these interactions and the extent to which there is parity between the two. The early work of Resnati and Metrangolo beginning around a similar time arose from a background in fluorine chemistry, which led to the development of highly fluorinated organic building blocks that could be deployed in halogen-bonded self-assembly in the solid state. Their report in 2000 showed that halogen bonding could be competitive with or even out-compete hydrogen bonding in the self-assembly of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystals from the solution phase (Fig. 2),22 and brought to the attention of a wider community the possibility that halogen bonding might be used as an alternative to the more well-known hydrogen bonding in self-assembly. Very recently we have returned to this competition between hydrogen bonds and halogen bonds and shown that the choice of solvent affects the outcome (Fig. 3).23 Generally speaking, less polar solvents favour association via hydrogen bonds over halogen bonds in forming the co-crystals, whereas the reverse is true in polar solvents. The cross-over point is related to the relative strengths of the hydrogen bonds and halogen bonds that are in competition.
:1 co-crystals from the solution phase (Fig. 2),22 and brought to the attention of a wider community the possibility that halogen bonding might be used as an alternative to the more well-known hydrogen bonding in self-assembly. Very recently we have returned to this competition between hydrogen bonds and halogen bonds and shown that the choice of solvent affects the outcome (Fig. 3).23 Generally speaking, less polar solvents favour association via hydrogen bonds over halogen bonds in forming the co-crystals, whereas the reverse is true in polar solvents. The cross-over point is related to the relative strengths of the hydrogen bonds and halogen bonds that are in competition.
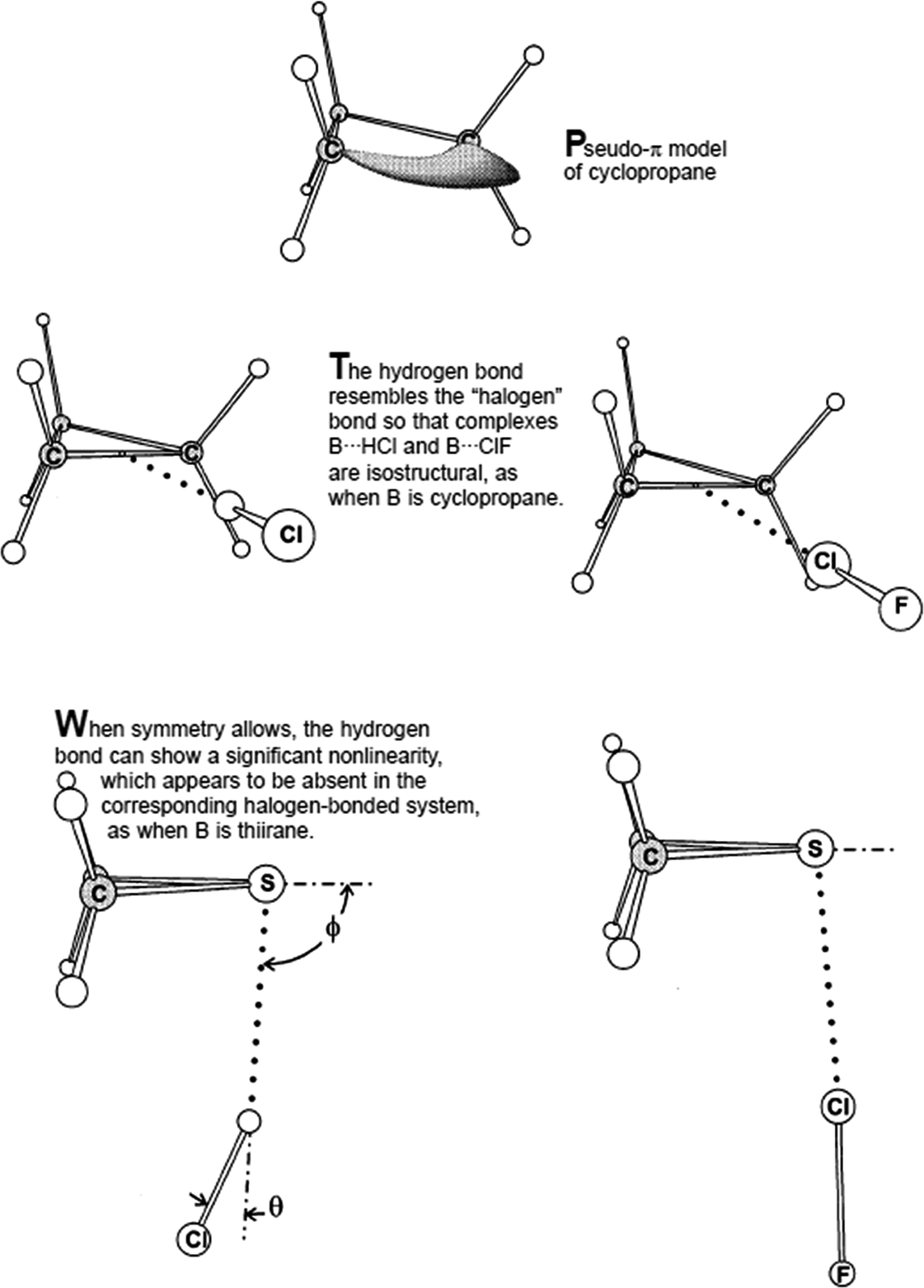 | ||
| Fig. 1 Experimentally characterised gas-phase hydrogen-bonded and halogen-bonded complexes. Reproduced from ref. 21 with permission from Wiley. | ||
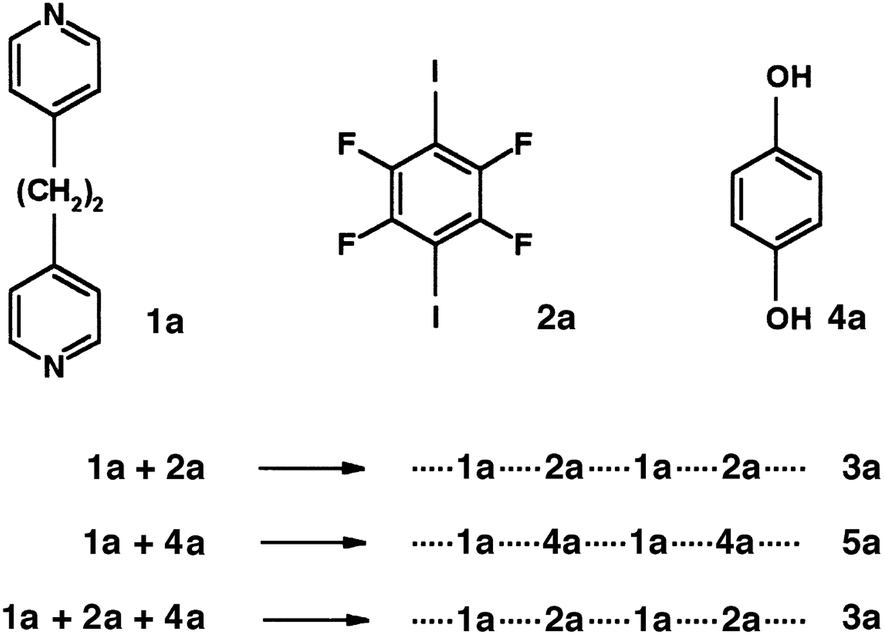 | ||
| Fig. 2 Halogen-bonded self-assembly can out-compete hydrogen-bonded self-assembly in forming co-crystals from solution. Reproduced from ref. 22 with permission from Wiley. | ||
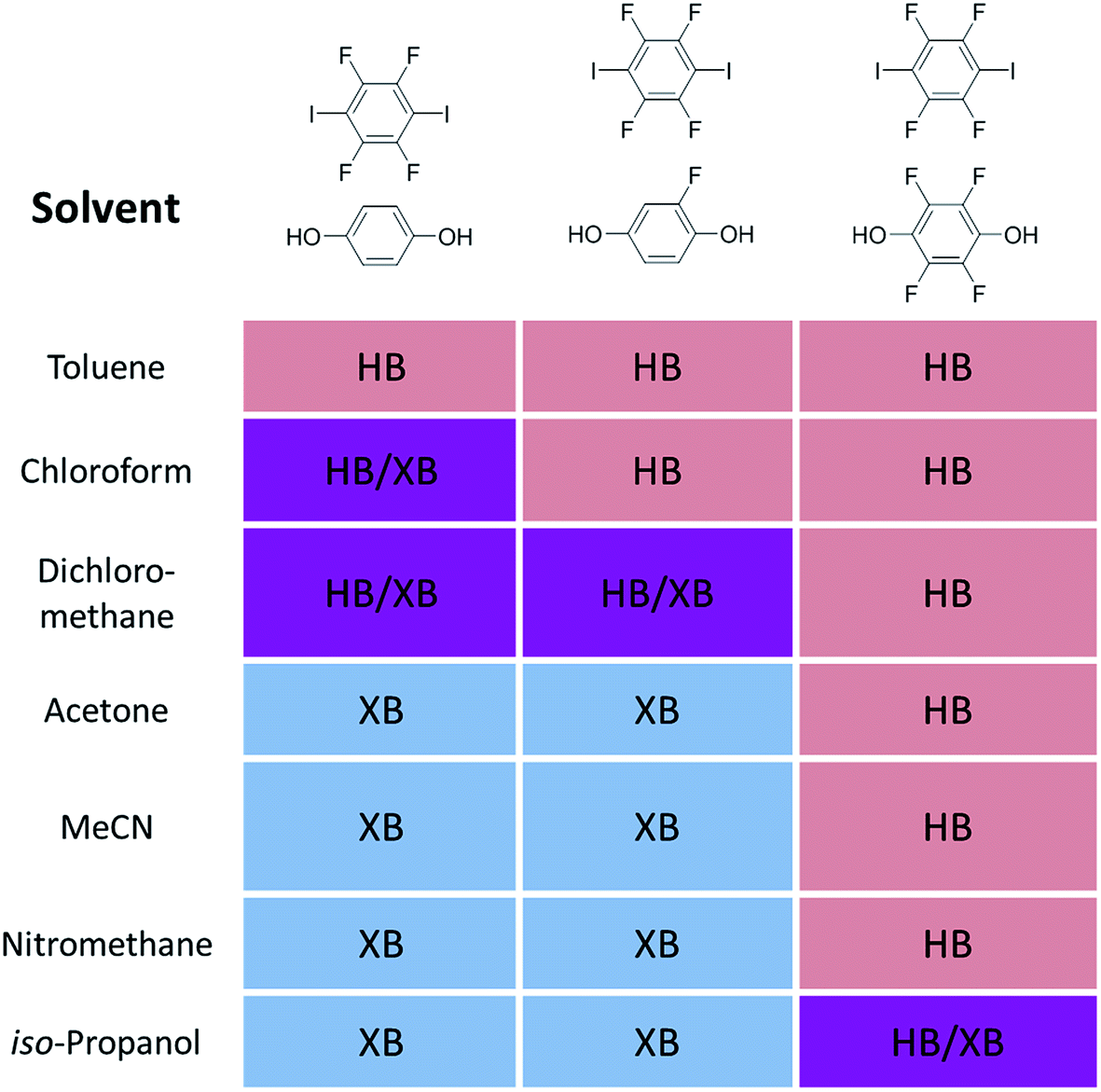 | ||
| Fig. 3 Solvent effect on self-assembly of 1:1 co-crystals of 4,4′-dipyridylethane with the ditopic hydrogen bond and halogen bond donors shown above each column (HB = hydrogen-bonded co-crystal formed; XB = halogen-bonded co-crystal formed). | ||
2. Fundamentals
A number of the papers presented in the Discussion focused on fundamental aspects of halogen bonding and related interactions. The discussion of the papers centred on understanding the bonding, and being able to use this understanding in a predictive manner. In his opening lecture, and accompanying article (DOI: 10.1039/c7fd00058h), Tim Clark emphasised the fact the chemistry is based on models and proposed a lex pasimoniae (Occam’s razor) for bonding theory as follows:• An analysis is adequate when it explains all the known facts.
• Try to stick with physically unique and defined phenomena wherever possible.
• Do not define new interactions where old ones will do.
The paper emphasised an electrostatic description of bonding, which in principle can account for all bonding interactions, but recognised the value of orbital models which have been embedded in the description of bonding in chemistry over many years. The electrostatic model includes:
(i) The Coulomb interaction, which is the foundation of the description of halogen bonding in terms of the interaction of the σ-hole of the halogen-bond donor and the electron-rich region of the halogen-bond acceptor.
(ii) Polarisation, because it is important for describing the interaction between unperturbed molecules where this is not otherwise obvious.
(iii) Dispersion, because otherwise the attraction of nonpolar species is not explained.
Co-operativity in halogen bonding was demonstrated in calculations by Del Bene and co-workers on a series of binary and ternary complexes (DOI: 10.1039/c7fd00048k). As shown in Fig. 4, the halogen bond between ClF and PH2F is enhanced slightly by the cooperative polarising effect from the formation of a second halogen bond involving HCl. This polarisation of the central ClF molecule is greater when a stronger halogen-bond donor (a second ClF molecule) is used to form the ternary complex, so much so that it leads to Cl+ transfer. Co-operativity has long been established in hydrogen bonding, as illustrated in the study by Hermansson of co-operativity in hydrogen-bonded water chains,24 in which the calculated difference electron density of the water molecules clearly shows the most polarised molecule to be the central one of the chain (Fig. 5). Polarisation in halogen bonding was further discussed in the computational study by Riley and Tran on charged halogen bonds between cationic halogen-bond donors and anionic acceptors (DOI: 10.1039/c7fd000106a), indicating a greater polarisation effect in interactions between charged species than neutral ones. Such interactions have also been studied extensively in experimental work involve halides, halometallate ions and other anions in the halogen-bond acceptor role with either cationic or neutral halogen bond donors.18,25–34 Kennepohl and co-workers (DOI: 10.1039/c7fd0076f) used X-ray Absorption Spectroscopy (XAS) to demonstrate and quantify the charge-transfer from chloride ions to I2 halogen-bond donors, by observation of the Cl1s → Cl3p transition, which cannot arise in the absence of Cl− to I2(σ*) charge-transfer (Fig. 6). Fourmigué and co-workers (DOI: 10.1039/c7fd00067g) further developed the discussion of covalency in halogen bonding in experimental and computational charge density studies of N–X⋯N halogen bonds (X = I, Br), which suggest greater covalency for X = Br than for X = I (Fig. 7), a reverse of what might have been anticipated. The nature of N–X⋯N halogen bonds have also been extensively studied in solution-phase using NMR spectroscopy by Erdelyi and co-workers.35–38
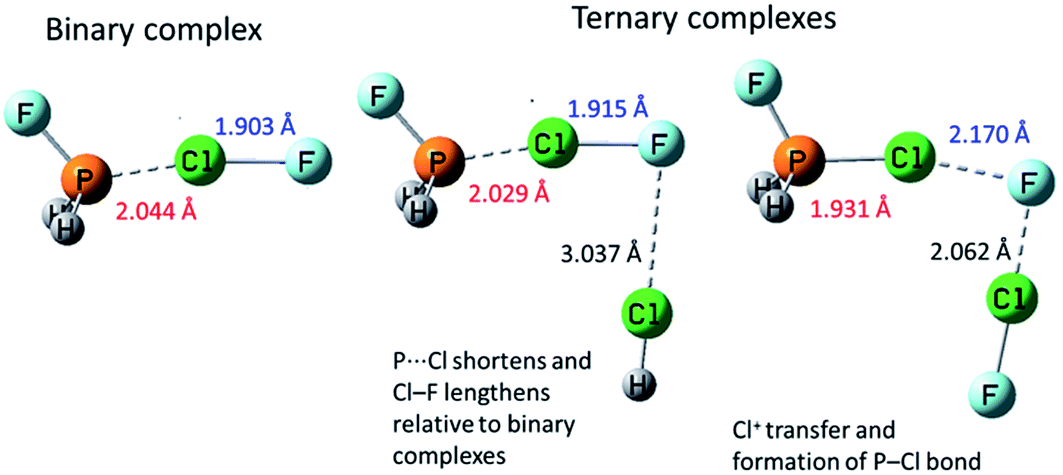 | ||
| Fig. 4 Computational models of complexes H2FP⋯ClF, H2FP⋯ClF⋯ClH and H2FP⋯ClF⋯ClF, illustrating co-operativity in halogen bonding interactions. Adapted from DOI: 10.1039/c7fd00048k with permission from The Royal Society of Chemistry. | ||
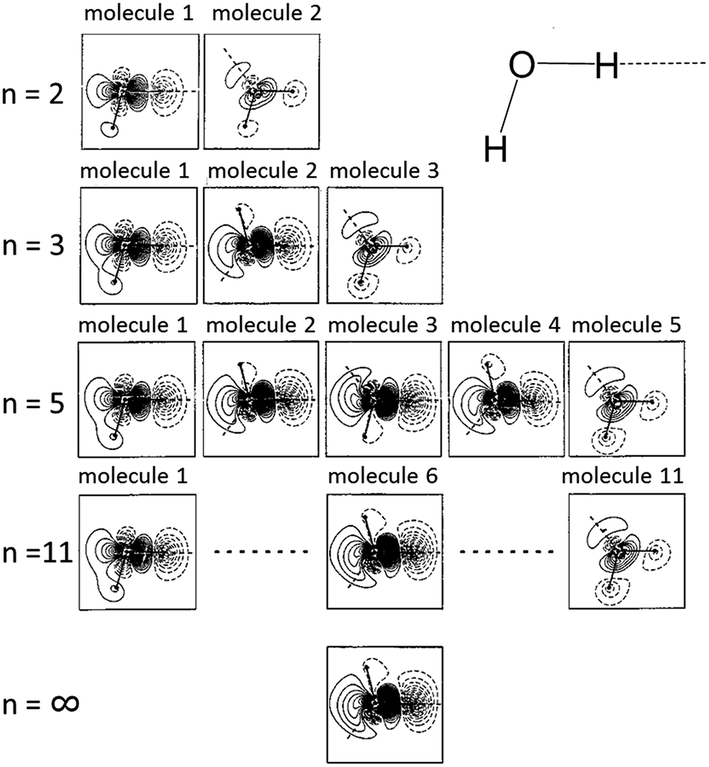 | ||
| Fig. 5 Calculated difference electron density of hydrogen-bonded water chains of length n molecules, showing co-operativity in hydrogen bonding, which leads to the central water molecule being the most polarised. Solid contours represent increased electron density and dashed contours decreased electron density relative to the pro-molecule. All electron densities are shown in the plane of the three atoms in each molecule for ease of comparison (note: only the central molecule is shown for n = ∞). Adapted from ref. 24 with permission from AIP Publishing. | ||
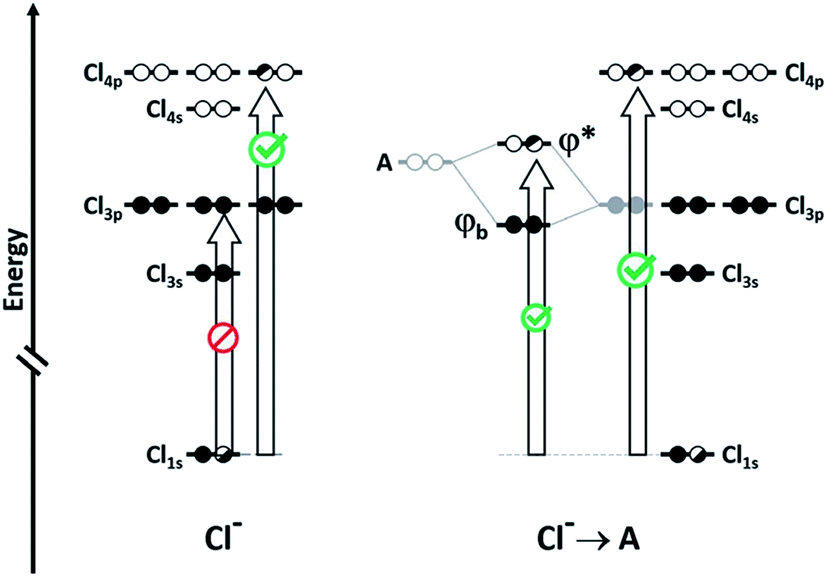 | ||
| Fig. 6 Cl1s to Cl3p transition, which can be probed by XAS, arises only if Cl3p to I2(σ*) charge-transfer is present (A represents a halogen-bond acceptor, here I2). The intensity of the transition is proportional to the Cl3p character in the ψ* orbital. Reproduced from DOI: 10.1039/c7fd0076f with permission from The Royal Society of Chemistry. | ||
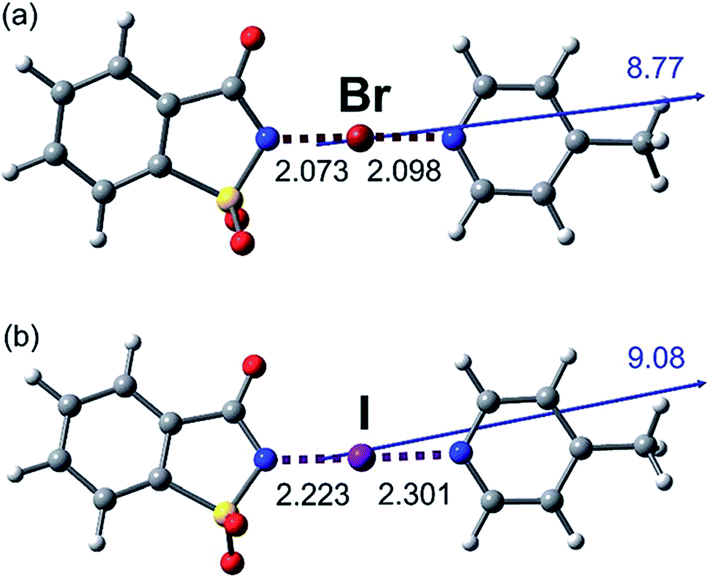 | ||
| Fig. 7 Halogen bonding interactions in (a) NBSac·Pic and (b) NISac·Pic (distances in Å; NBSac = N-bromosaccharin, NISac = N-iodosaccharin, Pic = 4-picoline). Arrows and values correspond to the orientation and the magnitude (in Debye) of the dipolar moments of the adducts calculated at frozen experimental geometries. Geometries indicate the greater asymmetry is in the N–I⋯N halogen bond. Reproduced from DOI: 10.1039/c7fd00067g with permission from The Royal Society of Chemistry. | ||
Alavi and co-workers (DOI: 10.1039/c7fd00064b) described their computational studies of chlorine and bromine hydrates, clathrates based on polyhedral hydrogen-bonded water cages with the dihalogens occupying the cage voids. These materials have been known for some 200 years, but are still revealing structural and bonding details.39–42 The study demonstrates the halogen-bonding interaction between the dihalogen and the water molecules, and highlights the bifurcation that occurs involving one of the water molecule lone pairs, which accepts both a hydrogen bond and a halogen bond. Bifurcation of interactions also featured in a number of the calculations presented by Politzer (DOI: 10.1039/c7fd00062f), which focused on σ-hole interactions across the p-block. Specifically, examples were presented in which the site of the most positive electrostatic potential was calculated to be located between atomic sites at which σ-holes might have been expected, e.g. S and C in octafluorothiophane and two hydrogens in dimethylurea (see Fig. 8). An alternative to the more conventional description of a bifurcated interaction was proposed, by connecting the sites of most positive and most negative potential on neighbouring molecules via a single interaction. Bifurcation remains an appropriate description if presented in terms of an atom–atom interaction framework, and its interpretation is well understood and in common use. Such a bifurcated description has been used previously where sites of maxima or minima in electrostatic potential have been determined to lie between atoms.43
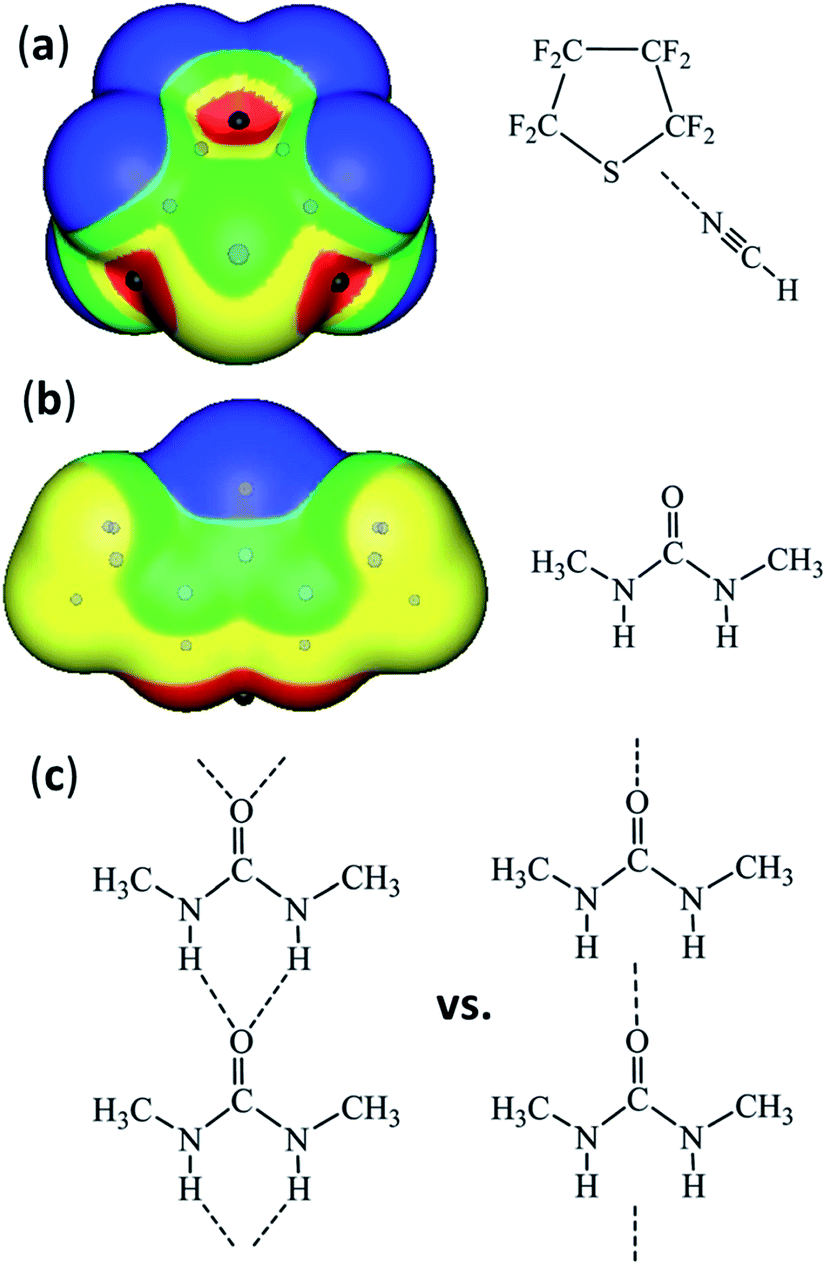 | ||
| Fig. 8 Maxima in positive electrostatic potential (shown as black dots) that lie between atoms: (a) between ring atoms S and C, providing the site for interaction with a Lewis base, and (b) between hydrogen atoms providing a site for interaction with the most negative region of the carbonyl oxygen leading to the hydrogen-bonded urea tape. (c) Two alternative descriptions of hydrogen-bonding between urea molecules – the conventional bifurcated interaction in an atom–atom interaction framework (left) and the interaction between sites of electrostatic potential minima and maxima (right). Adapted from DOI: 10.1039/c7fd00062f with permission from The Royal Society of Chemistry. | ||
The emphasis so often placed on drawing (dotted/dashed) lines between atoms to represent intermolecular interactions is reflected upon in the paper by Edwards, Spackman and co-workers (DOI: 10.1039/c7fd00072c). Their analysis of intermolecular interactions in crystals is based on the calculated molecular charge densities and enables molecule–molecule interactions to be partitioned into electrostatic and dispersion contributions, which can the be visualised in a quantitative manner by representing the strength of an interaction between two molecules by a line, the thickness of which is proportional to the interaction strength (Fig. 9). Spackman refers to this model of intermolecular interactions as an energy framework.44–46 Their analyses, and similar analyses by Gavezzotti, using his PIXEL calculation approach,47–49 show that the strongest interactions between molecules are not always those that conform to named interactions such as hydrogen bonds, halogen bonds etc., i.e. the interactions which are conveniently depicted as dotted lines between the atoms of the interacting molecules. The paper (DOI: 10.1039/c7fd00072c) also questions the need for so many specific names for many different types of atom–atom intermolecular interactions when intermolecular interactions may be better interpreted in terms of complementary electrostatic interactions between molecular surfaces combined with dispersion forces.
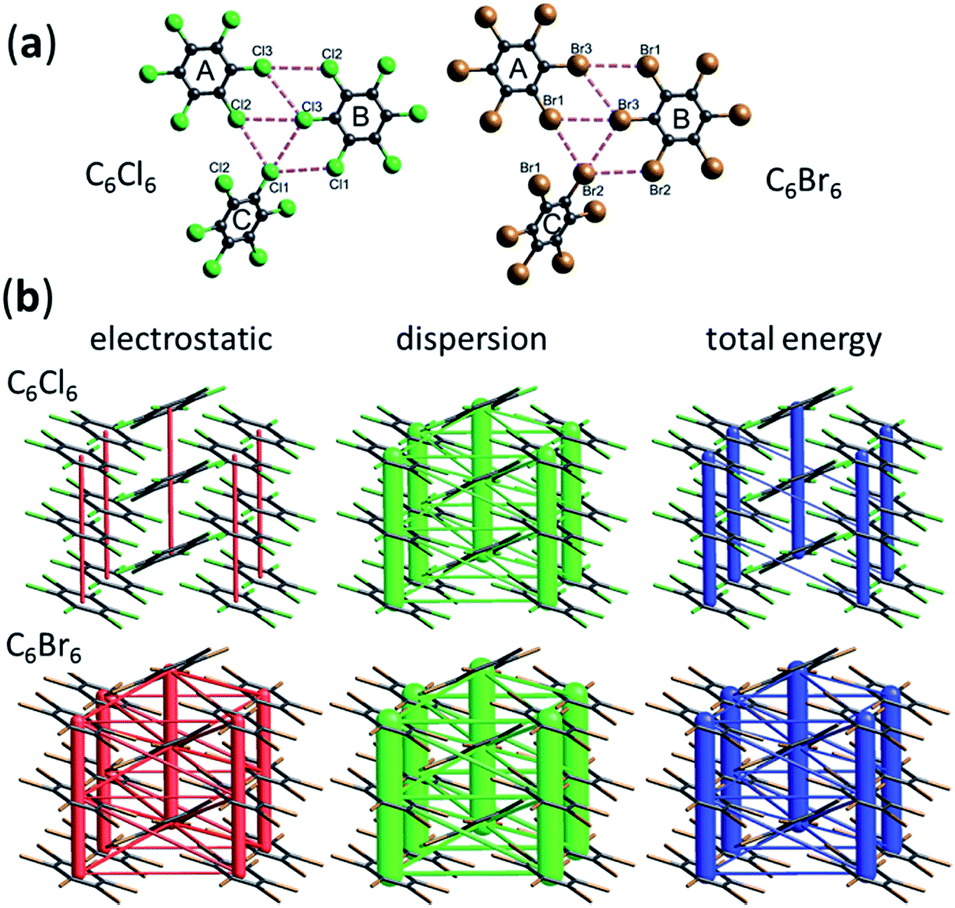 | ||
| Fig. 9 (a) Crystal structures of C6Cl6 and C6Br6, shown with dashed lines representing intermolecular halogen–halogen interactions. (b) Energy frameworks for crystal structures of C6Cl6 and C6Br6, showing that the largest contribution to the lattice energy comes from dispersion interactions between the stacked molecules and that halogen–halogen interactions (type I and type II) provide a much smaller contribution to the overall energy, which might be interpreted as providing a steering role for the orientation of adjacent molecular stacks. Adapted from DOI: 10.1039/c7fd00072c with permission from The Royal Society of Chemistry. | ||
3. Beyond the halogen bond
Although the study of halogen bonding has expanded dramatically over the past 15–20 years,50–54 analogous interactions involving other main group elements in a Lewis acidic role have seen far less attention over this same period. More recently, with the advent of the σ-hole terminology as a framework for describing such interactions,13 there has been a growing interest in the study of such interactions, which like halogen bonding have the potential to lead to a variety of applications in supramolecular chemistry, crystal engineering, materials chemistry and in medical/biological areas. The terms triel bond (group 13), tetrel bond (group 14), pnicogen/pnictogen bond (group 15), chalcogen bond (group 16) and aerogen bond (group 18) have begun to be used by analogy to the term halogen bond for the now well-established interactions involving group 17 elements. As noted in the Introduction, such interactions across the p-block were well established by crystallographic studies in the 1960s and 70s.10,11 The interactions were often described within a bonding framework of donor–acceptor interactions, in which a significant covalent or charge-transfer component to the bonding was inferred, rather than via an electrostatic (e.g. σ-hole) description. Early studies often focused on systems in which the main group element involved in the Lewis acidic role is bound to fluorine (often perfluorinated), leading to strong intermolecular interactions (Fig. 10). There is now scope to develop this palette of interactions in a similar manner to the modern development of halogen bonds through use of different electron-withdrawing groups (perfluoroaryl, perfluoroalkyl, alkynyl, cationic, etc.) that can be engaged through established synthetic organic and main group chemistry. Such development is necessary to enable opportunities for new applications of supramolecular chemistry across a variety of fields. Examples are known. Indeed in work with Braddock-Wilking and co-workers, we reported, in 1995, examples of intramolecular tetrel bonding, although not then named as such, involving SiF2(2,4,6-C6H2(CF3)3)2 and SiHF(2,4,6-C6H2(CF3)3)2 (Fig. 11).57 The interactions involving fluorine substituents of the ortho-CF3 groups interact with the Si centres trans to the primary bonding (to F, H and C) and are evident from the crystal structures and from solution-phase NMR spectroscopy in which JSiF coupling for the secondary (tetrel) bonds has a magnitude of approximately 3 Hz in comparison with the 1JSiF coupling of 300 Hz observed for primary Si–F bonds. The tetrel bond is shorter for interactions trans to F than trans to C, consistent with electrostatic or orbital (charge-transfer) arguments. We noted in our report that secondary F⋯E interactions have also been reported for Ar2Pb,58 Ar2Sn,59,60 Ar3Ga61 and for related filled-shell transition metal complexes Ar2Zn, Ar2Hg and Ar2Cd(NCMe),62 although not all adopt the linear arrangement observed in our examples. Thus, care is needed in interpreting the nature of these interactions.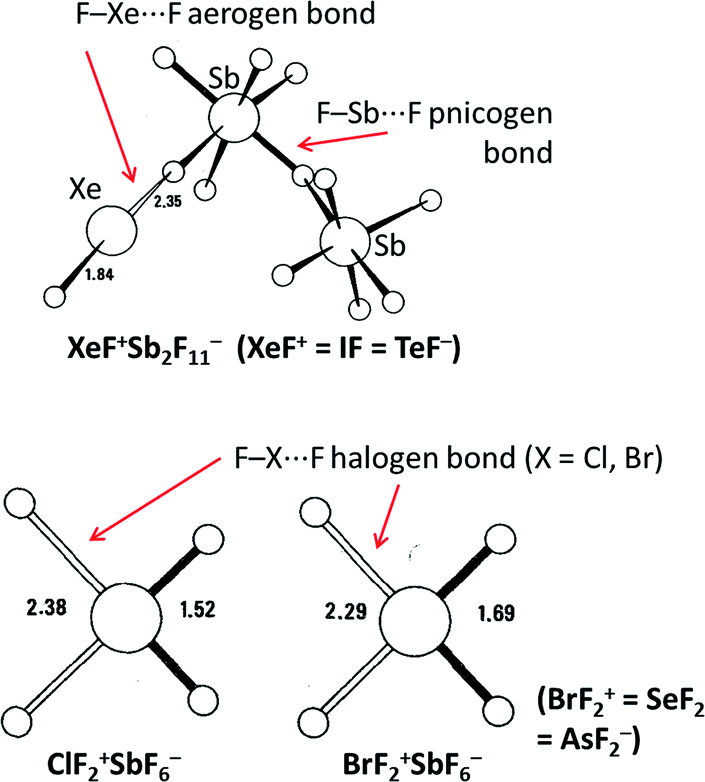 | ||
| Fig. 10 Examples of secondary bonding interactions involving p-block elements in Lewis acid roles.55,56 Equivalent isoelectronic species to secondary bond donors are noted in parentheses. Adapted from ref. 11 with permission from Elsevier. | ||
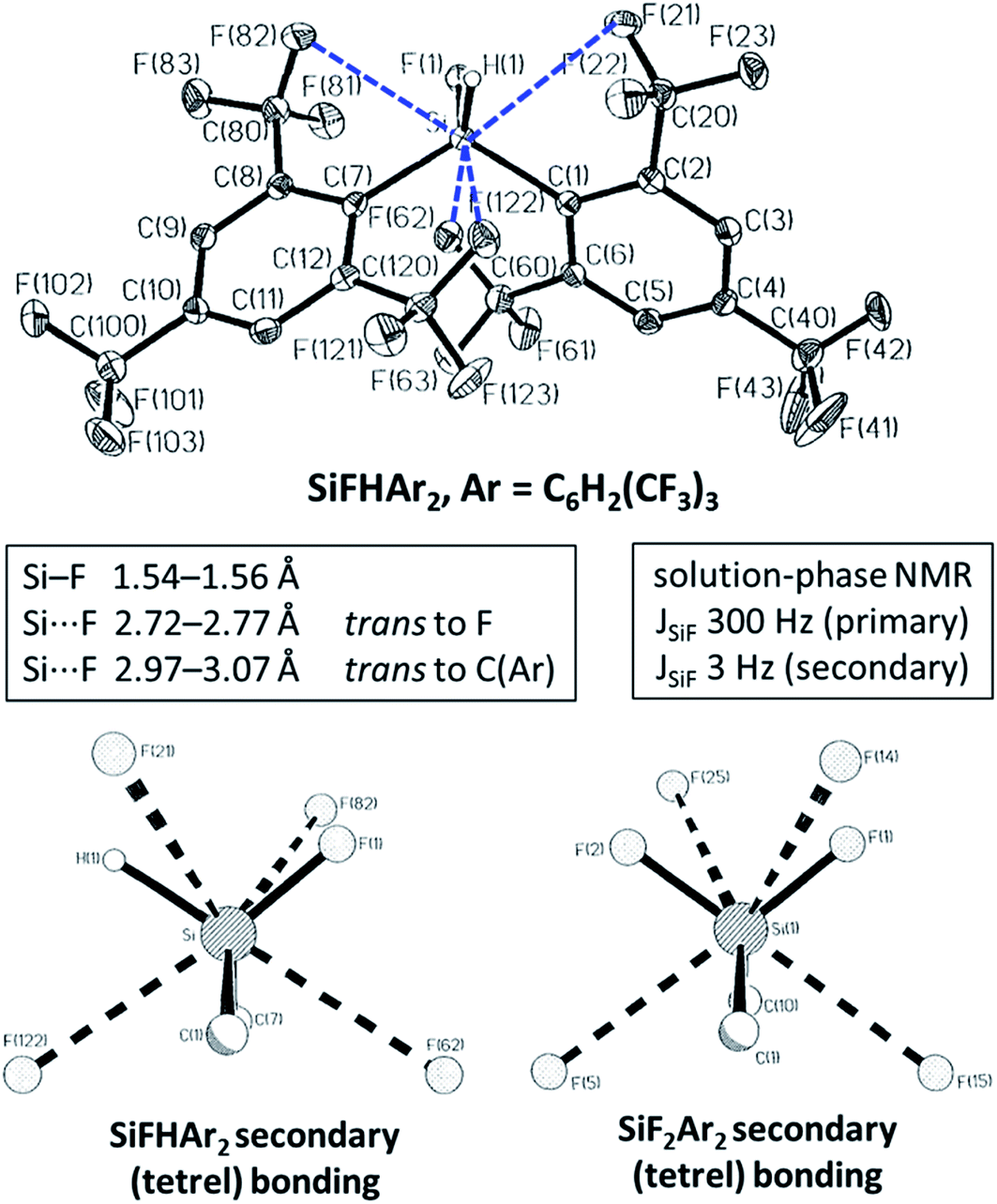 | ||
| Fig. 11 Crystal structure of SiFHAr2, Ar = C6H2(CF3)3 (top), with secondary (tetrel) bonding shown as dashed bonds. Primary bonding (solid lines) and secondary bonding (dashed lines) at Si in SiFHAr2 and SiF2Ar2 (bottom). Secondary (tetrel) Si⋯F bonds are longer than primary (covalent) Si–F bonds. Secondary bonds trans to F are shorter than those trans to C. Adapted from ref. 57 with permission from Elsevier. | ||
In an extensive computational study, Scheiner (DOI: 10.1039/c7fd00043j) reported binding energies and geometries for halide ion binding in a series of isostructural ditopic receptors in which hydrogen bonding, tetrel bonding, pnictogen bonding, chalcogen bonding and halogen bonding were compared (Fig. 12). As expected, heavier p-block elements form stronger interactions than their lighter congeners. Of particular interest is the fact that tetrel bonding from RSnH3 groups to the halides is stronger than that for corresponding halogen bond (RI) and at least as strong as the corresponding hydrogen bond (RH). Vargas-Baca and co-workers (DOI: 10.1039/c7fd00075h) presented a series of cyclic tellurazole N-oxides, which can form supramolecular assemblies via chalcogen bonds involving Te as the chalcogen bond donor. The N-oxide has an electron-withdrawing role that enhances the strength of the chalcogen bond. This role can be further amplified by coordination of the oxygen to Lewis acids such as BF3 (Fig. 13). Southern, Bryce and co-workers (DOI: 10.1039/c7fd00087a) described the use of solid-state 207Pb NMR spectroscopy to explore the formation of tetrel bonds in hemihedrally coordinated Pb(II) complexes. Although not a commonly used NMR nucleus, 207Pb is 22.1% abundant, has I = 1/2 and a very a large chemical shift range, offering the potential of great sensitivity to the nature of intermolecular interactions involving Pb centres. It is further noted that group 14 elements provide considerable opportunities for the investigation of tetrel bonding by NMR spectroscopy (Table 1).
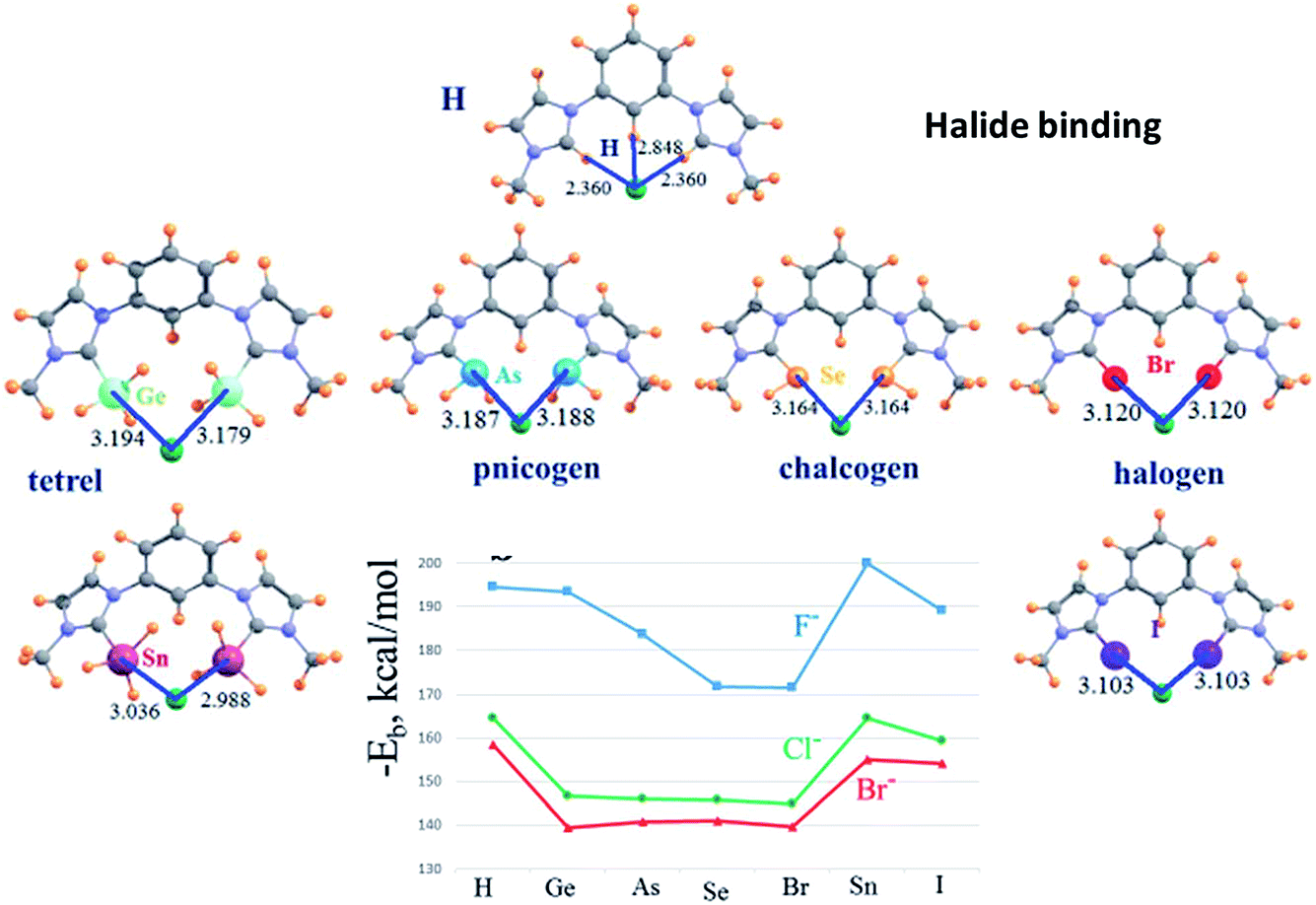 | ||
| Fig. 12 Optimized geometries of Cl− anion with ditopic receptors (distances in Å). Binding energies for halide ion binding by receptors shown. Adapted from DOI: 10.1039/c7fd00043j with permission from The Royal Society of Chemistry. | ||
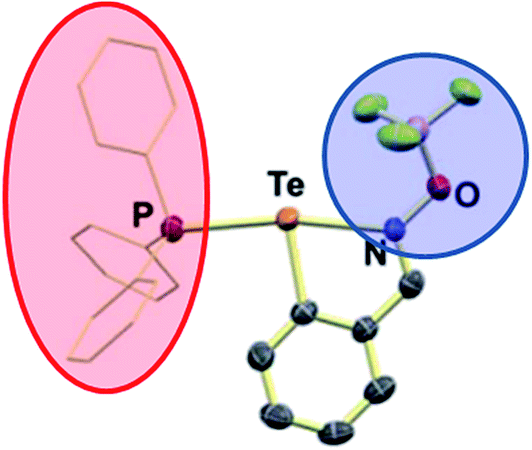 | ||
| Fig. 13 N–Te⋯P chalcogen bond. Strong chalcogen bonding capability of Te is activated by coordination of N-oxide, and further enhanced by Lewis acid coordination of the BF3 group to the oxygen (blue circle). Phosphine (PPh3, red ellipse) serves as the (Lewis basic) chalcogen bond acceptor. Adapted from DOI: 10.1039/c7fd00075h with permission from The Royal Society of Chemistry. | ||
| NMR-active isotope | I | Natural abundance (%) | Magnetogyric ratio (107 rad T−1 s−1) | Relative receptivity | Chemical shift range (ppm) |
|---|---|---|---|---|---|
| 13C | 1/2 | 1.07 | 6.728284 | 1.00 | 250 |
| 29Si | 1/2 | 4.6832 | −5.3190 | 2.16 | 900 |
| 73Ge | 9/2 | 7.73 | −0.9360303 | 0.642 | 1200 |
| 115Sn | 1/2 | 0.34 | −8.8013 | 0.711 | 2600 |
| 117Sn | 1/2 | 7.68 | −9.58879 | 20.8 | 2600 |
| 119Sn | 1/2 | 8.59 | −10.0317 | 26.6 | 2600 |
| 207Pb | 1/2 | 22.1 | 5.58046 | 11.8 | 17000 |
4. Characterisation
A number of papers in this Discussion showcased characterisation methods that provide insight into secondary bonding interactions, including halogen bonding. In many examples crystallographic studies were used to establish the geometries of these interactions, including the contributions by Riley and Tran (DOI: 10.1039/c7fd000106a), Alavi et al. (DOI: 10.1039/c7fd00064b), Kennepohl et al. (DOI: 10.1039/c7fd0076f), Edwards et al. (DOI: 10.1039/c7fd00072c), Politzer et al. (DOI: 10.1039/c7fd00062f), Southern et al. (DOI: 10.1039/c7fd00087a), Vargas-Baca et al. (DOI: 10.1039/c7fd00075h), Guru Row et al. (DOI: 10.1039/c7fd00084g), Mosquera et al. (DOI: 10.1039/c7fd00079k), Aakeröy et al. (DOI: 10.1039/c7fd00080d), Fourmigué et al. (DOI: 10.1039/c7fd00067g), Lloyd et al. (DOI: 10.1039/c7fd000108h), and Friščić et al. (DOI: 10.1039/c7fd00114b). Guru Row and co-workers (DOI: 10.1039/c7fd00084g) studied the structures of 2-bromo-4-chloro-benzoic acid and its isomer 4-bromo-2-chloro-benzoic acid. Both form structures that involve hydrogen-bonded carboxylic acid dimers but differ in the arrangement interactions involving the halogens. Solid solutions of the two compounds could be formed at a variety of different compositions. The resultant crystal structures were found to adopt that of the 4-bromo isomer until high concentrations of the 2-bromo isomer led its structure directing the final arrangement. The intermolecular interactions in crystals of each of the pure compounds were quantified by a topological analysis of the experimental electron density distributions.Erdelyi and co-workers (DOI: 10.1039/c7fd00107j) investigated natural abundance 15N NMR spectroscopy as a probe for weak halogen bond formation in the solution phase. Nonfluorinated halogen-bond donors involving I, Br and Cl were investigated as were substituent effects and solvent effects (Fig. 14). The study concluded that 15N NMR spectroscopy is suitable for the characterization of strong secondary interactions in solution, but not sufficiently accurate to detect weak halogen-bonded complexes due to the low mole fraction of the complex present. DFT calculations show that the 15N chemical shift does reflect the diamagnetic deshielding associated with the formation of a weak halogen bond, but that experimental detection was unreliable (Fig. 14a). Hydrogen-bonding interactions with the same acceptors show a 10-fold larger change in chemical shift (Δδ15N) and can readily be detected and correlated with the electronic properties of the hydrogen bond donor (Fig. 14b). The effect of solvent upon halogen bond formation is also apparent (Fig. 14c) wherein less polar solvents result in larger deshielding of the 15N nuclei (Δδ15N < 0), the Δδ15N having the largest magnitude for I > Br > Cl. In less polar solvents, the resulting positive Δδ15N is suggested to arise from the altered polarity of these mixtures as compared to the reference, i.e. this effect opposes the very small negative Δδ15N that results from the formation of the weak halogen bond.
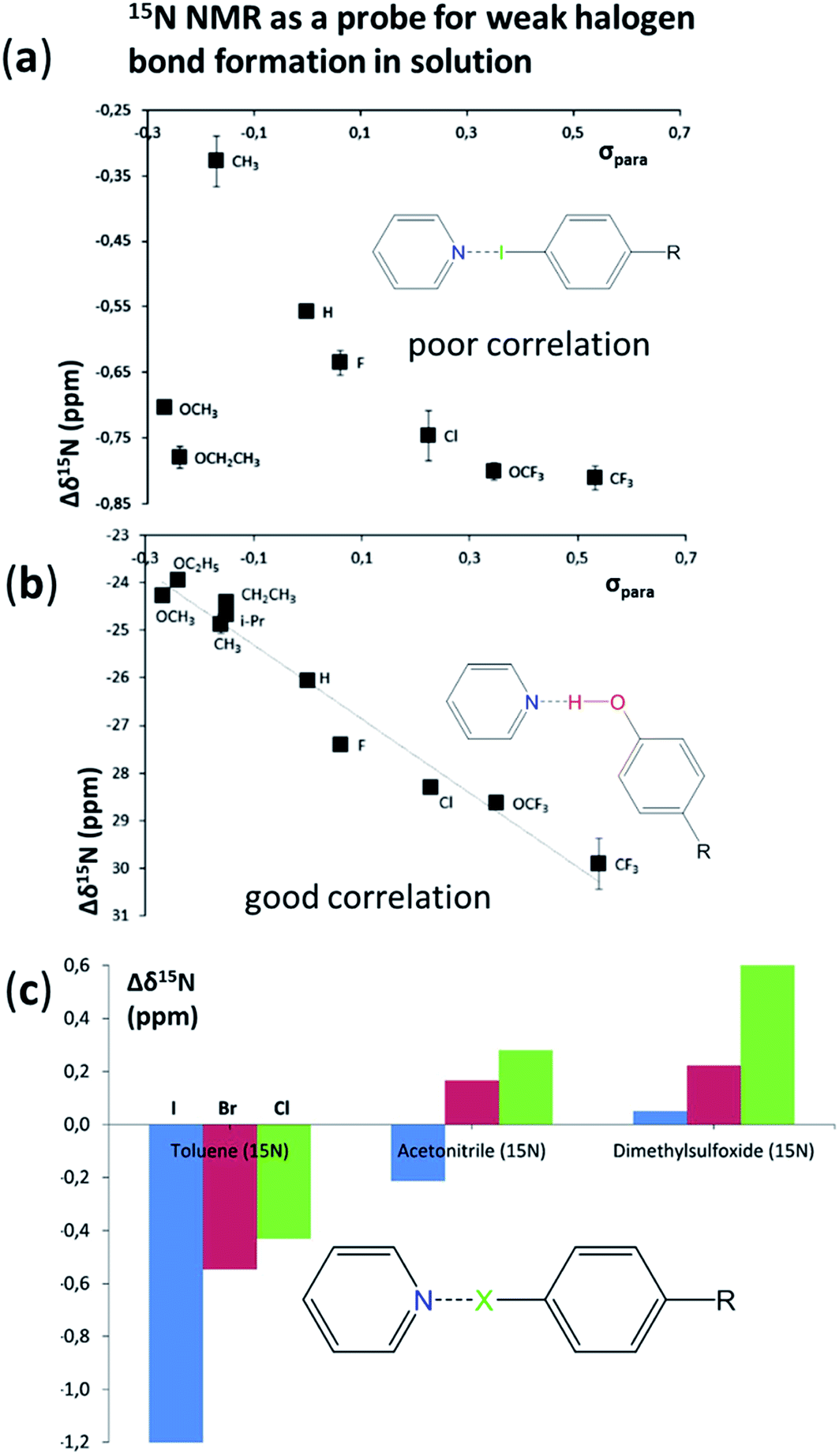 | ||
| Fig. 14 (a) (Poor) correlation of Δδ15N with (weak) halogen-bond donor strength expressed in terms of Hammett σpara constant for substituent R. (b) Good correlation of Δδ15N with hydrogen-bond donor strength expressed in terms of Hammett σpara constant for substituent R. All measurements for (a) and (b) are in toluene-d8. Standard error on δ15N < 0.02 ppm. (c) Average chemical shift changes for (weak) halogen bonds C–I⋯N (blue), C–Br⋯N (red), C–Cl⋯N (green), as a function of solvent. Adapted from DOI: 10.1039/c7fd00107j with permission from The Royal Society of Chemistry. | ||
Electrochemical methods were used in conjunction with spectroscopic measurements by Rosokha and by Branca, Schöllhorn and co-workers to investigate halogen bond formation in solution. Rosokha (DOI: 10.1039/c7fd00074j) used a combination of electrochemical and UV/Vis spectroscopic measurements to examine charge-transfer in halogen bonds formed by R–CBr3 halogen-bond donors with tetramethyl-p-phenylenediamine (TMPD) or iodide as the acceptor. The work established from kinetics determinations that electron transfer from diamine to R–CBr3 occurs too rapidly to be via an outer-sphere mechanism. The resulting fast redox process implicates transient halogen-bonded complexes in which barriers for electron transfer are lowered by the strong electronic coupling the two molecules. The study also suggests that halogen bonding should be taken into account in the mechanistic analysis of the reactions of halogenated species. In their study of cationic viologen-substituted halide receptors, Branca, Schöllhorn and co-workers (DOI: 10.1039/c7fd00082k) used a combination of electrochemical and NMR titrations to establish binding strengths and binding sites.
5. Applications
In the final section we will focus on applications of halogen bonding, with papers presented on anion binding, supramolecular synthesis, micelle formation in polymers, gel formation, dichroism and light-controlled ion transport. Beer and co-workers have developed rotaxane systems with a goal of selective binding of anions in water.63–65 Their use of halogen bonding for anion binding has enabled marked progress towards this aim. In their Discussion paper (DOI: 10.1039/c7fd00077d), they report the tuning of the binding site to enable excellent selectivity of Br− over I− and Cl− in a polar d6-acetone/D2O (9:1) solution. The anion binding site comprises three C–I halogen bond donors from triazole units (Fig. 15), for which the halogen-bond donor capability is enhanced by cationic charge or ligation to Re. Chelation of the Re centre also influences the spatial arrangement of the two C–I groups on the bistriazole unit improving preorganisation of the rotaxane for binding.
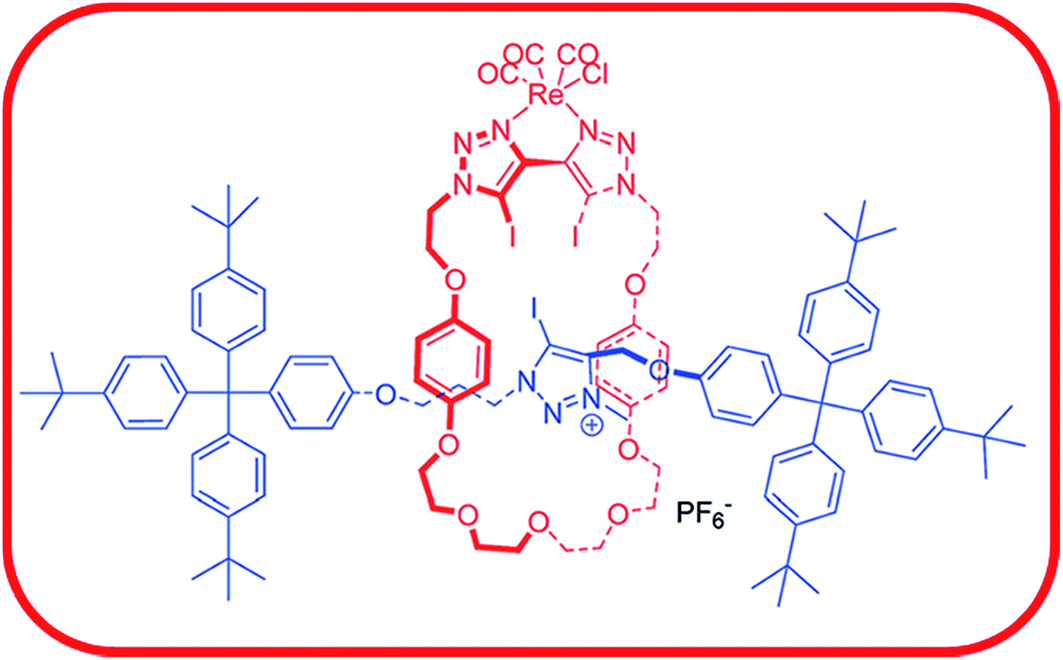 | ||
| Fig. 15 Cationic rotaxane that exhibits selective binding of Br− (K = 436(25) M−1) over Cl− and I−, for which binding is too weak to measure by NMR titration. All measurements are in d6-acetone/D2O at 298 K. Adapted from DOI: 10.1039/c7fd00077d with permission from The Royal Society of Chemistry. | ||
As part of a continuing programme of study of the competition between intermolecular interactions in supramolecular synthesis of crystals,66–69 Aakeröy and co-workers have designed a series of 1-(pyridylmethyl)-2,2′-biimidazoles, which contain three different sites capable of being hydrogen bond or halogen bond acceptors (Scheme 1). Co-crystallisation with a series of ditopic hydrogen bond donors and halogen bond donors allowed selectivity patterns for intermolecular interaction formation to be examined (DOI: 10.1039/c7fd00080d). Halogen-bond donors consistently led to self-association of the biimidazoles through R22(10) N–H⋯N hydrogen-bonded motifs, with halogen-bond donors selecting the pyridyl acceptor site over the remaining imidazole site. Carboxylic acid hydrogen-bond donors showed less consistency. In some cases biimidazole self-association occurred with carboxyl hydrogen bonding to pyridyl groups, but in others the carboxyl group disrupted the self-association, leading to a variety of outcomes. Mosquera, Gómez-Sal and co-workers (DOI: 10.1039/c7fd00079k) have carried out a mechanistic investigation that examines in detail the role of halogen bond formation on halide ligand substitution in the octahedral complex [RuCl2(CNR)4]. Their study implicates halogen bond formation, initially between dihalogens (I2, Br2) and the chloride ligands, in the mechanism leading to substitution of chloride by iodide or bromide. The rate of substitution depends on the electron-donating ability of the isocyanide (CNR) ligands. A number of intermediate stages of the reaction have been characterised crystallographically and studied by Raman spectroscopy. An unusual observation from these structures is that formation of Ru–X⋯I2 leads to Ru–X bond lengthening, whereas formation of Ru–Br⋯Br2 leads to Ru–Br bond shortening.
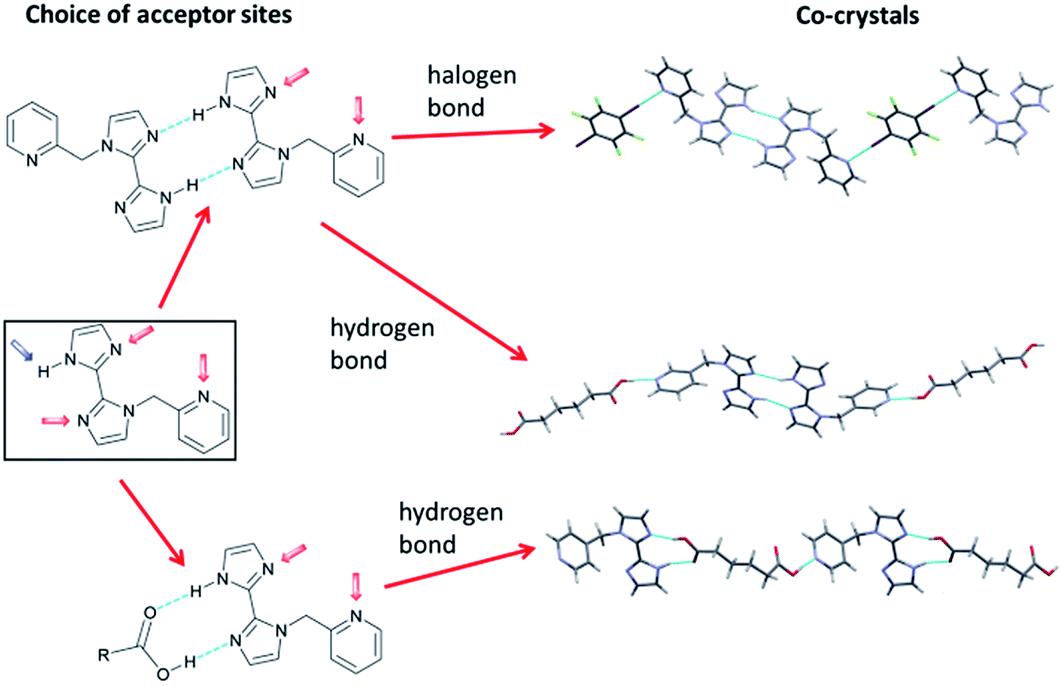 | ||
| Scheme 1 Competition between acceptor sites for hydrogen-bond or halogen-bond donors in co-crystal formation. Adapted from DOI: 10.1039/c7fd00080d with permission from The Royal Society of Chemistry. | ||
Vanderkooy and Taylor (DOI: 10.1039/c7fd00111h) have appended C6F4I halogen-bond donor groups to a polymer backbone enabling the use of halogen bonding in forming multicompartmental micelles from complementary macromolecules. The micelles were able to be imaged by TEM (Fig. 16). Lloyd, Khimyak and co-workers (DOI: 10.1039/c7fd000108h) have examined the effect of halogen bonding, and more generally the influence of intermolecular interactions involving halogen substituents, on a series of phenylalanine (Phe) derivatives with halogen substituents on the aromatic ring. Friščić, Barrett and co-workers (DOI: 10.1039/c7fd00114b) were able to synthesise and demonstrate dichroism in crystals comprising halogen-bonded chains of trans-4,4′-diiodooctafluoroazobenzene as chromophore and ditopic halogen-bond donor and dicyanoaurate(I) as ditopic halogen-bond acceptor (Fig. 17). The anionic assembly is charge-balanced by 15-crown-5-encapsulated K+ ions. The halogen bonding is used to provide alignment of the chromophores. Finally, remaining with the theme of optical properties, Cavallo, Dichiarante and co-workers (DOI: 10.1039/c7fd00120g) have prepared halogen-bonded lamellar liquid crystals (LC), which include an iodide-containing charged region as well as a lipophilic region. The halogen-bond donor unit contains a photo-switchable diazo group. A reversible cis–trans photo-isomerization of this group leads to a LC-to-isotropic phase transition, which the authors envision is a first step towards light-induced control over ion transport and conductance in these supramolecular complexes.
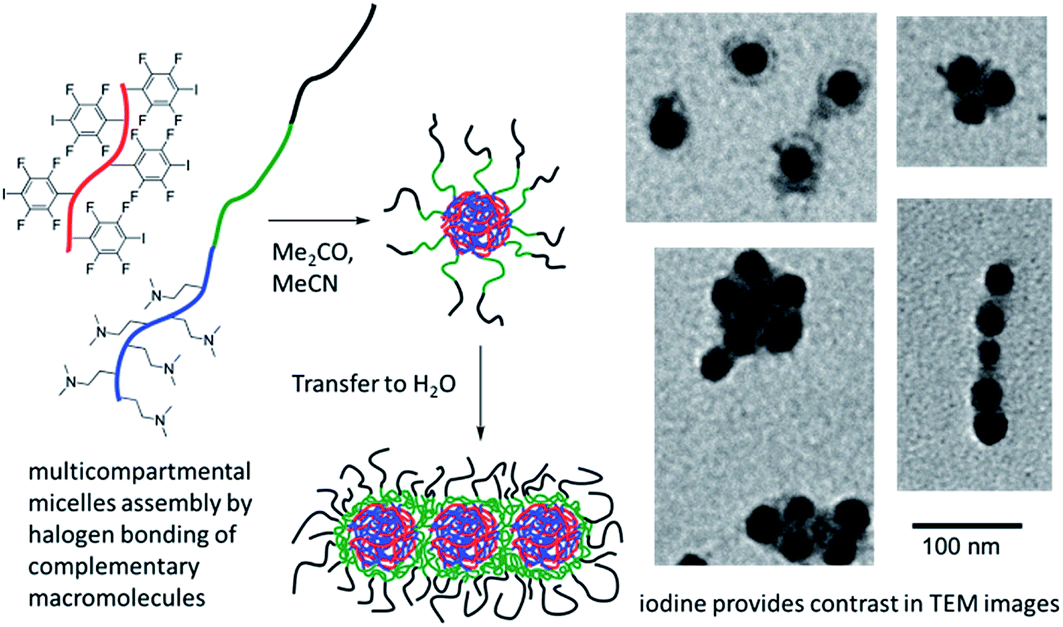 | ||
| Fig. 16 Micelles formed via halogen bonding between triblock copolymer and halogen-bond donor polymer. Adapted from DOI: 10.1039/c7fd00111h with permission from The Royal Society of Chemistry. | ||
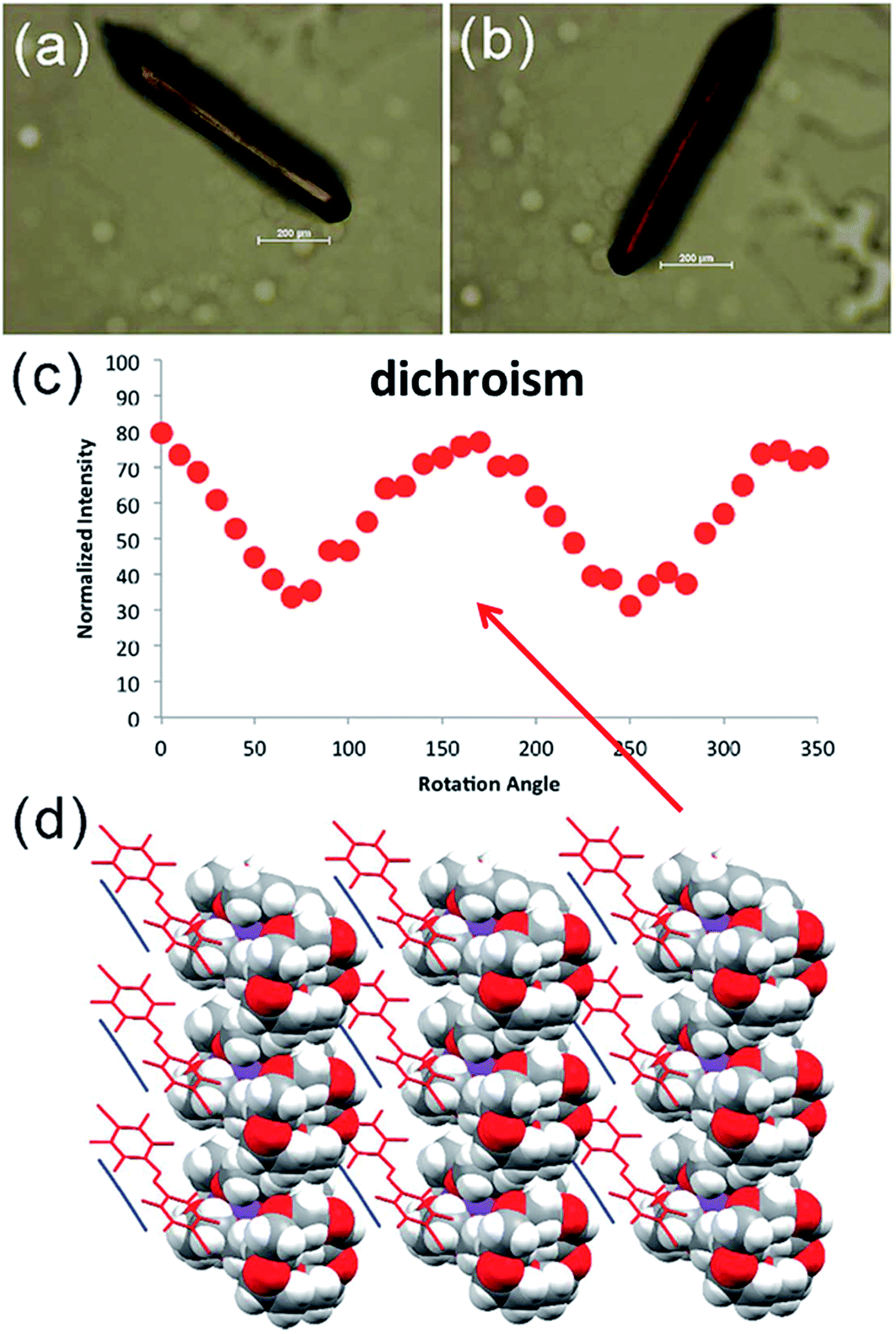 | ||
| Fig. 17 Dichroic crystals of [K(15-crown-5)][Au(CN)2]·(DIFAB)·(solvent) (DIFAB = 4,4′-diiodooctafluoroazobenzene) containing aligned linear halogen-bonded assemblies of DIFAB and [Au(CN)2]−. (a) and (b) Crystals presented at different rotation angles exhibit different colours in polarised light. (c) Light intensity as a function of crystal rotation. (d) Crystal structure [K(15-crown-5)][Au(CN)2]·(DIFAB)·(solvent). Reproduced from DOI: 10.1039/c7fd00114b with permission from The Royal Society of Chemistry. | ||
6. Conclusions and future outlook
Among the main themes that emerged in the Discussion (DOI: 10.1039/c7fd90061a, DOI: 10.1039/c7fd90062g, DOI: 10.1039/c7fd90063e and DOI: 10.1039/c7fd90064c) were those that involved the bonding model for describing halogen bonds and related interactions. It is clear that the electrostatic model is central to describing halogen bonds and their p-block analogues, but that care is needed in interpreting solid-state arrangements, as while it may be convenient to assign (on the basis of geometry) halogen bonds or hydrogen bonds as structure directing, these may not be the main cohesive intermolecular forces. Experimental measurements were presented to demonstrate charge transfer in halogen bonds indicating the importance that such a description can have in understanding the interactions.Although computational studies highlighted some differences between halogen bonds, chalcogen bonds, pnictogen bonds and tetrel bonds, more experimental studies are needed to quantify and exploit such differences. A wide variety of characterisation methods were employed in the studies presented, including, X-ray crystallography, multinuclear NMR spectroscopy (solution-phase and solid-state), UV/Vis spectroscopy, IR/Raman spectroscopy, cyclic voltammetry, X-ray absorption spectroscopy (XAS) and electron microscopy. Experimental studies were often complemented by computational studies, which enabled the calculation of structures, spectroscopic parameters, bonding models and energetics. It is also noticeable that studies of halogen bonding spanned a very wide range of molecular sizes from very small, e.g. diatomics (I2, ClF, etc.) to very large, e.g. rotaxanes and polymers.
A number of applications of halogen bonding were described, including supramolecular and covalent synthesis, anion binding, micelle formation in polymers, gel formation, dichroism and light-controlled ion transport. These represent examples from among the many areas in which halogen bonding is now being applied, such as medicine and biology, including pharmaceuticals, many aspects of molecule-based materials chemistry, encompassing crystal engineering, and in synthesis and catalysis. It should be anticipated that the recent development of interest in related chalcogen, pnictogen and tetrel bonds will also lead to opportunities for a wide array of applications that are underpinned by an improved understanding and an expanded “toolbox” for supramolecular chemistry.
I will finally return to the question of nomenclature, which was raised in the Introduction. The definition of halogen bonding70 (and re-definition of hydrogen bonding71) has recently been approved through IUPAC projects and the definitions of analogous interactions, commonly referred to as tetrel bonds (group 14), pnictogen bonds (group 15), chalcogen bonds (group 16) and aerogen bonds (group 18) are soon to be determined via a similar IUPAC project. Although the names of these interactions convey valuable precision in description and permit distinction to be made between these related interactions, it seems clear that a collective name for such interactions is also needed. Although not formally approved by IUPAC, that collective name already exists. The term secondary bond, coined by Alcock,11 fulfils the role and allows for the necessary derivative terms secondary bonding, secondary bond donor, secondary bond acceptor, etc. to convey meaning with clarity for this collection of interactions in the same way that more specific terms, e.g. halogen bond, chalcogen bonding, pnictogen bond donor and tetrel bond acceptor, can for the more limited subset of cases to which they apply. Some might argue that hydrogen bonds should remain separate, but this seems unnecessary as they can be described in the same manner (either by σ-hole electrostatic model72 or charge-transfer orbital model73,74) as the interactions that involve p-block elements in the Lewis acidic role, although forming less directional interactions. Indeed it can be anticipated that analogous classes of interactions involving elements of group 13 (triel) or s-block groups 1 and 2 can be similarly encompassed in the definition of secondary bonding. Thus, it seems appropriate to adopt the term secondary bonding as an umbrella term for all classes of intermolecular interaction involving s- and p-block elements in a Lewis acidic role.
Acknowledgements
Thanks are due to the Royal Society of Chemistry, the organising committee and particularly its Chair, David Bryce, who also served as an excellent host for the Discussion meeting. I would like to thank the PhD students and postdocs, Fiorenzo Zordan, Guillermo Mínguez Espallargas, Stefano Libri, Iñigo Vitórica-Yrezábal, Paul Smart, Johnathan Ormond-Prout, Adrián Pérez Redondo, James Wright, Craig Robertson and Tom Roseveare, as well as a number of undergraduate research students, who have worked on halogen-bond related projects in my group since 2001. I also value a number of enriching collaborations that have broadened our research and hastened our progress, particularly those with Robin Perutz (York), Chris Hunter (Cambridge), Paul Sherwood (Daresbury) and Anthony Meijer (Sheffield). I am grateful to the EPSRC, STFC, CCDC, the White Rose Consortium and University of Sheffield for support of work involving the aforementioned students.References
- M. Colin, Ann. Chim., 1814, 91, 252–272 Search PubMed.
- F. Guthrie, J. Chem. Soc., 1863, 16, 239–244 RSC.
- For example, see: (a) G. C. Pimental and A. L. McClellan, The Hydrogen Bond, W. H. Freeman, San Fransisco, 1960 Search PubMed; (b) The Hydrogen Bond: Recent Developments in Theory and Experiment, ed. P. Schuster, G. Zundeland C. Sandorfy, North-Holland, Amsterdam, 1976 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, Oxford University Press, Oxford, 1999 Search PubMed.
- O. Hassel and J. Hvoslef, Acta Chem. Scand., 1954, 8, 873 CrossRef CAS.
- O. Hassel and K. O. Stromme, Acta Chem. Scand., 1958, 12, 1146 CrossRef CAS.
- O. Hassel and K. O. Stromme, Acta Chem. Scand., 1959, 13, 1781–1786 CrossRef CAS.
- O. Hassel, Science, 1970, 170, 497–502 CAS.
- H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- N. W. Alcock, Adv. Inorg. Chem. Radiochem., 1972, 15, 1–58 CrossRef CAS.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS.
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725–8726 CrossRef CAS.
- V. R. Pedireddi, D. S. Reddy, B. S. Goud, D. C. Craig, A. D. Raeb and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1994, 2353–2360 RSC.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CAS.
- L. Brammer, G. Mínguez Espallargas and H. Adams, CrystEngComm, 2003, 5, 343–345 RSC.
- L. Brammer, F. Zordan, G. Mínguez Espallargas, S. L. Purver, L. Arroyo Marín and H. Adams, Trans. Am. Crystallogr. Assoc., 2004, 39, 114–122 CAS.
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef PubMed.
- E. Corradi, S. V. Meille, M. T. Messina, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2000, 39, 1782–1786 CrossRef CAS PubMed.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8, 5392–5398 RSC.
- K. Hermansson and M. Alfredsson, J. Chem. Phys., 1999, 111, 1993–2000 CrossRef CAS.
- R. D. Willett, F. Awwadi, R. Butcher, S. Haddad and B. Twamley, Cryst. Growth Des., 2003, 3, 301–311 CAS.
- F. Zordan and L. Brammer, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 512–519 Search PubMed.
- G. Mínguez Espallargas, L. Brammer and P. Sherwood, Angew. Chem., Int. Ed., 2006, 45, 435–440 CrossRef PubMed.
- F. Zordan, G. Mínguez Espallargas and L. Brammer, CrystEngComm, 2006, 8, 425–431 RSC.
- F. F. Awwadi, R. D. Willett and B. Twamley, Cryst. Growth Des., 2007, 7, 624–632 CAS.
- G. Mínguez Espallargas, F. Zordan, L. Arroyo Marín, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Chem.–Eur. J., 2009, 15, 7554–7568 CrossRef PubMed.
- P. Metrangolo, T. Pilati, G. Terraneo, S. Biella and G. Resnati, CrystEngComm, 2009, 11, 1187–1196 RSC.
- P. Cauliez, V. Polo, T. Roisnel, R. Llusar and M. Fourmigué, CrystEngComm, 2010, 12, 558–566 RSC.
- A. Abate, J. Marti-Rujas, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2011, 11, 4220–4226 CAS.
- J. E. Ormond-Prout, P. Smart and L. Brammer, Cryst. Growth Des., 2012, 12, 205–216 CAS.
- A.-C. C. Carlsson, J. Grafenstein, J. L. Laurila, J. Bergquist and M. Erdelyi, Chem. Commun., 2012, 48, 1458–1460 RSC.
- A.-C. C. Carlsson, J. Grafenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Grafenstein and M. Erdelyi, Chem. Sci., 2014, 5, 3226–3233 RSC.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Grafenstein, K. Rissanen and M. Erdelyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- L. Pauling and R. E. Marsh, Proc. Natl. Acad. Sci. U. S. A., 1952, 38, 112 CrossRef CAS.
- K. W. Allen and G. A. Jeffrey, J. Chem. Phys., 1963, 38, 2304 CrossRef CAS.
- K. A. Udachin, G. D. Enright, C. I. Ratcliffe and J. A. Ripmeester, J. Am. Chem. Soc., 1997, 119, 11481–11486 CrossRef CAS.
- K. A. Udachin, S. Alavi and J. A. Ripmeester, J. Phys. Chem. C, 2013, 117, 14176–14182 CAS.
- L. Brammer, J. K. Swearingen, E. A. Bruton and P. Sherwood, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4956–4961 CrossRef CAS PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154 CrossRef CAS.
- A. Gavezzotti, J. Phys.Chem. B, 2003, 107, 2344–2353 CrossRef CAS.
- J. D. Dunitz and A. Gavezzotti, Angew. Chem., Int. Ed., 2005, 44, 1766–1787 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- R. K. Rowe and P. S. Ho, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 255–264 CrossRef CAS PubMed.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- V. M. McRae, R. D. Peacock and D. R. Russell, Chem. Commun., 1969, 62–63 RSC.
- A. J. Edwards and R. J. C. Sills, J. Chem. Soc. A, 1970, 2697–2699 RSC.
- J. Braddock-Wilking, M. Schieser, L. Brammer, J. Huhmann and R. Shaltout, J. Organomet. Chem., 1995, 499, 89–98 CrossRef CAS.
- S. Brooker, J.-K. Buijink and F. T. Edelmann, Organometallics, 1991, 10, 25–26 CrossRef CAS.
- H. Grützmacher, H. Pritzkow and F. T. Edelmann, Organometallics, 1991, 10, 23–24 CrossRef.
- U. Lay, H. Pritzkow and H. Grützmacher, J. Chem. Soc., Chem. Commun., 1992, 260–262 RSC.
- R. D. Schluter, H. S. Isom, A. H. Cowley, D. A. Atwood, R. A. Jones, F. Olbrich, S. Corbelin and R. J. Lagow, Organometallics, 1994, 13, 4058–4063 CrossRef CAS.
- S. Brooker, N. Bertel, D. Stalke, M. Noltemeyer, H. W. Roesky, G. M. Sheldrick and F. T. Edelmann, Organometallics, 1992, 11, 192–195 CrossRef CAS.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499–507 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- L. C. Gilday and P. D. Beer, Chem.–Eur. J., 2014, 20, 8379–8385 CrossRef CAS PubMed.
- C. B. Aakeröy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772–13773 CrossRef PubMed.
- C. B. Aakeröy, T. K. Wijethunga, M. A. Haj, J. Desper and C. Moore, CrystEngComm, 2014, 16, 7218–7225 RSC.
- C. B. Aakeröy, C. L. Spartz, S. Dembowski, S. Dwyre and J. Desper, IUCrJ, 2015, 2, 498–510 Search PubMed.
- M. D. Perera, J. Desper, A. S. Sinha and C. B. Aakeröy, CrystEngComm, 2016, 18, 8631–8636 RSC.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- H. Ratajczak, J. Phys. Chem., 1972, 76, 3000–3004 CrossRef CAS.
- L. C. Allen, J. Am. Chem. Soc., 1975, 97, 6921–6940 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |