
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddressing a bottle neck for regulation of nanomaterials: quantitative read-across (Nano-QRA) algorithm for cases when only limited data is available
A.
Gajewicz
 a,
K.
Jagiello
a,
M. T. D.
Cronin
b,
J.
Leszczynski
c and
T.
Puzyn
*a
a,
K.
Jagiello
a,
M. T. D.
Cronin
b,
J.
Leszczynski
c and
T.
Puzyn
*a
aLaboratory of Environmental Chemometrics, Institute for Environmental and Human Health Protection, Faculty of Chemistry, University of Gdansk, Wita Stwosza 63, 80-308 Gdansk, Poland. E-mail: t.puzyn@qsar.eu.org; Fax: +48 58 523 50 12; Tel: +48 58 523 5248
bSchool of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Byrom Street, Liverpool L3 3AF, UK
cInterdisciplinary Nanotoxicity Center, Department of Chemistry and Biochemistry, Jackson State University, Jackson, MS, USA
First published on 1st December 2016
Abstract
The number and variety of engineered nanoparticles have been growing exponentially. Since the experimental evaluation of nanoparticles causing public health concerns is expensive and time consuming, efficient computational tools are amongst the most suitable approaches to identifying potential negative impacts, to the human health and the environment, of new nanomaterials before their production. However, developing computational models complimentary to experiments is impossible without incorporating consistent and high quality experimental data. Although there are limited available data in the literature, one may apply read-across techniques that seem to be an attractive and pragmatic alternative way of predicting missing physico-chemical or toxicological data. Unfortunately, the existing methods of read-across are strongly dependent on the expert's knowledge. In consequence, the results of estimations may vary dependently on personal experience of expert conducting the study and as such cannot guarantee the reproducibility of their results. Therefore, it is essential to develop novel read-across algorithm(s) that will provide reliable predictions of the missing data without the need to for additional experiments. We proposed a novel quantitative read-across approach for nanomaterials (Nano-QRA) that addresses and overcomes a basic limitation of existing methods. It is based on: one-point-slope, two-point formula, or the equation of a plane passing through three points. The proposed Nano-QRA approach is a simple and effective algorithm for filling data gaps in quantitative manner providing reliable predictions of the missing data.
Environmental significanceThough nanomaterials have been intensively studied for the last 25 years, there are still gaps in the reliable experimental data that would provide comprehensive information related to their structures, properties and environmental impacts. Such gaps hamper safe developments and applications of new nanomaterials. Concerning the increasing number of existing and newly synthetized nanomaterials and the serious health risk that they may introduce, developing new read-across algorithms for filling data gaps, without the necessity of performing time consuming and expensive experimental studies on large set of nanomaterials, is of very high importance for the whole society, including companies designing new chemicals, end-users and the administration regulatory bodies. We believe that computational technique developed by us and reported in this manuscript allows to overcome such a bottle neck providing novel tool for the general use in risk assessment of new species. |
Introduction
Apart from providing a wide range of potential benefits, the use of engineered nanoparticles (NPs) may also endanger human health through the potential induction of cytogenetic, mutagenic and/or neurotoxic health effects.1–3 Since engineered nanoparticles may introduce the effect of ageing, transformation, and biomolecule coating under environmental or biological conditions, it is still largely unknown which properties govern their toxicity.4 Therefore, a great effort should be put into defining and developing methods for NPs characterization as well as the assessment of exposure, engineering controls, potential toxicity, fate and transport, and their life cycle.5The great need for the development of novel, fast, and inexpensive procedures for risk assessment that would not only reduce the necessity of extensive animal testing but also provide details on the potential mechanisms of toxicity at the molecular level was stressed multiple times, in the REACH legislation,6 the European Chemicals Agency,7–10 United States – Canadian Regulatory Cooperation Council,11 as well as the Organization for Economic Co-operation Development (OECD).12,13 In addition, the idea of developing intelligent testing strategies (ITS)14 also known as alternative testing strategies (ATS)15 or risk assessment strategies (RAS)16 has also been the topic of extensive discussions for over three years, through many national or international political incentives or scientific research projects. Regardless of the names used for this strategy, the main idea standing behind refers to specific challenges for engineered nanoparticles and allows the risks assessment of nanomaterials to be performed accurately, effectively and efficiently.17 Some of the key-priority research components of such strategy can be defined as follows: (1) grouping/categorization based on: variations in chemical structure and physico-chemical properties, possible mechanisms of metabolism and/or mode of action, (2) identifying data gaps in physico-chemical characterization, exposure assessment, and hazard assessment within the defined groups/categories, (3) using modeling approaches (i.e. computational methods such as: quantitative structure–activity/property relationships (QSAR/QSPR) and read-across) for the prediction of missing data for specific NPs within the defined groups/categories; and finally (4) using research outcomes for the prioritization of hazardous NPs, regulatory decision-making, and in safe-by-design principle along the value chain of an innovation.
All of the methods listed above, i.e.: grouping, chemical category formation, QSAR/QSPR and read-across form a group of non-testing approaches. They are often mismatched and some of them are incorrectly used as synonyms. This “glossary problem” is a source of many misunderstandings and false explanations. Table 1 provides definitions of many of the terms used in the field of non-testing approaches.
| Term | Definition |
|---|---|
| Analogue approach | OECD defines an analogue as “a chemical whose intrinsic physicochemical, environmental or toxicological properties are likely to be similar to those of another chemical based upon a number of potential properties including structural and physicochemical properties”.13 The term analogue approach is used when “the grouping involves a very limited number of chemicals (typically two chemicals) and trends or regular patterns in properties are not apparent”.9,13 |
| Categorization | “Categorization describes the general approach to the grouping of chemicals. Categorization strategies may include grouping, ranking, and read-across as examples of types of categorization”.15 |
| Category approach | OECD defines a chemical category as “a group of chemicals whose physicochemical and human health and/or environmental toxicological properties and/or environmental fate properties are likely to be similar or follow a regular pattern as a result of structural similarity”.13 The term category approach is used when “read-across is employed between several substances that have structural similarity. These substances are grouped together on the basis of defined structural similarity and differences between the substances”.9,13 |
| Grouping | “Grouping describes the general approach to assessing more than one chemical at the same time. It can include formation of a chemical category or identification of a chemical analogue for which read-across may be applied. Substances that are structurally similar with physicochemical, toxicological, ecotoxicological and/or environmental fate properties that are likely to be similar or to follow a regular pattern may be considered as a group of substances. These similarities may be due to a number of factors: (i) common functional group (i.e. chemical similarity within the group), (ii) common precursors and/or likely common breakdown products via physical and/or biological processes which result in structurally-similar degrading chemicals, (iii) a constant pattern in the changing of the potency of the properties across the group (i.e. of physico-chemical and/or biological properties)”.9,13 For some time now, there has been an ongoing discussion on how to develop scientifically based categorization strategies, how to identify the grouping needs and possibilities as well as on how to define the key physico-chemical features and toxicological responses allowing the effect-driven grouping of nanomaterials. The overview on the existing concepts, schemes and various criteria for grouping nanomaterials can be found in the literature.18–21 |
| [Q]SARs | Under OECD, qualitative or quantitative structure–activity relationships is a mathematical technique that relates a (sub)structure to the presence or absence of a property or activity of interest. [Q]SAR is based on dependencies defined between the variance in molecular structures, encoded by so-called ‘descriptors’, and the variance in biological activity in a set of similar chemicals.13 |
| Read-across | Read-across is a data gap filling technique within an analogue or category approach. Can be qualitative or quantitative.9 Under REACH, read-across is a technique for predicting endpoint information for one substance (target substance), by using data from the same endpoint from (an) other substance(s), (source substance(s)).6 |
In light of the definitions presented in the Table 1, it should be clearly stated that grouping is not the same as read-across. According to OECD the concept of grouping includes: (1) chemical category formation and/or (2) chemical analogue(s) identification. Thus, it is restricted to techniques for creating groups of somehow similar chemicals. At the same time, the term read-across is reserved for one of the techniques available for filling data gaps.13 In other words, when groups (or categories) of NPs are already established one may start fill the data gaps within the groups/categories by using read-across or other techniques.
Additionally, quantitative structure–activity relationship approach (QSAR) has been also pointed out as a promising approach for filling data gaps within the groups/categories. However, this approach could be only used when there is already a large experimental dataset – unfortunately, it can not be applied when is a limited amount of experimental data. Nevertheless, the successful concept and application of Nano-QSAR to predict toxicity of NPs has been already demonstrated.22–34 However, there are serious limitations related to the development of Nano-QSARs.35,36 The limited size of the experimental dataset available for modeling37 remains the large obstacle for progress in this area. In the absence of relevant and sufficient data to build an appropriately validated Nano-QSAR model, one needs apply a method based on limited amount of data – read-across approach.
In principle, the read-across approach is based on the assumption that chemicals that are structurally similar, or follow a regular pattern as a result of structural similarity, should exhibit similar physico-chemical, toxicological and eco-toxicological properties.12 Once similar chemicals have been grouped together (at the stage of grouping), endpoint information (e.g. toxicity) for one, or more, chemical(s) (the so-called “source chemical(s)”) can be used to make predictions of the same endpoint for another chemical (the “target chemical”).12,13 Read-across can be carried out in one of the four schemes: one-to-one, one-to-many, many-to-one and many-to-many. In the first two cases, the use of the endpoint value for source substance as the estimated value of the target substance is the only possible way to make the prediction. However, when sufficient data allow the endpoint values from two or more source chemicals can be used to predict the same endpoint for target substance by averaging or taking the most conservative value among the source chemicals within the whole category of similar substances.12,13
Some studies have investigated the possibility of grouping and read-across predictions for nanomaterials based on methods of similarity analysis. In the work of Xia et al.38 grouping of nanomaterials was carried out using principal components analysis (PCA). Recently Gajewicz et al.39 have employed a two-dimensional hierarchical cluster analysis to identify groups of nanoparticles based on similarity in their structural features and then use the activity data for such defined groups (i.e. source chemical(s)) to assess the biological activity for empirically untested nanomaterials (i.e. target chemical(s)). However, the mentioned methods provide only the qualitative information and may be used exclusively to obtain a ‘yes/no’ answer for the presence (or absence) of the same property/activity for one or more target chemical(s).
Following the OECD official guidance documents13 and other10,40 the prediction with quantitative read-across can be conducted with one of four main concepts such as:
• Reading across from the endpoint value of a similar chemical (e.g., the closest source chemical);
• Applying a mathematical scale to the trend in available experimental results from two or more chemicals similar to the target chemical (e.g., trend analysis or structure–activity relationships);
• Processing the endpoint values from two or more source chemicals (e.g., by averaging, by taking the most representative value), o.
• When sufficient data allow, taking the most conservative value among the source chemicals within the whole category.
Unfortunately, despite a broad consultations at international level up to the date the existing international principles and guidelines on read-across do not provide clear recommendations on how to apply these concepts, and the existing methods of read-across have not been sufficiently standardized yet.41 Consequently, as evidenced by the results of a round-robin exercise on read-across42 very often the results of estimations with read-across are “expert-dependent” i.e. they may vary dependently on personal experience of expert conducting the study. Moreover, the level of uncertainty in predictions as well as the reproducibility of the results from the read-across evaluation depends on the approach selected. Thus, in order to reduce the differences in expert judgment results, there is clearly a need to proceed towards a new concept, namely the development of quantitative read-across that will be widely accepted by regulatory bodies as a “golden standard”.
This paper describes development and application of novel, effective algorithms for filling data gaps in quantitative manner. We rationalize that this approach provides reliable predictions of the missing data without the need for additional experiments.
Novel approach: Nano-QRA algorithm
Within a group/category, the compounds are often related to each other by a trend in the empirical data for a given endpoint. Thus, their property changes in a predictable manner. When a consistent trend (i.e. increasing, or decreasing pattern in the changing potency of the properties of member(s) across the group) is observed, the missing values can be estimated by simple scaling from the empirical data for a given endpoint to fill in the data gaps. The data gaps can be filled in a number of ways, including interpolation and extrapolation from one or more other group members.13 Interpolation refers to the estimation of the activity/property of a chemical using empirically measured values from two chemicals on “both sides” of that chemical within a defined group. When the estimated activity/property is bracketed on only one side by empirical data (i.e. beyond the range of the measurements), then read-across through extrapolation may be used to fill data gaps. However, it needs to be emphasized that extrapolation will become increasingly uncertain and potentially more unreliable the further from the source chemical the target is. Therefore there is a preference for the use of interpolation rather than extrapolation.13It should also be noted that for both interpolation and extrapolation approaches, there will be an increase in uncertainty if a trend (linear or otherwise) in the empirical data for a given endpoint is poorly defined or missing. Since the quality of the relationship influences the uncertainty, it is intuitive that interpolation/extrapolation based on a large number of data, leads to lesser uncertainty than interpolation/extrapolation based on few data points adopted from different sources. In addition, since the relationship depends on the quality of the independent variable (i.e. descriptor), thus a key aspect of the approach presented here is to ensure that the structural property is – in some way – related to the activity being modeled.
Ideally, the independent variable(s) should be selected arbitrarily, based on the well-known mechanism of action. However, very often one does not know which chemical properties or structural features are associated with biological activity. In such a case, the selection of descriptor(s) is performed on a statistical, rather than mechanistic, basis. Regardless the method used to select the descriptor(s), it is important to provide a mechanistic interpretation of a given model and to ensure that some consideration is given to the possibility of a mechanistic association between the descriptor(s) and the endpoint being modeled.
Since the arrangement of the species in a matrix of selected descriptor(s) and endpoint should ideally reflect any trend within the group of similar compounds, the considered chemicals should be arranged in a suitable order, i.e. according to increasing or decreasing value of selected independent variable. Thus, by plotting the selected descriptor with empirical data, any trends can be easily verified. If the trend has been found to be statistically sound and the investigated property of compounds display a notable trend, then the missing values can be estimated by simple scaling the available experimental data from one source compound (i.e. extrapolation) or two and more source compounds (i.e. interpolation) to the target compound.
In the simplest case, when only one descriptor is selected, we have proposed to use the two-point slope formula (Fig. 1A) to perform interpolation and find intermediate point(s) in the data.
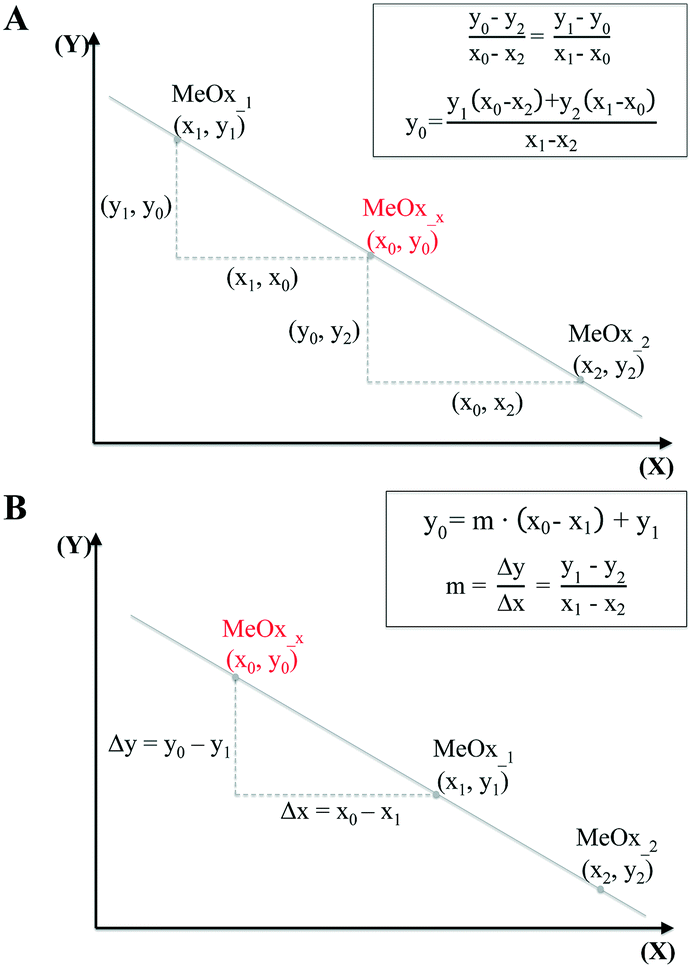 | ||
| Fig. 1 Predicting missing data: interpolation using one-point-slope formula (A), and extrapolation using two-point formula (B). | ||
The equation that goes through the two given points (MeOx_1, MeOx_2) represented as (x1, y1) and (x2, y2) respectively, was computed according to eqn (1):
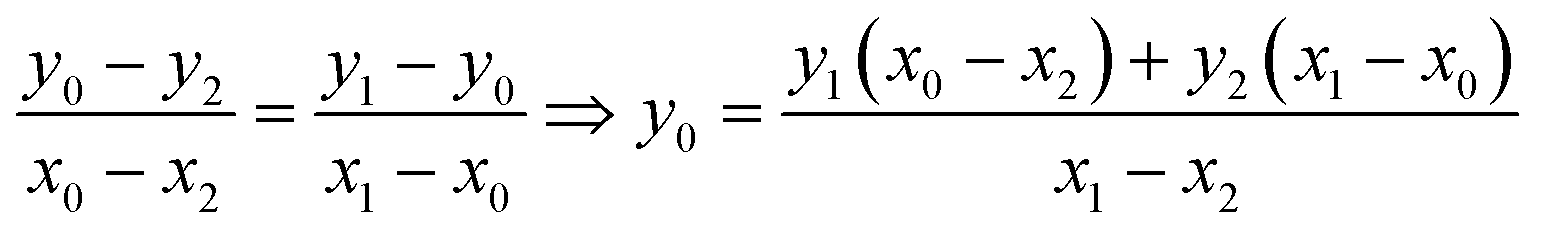 | (1) |
To extrapolate the predicted value for chemicals beyond the range of the measurements, we employed a one-point-slope formula (Fig. 1B). Thus, in order to estimate the missing value of a given endpoint (y0) for MeOx_x from source chemical MeOx_1 represented as (x1, y1) we applied eqn (2):
y0 = m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ·(x0 − x1) + y1 ·(x0 − x1) + y1 | (2) |
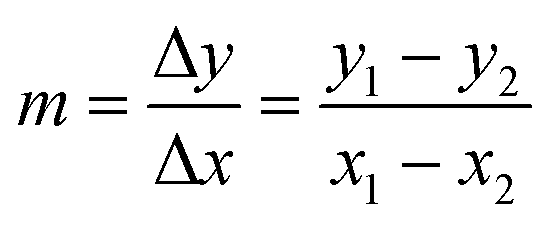 | (3) |
It should be noted, however, that in some cases the trend in the empirical data will be difficult to establish based only on one descriptor. Therefore, if the studied activity/property is expected to follow a trend based on two relevant structural characteristics (i.e. descriptors) then the use of the equation of a plane passing through three points would be required. To determine the equation of a plane in three-dimensional space, three points represented as (x1, y1, z1), (x2, y2, z2) and (x3, y3, z3) are required. To compute the third-order determinants (i.e. 3 × 3 matrix) we used Sarrus's rule, presented graphically in Fig. 2. By assuming that the resolved equation of a plane in three-dimensional space has the form as shown in eqn (4) and, if the chemicals belong to the plane, it is possible to interpolate the missing value of a given endpoint (y0) for desired point (MeOx_x):
| b0 + b1xMeOxx + b2yMeOxx + b3zMeOxx = 0 | (4) |
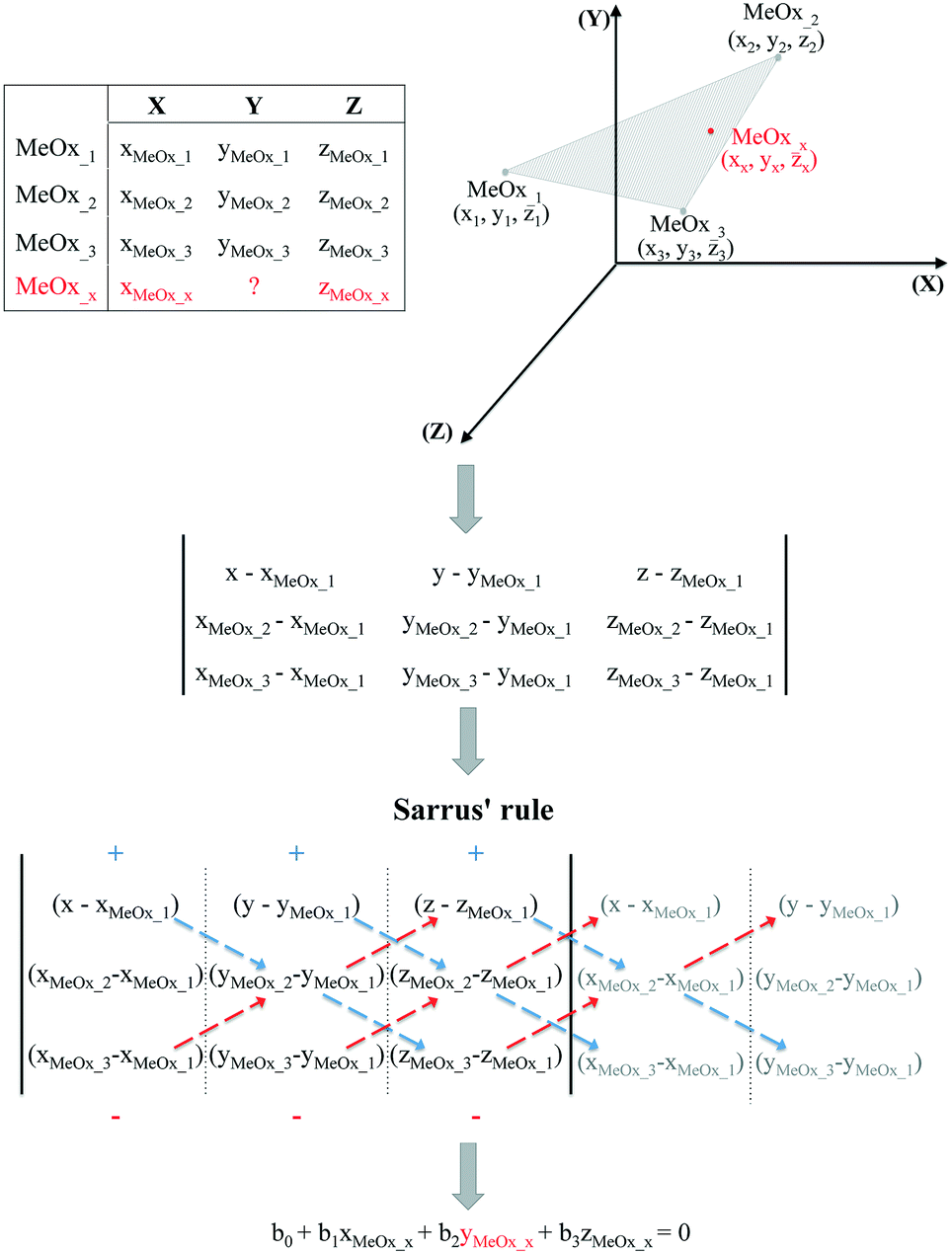 | ||
| Fig. 2 Workflow of estimating missing data using the equation of a plane passing through three points. | ||
Even if a point does not lie directly on the plane, the proposed approach may also be acceptable in certain cases for extrapolation.
Methods
The methodology applied in this study involved the following steps: (i) exploring the multidimensional space of molecular descriptors in order to select the independent variable(s) that may reflect the endpoint of interest; (ii) identifying a trend in the experimental data for a given endpoint across chemicals; (iii) undertaking read-across analysis, i.e. predicting a particular activity for an unknown chemical(s) using interpolation and extrapolation approaches: one-point-slope, two-point formula, or the equation of a plane passing through three points.Experimental data
As metal oxide nanoparticles (MeOx NPs) become more commonly used in everyday products, they were selected as a group case study. Data on cytotoxicity to bacteria Escherichia coli, for 17 metal oxides nanoparticles, expressed in terms of the concentration of metal oxide nanoparticles that reduces bacteria viability of 50% (EC50), were taken from our previous study.23 These toxicity data were supplemented by additional information on the toxicity of 18 metal oxide nanoparticles to the human keratinocyte (HaCaT) cell line, expressed in terms of the concentration of metal oxide nanoparticles that caused a 50% reduction of the cells after 24 h exposure (LC50).33To ensure that the outcome of the current study is directly comparable with the results obtained from the Nano-QSAR modeling,23,33 we employed the same method of data splitting into training and validation sets. The training sets (i.e. training source compounds) were later used to identify the trends in the experimental data for a given endpoint across chemicals and to predict a particular activity for compounds from validation and prediction sets, whereas the external validation sets (i.e. validation source compounds) were applied to evaluate the predictive ability of Nano-QRA models. Finally, the proposed here predictive tools were applied to predict the toxicity towards bacteria E. coli and human keratinocyte (HaCaT) cell line for untested metal oxide nanoparticles (i.e. target compounds) from the prediction sets.
Descriptors
The parameters quantitatively describing variability of the nanoparticles' structure (i.e. structural descriptors) were taken from our previous studies.23,33 The descriptors have been auto-scaled, which means that the average value was subtracted from the descriptors and the resultant values divided by the standard deviation to ensure the same scale and range of all variables (eqn (5)):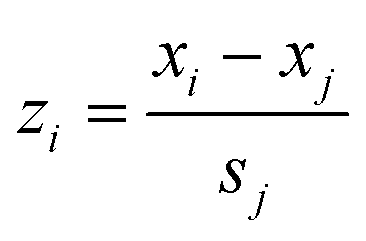 | (5) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) j – is the mean value of a given variable for all studied compounds; sj – is the standard deviation of a given variable for all studied compounds.
j – is the mean value of a given variable for all studied compounds; sj – is the standard deviation of a given variable for all studied compounds.
Statistical analysis and computational modeling methods
To verify how well the read-across algorithm based on the equations of lines and planes accounts for the variance of the response in the training set, the determination coefficient (R2) was calculated according to the formula (eqn (6)):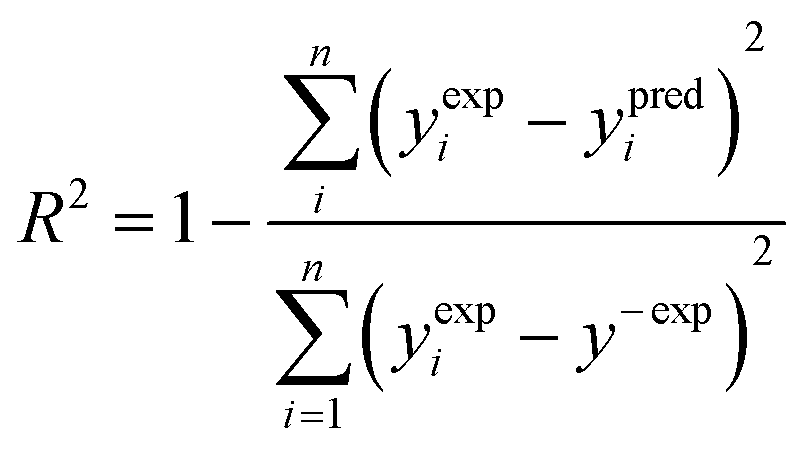 | (6) |
The predictive power (i.e. prognostic ability) of the proposed approach was additionally confirmed by employing the external validation coefficient (QF22) defined as (eqn (7)):
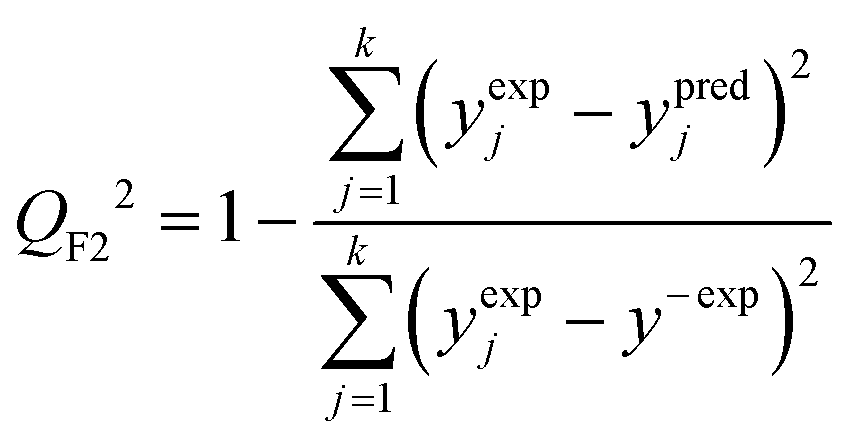 | (7) |
Finally, in order to assess the uncertainty of the read-across predictions, the error propagation analysis was performed. For quantifying the uncertainty of prediction the following metrics have been employed:43
(1) The average absolute percentage error (AAPE) (eqn (8)):
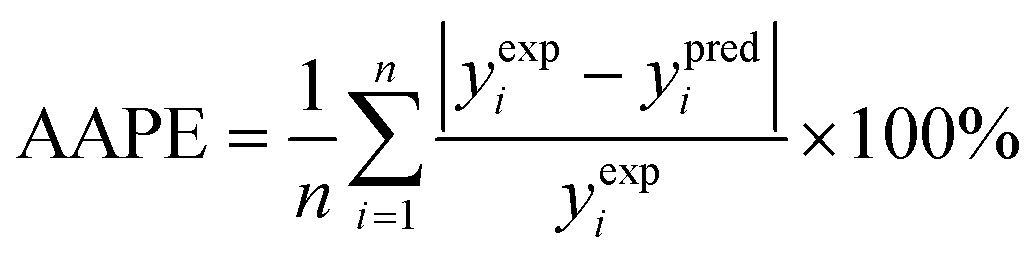 | (8) |
(2) The average absolute error (AAE) (eqn (9)):
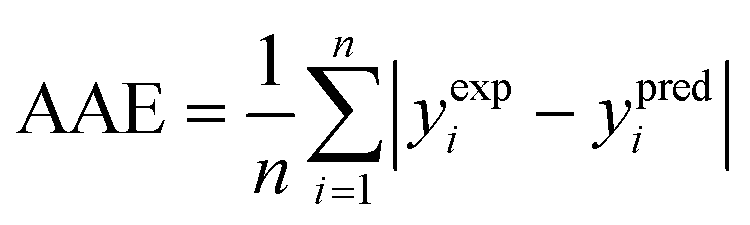 | (9) |
(3) The root mean square deviation (RMSD) (eqn (10)):
 | (10) |
In addition, we assumed that the predicted toxicity of metal oxide nanoparticles to bacteria E. coli and the human keratinocyte (HaCaT) cell line obtained through the Nano-QRA approach presented above should not be substantially different from the values obtained experimentally, as well as those predicted from the Nano-QSAR models. To verify whether the hypothesis and conclusions can be extended to other activities/properties and groups of nanoparticles, a pairwise comparison was performed. To this end, we employed a pairwise t-Student's test to verify whether the average residuals from the predictions from Nano-QRA technique differed significantly from the experimental values, as well as those predicted from the Nano-QSAR modeling.
Results
Case study 1: one dimensional read-across
Our previous study on the cytotoxicity of 17 metal oxides nanoparticles to E. coli23 utilized a single descriptor; consequently the same descriptor was used in the current study to perform read-across. The selected descriptor i.e. the enthalpy of formation of a gaseous cation with the same oxidation state as that in the metal oxide structure (ΔHMe+) refers to the detachment of metal cations from the surface of MeOx NPs. ΔHMe+ was well correlated to toxicity (r = −0.92). This indicates that the descriptor, describing the ionization enthalpy of the (detached) metal atoms, explains approximately 85% of the variability of the cytotoxicity the metal oxide nanoparticles.23To determine the trend in the empirical data, all training source compounds were sorted according to the decreasing value of the standardized descriptor (ΔHMe+). Subsequently, the distributions of independent and dependent variables for the training set were plotted to investigate any trends in the data (Fig. 3).
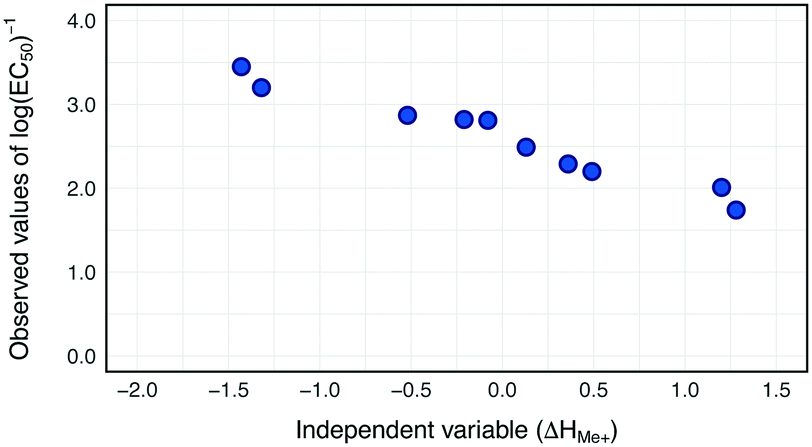 | ||
| Fig. 3 Two-dimensional trend analysis performed for 10 metal oxide nanoparticles from training set. | ||
Since all training source compounds are well correlated, as indicated by the strength of the correlation coefficient between dependent and independent variables (r > |0.7|),44 we assumed that the trend is statistically significant. Before starting the interpolation and extrapolation of missing E. coli cytotoxicity data using the two-point formula and one-point-slope respectively, all validation source compounds and target compounds (i.e. from prediction set) were incorporated to the training set. To achieve this, the theoretical values of the selected descriptor for compounds from the validation and prediction sets were rescaled using the mean and standard deviation values from the training set. Then, to ensure that all validation source compounds and target compounds (i.e. experimentally untested NPs) were arranged in a suitable order, all nanoparticles were once again sorted along with the decreasing values of rescaled ΔHMe+. Finally by using eqn (1) (for interpolation) and eqn (2) (for extrapolation) we estimated missing values of cytotoxicity data for training and validation source compounds. The predicted results together with the calculated residuals are presented in Table 2.
| MeOx | Calculated ΔHMe+ [kcal mol−1] | Rescaled ΔHMe+ [kcal mol−1] | Experimental values of log(EC50)−1 [molar] | Set | Predicted values of log(EC50)−1 [molar] | Residuals | |
|---|---|---|---|---|---|---|---|
| TiO2 | 1575.73 | 1.28 | 1.74 | T | Extrapolation | 1.71 | 0.03 |
| SnO2 | 1717.32 | 1.20 | 2.01 | T | Interpolation | 1.79 | 0.22 |
| ZrO2 | 1357.66 | 0.92 | 2.15 | V | Interpolation | 2.08 | 0.07 |
| SiO2 | 1686.38 | 0.49 | 2.20 | T | Interpolation | 2.25 | −0.05 |
| Ga2O3 | 1384.15 | 0.43 | N/A | P | Interpolation | 2.24 | — |
| Fe2O3 | 1408.29 | 0.36 | 2.29 | T | Interpolation | 2.30 | −0.01 |
| Tl2O3 | 1341.37 | 0.32 | N/A | P | Interpolation | 2.32 | — |
| Au2O3 | 1302.95 | 0.22 | N/A | P | Interpolation | 2.41 | — |
| Al2O3 | 1187.83 | 0.13 | 2.49 | T | Interpolation | 2.56 | −0.07 |
| Cr2O3 | 1268.70 | 0.13 | 2.51 | V | Interpolation | 2.49 | 0.02 |
| Sb2O3 | 1233.06 | 0.04 | 2.64 | V | Interpolation | 2.63 | 0.01 |
| In2O3 | 1271.13 | −0.08 | 2.81 | T | Interpolation | 2.69 | 0.12 |
| Bi2O3 | 1137.40 | −0.21 | 2.82 | T | Interpolation | 2.83 | −0.01 |
| La2O3 | 1017.22 | −0.31 | 2.87 | V | Interpolation | 2.84 | 0.03 |
| Yb2O3 | 1039.03 | −0.46 | N/A | P | Interpolation | 2.86 | — |
| Er2O3 | 1016.15 | −0.52 | N/A | P | Interpolation | 2.87 | — |
| Mn2O3 | 1017.99 | −0.52 | N/A | P | Interpolation | 2.87 | — |
| Y2O3 | 837.15 | −0.52 | 2.87 | T | Interpolation | 2.93 | −0.06 |
| Ho2O3 | 1009.60 | −0.54 | N/A | P | Interpolation | 2.88 | — |
| Eu2O3 | 1006.60 | −0.55 | N/A | P | Interpolation | 2.88 | — |
| Tb2O3 | 999.00 | −0.57 | N/A | P | Interpolation | 2.89 | — |
| Gd2O3 | 991.37 | −0.59 | N/A | P | Interpolation | 2.90 | — |
| Sm2O3 | 974.40 | −0.63 | N/A | P | Interpolation | 2.92 | — |
| Nd2O3 | 962.80 | −0.66 | N/A | P | Interpolation | 2.93 | — |
| Ag2O3 | 831.56 | −0.97 | N/A | P | Interpolation | 3.06 | — |
| V2O3 | 1097.73 | −0.99 | 3.14 | V | Interpolation | 3.06 | 0.08 |
| FeO | 748.98 | −1.21 | N/A | P | Interpolation | 3.15 | — |
| AuO | 712.50 | −1.30 | N/A | P | Interpolation | 3.19 | — |
| CuO | 706.25 | −1.32 | 3.20 | T | Interpolation | 3.38 | −0.18 |
| ZnO | 662.44 | −1.43 | 3.45 | T | Extrapolation | 3.22 | 0.23 |
| NiO | 596.70 | −1.59 | 3.45 | V | Extrapolation | 3.81 | −0.36 |
| CoO | 601.80 | −1.60 | 3.51 | V | Extrapolation | 3.84 | −0.33 |
| MnO | 548.13 | −1.73 | N/A | P | Extrapolation | 4.13 | — |
| MgO | 543.10 | −1.74 | N/A | P | Extrapolation | 4.15 | — |
| PbO | 499.19 | −1.85 | N/A | P | Extrapolation | 4.40 | — |
The quantitative assessment of the uncertainty of the Nano-QRA model was expressed by the: AAPET = 3.71%; AAET = 0.10; RMSDT = 0.13 in training set and AAPEV = 3.96%; AAEV = 0.13; RMSDV = 0.19 in validation set, respectively. All metrics for evaluating performance and uncertainty of Nano-QRA model were low (<5%). Additionally, the determination coefficient in the training set (R2 = 0.94) as well as the external validation coefficient (QF22 = 0.83) in the validation set were high and close to 1. Consequently, we can conclude that the developed model is well-fitted and has satisfactory predictive capabilities.
To compare log(EC50)−1 values calculated with the Nano-QRA approach with experimental ones (graphically presented in Fig. 4A)), as well as with those obtained with Nano-QSAR model, we have performed a pairwise t-Student's test for each pair in the data sets. Differences were not considered to be statistically significant (p > 0.001) (Table 3). The observed differences between the experimentally measured and values predicted from both modelling methods (i.e. Nano-QRA and Nano-QSAR) were comparable and did not exceed 0.40 of a log unit, which was consistent with our assumption. Additionally, the results of a comparison between the statistical quality of Nano-QSAR model (R2 = 0.85, RMSDT = 0.20, QP2 = 0.83, RMSDV = 0.19)23 and Nano-QRA model (R2 = 0.94, RMSDT = 0.13, QF22 = 0.83, RMSDV = 0.19) indicate that both models have the same and very high predictive capabilities. Furthermore the Nano-QRA model has a slightly better goodness of fit (>R2). In addition, in both cases the difference between R2 and Q2 value is small (<0.3) indicating stability of the models.
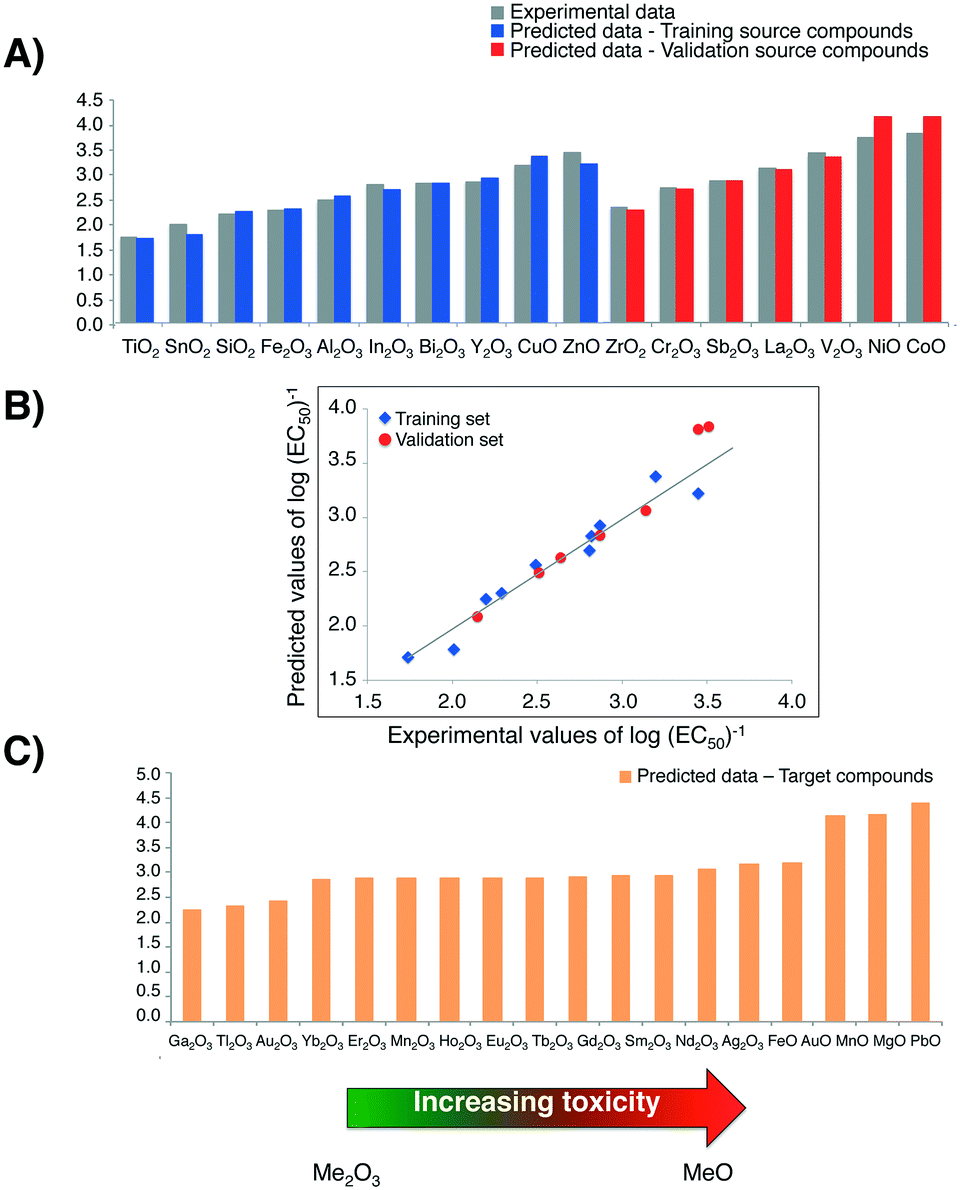 | ||
| Fig. 4 A) Predicted values of cytotoxicity (in log units) to the bacteria E. coli for 17 metal oxide nanoparticles (i.e. training/validation source compounds); B) experimentally determined (observed) versus predicted log values of (EC50)−1. The straight line represents perfect agreement between experimental and calculated values; C) predicted values of cytotoxicity to the bacteria E. coli for 18 MeOx NPs, for which the experimental data have been unavailable target compounds. | ||
| Statistics | Nano-QRA | |
|---|---|---|
| vs. | ||
| Experiment | Nano-QSAR | |
| Case study 1: one dimensional read-across | ||
| t-Test statistic | 0.396 | 0.669 |
| t-Test critical value (α 0.001) | 4.015 | 4.015 |
| p-Value | 0.694 | 0.513 |
| Case study 2: two dimensional read-across | ||
| t-Test statistic | 0.186 | 0.201 |
| t-Test critical value (α 0.001) | 3.965 | 3.965 |
| p-Value | 0.852 | 0.843 |
Furthermore, the strong linear correlation between experimental data describing the cytotoxicity of MeOx NPs to bacteria E. coli and the values predicted with Nano-QRA algorithm (Fig. 4B)), additionally confirms validation results.
Finally, after detailed validation with external set of compounds (i.e. validation source compounds), in the next step we applied the eqn (1) (for interpolation) and eqn (2) (for extrapolation) to estimate the values of cytotoxicity to the bacteria E. coli for 18 target compounds (i.e. MeOx NPs, for which the experimental data have been unavailable). The obtained results, presented graphically in Fig. 4C), show a very good agreement with those predicted with Nano-QSAR model. They also correspond to the mechanism of cytotoxicity previously discussed and described in detail by Puzyn et al.23
Case study 2: two dimensional read-across
The results presented in the previous section proved that the predictions of cytotoxicity reported for a series of metal oxide nanoparticles are characterized by acceptable levels of uncertainty. However, we must not forget that the read-across predictions were made from only one theoretical structural feature (i.e. descriptor) and sometimes a single parameter may be not enough to cope with the complexity of NPs. Thus, the next logical step for the development of read-across methodologies should be an attempt to extend the proposed approach for predicting of various properties (physico-chemical as well as toxicological) based on two independent variables, which individually may have a lower correlation with activity, but in combination provide the mechanistic interpretation of the toxicity source. Therefore, in the current study, we have illustrated an efficient Nano-QRA approach predicting the toxicity of metal oxide nanoparticles to a human keratinocyte cell line (HaCaT) based on two descriptors that were recently employed in a Nano-QSAR model.33 These descriptors, derived from quantum-mechanical calculations, i.e. the enthalpy of formation of metal oxide nanocluster representing a fragment of the surface (ΔHcf) and Mulliken's electronegativity of the cluster (χc) are connected closely with the modeled activity and have been demonstrated to be associated with the toxicity of the metal oxide nanoparticles to the HaCaT cell line. These descriptors refer to the two types of processes related to (or properties of) the NPs: the first process concerns the release of metal cations from the surface of nanoparticles, whereas the second one is related to the surface redox activity of nanoparticles. Consequently, both processes lead to the formation of highly reactive hydroxyl radicals, which are mainly responsible for the induction of oxidative stress in the cells and thus can be considered as a pre-cursor to toxicity.33The selected descriptors (i.e. ΔHcf and χc) have the values of Pearson correlation coefficient (r) with toxicity of 0.48 and 0.81 respectively, as well as a very low correlation coefficient with each other (r = −0.05). In order to visualize the relationship between the three variables (i.e. both descriptors (ΔHcf and χc) and the endpoint (log(LC50)−1) and to reveal the distribution trend in the training data set in the most efficient way, we have created a 3D scatterplot (Fig. 5). Moreover, an additional data dimension represented by a gradual colour change was used to highlight the changing potency of toxicity among the training set compounds. Colours represent the logarithmic values of the toxicity to human keratinocyte cell line (HaCaT) measured for metal oxide nanoparticles: dark blue means the lowest value of the endpoint, whereas dark red – the highest cytotoxicity. One can notice that the trend in the experimental data across the chemicals from the training set has been confirmed.
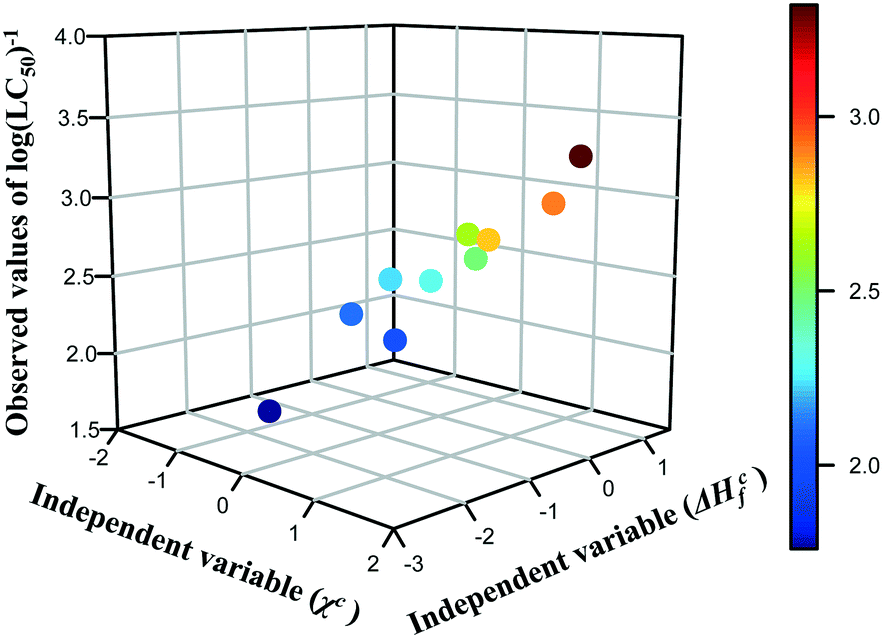 | ||
| Fig. 5 Three-dimensional trend analysis performed for 10 metal oxide nanoparticles from training set. | ||
Since a consistent trend in the properties within the chemicals in the training set was observed, we assumed that data gaps can be filled by interpolation and extrapolation to other group members, using the equation of a plane passing through three points. To achieve this, the descriptors for nanoparticles from the validation set (i.e. validation source compounds) and prediction set (i.e. target compounds) were rescaled using the mean and standard deviation values from the training set. Finally, all compounds were sorted by decreasing values of rescaled χc, since this descriptor has higher correlation coefficient with the endpoint. As a result, we estimated the missing values of toxicity for 18 MeOx NPs (i.e. training source compounds and validation source compounds) to the HaCaT cell line, utilizing the workflow presented in Fig. 2. The results we obtained are summarized in Table 4.
| MeOx | Calculated values of χC [eV] | Calculated values of ΔHcf [eV] | Rescaled values of χC [eV] | Rescaled values of ΔHcf [eV] | Experimental values of log(LC50)−1 [molar] | Set | Predicted values of log(LC50)−1 [molar] | Residuals |
|---|---|---|---|---|---|---|---|---|
| ZnO | 8.33 | −449.4 | 1.80 | 0.03 | 3.32 | T | 3.19 | 0.13 |
| CoO | 7.44 | −786.8 | 1.25 | −0.73 | 2.83 | T | 3.03 | −0.20 |
| In2O3 | 6.78 | −52.1 | 0.84 | 0.92 | 2.92 | T | 3.03 | −0.11 |
| WO3 | 6.73 | −715.4 | 0.81 | −0.57 | 2.56 | V | 2.67 | −0.11 |
| La2O3 | 6.45 | −157.7 | 0.64 | 0.68 | 2.87 | V | 2.76 | 0.11 |
| PbO2 | 6.13 | −269.5 | 0.44 | 0.43 | N/A | P | 2.67 | — |
| Gd2O3 | 5.91 | −234.1 | 0.30 | 0.51 | N/A | P | 2.62 | — |
| FeO | 5.88 | −883.2 | 0.28 | −0.94 | N/A | P | 2.38 | — |
| Bi2O3 | 5.34 | −148.5 | −0.05 | 0.70 | 2.50 | T | 2.68 | −0.18 |
| PbO | 5.12 | −306.3 | −0.19 | 0.35 | N/A | P | 1.99 | — |
| Mn2O3 | 5.00 | −96.3 | −0.26 | 0.82 | 2.64 | T | 2.63 | 0.01 |
| ZrO2 | 4.95 | −638.1 | −0.30 | −0.39 | 2.02 | T | 2.31 | −0.29 |
| TiO2 | 4.91 | −1492.0 | −0.32 | −2.31 | 1.76 | T | 1.07 | 0.70 |
| SnO2 | 4.57 | −266.6 | −0.53 | 0.44 | 2.67 | V | 2.26 | 0.41 |
| NiO | 4.47 | 68.0 | −0.59 | 1.19 | 2.49 | V | 2.39 | 0.10 |
| Sb2O3 | 4.46 | −206.7 | −0.60 | 0.57 | 2.31 | T | 2.19 | 0.12 |
| Cr2O3 | 4.36 | −235.3 | −0.66 | 0.51 | 2.30 | V | 2.30 | 0.00 |
| CuO | 4.25 | −76.3 | −0.73 | 0.87 | N/A | P | 2.37 | — |
| Fe2O3 | 4.21 | −378.5 | −0.76 | 0.19 | 2.05 | V | 2.23 | −0.18 |
| SiO2 | 3.81 | −618.3 | −1.00 | −0.35 | 2.12 | T | 2.07 | 0.05 |
| Al2O3 | 3.44 | −600.0 | −1.23 | −0.31 | 1.85 | V | 2.10 | −0.25 |
| Y2O3 | 3.35 | −135.3 | −1.29 | 0.73 | 2.21 | V | 2.25 | −0.04 |
| V2O3 | 3.24 | −139.5 | −1.36 | 0.72 | 2.24 | T | 2.32 | −0.08 |
Analogously to the first case study, the uncertainty of the result of read-across predictions was evaluated by the: AAPET = 8.71%; AAET = 0.19; RMSDT = 0.26 in training set and AAPEV = 6.48%; AAEV = 0.15; RMSDV = 0.19 in validation set, respectively. Also in this case, low value of all metrics for evaluating the uncertainty of Nano-QRA model and simultaneously high value of the determination coefficient in the training set, and the external validation coefficient in the validation set indicated model's goodness-of-fit and it good predictive ability.
By the means of the pairwise t-Student's test we confirmed that the values of log(LC50)−1 obtained from Nano-QRA approach did not differ significantly from those measured experimentally (p = 0.852), as well as those predicted from the Nano-QSAR model (p = 0.843) (Table 3). In addition, we compared the differences in values calculated between the experimentally measured toxicity and that predicted with Nano-QRA (Fig. 6A)). However, it should be noted that one metal oxide, namely TiO2, is characterized by higher residual compared to the rest of the other training/validation source compounds, which might be due to a fact that it has the lowest toxicity to human keratinocyte (HaCaT) cell line whilst having the highest value of the rescaled value of second descriptor (ΔHcf). Additionally, we compared the statistical quality of Nano-QSAR model (R2 = 0.93, RMSDT = 0.12, QP2 = 0.83, RMSDV = 0.13)33 and Nano-QRA model (R2 = 0.65, RMSDT = 0.26, QF22 = 0.62, RMSDV = 0.19). The obtained results indicate that Nano-QSAR model yields a better statistical fit and predictive capability than the Nano-QRA model. However, after removing TiO2, which shows the lowest toxicity to human keratinocyte (HaCaT) cell line the statistical quality of Nano-QRA model (R2 = 0.86, RMSDT = 0.14) is significantly increasing and is being comparable to the statistical quality of Nano-QSAR model.
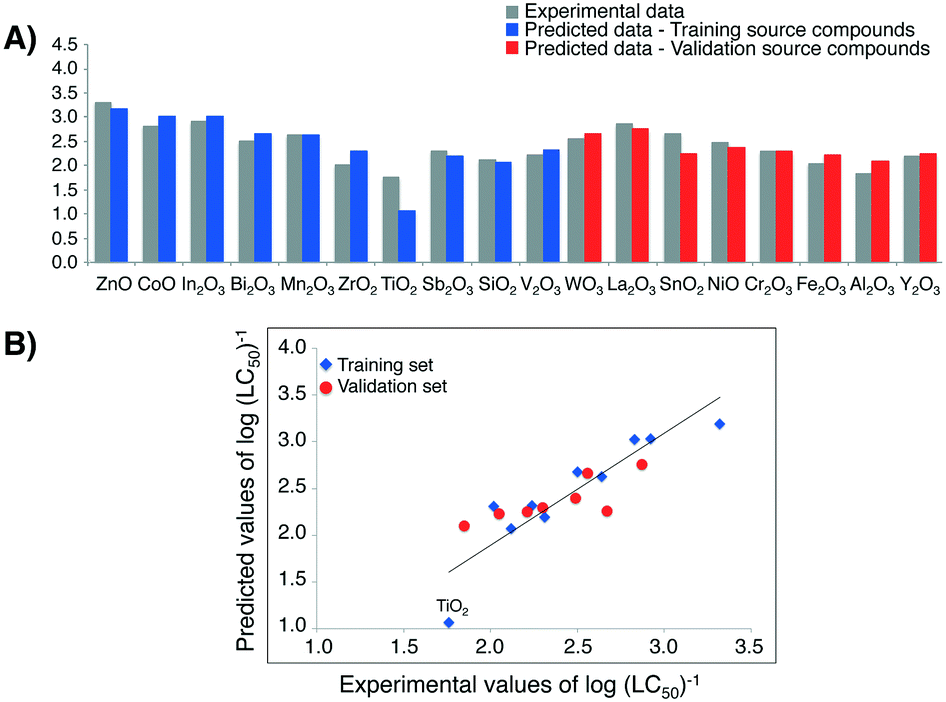 | ||
| Fig. 6 A) Predicted values of cytotoxicity (in log units) to HaCaT cell line for 18 MeOx NPs (i.e. training/validation source compounds); B) experimentally determined (observed) versus predicted values of log(LC50)−1. The straight line represents perfect agreement between experimental and calculated values. | ||
High correlation between the observed and estimated values of log(LC50)−1 for MeOx NPs to HaCaT cell line in case of both training source compounds and validation source compounds (Fig. 6B)) provides another proof of the model's quality (i.e. very good fit and the high predictive ability).
Finally by utilizing the workflow presented in Fig. 2, we estimated the missing values of toxicity towards HaCaT cell line for five experimentally untested metal oxides (i.e. target compounds) (Table 4).
Nano-QRA versus the existing Nano-QSAR models
It was also interesting to compare performance of the presented Nano-QRA models with previously reported Nano-QSAR studies related to the same endpoints. Whenever one wants to compare the performance of in silico models each other, one usually starts from evaluating their statistical characteristics. Without doubt, in case study 1 the measures of goodness-of-fit and predictivity (Table 5) favour the Nano-QRA approach. Higher correlation coefficients and lower values of the root mean square errors for both the training and the validation sets proved that Nano-QRA algorithm can be used as more efficient tool for filling data gaps in quantitative manner then the previously developed Nano-QSAR models. Nano-QRA provides more reliable predictions of the missing data.| Ref. | Training set | Validation set | ||||
|---|---|---|---|---|---|---|
| R 2 | RMSE | n | R 2 | RMSE | n | |
| Case study 1 | ||||||
| Nano-QRA | 0.94 | 0.13 | 10 | 0.83 | 0.19 | 7 |
| Puzyn et al.23 | 0.85 | 0.20 | 10 | 0.83 | 0.19 | 7 |
| Toropov et al.30 | 0.74–0.84 | 0.17–0.26 | 11 | 0.83–0.96 | 0.14–0.34 | 6 |
| Kar et al.28 | 0.82 | 0.23 | 11 | 0.78 | 0.22 | 6 |
| Sizochenko et al.32 | 0.93 | 0.13 | 13 | 0.78 | 0.32 | 3 |
| Pan et al.34 | 0.89 | 0.18 | 10 | 0.82 | 0.26 | 7 |
| Singh et al.45 | 0.91 | 0.20 | 14 | 0.86 | 0.29 | 3 |
| Case study 2 | ||||||
| Nano-QRA (after removing TiO 2 ) | 0.86 | 0.14 | 9 | 0.82 | 0.14 | 8 |
| Gajewicz et al. | 0.93 | 0.12 | 10 | 0.83 | 0.13 | 8 |
| Sizochenko et al.32 | 0.96 | 0.10 | 14 | 0.92 | 0.12 | 3 |
| Pan et al.34 | 0.96 | 0.08 | 13 | 0.83 | 0.25 | 5 |
Moreover, considering the statistical parameters (Table 5) one can observe that the presented Nano-QRA model is comparable to the previous Nano-QSAR models.
Nano-QRA versus the existing concepts of read-across
As mentioned above, according to the OECD official guidance documents13 several concepts for quantitative read-across may be applied. When the experimental enpoint has been measured for at least two compounds, the endpoint values for those two or more co-called “source chemicals” can be used to predict the endpoint value for untested “target substance(s)”. This can be done either by averaging or taking the most conservative value among the source chemicals. Another option is to use mode or median value calculated for the group of source compounds.In order to make a detailed comparison between the proposed Nano-QRA approach and commonly used read-across concepts, we estimated log(EC50)−1 of 7 validation source compounds to bacteria E. coli (case study 1) and log(LC50)−1 of 8 validation source compounds to HaCaT cell line (case study 2), respectively. For this purpose, we used following types of approximation: (i) average, (ii) most conservative, (iii) median, (iv) lower-median, and (v) higher-median value among the source chemicals (i.e. training source compounds). Subsequently, we calculated the external validation coefficient (QP2) as a measure of the predictive power, as well as: the average absolute percentage error (AAPE), the average absolute error (AAE) and the root mean square deviation (RMSD) as the measures of reliability of predictions (Table 6).
| Statistics | Nano-QRA | Read-across using following types of approximation | ||||
|---|---|---|---|---|---|---|
| Average | Most conservative | Median | Lower median | Higher median | ||
| Case study 1: one dimensional read-across | ||||||
| Q F2 2 | 0.83 | −0.43 | −1.42 | −0.28 | −0.76 | −0.03 |
| AAPEV | 3.96% | 14.85% | 22.91% | 14.31% | 16.16% | 14.31% |
| AAEV | 0.13 | 0.45 | 0.57 | 0.43 | 0.50 | 0.41 |
| RMSDV | 0.19 | 0.56 | 0.72 | 0.53 | 0.62 | 0.47 |
| Case study 2: two dimensional read-across | ||||||
| Q F2 2 | 0.62 | −0.09 | −9.00 | −0.01 | −0.04 | −0.16 |
| AAPEV | 6.48% | 12.36% | 42.38% | 12.06% | 11.56% | 12.61% |
| AAEV | 0.15 | 0.27 | 0.95 | 0.27 | 0.27 | 0.28 |
| RMSDV | 0.19 | 0.33 | 1.00 | 0.32 | 0.32 | 0.34 |
The obtained results reveal that Nano-QRA approach far surpasses the other considered read-across methods with different types of approximation, in both: predictive power (≫QF22), and reliability of predictions (≪AAPEV, AAEV, RMSDV).
Discussion
One can notice that the philosophy standing behind Nano-QRA is somewhat similar to that of Nano-QSAR modelling based on the k-nearest neighbour principle (kNN-QSAR).46 Both methods rely upon the analogue approach, which implies that similar compounds display similar biological activity. In effect, the activity of an untested compound can be predicted by using the activities of k the most similar compound(s). Along with the kNN-QSAR algorithm the activity of each compound is predicted as the average activity of k most chemically similar compounds from the data set.46 However, as it was highlighted by the same authors in their previous paper, the kNN-QSAR method was designed for analyzing relatively large data sets, where a multitude of different classes of compounds is represented in the training set.47 In effect, no reliable kNN-QSAR model can be developed for the limited data set. In contrast to kNN-QSAR method, and taking a step beyond the currently used types of value approximations (e.g. average), we have introduced new Nano-QRA approach for filling data gaps in quantitative manner for cases when only limited data is available. In case of Nano-QRA, the prediction is based on the two-point formula, which overcomes the problem of kNN.Based to the presented results, we may draw the conclusion that proposed Nano-QRA approach is an simple and effective algorithm for filling data gaps in quantitative manner that provides reliable predictions of the missing data. However, the logical question that appears here is about the uncertainty as well as the acceptable level of uncertainty of the read-across prediction to fill the data gaps for a specific regulatory purpose? Over the last few years a number of initiatives have been taken to determine the areas/opportunities for making read-across more robust, reliable, less uncertain and more available to a broader array of stakeholders.48,49 Along with the most comprehensive scheme for addressing the various facets of uncertainty for read-across, the following key issues should be taken into consideration: (1) data uncertainty should be separated from the read-across prediction uncertainty, (2) the method of modeling itself should be as transparent as possible, (3) the predictive ability of a read-across should be examined with appropriate measures of goodness-of-fit and predictivity.50,51
Certainly, the degree of uncertainty and the predictive accuracy of the in silico predictions depend on the reliability of the experimental data. It is also obvious that the experimental data always have an uncertainty of their own. Thus, in order to reduce the epistemic uncertainty of experimental values as much as possible, in the presented case studies we have used only high-quality experimental data measured by a standardized experimental protocol within one laboratory at the same conditions. In addition following the current guidelines for reporting nanotoxicology research, all tested nanomaterials have been fully characterized by transmission electron microscopy (TEM). It needs to be also acknowledged that in order to demonstrate the reliability and the scientific robustness of the read-across predictions, the uncertainty of model was assessed by estimation of the average absolute percentage error (AAPE), the average absolute error (AAE) and the root mean square deviation (RMSD). By analyzing obtained results, we found that uncertainty (in both presented case studies) was relatively low. This implies that the accuracy of the read-across prediction is high and the overall encouraging performance of Nano-QRA approach assures that this algorithm could be used as an attractive and pragmatic technique to fill data gaps.
When dealing with in silico methods, one has to keep in mind that the method of modeling itself should be as transparent as possible. Therefore to ensure the transparency in the read-across algorithm for filling data gaps we have used basic equations of lines and planes (i.e. one/two-point-slope formula and equation of a plane passing through three points). As such, the read-across model's predictions may be independently reproduced by others at any time in the future regardless of the scientific expertise.
It is well recognized that, the only way to determine the true predictive power of any in silico model is its external validation. Thus, in order to measure how well the model predicts the endpoint for new compounds the external validation was carried out using an external set of compounds. Since this type of assessment requires the use of an independent set of compounds (i.e. compounds that were not previously used for the identification of a trend in the experimental data for a given endpoint across chemicals), one can be sure that they do not affect the model development. In the presented study the predictive ability of the developed read-across model was examined with the set of seven and eight MeOx NPs from the validation set, respectively. We received relevant estimation for all of validation source compounds what was finally confirmed by high value of QF22 and low value of error. The predictive power of proposed approach was additionally confirmed by employing pairwise t-Student's. It was found that the values obtained from the read-across technique do not differ significantly from those measured experimentally.
Using the pairwise t-Student's test, we have confirmed that the values obtained from the proposed read-across techniques do not differ significantly from those measured experimentally. However, the above-mentioned algorithms are limited by the possibility of using only one descriptor in case of one/two-point formula, and maximum two descriptors in case of the equation of the plane passing through three points. It needs to be emphasised, however, that in some cases a one or even two descriptors may be not enough to cope with the complexity of the expected mode of toxic action. In addition, the methods proposed within this study require the existence of a visible trend between the endpoint and descriptor(s). One should be aware that, in some cases, the linear trend in the empirical data would be difficult to observe. Thus, the next logical step is to develop new techniques employing more than two descriptors and working also when the linear trend is not present. In addition, future directions for an increasing acceptance of read-across in the hazard assessment of nanomaterials should include design of novel and suitable numerical algorithms that would be transparent, reproducible and clearly documented. The feasibility and predictive ability of newly developed read-across algorithms should be verified and validated. Therefore, it would be very practical to establish the principles for the validation of read-across approaches by means of suitable case-studies (i.e. using external data obtained from regulatory (eco)toxicity tests). Furthermore, the recommendations on existing read-across approaches, which are the most relevant for filling data gaps for nanomaterials, should be delivered. In a further perspective, the acceptable and sufficiently standardized algorithm(s) should be implemented into the user-friendly software (e.g. OECD QSAR Toolbox).
Conclusions
The present study provides novel and effective algorithms for filling data gaps in quantitative manner. The interpolation and extrapolation of data performed here allowed us to estimate a particular activity for a series of unknown metal oxide nanoparticles with acceptable levels of uncertainty. The proposed method is the first published example of the use of one-point-slope, two-point formula as well as equation of a plane passing through three points approaches to estimate the biological activity for empirically untested metal oxide nanomaterials. The case study performed with MeOx NPs serves as the proof-of-the-concept of the proposed new read-across algorithms. It could be easily adapted to any group of species. Thus, in future, the concept will be extended to other materials and substances. A great advantage of presented method is the fact that it is based on information extracted from very few known species (i.e. 10 training source compounds) and enables the prediction for groups of NPs, for which the number of experimental data is insufficient to develop appropriate Nano-QSAR model(s). It opens new opportunities for research groups and industrial labs to evaluate the potential negative impact of nanomaterials to the human health and the environment without the necessity of performing time consuming and expensive experimental studies on large set of NPs. In addition, in the light of the different regulatory frameworks and guidance published in the EU, USA, Canada, Japan, and Australia as well as in the context of current and future efforts to develop the Alternative Testing Strategies, the proposed method provides an efficient tool to support the risk assessment of nanomaterials.Author contributions
A. G. performed statistical analysis, developed and validated the Nano-QRA models. A. G., K. J., M. T. D. C., J. L., and T. P. discussed the results.Acknowledgements
This material is based on research sponsored by the Polish National Science Center (grant number UMO-2011/01/M/NZ7/01445). J. L. thanks for support of the NSF-CREST “Interdisciplinary Center for Nanotoxicity” - grant number NSF HRD # 1547754.References
- F. S. Hansen, A. Maynard, A. Baun and J. A. Tickner, Nat. Nanotechnol., 2008, 3, 444 CrossRef PubMed.
- A. G. Cattaneo, R. Gornati, E. Sabbioni, M. Chiriva-Internati, E. Cobos, M. R. Jenkins and G. Bernardini, J. Appl. Toxicol., 2010, 30, 730 CrossRef CAS PubMed.
- A. Gajewicz, B. Rasulev, T. Dinadayalane, P. Urbaszek, T. Puzyn, D. Leszczynska and J. Leszczynski, Adv. Drug Delivery Rev., 2012, 64, 1663 CrossRef CAS PubMed.
- I. Lynch, C. Weiss and E. Valsami-Jones, Nano Today, 2014, 9, 266 CrossRef CAS.
- J. Morris, J. Willis, D. De Martinis, B. Hansen, H. Laursen, J. R. Sintes, P. Kearns and M. Gonzalez, Nat. Nanotechnol., 2011, 6, 73 CrossRef CAS PubMed.
- Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. Official Journal of the European Union, L 396/1 of 30.12.2006. Office for Official Publications of the European Communities (OPOCE). 2006.
- ECHA, 2012a. Background Paper. An introduction to the assessment of read-across. Experts workshop on read-across assessment with the active support of Cefic LRI, 2–3 October 2012, European Chemicals Agency, 8 p. Available at: http://echa.europa.eu/documents/10162/5649897/ws_raa_20121003_background_paper_an_introduction_to_the_assessment_of_read-across_in_echa_en.pdf.
- ECHA, 2012b. Assessment of Read-Across in REACH, European Chemicals Agency, 58p. Available at: http://echa.europa.eu/documents/10162/5649897/ws_raa_20121003_assessment_of_read-across_in_echa_de_raat_en.pdf.
- ECHA, 2013. Grouping of Substances and Read-Across Approach. Part I. Introductory Note, European Chemicals Agency ECHA-13-R-02-EN, 11 p. Available at: http://echa.europa.eu/documents/10162/13628/read_across_introductory_note_en. pdf.
- ECHA, 2015. Read-Across Assessment Framework (RAAF). European Chemicals Agency ECHA-15-R-07-EN, 38 p. Available at: http://echa.europa.eu/documents/10162/13628/raaf_en.pdf.
- RCC-NI, Regulatory Cooperation Council – Nanotechnology Initiative. Work element 2. Development of a classification scheme for nanomaterials regulated under the new substances programs of Canada and the United States, 17 p. Available at: http://www.ec.gc.ca/scitech/default.asp?lang=En&n=6A2D63E5-1&xsl=privateArticles2,viewfull&po=08C45DB6.
- OECD, Guidance of gruping of chemicals, Organisation of Economic Cooperation and Development, Paris, France, 2007 Search PubMed.
- OECD, Guidance on grouping of chemicals, Series on testing and assessment No. 194 2nd edn, Organisation of Economic Cooperation and Development, Paris, France, 2014 Search PubMed.
- V. Stone, S. Pozzi-Mucelli, L. Tran, K. Aschberger, S. Sabella, U. Vogel, C. Poland, D. Balharry, T. Fernandes, S. Gottardo, S. Hankin, M. G. Hartl, N. Hartmann, D. Hristozov, K. Hund-Rinke, H. Johnston, A. Marcomini, O. Panzer, D. Roncato, A. T. Saber, H. Wallin and J. J. Scott-Fordsmand, Part. Fibre Toxicol., 2014, 11, 9 CrossRef PubMed.
- H. Godwin, C. Nameth, D. Avery, L. L. Bergeson, D. Bernard, E. Beryt, W. Boyes, S. Brown, A. J. Clippinger, Y. Cohen, M. Doa, C. O. Hendren, P. Holden, K. Houck, A. B. Kane, F. Klaessig, T. Kodas, R. Landsiedel, I. Lynch, T. Malloy, M. B. Miller, J. Muller, G. Oberdorster, E. J. Petersen, R. C. Pleus, P. Sayre, V. Stone, K. M. Sullivan, J. Tentschert, P. Wallis and A. E. Nel, ACS Nano, 2015, 9, 3409 CrossRef CAS PubMed.
- A. E. Nel, J. Intern. Med., 2013, 274, 561 CrossRef CAS PubMed.
- A. E. Nel, E. Nasser, H. Godwin, D. Avery, T. Bahadori, L. Bergeson, E. Beryt, J. C. Bonner, D. Boverhof, J. Carter, V. Castranova, J. R. DeShazo, S. M. Hussain, A. B. Kane, F. Klaessig, E. Kuempel, M. Lafranconi, R. Landsiedel, T. Malloy, M. B. Miller, J. Morris, K. Moss, G. Oberdorster, K. Pinkerton, R. C. Pleus, J. A. Shatkin, R. Thomas, T. Tolaymat, A. Wang and J. Wong, ACS Nano, 2013, 7, 6422 CrossRef CAS PubMed.
- J. H. E. Arts, M. Hadi, A. M. Keene, R. Kreiling, D. Lyon, M. Maier, K. Michel, T. Petry, U. G. Sauer, D. Warheit, K. Wiench and R. Landsiedel, Regul. Toxicol. Pharmacol., 2014, 70, 492 CrossRef CAS PubMed.
- J. H. E. Arts, M. Hadi, M. A. Irfan, A. M. Keene, R. Kreiling, D. Lyon, M. Maier, K. Michel, T. Petry, U. G. Sauer, D. Warheit, K. Wiench, W. Wohlleben and R. Landsiedel, Regul. Toxicol. Pharmacol., 2015, 71, S1 CrossRef CAS PubMed.
- T. Gebel, H. Foth, G. Damm, A. Freyberger, P. J. Kramer, W. Lilienblum, C. Rohl, T. Schupp, C. Weiss, K. M. Wollin and J. G. Hengstler, Arch. Toxicol., 2014, 88, 2191 CrossRef CAS PubMed.
- A. G. Oomen, P. M. J. Bos, T. F. Fernandes, K. Hund-Rinke, D. Boraschi, H. J. Byrne, K. Aschberger, S. Gottardo, F. von der Kammer, D. Kuhnel, D. Hristozov, A. Marcomini, L. Migliore, J. Scott-Fordsmand, P. Wick and R. Landsiedel, Nanotoxicology, 2014, 8, 334 CrossRef PubMed.
- D. Fourches, D. Pu, C. Tassa, R. Weissleder, S. Y. Shaw, R. J. Mumper and A. Tropsha, ACS Nano, 2010, 4, 5703 CrossRef CAS PubMed.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H. M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175 CrossRef CAS PubMed.
- D. Fourches, D. Pu and A. Tropsha, Comb. Chem. High Throughput Screening, 2011, 217-25, 217 CrossRef.
- R. Liu, R. Rallo, S. George, Z. Ji, S. Nair, A. E. Nel and Y. Cohen, Small, 2011, 7, 1118 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y. P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, Y. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349 CrossRef CAS PubMed.
- V. C. Epa, F. R. Burden, C. Tassa, R. Weissleder, S. Shaw and D. A. Winkler, Nano Lett., 2012, 12, 5808 CrossRef CAS PubMed.
- S. Kar, A. Gajewicz, T. Puzyn, K. Roy and J. Leszczynski, Ecotoxicol. Environ. Saf., 2014, 107, 162 CrossRef CAS PubMed.
- S. Kar, A. Gajewicz, T. Puzyn and K. Roy, Toxicol. In Vitro, 2014, 2, 600 CrossRef PubMed.
- A. A. Toropov and A. P. Toropova, Chemosphere, 2014, 104, 262 CrossRef CAS PubMed.
- K. Pathakoti, M. J. Huang, J. D. Watts, X. He and H. M. Hwang, J. Photochem. Photobiol., B, 2014, 130, 234 CrossRef CAS PubMed.
- N. Sizochenko, B. Rasulev, A. Gajewicz, V. Kuz'min, T. Puzyn and J. Leszczynski, Nanoscale, 2014, 6, 13986 RSC.
- A. Gajewicz, N. Schaeublin, B. Rasulev, S. Hussain, D. Leszczynska, T. Puzyn and J. Leszczynski, Nanotoxicology, 2015, 9, 313 CrossRef CAS PubMed.
- Y. Pan, T. Li, J. Cheng, D. Telesca, J. I. Zink and J. Jiang, RSC Adv., 2016, 6, 25766 RSC.
- T. Puzyn, D. Leszczynska and J. Leszczynski, Small, 2009, 5, 2494 CrossRef CAS PubMed.
- D. A. Winkler, E. Mombelli, A. Pietroiusti, L. Tran, A. Worth, B. Fadeel and M. J. McCall, Toxicology, 2013, 313, 15 CrossRef CAS PubMed.
- L. Lubinski, P. Urbaszek, A. Gajewicz, M. T. D. Cronin, S. J. Enoch, J. C. Madden, D. Leszczynska, J. Leszczynski and T. Puzyn, SAR QSAR Environ. Res., 2013, 24, 995 CrossRef CAS PubMed.
- X. R. Xia, N. A. Monteiro-Riviere, S. Mathur, X. Song, L. Xiao, S. J. Oldenberg, B. Fadeel and J. E. Riviere, ACS Nano, 2011, 11, 9074 CrossRef PubMed.
- A. Gajewicz, M. T. D. Cronin, B. Rasulev, J. Leszczynski and T. Puzyn, Nanotechnology, 2015, 2, 015701 CrossRef PubMed.
- ECETOC, 2012. Category approaches, Read-across, (Q)SAR. Technical Report No 116. European Centre for Ecotoxicology and Toxicology of Chemicals. Available at: http://www.ecetoc.org/wp-content/uploads/2014/08/ECETOC-TR-116-Category-approaches-Read-across-QSAR.pdf.
- S. Gottardo, L. Q. Pesudo, S. Totaro, J. R. Sintes and H. Crutzen, EUR 27808, 2016, DOI:10.2788/71213.
- E. Benfenati, M. Belli, T. Borges, E. Casimiro, J. Cester, A. Fernandez, G. Gini, M. Honma, M. Kinzl, R. Knauf, A. Manganaro, E. Mombelli, M. I. Petoumenou, M. Paparella, P. Paris and G. Raitano, SAR QSAR Environ. Res., 2016, 27, 371 CrossRef CAS PubMed.
- K. Müller, Chem. Eng. Technol., 2016, 39, 365 CrossRef.
- A. G. Asuero, A. Sayago and A. G. Gonzalez, Crit. Rev. Anal. Chem., 2006, 36, 41 CrossRef CAS.
- K. P. Singh and S. Gupta, RSC Adv., 2014, 4, 13215 RSC.
- Z. Xiao, S. Varma, Y.-D. Xiao and A. Tropsha, J. Mol. Graphics Modell., 2004, 23, 129 CrossRef CAS PubMed.
- W. Zheng and A. Tropsha, J. Chem. Inf. Comput. Sci., 2000, 40, 185 CrossRef CAS PubMed.
- N. Ball, M. T. D. Cronin, J. Shen, K. Blackburn, E. D. Booth, M. Bouhifd, E. Donley, L. Egnash, C. Hastings, D. R. Juberg, A. Kleensang, N. Kleinstreuer, E. D. Kroese, A. C. Lee, T. Luechtefeld, A. Maertens, S. Marty, J. M. Naciff, J. Palmer, D. Pamies, M. Penman, A. N. Richarz, D. P. Russo, S. B. Stuard, G. Patlewicz, B. van Ravenzwaay, S. Wu, H. Zhu and T. Hartung, ALTEX Altern. Anim. Ex., 2016, 33, 149 Search PubMed.
- G. Y. Patlewicz and J. Fitzpatrick, Chem. Res. Toxicol., 2016, 29, 438 CrossRef CAS PubMed.
- K. Blackburn and S. B. Stuard, Regul. Toxicol. Pharmacol., 2014, 68, 353 CrossRef PubMed.
- T. W. Schultz, P. Amcoff, E. Berggren, F. Gautier, M. Klaric, D. J. Knight, C. Mahony, M. Schwarz, A. White and M. T. D. Cronin, Regul. Toxicol. Pharmacol., 2015, 72, 586 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |