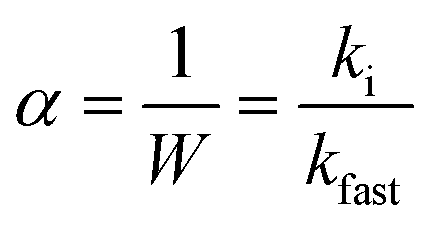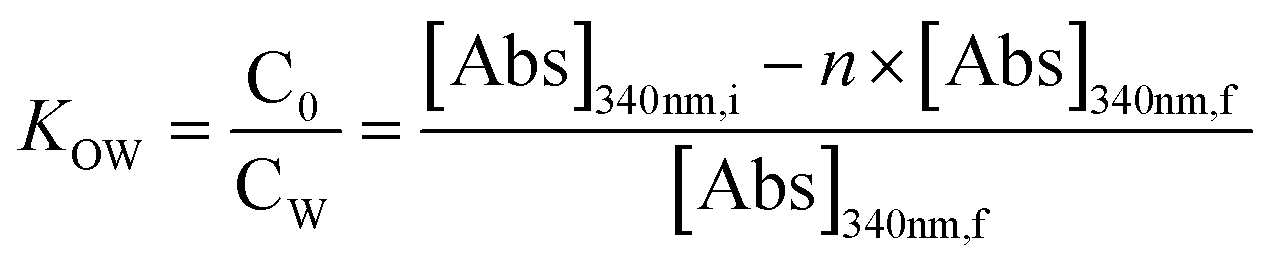Photoenhanced oxidation of C60 aggregates (nC60) by free chlorine in water†
Received
1st July 2016
, Accepted 17th October 2016
First published on 19th October 2016
Abstract
While there have been a number of fundamental studies focused on the physical and biological behaviors of C60 aggregates (nC60) in model environmental and engineered systems, the aqueous reactivity of C60 (as nC60) is much less understood and remains a critical gap in accurate life cycle modeling. In this study, facile, photo-enhanced physicochemical transformations of nC60 in the presence of free chlorine, a globally applied disinfectant, are quantitatively described. Batch reaction kinetics between nC60 and free chlorine are explored and modeled for both the ground state (dark conditions) and the photo-excited state (light irradiation conditions, UVA) over a matrix of environmentally relevant conditions (e.g., redox potentials, dissolved oxygen, light irradiation). The resulting products are described using a suite of spectroscopy techniques, including FTIR, XPS, and UV-vis, demonstrating oxidized fullerene products with varied covalent addition(s) of oxygen and chlorine atoms depending on the reaction conditions. For all conditions, UVA light irradiation (351 nm, 2 mW cm−2) significantly enhances the oxidation of nC60 by free chlorine, as indicated by both higher reaction rates and product(s) oxidation extent. Aggregation kinetic studies indicate that the resulting oxidized products, as soft clusters, are relatively more stable, compared to the parent nC60, upon photo-oxidation in the presence of monovalent (NaCl) and divalent (MgCl2) ions (via critical coagulation concentration evaluations). Further, increased product surface hydrophilicity is also demonstrated through classic water–octanol partition (Kow) experiments.
Environmental significance
As the production of fullerene-based materials approaches industrial scale, there are increasing concerns regarding their potential environmental impacts upon release. To date, aqueous-based fullerene transformation pathways under environmentally relevant conditions have remained poorly understood. In this work, photo-enhanced transformations of C60 aggregates in the presence of free chlorine (a global disinfectant/oxidant) is fundamentally explored and described over a matrix of relevant scenarios. The results highlight the importance/significance of characterizing likely reaction pathways for these and other carbon-based nanomaterials, which is crucial for accurate life cycle and risk assessments needed to eventually realize a sustainable (nano)materials industry.
|
Introduction
Fullerene production and application have been steadily expanding over the past decade and now include photo-inducible electron transfer agents (now an industry standard) in organic-based, solar voltaics, solid-state hydrogen storage, medicinal chemistry platforms, cosmetics, advanced lubricants, broad insulators, and antimicrobial agents, among others.1–10 Based on the combination of commercial applicability and economies of scale, fullerene environmental exposure is near certain through intentional (e.g., engineered waste management) and/or unintentional (e.g., unplanned release) scenarios. While there has been significant research focused on the transport, fate and biological impacts of C60, significantly less attention has been paid to the potential chemical reactions and associated transformations that fullerenes will undergo in both natural and engineered systems. With regard to aqueous-based reactions, C60, as a model fullerene, is limited by its negligible solubility in water.11,12 However, C60 as a stable, nanoscale suspension (termed as “nC60”) can be effectively water available, with effective concentrations >11 orders of magnitude (up to ca. 100 mg L−1) compared to the estimated molecular solubility of C60 in water.13–15 nC60 was first observed to be produced via solvent exchange protocols, by which solid C60 was first dissolved in an organic solvent (e.g., toluene, tetrahydrofuran, acetone, ethanol, etc.). After mixing with water as a heterogeneous system, the mediating organic solvent was subsequently removed through distillation or sonication.13,14,16–18 Moreover, without a transferring organic solvent(s), aqueous nC60 (termed as “aqu/nC60”) has also been widely reported via simple mixing of C60 solid powder in water over a longer time scale (days to months).19–27 The physicochemical properties of nC60 have been widely characterized and studied, including its transport behaviors28–32 and biological responses.18,33–35
Due to the net nucleophilicity of the conjugated π systems, C60 readily reacts with various electrophiles (e.g., oxygen, ozone, halogens, osmium tetroxide, etc.).36–43 Among these, fullerene halogenation has been studied with particular interest due to its potential broad applications (e.g., superlubricants, lithium batteries, insecticides, etc.), or as the starting/platform reactant material for further derivatization via substitution reactions.38,44–48 Chlorofullerenes (C60Clx), typically C60 based, can be synthesized via different reaction routes, including photo-chlorination.38,49,50 Upon solvation in water, C60 aggregates (nC60) are also reactive towards environmentally relevant oxidants.51–60 Reactions between nC60 and ozone led to highly oxidized, water-soluble products with ca. 29 oxygen additions per C60 molecule.61 Lee et al. demonstrated that ground-state (dark) nC60 was susceptible to oxidation in the presence of high concentrations of hydroxyl radicals, resulting in oxidized products with ca. 30% (mono)oxidized carbons.55 Additionally, C60 is known to be highly photosensitive and easily excited under UV irradiation (especially in the UVA range) to 1C60* from the ground singlet state (1C60), as well as being transformed to an excited triplet state (3C60*) through intersystem crossing.62,63 The triplet excited state is susceptible to various quenching processes, including ground-state quenching, triplet–triplet annihilation, and self-quenching.36 Singlet oxygen (1O2) can be produced via quenching the triplet excited state 3C60* in the presence of O2, which is also capable of accepting electrons from other electron donors, leading to the formation of superoxide radicals (O2˙−).63,64 Subsequently, hydroxyl radicals were demonstrated as secondary reactive oxygen species (ROS) via fullerene photo-excitation in water.65 nC60 exposed to UVC irradiation (λ 254 nm) in the presence of oxygen for 110 hours was reported to undergo significant oxygen-based transformation.57 Under UVA (λ 300–400 nm) exposure, nC60 was also observed to undergo chemical transformations in the presence of dissolved oxygen, resulting in surface oxidized products, including hydroxyl functionalities.53,58 Interestingly, under natural sunlight, photo-oxidation of nC60 was also observed, with longer wavelengths identified to play a key role in 1O2 production.51
In engineered water treatment systems, free chlorine is widely applied at concentrations ranging from 1–70 mg L−1, depending on system requirements.66–69 For water treatment systems with organics (e.g., dissolved organic pollutants and natural organic matter, etc.) exposed to free chlorine, unwanted halogenated disinfection by-products (DBPs) can be produced, as widely described by others.70–73 The redox potential of C60 (E0(C60/C60−/6−) = −0.98/−3.26 V) compared with that of free chlorine (E0(OCl−/Cl−) = 0.89 V) suggests the possibility of free chlorine-based oxidation of C60, which we have demonstrated by our previous study (in the ground state), leading to products with oxygen/chlorine atom addition and significantly enhanced water stability.59 Interestingly, under light irradiation, the photolysis of free chlorine leads to the generation of various reactive radicals (e.g., ˙OH, Cl˙, ClO˙, Cl2˙−, O(3P), etc.),74–76 which can potentially react with nC60 in water systems. In addition, the triplet-excited C60 (3C60*) is more reactive towards various oxidants (e.g., oxygen, reactive radicals) via ground-state quenching or triplet–triplet annihilation23 (whereas C60 in the ground state shows little reactivity to singlet oxygen).77 In a high salinity solution (15 g NaCl per L), disaggregation of nC60 aggregates was previously reported in the presence of free chlorine under fluorescent light irradiation (400–700 nm, 245 μW cm−2) with surface oxychlorination observed.60 To date, a comprehensive description of nC60 photochemical transformation(s) in the presence of free chlorine at relevant ionic strengths under environmentally relevant UV irradiation has not been reported.
For sunlight-based ultraviolet light irradiation reaching the troposphere, UVA (315–400 nm) is the primary fraction (ca. 90% of total tropospheric UV). Further, UVA can also readily excite C60 to a triplet state (3C60*) with two maximum absorbance peaks at 260–270 and 336–350 nm (depending on the solvent and/or aggregation status).62,63,78 Thus, reactions between nC60 and free chlorine were explored and compared for both photo-excited state (under sunlight intensity calibrated UVA irradiation at 351 nm) and ground state (dark conditions) with batch reaction kinetics modeled for a matrix of environmentally relevant conditions (e.g., redox potentials, dissolved oxygen, light irradiation). The resulting oxidized products are described using a suite of complementary spectroscopy techniques, including FTIR, XPS, and UV-vis, demonstrating oxidized fullerene products with varied covalent addition(s) of oxygen and chlorine atoms depending on the reaction conditions. Lastly, the fundamental aqueous behaviors of the parent and resulting product materials are compared and contrasted through aggregation kinetic studies, as a function of ionic strength and type, along with classic water–octanol partition (Kow) experiments.
Experimental
Materials
nC60 suspensions were prepared by a solvent exchange method with tetrahydrofuran (THF) as an intermediate organic solvent, as described extensively in previous studies by our lab and others.13,59 Suspensions were filtered using a 0.22 μm PES filtration system (Corning, NY) and then purified using a stirring cell membrane unit (Amicon, molecular weight cut-off (MWCO) of 10k) until >99.5% (volume) water replacement (exchange) was reached to eliminate residual organics, as described previously for nC60 reactivity in water.13,52–55,61,79 Parent nC60 clusters were identified to be 89.2 ± 1.5 nm (diameter) in size with a net negative surface charge (ζ = −30.1 ± 0.5 mV), as measured by dynamic light scattering and zeta potential measurements using a Zetasizer Nano ZS (ZEN3600, Malvern, UK).
Dibasic potassium phosphate (K2HPO4), sodium thiosulfate pentahydrate (NaS2O3·5H2O), sodium chloride (NaCl), magnesium chloride (MgCl2), tert-butanol (99.7%, ACS reagent), and 1-octanol were all purchased from Sigma-Aldrich (St. Louis, MO). Sodium hypochlorite solution (NaClO, laboratory grade, 5.65–6.00%), phosphate buffer solution (for free chlorine concentration determination via the DPD method), and disodium ethylenediaminetetraacetic acid (disodium EDTA) were purchased from Fisher Scientific (Pittsburgh, PA). Sodium hypochlorite (NaOCl) solution used in this study as the chlorine source was quantified as 39.92 g L−1 as Cl2 through the standard iodometric method.69 All of the aqueous solutions in this study were prepared in ultrapure water (>18.2 MΩ cm resistivity, Milli-Q, Millipore Corp.)
(Photo)chlorination batch experiments
nC60 photo-oxidation reactions with free chlorine were conducted as batch experiments by mixing ca. 7 mg L−1 nC60 (as total organic carbon concentration, TOC) with free chlorine at different (initial) levels (0, 10, 50, and 100 mg L−1) in 100 mL customized quartz reactors (Technical Glass Products). The photo-reactor was designed with two monochromatic UVA lamps (wavelength at 315 nm, BHK, CA) on the opposing walls of the reactor, and fans were installed on each side for heat dissipation and air fluting during the reaction, as described previously.80 To mimic relevant sunlight conditions (within the UVA), light intensity of 2 mW cm−2 (calibrated to 2 ± 0.05 mW cm−2 using a radiometer (UVP, Inc.)), was regularly monitored and adjusted during batch reactions. The initial pH of the reaction solutions was adjusted with HNO3 or NaOH to 6.5 ± 0.2 in the presence of 10 mM phosphate buffer. For reactions under anaerobic conditions, solutions were purged with nitrogen gas (Airgas, Bowling Green, KY) for at least 1 hour in order to lower the oxygen concentration to below 0.5 ppm, which was monitored using a DO meter with a fiber-probe (1000-micron OD) oxygen sensor (Neofox system, Oceanoptics, FL). Free chlorine was injected after the deoxygenation process (to avoid any reaction initiation with free chlorine during the deoxygenation process). Control experiments consisting of nC60 reacting with varied free chlorine concentrations (0, 10, 50, and 100 mg L−1) in dark conditions were also conducted in 100 mL borosilicate glass vials coated with aluminum foil to exclude light irradiation. Sample aliquots were taken and measured immediately via a UV-vis spectrophotometer over the wavelength range of 190–800 nm (Varian Cary Bio50). For relative reaction kinetic analyses, absorbance at 340 nm (a primary C601T1u–1Ag transition peak) was used to determine the concentration of C60 (with a molar adsorption coefficient of 4.486 × 104 M−1 cm−1), which is linearly proportional to the molecular C60 concentrations measured by HPLC upon solvent extraction, as described by others.55,58 The authors do note the technical challenges when considering/discerning a mixture of oxidized fullerene products, which is hindered by the current lack of effective chromatographic protocols (for these and other fullerene-based mixtures in water). Thus, relative reaction rates are described using 340 nm absorbance to provide a reproducible yet quasi-quantitative reference for reaction kinetics modelling and comparisons. A number of reports have also employed a similar method to describe nC60 reaction kinetics in water.13,57,59,61 Immediately upon sampling, residual free chlorine was quenched using excess sodium thiosulfate with a Na2S2O3/Cl2 molar ratio of 1/4.81 Free chlorine concentrations were also monitored during the reaction, using the standard DPD colorimetric method (Fig. S1, ESI†).69 All experiments were conducted in triplicate at room temperature (21.0 ± 1.0 °C). The reaction products under varied experimental conditions were collected at the end of each reaction for further characterization using FTIR, XPS, TEM analyses, aggregation kinetics studies, and water–octanol partition experiments, accordingly.
Total organic carbon
Total carbon measurements over longer exposure times (12 hours) within an intensively oxidizing reaction system were also conducted to monitor for any mass loss via precipitation and/or mineralization. Semi-batch experiments were conducted under UVA irradiation (351 nm, 2 mW cm−2) with free chlorine injected into the reacting system every two hours to reach a concentration of 100 mg L−1 Cl2 (since 100 mg L−1 free chlorine was consumed after 2 hours, Fig. S1, ESI†) with TOC concentrations of the reacting solutions monitored (TOC-L total carbon analyzer, Shimadzu Scientific Instrument, Inc., MD) over a time period of 12 hours. The pH of the solutions was maintained at ∼6.5 with 10 mM phosphate buffer.
Fourier transform infrared (FTIR) spectroscopy
For FTIR analyses, solid reaction products were prepared from 200 mL of 7 mg L−1 nC60 at 100 mg L−1 Cl2 either in the presence or in the absence of light irradiation (2 hours). nC60 under UVA irradiation (2 hours) in the absence of free chlorine under ambient conditions was also prepared as a control sample. Residual NaOCl (background) for reacted nC60 samples was removed as described previously via a stirred-cell membrane unit (MWCO of 1k Da), resulting in a 99.9% water exchange, with the effluent monitored by UV-vis and for pH.59 The MWCO of the membrane was selected as 1k Da to avoid carbon (parent/product) loss during purification. The purified products and parent nC60 suspensions were then dried in a vacuum oven (Thermo Nicolet, NC) at room temperature (>24 h). Diffuse reflectance infrared spectroscopy (DRIFTS) of the solid sample was conducted using a DRIFTS optical accessory (Praying Mantis, Harrick Scientific) coupled to a Nexus 470 FTIR instrument (Thermo Nicolet, NC). Unreacted nC60 or reacted products (purified) were mixed with KBr powder (Spectrograde, International Crystal Lab, NJ) at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 w/w, for a signal-optimized ratio, as done in our previous work.59 Solid mixtures were homogenized using an agate mortar with a pestle. The KBr background spectrum was collected for background subtraction before every sample spectrum was taken.
:200 w/w, for a signal-optimized ratio, as done in our previous work.59 Solid mixtures were homogenized using an agate mortar with a pestle. The KBr background spectrum was collected for background subtraction before every sample spectrum was taken.
X-ray photoelectron spectroscopy.
Samples for X-ray photoelectron spectroscopy (XPS) were prepared under parallel experimental conditions as described for FTIR above. Purified sample suspensions were dropped continuously on indium wafers (0.5 mm, 99.99% trace metal, Sigma Aldrich, MO) in a vacuum oven at room temperature to achieve ca. 1 mL total volume. XPS analyses were conducted using a Physical Electronic 5000 Versa Probe II Scanning ESCA Microprobe with an Al Kα X-ray source at 23.5 eV pass energy at a 100 μm X-ray spot size.
Transmission electron microscopy.
The TEM images of parent nC60 and reacted nC60 products were taken using an FEI Spirit transmission electron microscope at 120 kV voltage. TEM specimens were prepared by evaporating ca. 30 μL of parent or reacted nC60 suspension on 400 mesh carbon-coated copper grids (Electron Microscopy Sciences, PA).
Aggregation kinetics
Aggregation kinetics of nC60 and reacted products were evaluated via critical coagulation concentration (CCC) determination in the presence of varied electrolytes (NaCl and MgCl2), which is a widely used technique to assess aqueous nanoparticle stability.59,82–89 Briefly, the initial aggregation rates (ki) were calculated via linear regression of the initial slope for increasing hydrodynamic sizes (determined with DLS) as a function of NaCl or MgCl2 concentration. The aggregation rate in the diffusion-limited regime (kfast), which is independent of electrolyte concentrations, was used to normalize the attachment efficiency within different aggregation regimes and is expressed as eqn (1):82,88,90–93| | 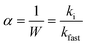 | (1) |
The detailed methodology for CCC determination is described in our previous study.59
Octanol–water partition coefficient measurements
To compare phase partitioning behaviors, classic water–octanol partition coefficients (Kow) were determined for the parent nC60 and reacted products, as done by others.94–96 The Kow of nC60 and reacted products were reported as the ratio of C60/product concentrations in the octanol phase:water phase according to eqn (2):| | 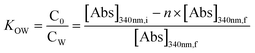 | (2) |
[Abs]340nm,i and [Abs]340nm,f are the nC60 absorbance in water at 340 nm before and after partitioning, respectively. A concentration factor, n, is used to correct for the partition coefficient as the volume of water dissolved in octanol is not negligible. Additional details of similar Kow fullerene partition experiments can be found in our previous studies.59,80,96 All experiments were conducted in triplicate at room temperature.
Results and discussion
Reaction kinetics
Reactions between nC60 and free chlorine occurred under both dark and light conditions, as indicated by the absorbance loss at 340 nm (Fig. 1).59,61 In the ground state (Fig. 1a), reaction rates increased as a function of free chlorine concentration due to a net increase in the system redox potential. As we assumed that the concentration of total chlorine species is in excess relative to that of the (reacting) fullerene species, a pseudo-first-order reaction rate model was employed (eqn (3)) with regard to nC60 UV adsorption at 340 nm.| | 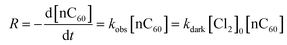 | (3) |
where [nC60] and [Cl2] represent the concentrations of nC60 and free chlorine during the reaction, respectively. Such an approach to modeling pseudo-first-order reactions (with observed rate constants, kobs) was adapted from previous studies on the chlorination of polycyclic aromatic hydrocarbons (PAHs) in water.97 The reaction rate constants (kdark) here were employed to evaluate the reaction regarding the nC60 and free chlorine concentrations ([Cl2]∼[Cl2]0, Fig. S1 in the ESI†) for further comparison with photo-irradiation reactions. In the presence of UVA irradiation (351 nm, 2 mW cm−2), all reaction rates are significantly enhanced (Fig. 1b), with rates eventually slowing as free chlorine is consumed (Fig. S1, ESI†). Batch reactions were also conducted under anaerobic conditions (dissolved oxygen <0.5 ppm) to evaluate the role of oxygen. As shown in Fig. 1c, anaerobic reaction rates between nC60 and free chlorine were comparably slower, implicating a direct role for oxygen in this photochemical transformation, which has been observed by others.53,57,58 Thus, for aerobic conditions (DO ∼9 ppm), the overall photo-oxidation process of nC60 is simplified here into two reactions: the reactions of nC60 with dissolved oxygen and free chlorine, as expressed in eqn (4) and (5), respectively:| | 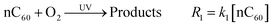 | (4) |
| | 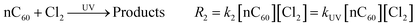 | (5) |
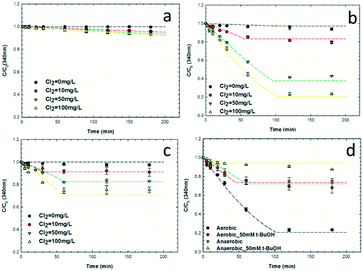 |
| | Fig. 1 UV adsorption of nC60 solution at 340 nm as a function of time under (pH ∼6.5, phosphate buffer = 10 mM) (a) dark aerobic conditions; (b) UV aerobic conditions; (c) UV anaerobic conditions; (d) in the presence of radical scavenger (100 mg L−1 Cl2, UVA). (Dotted lines in Fig. 1a–d represent the fitted reaction rates following eqn (8) and (9), R2 >0.80.) | |
Additionally, the photo-decomposition of free chlorine (Fig. S1, ESI†)75 independent of nC60 was considered as:
| |  | (6) |
k
1, kUV (k2) and k3 are rate constants for reactions (4)–(6), respectively. Detailed information for chlorine photolysis reactions (eqn (6)) in water are described in eqn S3–S10 in the ESI.† As the degradation of free chlorine is a zero-order reaction (rate constant k3), shown in Fig. S1 in the ESI,† the free chlorine concentration over the reaction time is expressed as eqn (7):
The reaction of nC60 photo-oxidation with oxygen was observed to be a pseudo-first-order reaction with regard to nC60, which was also demonstrated by others.53 The reaction rate constant k1 can be derived from the control experiments of nC60 oxidation reaction under aerobic conditions in the absence of free chlorine (Fig. 2b, Cl2 = 0 mg L−1). Thus, the overall reaction of nC60 chlorination/oxidation under aerobic conditions is expressed as:
| | 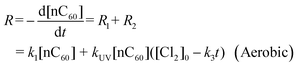 | (8) |
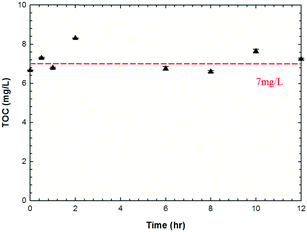 |
| | Fig. 2 TOC concentrations of nC60 solution over a semi-batch reaction under UV irradiation (pH ∼6.5, ca. 7 mg L−1 nC60 with free chlorine initial concentration of 100 mg L−1; free chlorine was injected into the reaction system every two hours to reach the concentration of 100 mg L−1). | |
However, for anaerobic conditions, the reaction of nC60 photo-oxidation by oxygen (R1) can be neglected and thus the overall reaction is presented as:
| | 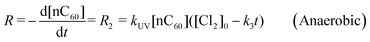 | (9) |
Rate constants (kdark/UV) for the reactions between nC60 and free chlorine are summarized in Table 1 (more detailed calculations are provided as eqn S1 and S2, Fig. S2, ESI†). Under photo-irradiation, the rate constants are at least one order of magnitude higher compared to dark conditions. We hypothesize that a variety of reactive radicals produced through photolysis of free chlorine, such as 1O2, O2˙−, ˙OH, Cl˙, ClO˙, Cl2˙−, O(3P), etc.,74–76 significantly enhance the oxidation rates of nC60, in addition to the photoexcitation of C60 itself (which is a more reactive state).36 Rate constants (kUV) for reactions under anaerobic conditions are relatively lower, likely due to the altered (lowered) ROS production equilibrium within the system. For example, in reducing reaction scenarios, more O2˙− and ˙OH can be produced via electron transfer to 1O2,98 and singlet oxygen has been identified to oxidize C60 to a large extent, inducing epoxide, oxa-bridged annulene, and/or cage-open dicarbonyl structures.99–101 Complicating the deconvolution of responsible (dominant) reactive species, ROS production is also enhanced by the presence of oxidized fullerene derivatives/products (fullerols) compared with pristine nC60 itself.74,76,102 Decreased rate constants (kdark/UV) were observed for both ground and photo-excited states as the initial free chlorine concentration increased, indicating diffusion limitations rather than a reaction-controlled oxidation process. In the presence of 50 mM t-BuOH (a strong hydroxyl and chlorine radical scavenger103,104) free chlorine reactions are shown in Fig. 1d. For these, rate constants decreased by ca. 70% for both aerobic and anaerobic conditions, implicating radical mediated transformation pathways. As t-BuOH did not completely inhibit reactivity, the other reactive oxygen species (e.g., singlet oxygen and superoxides), albeit to a lesser degree, likely participate in the overall reaction process. For the most oxidizing conditions evaluated, nC60 did not precipitate or mineralize, per TOC measurements (Fig. 2).
Table 1 Reaction rate constants (kdark/UV, L min−1 mg−1) under varied experimental conditions
| Cl2 (mg L−1) |
Aerobic |
Anaerobic |
| 10 |
Dark |
2.51 × 10−5 |
— |
| UVA |
2.02 × 10−4 |
1.42 × 10−4 |
| 50 |
Dark |
7.11 × 10−6 |
— |
| UVA |
1.40 × 10−4 |
4.34 × 10−5 |
| 100 |
Dark |
4.04 × 10−6 |
— |
| UVA |
9.43 × 10−5 |
3.71 × 10−5 |
| UVA(50 mM t-BuOH) |
2.93 × 10−5 |
1.33 × 10−5 |
Product characterization
FTIR spectra (primarily as surface analysis) of nC60 and reacted products under different reaction conditions are shown in Fig. 3. Besides pristine C60 characteristic peaks at 528, 576, 1180, 1480, and 1540 cm−1, which are attributed to C–C vibration modes, nC60 also exhibits peaks within 1830–1684 cm−1 (centered at 1726 cm−1, carbonyl group) and 1000–1250 cm−1 (centered at 1100 cm−1, C–O stretching),57,58,61,80,96 due to the minor surface oxidation of fullerene clusters, which has also been observed by others.23 After exposure to UV irradiation under ambient conditions for 2 hours (Fig. 3b), slight surface oxidation of nC60 clusters was observed, as indicated by the increased intensities of C–O stretching and carbonyl groups. Hou et al. and Lee et al. all observed high levels of oxygen moiety addition to nC60 clusters after long exposure times (974 hours and 110 hours, respectively) under photo-irradiation in the presence of ambient oxygen.51,57 In the presence of free chlorine (100 mg L−1) under dark conditions (Fig. 3c), oxygen-containing functional groups with higher intensities compared with the parent nC60 are observed. In the presence of free chlorine (100 mg L−1 Cl2, Fig. 3d) and UVA irradiation, extensive oxidation of nC60 clusters occurred, as indicated by significantly increased peak intensities at ca. 1700 cm−1 (carbonyl groups), 1400 cm−1 (C–OH in-plane bending), and 1100 cm−1 (C–O stretching).57,58,61 Besides oxygen-based moieties, an additional group of peaks is also observed in the range from 500 to 900 cm−1, which are attributed to typical C–Cl stretching bonds.38 It is likely that reactive chlorine radicals produced via chlorine decomposition under photo-irradiation75 can bond with nucleophilic C60via a free radical addition reaction. Such a reaction has also been demonstrated to be the primary mechanism during the photo-chlorination of graphene.105 In addition, photo-excited C60 in (3C60*) is more reactive to surrounding electrophiles (including free chlorine, oxygen, and chlorine/oxygen radicals) compared with that in the ground state.36 The significantly enhanced carbonyl groups (peak of 1620 cm−1 with shoulder at 1720 cm−1) for photo-oxidized products may also be the result of a subsequent reaction(s), whereby Cl elimination begets further oxygen additions (dehydrohalogenation) after C–Cl formation, which has also been demonstrated by others.97,106
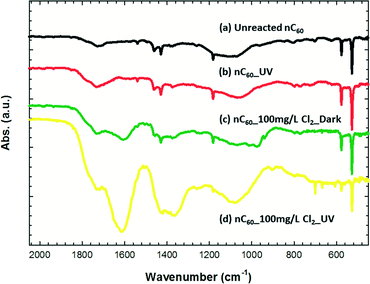 |
| | Fig. 3 FTIR DRIFTS spectra of unreacted nC60 and reacted products: (a) unreacted nC60; nC60 products after reaction (b) under UV irradiation for 2 hours; (c) with 100 mg L−1 Cl2 in the dark for 2 hours; (d) under UV irradiation with 100 mg L−1 Cl2 for 2 hours. | |
XPS spectra of parent nC60 and reacted products are compared in Fig. 4 for surface carbon derivatization (oxygenation/chlorination) quantification, and the carbon (1s) and chlorine (2p) oxidation state ratios are summarized in Table S1.† For pristine nC60 (Fig. 4a), underivatized carbon is observed to be dominant with a relative ratio of 89.3%, along with low percentages of mono-oxidized and di-oxidized carbons (286.52 and 289.03 eV, respectively), which is consistent with FTIR results (Fig. 3a), indicative of slight surface oxidation of parent nC60 before any reaction. Upon UV irradiation in the presence of oxygen (no free chlorine, Fig. 4b), the underivatized carbon ratio decreased to 72.4% coupled with increased mono-oxidized and di-oxidized carbons ratios of 18.5% and 9.2%, respectively. Reacted products with free chlorine (100 mg L−1 Cl2) under dark conditions (2 hour reaction), shown in Fig. 4c, also increased with regard to oxidized carbons to an overall ratio of 18.6%. It should be noted for dark chlorination conditions that di-oxidized carbons (carbonyl, ketone, ester, etc.) appeared with a relatively higher ratio (14.0%) compared with the mono-oxidized carbon, which was also observed by FTIR (Fig. 3). Upon exposure to both UV light and free chlorine (100 mg L−1) after 2 hours (Fig. 4d), a significantly higher level of oxidized carbons was observed in the product spectrum compared to those of the parent nC60 and control samples (Fig. 4b and c), with mono-oxidized carbon and di-oxidized carbon ratios increased to 21.8% and 22.5%, respectively. A similar level of nC60 chloro-oxidation was observed for ground-state reactions (photoreactions were not evaluated) with free chlorine for much longer exposure times (5 days) as demonstrated in our previous study.59 XPS analyses of chlorine state are also provided in Fig. 4e–h for differentiation of C–O versus C–Cl (both with binding energy at ca. 286–287 eV).107,108 For unreacted nC60 and products under UV irradiation without free chlorine (Fig. 4e and f), no detectable signal of chlorine was observed. In the presence of free chlorine (100 mg L−1, 2 hours) in the ground-state, chlorine peaks (Cl 2p3/2/Cl 2p1/2), shown as a classic split response (spin–orbit split peaks), located at 197.58/199.25 eV, which is attributed to residual chloride ions (the control XPS spectrum is shown in Fig. S3, ESI†) from reaction solutions,109 with no obvious C–Cl covalent bonds observed (Fig. 4g). However, under UV irradiation, besides chloride ion signal at 197.72/199.18 eV, another set of split peaks appeared at 199.85/201.21 eV (Fig. 4h), clearly indicating that chlorine atom addition to the carbon cage occurred,109 which is consistent with FTIR results.
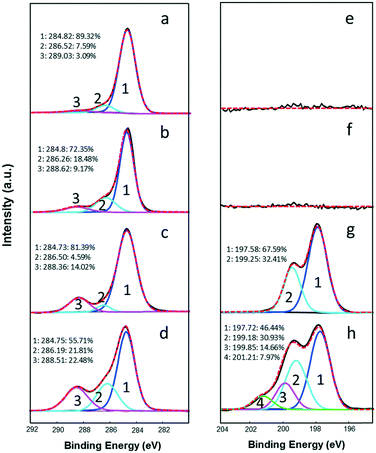 |
| | Fig. 4 XPS spectra and curve-fitting analyses of nC60 and reacted products: (a) unreacted nC60 (C1s); (b) nC60 products under UV irradiation for 2 hours (C1s); (c) nC60 products with 100 mg L−1 Cl2 in the dark for 2 hours (C1s); (d) nC60 products with 100 mg L−1 Cl2 under UV irradiation for 2 hours (C1s); (e) unreacted nC60 (Cl2p); (f) nC60 products under UV irradiation for 2 hours (Cl2p); (g) nC60 products with 100 mg L−1 Cl2 in the dark for 2 hours (Cl2p); (h) nC60 products with 100 mg L−1 Cl2 under UV irradiation for 2 hours (Cl2p). | |
Behavior evaluation
Relative (particle–particle) attachment efficiencies (α) of fullerene clusters, before and after reactions, were measured to determine the critical coagulation concentration (CCC) as a function of ionic strength and type (Fig. 5). For Na+ (NaCl), a model monovalent electrolyte, the hydrodynamic diameter of the unreacted nC60 (Fig. S4a, ESI†) remained stable (ca. 100 nm)13,110,111 below 50 mM. Due to electrical double layer repulsion screening effects,112 the hydrodynamic size of nC60 increased when NaCl concentrations were higher than 80 mM, with a calculated CCC value of 104 mM for unreacted nC60,59 which is within the range of the previously reported nC60 CCC values (84–260 mM, nC60 synthesized via different routes).82,90,91,113,114 After a 2 hour reaction with 100 mg L−1 Cl2 in the dark, the product CCC value increased to 142 mM but remained in the same range as that of parent nC60 (Fig. 5a). Similarly, in the absence of free chlorine, upon UV irradiation for 2 hours, aqueous stability was not significantly enhanced, with a similar CCC calculated to be 112 mM (NaCl). Interestingly, in the presence of both UV irradiation and free chlorine (2 hour reaction time), the water stability of nC60 was dramatically enhanced (CCC of 650 mM), which is comparable to a 5 day dark reaction in the presence of free chlorine.59 We hypothesize that the presence of both oxygen and chlorine functionalities enhance the electron repulsion forces as well as the hydration repulsion forces (i.e. decreased hydrophobic attraction forces) between product clusters in water, effectively enhancing the aqueous stability.115 In addition, TEM micrographs (Fig. S5, ESI†) of nC60 and reacted products clearly show a loss of ordered structure, appearing as soft, amorphous clusters upon reaction (Fig. S5d, ESI†). Oxidized fullerenes (also termed “fullerols”) in water have been observed in a number of other reports to exist as relatively soft aggregates in water.116 Mg2+ (MgCl2) was evaluated as a divalent cation for aggregation kinetics (Fig. 5b).117 Slightly increased CCC values for dark and UV control samples were observed to be 9.2 and 8.6 mM, respectively, compared with that for the parent nC60 (8.0 mM). Similar to NaCl, after photo-oxidation with free chlorine (100 mg L−1), the MgCl2 CCC increased relatively significantly to 15.4 mM, also indicating enhanced aqueous stability. Phase partitioning behavior was evaluated by water–octanol partition (Kow, pH ∼ 7.5) measurements (Fig. S6, ESI†), with increasing relative aqueous partitioning observed (more hydrophilic) for all products. In line with extensive polar functionalization, photo-oxidized products' (100 mg L−1 Cl2) Kow decreased ca. 90% compared with parent nC60.
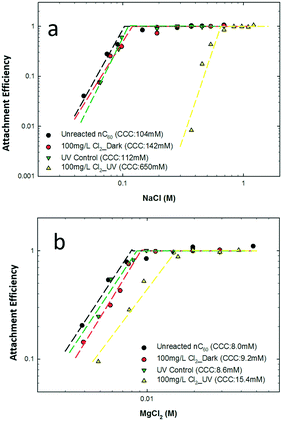 |
| | Fig. 5 Aggregation stability curves of pristine nC60 and reacted products under varied experimental conditions (pH ∼6.5): (a) attachment efficiency in NaCl solutions; (b) attachment efficiency in MgCl2 solutions. | |
Conclusions
This work systematically describes the photo-enhanced reactivity of nC60 with free chlorine. Relative reaction rates increase with free chlorine levels and are significantly enhanced in the presence of simulated sunlight UVA irradiation. The products were observed to contain varying degrees of oxygen and chlorine functionality depending on the reaction conditions, with no fullerene mineralization or precipitation. Under UVA, the products showed relatively enhanced surface-based functional derivatization. Further, for all products, physical behavior differed significantly compared to parent nC60. Through aggregation kinetic studies, the reaction products were considerably more stable in water. Changes in partitioning behavior, via octanol–water phase partitioning measurements, were also observed, indicating relatively more hydrophilic products. Taken together, as these reaction conditions are relevant to engineered water treatment systems, it is likely that fullerenes (here as C60) will be chemically transformed during chlorination treatment processes, especially in the presence of UV light (including sunlight). This work clearly highlights the importance/significance of characterizing reaction pathways for these and other carbon-based nanomaterials, which is crucial for accurate life cycle and risk assessments needed to eventually realize a sustainable (nano)materials industry.
Acknowledgements
This study is supported by Washington University in St. Louis faculty startup funding for Dr. John D. Fortner. We are grateful for the FTIR, TEM, and DLS facility support by Nano Research Facility (NRF) at Washington University in St. Louis, a member of the National Nanotechnology Infrastructure Network (NNIN), which is funded by the National Science Foundation (No. ECS-0335765). We also appreciate the facility support (XPS) from Institute of Materials Science and Engineering (IMSE) at Washington University in St. Louis. We sincerely thank Materials and Electrochemical Research (MER) Corporation (Tucson, AZ) for the help in fullerene material preparation.
References
- F. Gao and O. Inganas, Phys. Chem. Chem. Phys., 2014, 16, 20291–20304 RSC.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- B. C. Thompson and J. M. J. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- J. Tong, M. C. Zimmerman, S. Li, X. Yi, R. Luxenhofer, R. Jordan and A. V. Kabanov, Biomaterials, 2011, 32, 3654–3665 CrossRef CAS PubMed.
- S. W. Cho, L. F. J. Piper, A. DeMasi, A. R. H. Preston, K. E. Smith, K. V. Chauhan, P. Sullivan, R. A. Hatton and T. S. Jones, J. Phys. Chem. C, 2010, 114, 1928–1933 CAS.
- L. L. Bergeson and M. F. Cole, Nanotechnol. Law Bus., 2012, 9, 114–120 Search PubMed.
- R. M. Vallant, Z. Szabo, S. Bachmann, R. Bakry, M. Najam-ul-Haq, M. Rainer, N. Heigl, C. Petter, C. W. Huck and G. K. Bonn, Anal. Chem., 2007, 79, 8144–8153 CrossRef CAS PubMed.
- R. Bakry, R. M. Vallant, M. Najam-Ul-Haq, M. Rainer, Z. Szabo, C. W. Huck and G. K. Bonn, Int. J. Nanomed., 2007, 2, 639–649 CAS.
- L. Y. Huang, M. Terakawa, T. Zhiyentayev, Y. Y. Huang, Y. Sawayama, A. Jahnke, G. P. Tegos, T. Wharton and M. R. Hamblin, Nanomedicine, 2010, 6, 442–452 CAS.
- Y. Gao, X. Wu and X. C. Zeng, J. Mater. Chem. A, 2014, 2, 5910–5914 CAS.
- D. Heymann, Fullerene Sci. Technol., 1996, 4, 509–515 CrossRef CAS.
- C. T. Jafvert and P. P. Kulkarni, Environ. Sci. Technol., 2008, 42, 5945–5950 CrossRef CAS PubMed.
- J. D. Fortner, D. Y. Lyon, C. M. Sayes, A. M. Boyd, J. C. Falkner, E. M. Hotze, L. B. Alemany, Y. J. Tao, W. Guo, K. D. Ausman, V. L. Colvin and J. B. Hughes, Environ. Sci. Technol., 2005, 39, 4307–4316 CrossRef CAS PubMed.
- G. V. Andrievsky, M. V. Kosevich, O. M. Vovk, V. S. Shelkovsky and L. A. Vashchenko, J. Chem. Soc., Chem. Commun., 1995, 1281–1282 RSC.
- S. Deguchi, R. G. Alargova and K. Tsujii, Langmuir, 2001, 17, 6013–6017 CrossRef CAS.
- W. A. Scrivens, J. M. Tour, K. E. Creek and L. Pirisi, J. Am. Chem. Soc., 1994, 116, 4517–4518 CrossRef CAS.
- R. G. Alargova, S. Deguchi and K. Tsujii, J. Am. Chem. Soc., 2001, 123, 10460–10467 CrossRef CAS PubMed.
- A. Dhawan, J. S. Taurozzi, A. K. Pandey, W. Shan, S. M. Miller, S. A. Hashsham and V. V. Tarabara, Environ. Sci. Technol., 2006, 40, 7394–7401 CrossRef CAS PubMed.
- X. K. Cheng, A. T. Kan and M. B. Tomson, J. Chem. Eng. Data, 2004, 49, 675–683 CrossRef CAS.
- X. Chang, L. K. Duncan, J. Jinschek and P. J. Vikesland, Langmuir, 2012, 28, 7622–7630 CrossRef CAS PubMed.
- X. Chang and P. J. Vikesland, Environ. Pollut., 2009, 157, 1072–1080 CrossRef CAS PubMed.
- J. Brant, H. Lecoanet, M. Hotze and M. Wiesner, Environ. Sci. Technol., 2005, 39, 6343–6351 CrossRef CAS PubMed.
- J. Labille, A. Masion, F. Ziarelli, J. Rose, J. Brant, F. Villiéras, M. Pelletier, D. Borschneck, M. R. Wiesner and J.-Y. Bottero, Langmuir, 2009, 25, 11232–11235 CrossRef CAS PubMed.
- L. K. Duncan, J. R. Jinschek and P. J. Vikesland, Environ. Sci. Technol., 2007, 42, 173–178 CrossRef.
- X. Ma and D. Bouchard, Environ. Sci. Technol., 2009, 43, 330–336 CrossRef CAS PubMed.
- C. W. Isaacson and D. C. Bouchard, Environ. Sci. Technol., 2010, 44, 8971–8976 CrossRef CAS PubMed.
- P. Indeglia, V. Krishna, A. Georgieva and J.-C. Bonzongo, J. Nanopart. Res., 2013, 15, 1–16 CrossRef.
- L. Wang, Y. Huang, A. T. Kan, M. B. Tomson and W. Chen, Environ. Sci. Technol., 2012, 46, 5422–5429 CrossRef CAS PubMed.
- L. Zhang, L. Hou, L. Wang, A. T. Kan, W. Chen and M. B. Tomson, Environ. Sci. Technol., 2012, 46, 7230–7238 CrossRef CAS PubMed.
- Y. Wang, Y. Li, J. D. Fortner, J. B. Hughes, L. M. Abriola and K. D. Pennell, Environ. Sci. Technol., 2008, 42, 3588–3594 CrossRef CAS PubMed.
- L. Cai, M. Tong, H. Ma and H. Kim, Environ. Sci. Technol., 2013, 47, 5703–5710 CrossRef CAS PubMed.
- Y. Wang, Y. Li, J. Costanza, L. M. Abriola and K. D. Pennell, Environ. Sci. Technol., 2012, 46, 11761–11769 CrossRef CAS PubMed.
- D. Y. Lyon and P. J. J. Alvarez, Environ. Sci. Technol., 2008, 42, 8127–8132 CrossRef CAS PubMed.
- X. Tao, Y. He, J. D. Fortner, Y. Chen and J. B. Hughes, Chemosphere, 2013, 92, 1245–1252 CrossRef CAS PubMed.
- M. Kovochich, B. Espinasse, M. Auffan, E. M. Hotze, L. Wessel, T. Xia, A. E. Nel and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 6378–6384 CrossRef CAS PubMed.
-
A. Hirsch, M. Brettreich and F. Wudl, Fullerenes: Chemistry and Reactions, Wiley, 2006 Search PubMed.
- M. P. Anachkov, F. Cataldo and S. K. Rakovsky, Fullerenes, Nanotubes, Carbon Nanostruct., 2004, 12, 745–752 CrossRef CAS.
- F. Cataldo, Carbon, 1994, 32, 437–443 CrossRef CAS.
- D. Heymann, S. M. Bachilo, R. B. Weisman, F. Cataldo, R. H. Fokkens, N. M. M. Nibbering, R. D. Vis and L. P. F. Chibante, J. Am. Chem. Soc., 2000, 122, 11473–11479 CrossRef CAS.
- K. Kniaz, J. E. Fischer, H. Selig, G. B. M. Vaughan, W. J. Romanow, D. M. Cox, S. K. Chowdhury, J. P. McCauley, R. M. Strongin and A. B. Smith, J. Am. Chem. Soc., 1993, 115, 6060–6064 CrossRef CAS.
- J. M. Hawkins, T. A. Lewis, S. D. Loren, A. Meyer, J. R. Heath, Y. Shibato and R. J. Saykally, J. Org. Chem., 1990, 55, 6250–6252 CrossRef CAS PubMed.
- C. Bruno, I. Doubitski, M. Marcaccio, F. Paolucci, D. Paolucci and A. Zaopo, J. Am. Chem. Soc., 2003, 125, 15738–15739 CrossRef CAS PubMed.
- Y. Tajima and K. Takeuchi, J. Org. Chem., 2002, 67, 1696–1698 CrossRef CAS PubMed.
- D. M. Cox, S. D. Cameron, A. Tuinman, A. Gakh, J. L. Adcock, R. N. Compton, E. W. Hagaman, K. Kniaz and J. E. Fischer, J. Am. Chem. Soc., 1994, 116, 1115–1120 CrossRef CAS.
- F. N. Tebbe, J. Y. Becker, D. B. Chase, L. E. Firment, E. R. Holler, B. S. Malone, P. J. Krusic and E. Wasserman, J. Am. Chem. Soc., 1991, 113, 9900–9901 CrossRef CAS.
-
N. Watanabe, T. Nakajima and H. Touhara, Graphite fluorides, Elsevier, 1988 Search PubMed.
- G. A. Olah, I. Bucsi, R. Aniszfeld and G. K. Surya Prakash, Carbon, 1992, 30, 1203–1211 CrossRef CAS.
- F. Cataldo, Fullerene Sci. Technol., 1996, 4, 1041–1059 CAS.
- G. A. Olah, I. Bucsi, C. Lambert, R. Aniszfeld, N. J. Trivedi, D. K. Sensharma and G. K. S. Prakash, J. Am. Chem. Soc., 1991, 113, 9387–9388 CrossRef CAS.
- P. A. Troshin, O. Popkov and R. N. Lyubovskaya, Fullerenes, Nanotubes, Carbon Nanostruct., 2003, 11, 165–185 CrossRef CAS.
- W.-C. Hou, L. Kong, K. A. Wepasnick, R. G. Zepp, D. H. Fairbrother and C. T. Jafvert, Environ. Sci. Technol., 2010, 44, 8121–8127 CrossRef CAS PubMed.
- W.-C. Hou and C. T. Jafvert, Environ. Sci. Technol., 2009, 43, 5257–5262 CrossRef CAS PubMed.
- W.-C. Hou and C. T. Jafvert, Environ. Sci. Technol., 2008, 43, 362–367 CrossRef.
- J. Lee, Y. Yamakoshi, J. B. Hughes and J.-H. Kim, Environ. Sci. Technol., 2008, 42, 3459–3464 CrossRef CAS PubMed.
- J. Lee, W. Song, S. S. Jang, J. D. Fortner, P. J. J. Alvarez, W. J. Cooper and J.-H. Kim, Environ. Sci. Technol., 2010, 44, 3786–3792 CrossRef CAS PubMed.
- J. Lee, J. D. Fortner, J. B. Hughes and J.-H. Kim, Environ. Sci. Technol., 2007, 41, 2529–2535 CrossRef CAS PubMed.
- J. Lee, M. Cho, J. D. Fortner, J. B. Hughes and J. H. Kim, Environ. Sci. Technol., 2009, 43, 4878–4883 CrossRef CAS PubMed.
- Y. S. Hwang and Q. L. Li, Environ. Sci. Technol., 2010, 44, 3008–3013 CrossRef CAS PubMed.
- J. Wu, D. Benoit, S. S. Lee, W. Li and J. D. Fortner, Environ. Sci. Technol., 2016, 50, 721–731 CrossRef CAS PubMed.
- C. Wang, C. Shang, M. Ni, J. Dai and F. Jiang, Environ. Sci. Technol., 2012, 46, 9398–9405 CrossRef CAS PubMed.
- J. D. Fortner, D. I. Kim, A. M. Boyd, J. C. Falkner, S. Moran, V. L. Colvin, J. B. Hughes and J. H. Kim, Environ. Sci. Technol., 2007, 41, 7497–7502 CrossRef CAS PubMed.
- S. H. Hoke, J. Molstad, D. Dilettato, M. J. Jay, D. Carlson, B. Kahr and R. G. Cooks, J. Org. Chem., 1992, 57, 5069–5071 CrossRef CAS.
-
K. M. Kadish and R. S. Ruoff, Fullerenes: Chemistry, Physics, and Technology, Wiley, 2000 Search PubMed.
- Y. Yamakoshi, N. Umezawa, A. Ryu, K. Arakane, N. Miyata, Y. Goda, T. Masumizu and T. Nagano, J. Am. Chem. Soc., 2003, 125, 12803–12809 CrossRef CAS PubMed.
- Y. Yamakoshi, S. Sueyoshi, K. Fukuhara and N. Miyata, J. Am. Chem. Soc., 1998, 12363–12364 CrossRef CAS.
-
J. C. M. W. H. Crittenden, Water treatment principles and design, J. Wiley, Hoboken, N.J., 2005 Search PubMed.
- M. Deborde and U. von Gunten, Water Res., 2008, 42, 13–51 CrossRef CAS PubMed.
-
G. Tchobanoglous, F. L. Burton, H. D. Stensel and Metcalf & Eddy, Inc., Wastewater Engineering: Treatment and Reuse, McGraw-Hill, 2003 Search PubMed.
-
L. S. Clesceri, A. E. Greenberg and A. D. Eaton, Standard Methods for the Examination of Water and Wastewater, 20th Edition, APHA American Public Health Association, 1998 Search PubMed.
-
J. J. Rook, Formation of haloforms during chlorination of natural waters, 1974 Search PubMed.
- T. A. Bellar, J. J. Lichtenberg and R. C. Kroner, J. - Am. Water Works Assoc., 1974, 66, 703–706 CAS.
- S. W. Krasner, H. S. Weinberg, S. D. Richardson, S. J. Pastor, R. Chinn, M. J. Sclimenti, G. D. Onstad and A. D. Thruston, Environ. Sci. Technol., 2006, 40, 7175–7185 CrossRef CAS PubMed.
- S. E. Hrudey, Water Res., 2009, 43, 2057–2092 CrossRef CAS PubMed.
- K. D. Pickering and M. R. Wiesner, Environ. Sci. Technol., 2005, 39, 1359–1365 CrossRef CAS PubMed.
- L. H. Nowell and J. Hoigné, Water Res., 1992, 26, 599–605 CrossRef CAS.
- S.-R. Chae, E. M. Hotze and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 6208–6213 CrossRef CAS PubMed.
- L. Juha, V. Hamplova, J. Kodymova and O. Spalek, J. Chem. Soc., Chem. Commun., 1994, 2437–2438 RSC.
- X. Chang and P. J. Vikesland, Environ. Sci. Technol., 2011, 45, 9967–9974 CrossRef CAS PubMed.
- J. Lee, Y. Mackeyev, M. Cho, D. Li, J.-H. Kim, L. J. Wilson and P. J. J. Alvarez, Environ. Sci. Technol., 2009, 43, 6604–6610 CrossRef CAS PubMed.
- J. Wu, D. G. Goodwin, K. Peter, D. Benoit, W. Li, D. H. Fairbrother and J. D. Fortner, Environ. Sci. Technol. Lett., 2014, 1, 490–494 CrossRef CAS.
-
R. Chang, General chemistry, Random House, 1986 Search PubMed.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 22, 10994–11001 CrossRef CAS PubMed.
- A. R. Petosa, D. P. Jaisi, I. R. Quevedo, M. Elimelech and N. Tufenkji, Environ. Sci. Technol., 2010, 44, 6532–6549 CrossRef CAS PubMed.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS PubMed.
- S. Zhang, Y. Jiang, C.-S. Chen, J. Spurgin, K. A. Schwehr, A. Quigg, W.-C. Chin and P. H. Santschi, Environ. Sci. Technol., 2012, 46, 8764–8772 CrossRef CAS PubMed.
- L. T. Trinh, A.-L. Kjøniksen, K. Zhu, K. Knudsen, S. Volden, W. Glomm and B. Nyström, Colloid Polym. Sci., 2009, 287, 1391–1404 CAS.
- R. A. French, A. R. Jacobson, B. Kim, S. L. Isley, R. L. Penn and P. C. Baveye, Environ. Sci. Technol., 2009, 43, 1354–1359 CrossRef CAS PubMed.
- W. Li, D. Liu, J. Wu, C. Kim and J. D. Fortner, Environ. Sci. Technol., 2014, 48, 11892–11900 CrossRef CAS PubMed.
- W. Li, C. H. Hinton, S. S. Lee, J. Wu and J. D. Fortner, Environ. Sci.: Nano, 2016, 3, 85–93 RSC.
- X. Qu, Y. S. Hwang, P. J. J. Alvarez, D. Bouchard and Q. Li, Environ. Sci. Technol., 2010, 44, 7821–7826 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2009, 43, 7270–7276 CrossRef CAS PubMed.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- Y. Xiao and M. R. Wiesner, J. Hazard. Mater., 2012, 215–216, 146–151 CrossRef CAS PubMed.
- W.-C. Hou, B. Y. Moghadam, P. Westerhoff and J. D. Posner, Langmuir, 2011, 27, 11899–11905 CrossRef CAS PubMed.
- J. Wu, L. B. Alemany, W. Li, L. Petrie, C. Welker and J. D. Fortner, Environ. Sci. Technol., 2014, 48, 7384–7392 CrossRef CAS PubMed.
- A. R. Oyler, R. J. Liukkonen, M. T. Lukasewycz, K. E. Heikkila, D. A. Cox and R. M. Carlson, Environ. Sci. Technol., 1983, 17, 334–342 CrossRef CAS.
-
K. Krumova and G. Cosa, in Singlet Oxygen: Applications in Biosciences and Nanosciences, The Royal Society of Chemistry, 2016, vol. 1, pp. 1–21 Search PubMed.
- F. Diederich, R. Ettl, Y. Rubin, R. L. Whetten, R. Beck, M. Alvarez, S. Anz, D. Sensharma, F. Wudl, K. C. Khemani and A. Koch, Science, 1991, 252, 548–551 CAS.
- C. Taliani, G. Ruani, R. Zamboni, R. Danieli, S. Rossini, V. N. Denisov, V. M. Burlakov, F. Negri, G. Orlandi and F. Zerbetto, J. Chem. Soc., Chem. Commun., 1993, 220–222, 10.1039/C39930000220.
- J. P. Deng, D. D. Ju, G. R. Her, C. Y. Mou, C. J. Chen, Y. Y. Lin and C. C. Han, J. Phys. Chem., 1993, 97, 11575–11577 CrossRef CAS.
- L. J. Kong, O. Tedrow, Y. F. Chan and R. G. Zepp, Environ. Sci. Technol., 2009, 43, 9155–9160 CrossRef CAS PubMed.
- J. Staehelin and J. Hoigne, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef CAS PubMed.
-
A. Miller and P. H. Solomon, Writing Reaction Mechanisms in Organic Chemistry, Harcourt/Academic Press, 2000 Search PubMed.
- B. Li, L. Zhou, D. Wu, H. Peng, K. Yan, Y. Zhou and Z. Liu, ACS Nano, 2011, 5, 5957–5961 CrossRef CAS PubMed.
-
J. L. G. Wade, Organic Chemistry, Pearson Education, 2008 Search PubMed.
- J. M. Gonzalez-Dominguez, P. Castell, S. Bespin-Gascon, A. Anson-Casaos, A. M. Diez-Pascual, M. A. Gomez-Fatou, A. M. Benito, W. K. Maser and M. T. Martinez, J. Mater. Chem., 2012, 22, 21285–21297 RSC.
- J. Sui and X. Liu, Surf. Coat. Technol., 2013, 235, 469–474 CrossRef CAS.
- E. Papirer, R. Lacroix, J.-B. Donnet, G. Nansé and P. Fioux, Carbon, 1995, 33, 63–72 CrossRef CAS.
- M. Song, S. Liu, J. Yin and H. Wang, Int. J. Mol. Sci., 2011, 12, 4964–4974 CrossRef CAS PubMed.
- W. Zhang, U. S. Rattanaudompol, H. Li and D. Bouchard, Water Res., 2013, 47, 1793–1802 CrossRef CAS PubMed.
- P. Yi and K. L. Chen, Langmuir, 2011, 27, 3588–3599 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS PubMed.
- D. Bouchard, X. Ma and C. Isaacson, Environ. Sci. Technol., 2009, 43, 6597–6603 CrossRef CAS PubMed.
-
M. Elimelech, J. Gregory, X. Jia and R. A. F. Williams, Particle Deposition and Aggregation: Measurement, Modelling and Simulation, Elsevier Science, 2013 Search PubMed.
- J. A. Brant, J. Labille, C. O. Robichaud and M. Wiesner, J. Colloid Interface Sci., 2007, 314, 281–288 CrossRef CAS PubMed.
- W. B. Hardy, J. Phys. Chem. B, 1899, 4, 235–253 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information includes free chlorine concentrations, XPS spectrum of NaCl, aggregation kinetics profiles, TEM graphs, and water–octanol partition coefficients of nC60 and reacted products. See DOI: 10.1039/c6en00230g |
|
| This journal is © The Royal Society of Chemistry 2017 |