
A bifunctional solid state catalyst with enhanced cycling stability for Na and Li–O2 cells: revealing the role of solid state catalysts†
Hossein
Yadegari‡
 a,
Mohammad
Norouzi Banis‡
ab,
Andrew
Lushington
a,
Qian
Sun
a,
Ruying
Li
a,
Tsun-Kong
Sham
c and
Xueliang
Sun
*a
a,
Mohammad
Norouzi Banis‡
ab,
Andrew
Lushington
a,
Qian
Sun
a,
Ruying
Li
a,
Tsun-Kong
Sham
c and
Xueliang
Sun
*a
aDepartment of Mechanical and Materials Engineering, University of Western Ontario, London, Ontario N6A 5B9, Canada. E-mail: xsun@eng.uwo.ca
bCanadian Light Source, Saskatoon S7N 2V3, Canada
cDepartment of Chemistry, University of Western Ontario, London, Ontario N6A 5B7, Canada
First published on 2nd December 2016
Abstract
Solid state catalysts play a critical role in peroxide alkali metal–O2 cells. However, the underlying mechanism behind the catalytic activity remains controversial due to the different nature of oxygen reduction and evolutions reactions (ORR, OER) in non-aqueous cells compared to those in classic aqueous based reactions. In the present study, we reveal a detailed spectroscopic and electrochemical picture of the mechanism of catalytic activity in Na– and Li–O2 cells. We demonstrate that ORR and OER catalytic activity in alkali metal–O2 cells primarily originates from the stabilization of O2− intermediates on the catalyst surface during the electrochemical reaction. Monitoring the electronic state of the solid state catalyst during the ORR and OER revealed a dynamic interaction occurring between the catalyst and the discharge product. The morphology and composition of discharge products is also illustrated to be influenced by solid state catalysts. The findings of the present study suggest that catalysts with a higher oxygen-bonding capability may exhibit a higher catalytic activity in alkali metal–O2 cells.
Broader contextAlkali metal–oxygen (Li– and Na–O2) batteries have attracted a great deal of attention over the past decade. The high theoretical energy density of these battery systems which is comparable with that of gasoline makes them desirable candidates for potential applications in electrical transportation. However, multiple basic challenges associated with the working mechanisms of alkali metal–oxygen cells limit their cycle life and hinder them from further development. The large overpotential required for charging the cells with a peroxide discharge product is among the major challenges facing the alkali metal–oxygen batteries. An extensive amount of effort has been devoted to develop and employ solid-state catalysts in order to reduce the charging overpotential and improve the cycling stability of the cells. Nevertheless, a little is known about the mechanism of the catalytic activity in these cells which makes it a controversial topic in the field. The present study reveals detailed spectroscopic evidence toward the working mechanisms of solid-state catalysts in alkali metal–oxygen cells. The obtained results suggest a correlation between the ability of the catalyst surface for stabilizing superoxide (O2−) intermediates with the catalytic activity. |
Introduction
Alkali metal (Li and Na)–O2 cells are considered as the next generation of electrochemical energy storage technology with potential applications for electrical transportation.1–5 The high energy density produced by alkali metal–O2 cells is based on coupling a high energy alkali metal (negative electrode) with a breathing oxygen electrode (positive electrode). The resulting reaction, known as an oxygen reduction reaction (ORR), produces superoxide (O2−) ions which combine with alkali metal ions, from the negative electrode, to form a solid metal oxide complex as the discharge product.4,6 The superoxide then further reduces (chemically or electrochemically) to peroxide (O22−) in Li–O2 cells and produces lithium peroxide (Li2O2) as the major discharge product of the cell.7 In the case of a Na–O2 cell, however, the larger radii of the sodium ion stabilizes the superoxide intermediate and result in the formation of either sodium superoxide (NaO2) or peroxide (Na2O2) as the product of the cell.8–13 The formed solid discharge products of the cells are then forced to decompose back to molecular oxygen and alkali metal during the charge cycle. However, the oxygen evolution reaction (OER) in the air electrode of alkali metal–O2 peroxide cells requires a large overpotential, thereby significantly reducing the energy efficiency of the cell.1–3,5The large charging overpotential in Li− and Na–O2 peroxide cells originates from various factors including the irreversible electrochemistry of the O2/O22− redox pairs,6 poor electric conductivity of the discharge product,14–20 instability of the electrolyte and the air electrode,21,22 and formation of insulating parasitic products during the discharge and charge cycles of the cells.10,23,24 On the other side, Li– and Na–O2 cells with superoxide discharge products exhibit a lower charging overpotential compared to their peroxide counterparts.4,12,13,25 The low charging overpotential observed in superoxide cells stems from the reversible electrochemical reaction of the O2/O2− redox pair. In contrast, peroxide cells undergo a multi-step ECC mechanism during the discharge cycle, followed by a single-step decomposition reaction during the charge cycle.6 This large overpotential required to reverse the products produced in peroxide cells typically results in a significantly diminished cycle life.2,9,26 Critical components of the cell, such as the electrolyte and the air electrode, are unstable at high positive potentials and produce insulating parasitic side products in an oxidative decomposition process. Accumulation of parasitic side products at the electrode/electrolyte interface further increases the charging overpotential of the cell, resulting in capacity failure.10,24 Meanwhile, the cycle life of superoxide cells is also limited to a few tens of cycles.27–29 Chemical instability of the oxygen-rich discharge product against the cell electrolyte and/or mechanical detachment of the micrometer-sized crystalline superoxide product at the early stage of the charge process often results in a limited cycle life of the superoxide cells.27,30,31
Solid state catalysts have been widely employed in peroxide cells to reduce the charging overpotential and improve the cycle life of cells.32–35 However, the mechanism behind the catalytic activity in these alkali metal–O2 cells is still a point of controversy. On one side, formation of the very first layers of the solid products during the discharge cycle is argued to hinder the catalytic activity very quickly.36 On the other side, solid state catalysts have been proven to enhance the electro/chemical performance of the peroxide cells.32–35 Multiple interpretations have been proposed as the potential mechanism behind the function of solid state catalysts in Li–O2 cells. Bruce et al. showed that the catalytic activity in nanoporous gold and TiC based air electrodes is based on the suppression of side reactions occurring due to the air electrode and electrolyte degradation.32,33 Nazar et al. attributed the advanced performance of mesoporous pyrochlore catalysts to the presence of defect sites with oxygen vacancies on a conductive nanoporous network.34 Shao-Horn et al. also proposed that the Pt–Au nanoparticles boost the sluggish kinetics of the ORR and OER in non-aqueous electrolytes.35 In addition, catalysts may alter the chemical composition, size, structure, density and electronic conductivity of the metal oxide discharge products in alkali metal O2 cells by stabilizing various oxygen–metal intermediates during the ORR.5,37 Recently, growth of a stable LiO2 phase on iridium nanoparticles further highlighted the role of catalysts in controlling the chemical composition of the discharge product in Li–O2 cells.4 Similarly, catalytic activity is also expected to enhance the electrochemistry of Na–O2 superoxide and peroxide cells.
In the present paper, we reveal detailed spectroscopic evidence toward the mechanism governing the catalytic function in Na– and Li–O2 peroxide cells. Herein, a 3D structured air electrode with bifunctional catalytic activity was synthesized and employed in Na– and Li–O2 peroxide cells. The air electrode was composed of light weight graphene foam as a macroporous framework, facilitating rapid oxygen and metal ion transportation into and from the air electrode. Nitrogen doped carbon nanotubes (NCNTs) were also synthesized on the graphene foam substrate to increase the active surface area of the air electrode. Additionally, a mesoporous Mn3O4 network combined with atomically deposited Pd nanoclusters as ORR and OER catalysts was mounted on the carbonaceous framework. We show that the catalytic activity in alkali metal–O2 cells is a result of the dynamic interaction occurring between the catalyst and the discharge product. Besides, the observed synergetic effect between Mn3O4 and Pd is shown to originate based on an electron exchange between the noble metal and the metal oxide.
Results and discussion
The air electrode designed in this study is based on the 3D structure of Ni foam as the underlying skeleton. This 3D skeleton exhibits a macroporous structure which serves for fast oxygen and ion transportation into the air electrode. However, large void mass of the Ni foam dramatically decreases the specific capacity of the air electrode. Therefore, Ni foam was employed as both a template and CVD catalyst simultaneously to synthesize graphene. The Ni foam was then removed through an etching process, leaving hollow and light-weight graphene foam (Gr). The resultant Gr foam maintained the underlying 3D structure of the Ni foam following Ni removal, while decreasing the electrode mass (Fig. S1, ESI†). Nonetheless, Gr foam presents a limited surface area due to its macroporous structure. To increase the surface area of the electrode, nitrogen doped carbon nanotubes (NCNTs) were synthesized on the Gr foam. A modified spray pyrolysis chemical vapor deposition (SPCVD) method was employed to synthesize NCNTs.10 As depicted in Fig. 1a–c, the Gr foam is uniformly covered by randomly oriented NCNTs. The random orientation of the NCNTs provides increased void volume, exposing ample surface area for catalyst deposition as well as electrolyte accessibility and accommodation of discharge products. In the next synthesis step, Mn3O4 with a mesoporous structure was synthesized on the Gr/NCNT electrode using a chemical precipitation method. The obtained electrode is denoted as Gr/NCNT/MnO throughout the text. Mesoporous Mn3O4 not only serves as an ORR catalyst, but also increases the active surface area of the air electrode and provides appropriate pore sizes for accommodation of discharge products.9 The microstructure of the Gr/NCNT/MnO electrode (Fig. 1d–f) shows uniform deposition of mesoporous Mn3O4 all over the NCNT surface.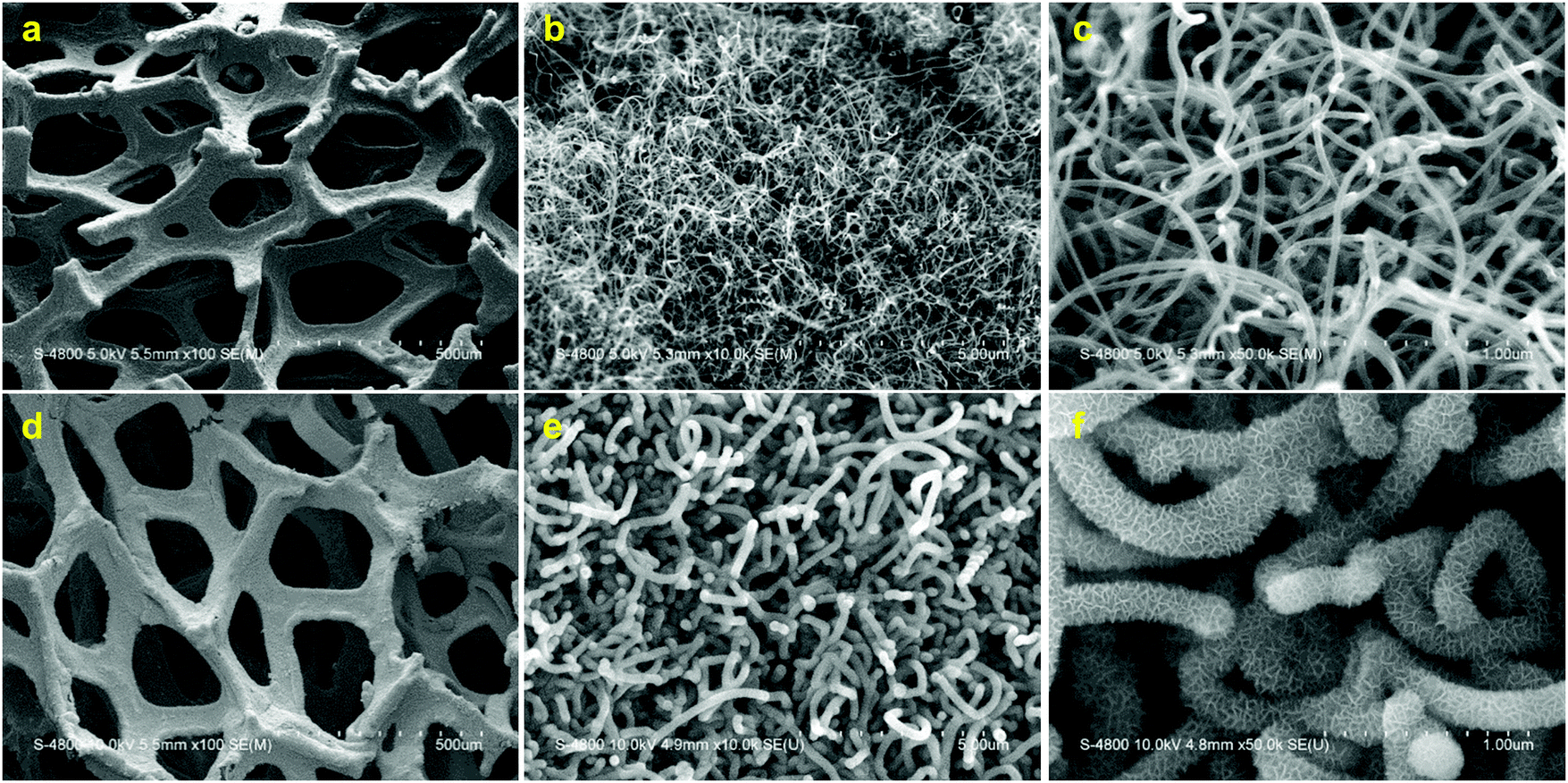 | ||
| Fig. 1 SEM micrographs of Gr/NCNT (a–c) and Gr/NCNT/MnO (d–f) electrodes with different magnifications. | ||
Subsequently, an ultrathin Pd film was deposited on the Gr/NCNT/MnO surface using atomic layer deposition (ALD). The thickness of the Pd film can be precisely controlled by controlling the number of ALD cycles employed. Cycle numbers of 50, 100 and 150 were applied to the final air electrode which is denoted as Gr/NCNT/MnO/Pd-(50C, 100C or 150C) throughout the text. To further clarify the synergetic effect between Mn3O4 and Pd, control air electrodes without Mn3O4 were also synthesized and are denoted as Gr/NCNT/Pd-(50C, 100C or 150C). Detailed synthesis procedures as well as physicochemical characterization of the electrode materials are provided in the ESI† (Fig. S1–S11). X-ray absorption spectroscopy (XAS) was employed to confirm the chemical state of Mn3O4 as well as the formation of Pd on Mn3O4. The normalized Mn K-edge X-ray absorption near-edge structure (XANES) spectra of Gr/NCNT/MnO and Gr/NCNT/MnO/Pd are compared to the standard sample of Mn3O4 in Fig. 2a. All XANES spectra display a weak pre-edge absorption peak which can be attributed to a forbidden 1s dipole transition to the unoccupied 3d orbitals.38 The main absorption edge of synthesized Mn3O4, correlated with the Mn 1s → 4p transition, appears at a slightly higher energy compared to the standard sample, indicating a higher oxidation state for the synthesized Mn3O4.
 | ||
| Fig. 2 Normalized Mn (a) and TEY O (b) K-edge XANES spectra of standard Mn3O4, Gr/NCNT/MnO and Gr/NCNT/MnO/Pd electrodes; (c) normalized Pd K-edge XANES spectra of standard Pd foil, Gr/NCNT/Pd and Gr/NCNT/MnO/Pd electrodes; (d) Fourier transform K3-weighted EXAFS spectra of standard Pd foil, Gr/NCNT/Pd and Gr/NCNT/MnO/Pd electrodes alongside with simulated results for Pd and PdO crystals; (e) schematic diagram for Pd ALD growth mechanisms on NCNT and Mn3O4 surfaces. | ||
In addition, no changes can be seen in the XANES spectra of Gr/NCNT/MnO before and after Pd deposition, suggesting that the Mn3O4 substrate is stable during the ALD process. However, a distinct change can be seen between the O K-edge spectra of Gr/NCNT/MnO and Gr/NCNT/MnO/Pd electrodes (Fig. 2b). The standard Mn3O4 sample as well as Gr/NCNT/MnO exhibit a feature around 531 eV. This excitation is split into two peaks due to the ligand field and has been attributed to the electron transition from core O 1s orbitals to 2p–3d hybridized Mn orbitals (t2g and eg symmetry bands).39 Meanwhile, the broad feature around 534 eV is related to unoccupied oxygen p states mixed with the manganese 4sp band. The O K-edge spectra of MnO2, Mn2O3 and Mn3O4 standard samples are also depicted in Fig. S12 (ESI†) for comparison. In the case of the Gr/NCNT/MnO/Pd electrode, however, the pre-edge feature (t2g) is followed by an intense and broad feature which overlaps the eg peak. Appearance of this broad feature after Pd deposition is a result of overlapping of O 2p with Mn 3d and Pd 4d orbitals. In fact, the O K-edge XANES results illustrate that O 2p orbitals bridge between Mn 3d and Pd 4d orbitals. Further, similar features appear in the O K-edge spectra recorded under the FLY mode (Fig. S13, ESI†), confirming that both mesoporous Mn3O4 and Pd nanoclusters are composed of thin layers with a uniform surface and bulk properties.
The normalized Pd K-edge XANES spectra of Gr/NCNT/Pd and Gr/NCNT/MnO/Pd electrodes, along with Pd foil are presented in Fig. 2c. Gr/NCNT/Pd shows the same characteristic features as the Pd foil, indicating that the ALD Pd nanoparticles on the NCNT surface exhibit a similar oxidation state, as well as a local structure, compared to bulk Pd. The Gr/NCNT/MnO/Pd electrode also demonstrates the same adsorption edge as Pd foil, confirming the presence of metallic Pd at the zero-valence state on the Mn3O4 surface. However, in contrast to the Pd nanoparticles on NCNTs, the local structure of the deposited Pd on Mn3O4 is obviously different from the bulk Pd (evident from the features in the multiple scattering region). Fig. 2d shows the Fourier transform K3-weighted extended X-ray absorption fine structure (EXAFS) spectra of the electrode materials. The simulated results for Pd and PdO crystals are also shown for comparison. The feature around 2.6 Å, existing for all electrodes, represents the Pd–Pd bond. In addition, comparing the EXAFS data of Gr/NCNT/Pd with Pd foil confirms the metallic nature of Pd nanoparticles on NCNTs. In contrast, the R-space analysis of Gr/NCNT/MnO/Pd shows a significant decrease in the relative intensity of Pd–Pd along with an additional feature at 1.5 Å which can be correlated with a Pd–O bond.40,41 Meanwhile, the Pd–Pd feature appearing at 2.6 Å (compared with the Pd–Pd in PdO crystals at 3 Å) contradicts the formation of PdO. The low intensity of the Pd–Pd peak implies low coordination of Pd atoms, indicating that the Pd layer on Mn3O4 is composed of nanoclusters.41 In addition, the presence of the first shell Pd–O peak in the EXAFS spectra of the Gr/NCNT/MnO/Pd electrode illustrates the presence of an oxygen layer within the vicinity of the Pd layer, bridging between the Pd and Mn atoms. To determine the structural parameters of Pd on various substrates, the experimental data were fitted using a non-linear least-squares curve fitting procedure (Fig. S14, ESI†). The obtained structural parameters from curve fitting are presented in Table 1. The comparison of the Pd–Pd coordination number (CN) for Gr/NCNT/Pd with Pd foil indicates that the Pd nanoparticles on the NCNT surface are slightly oxidized. In the case of Gr/NCNT/MnO/Pd, however, the Pd–Pd CN is significantly decreased while the Pd–O CN is increased, confirming the formation of planar nanoclusters of Pd on the Mn3O4 surface.
| Pd–Pd coordination | Pd–O coordination | Pd–Pd Debye–Waller factor (Å2) | Pd–O Debye–Waller factor (Å2) | |
|---|---|---|---|---|
| Gr/NCNT/MnO/Pd | 3.581 | 2.188 | 7.52 × 10−3 | 4.91 × 10−3 |
| Gr/NCNT/Pd | 9.56 | 0.332 | 6.14 × 10−3 | 4.92 × 10−3 |
| Pd foil | 12 | — | 5.41 × 10−3 | — |
The XAS results alongside SEM, TEM and XRD illustrate that the structure of Pd deposited on Mn3O4 is different from the Pd nanoparticles on the NCNT surface. This structural difference originates from the distinct ALD growth mechanisms on these surfaces. The half-reaction of Pd on the surface of a substrate has preferential deposition toward high-energy defect sites on a non-uniform substrate like NCNT, resulting in nucleation of Pd. With increasing number of ALD cycles, the initially formed nucleation sites grow in size to form islands of Pd on the NCNT surface. This island growth regime is also known as the Volmer–Weber mechanism. In contrast to NCNTs, Mn3O4 provides a more uniform surface which leads to formation of Pd nanoclusters on the oxide surface.42 Increasing the ALD cycles on the Mn3O4 surface results in planar expansion of Pd nanoclusters. These nanoclusters may grow and coalesce together after multiple ALD cycles to form a continuous film. Based on the XAS analysis, formation of Pd nanoclusters on Mn3O4 is highly likely. The Pd deposited on Mn3O4 demonstrates a significantly lower Pd–Pd CN than Pd nanoparticles on NCNTs (see Table 1). This low Pd–Pd CN suggests that Pd nanoclusters on Mn3O4 have a thickness of a few atomic layers. The ALD growth mechanisms on NCNTs and Mn3O4 surfaces are also schematically outlined in Fig. 2e.
The electrochemical responses of the prepared electrodes were examined in both Na– and Li–O2 cells. The discharge/charge curves of the prepared electrodes in Na– and Li–O2 cells are presented in Fig. 3a and b, respectively (see also Fig. S15, ESI†). Gr/NCNT electrodes exhibit the typical discharge/charge behavior seen for carbonaceous materials with large charging overpotentials occurring for both cells.2,10 A noticeable increase in the discharge potential of both Na– and Li–O2 cells can be seen for Gr/NCNT/MnO electrodes. This increase in discharge potential is greater in Li–O2 cells. Similar electrocatalytic effects for Mn3O4 on the ORR in alkaline solutions43–45 as well as Li–O2 cells46 have been reported previously. The presence of surface defects and hydroxyl (–OH) functional groups on the surface of manganese oxide facilitates the adsorption of oxygen onto the reaction sites and decreases the ORR activation energy. Moreover, the redox reaction involving Mn species is also believed to contribute to the high electrocatalytic activity of manganese oxide.47 The charging overpotential in both Na– and Li–O2 cells is also diminished by either the addition of mesoporous Mn3O4 or Pd nanoparticles onto the NCNT surface. The discharge/charge curves of Gr/NCNT/Pd electrodes with various ALD cycles in both Na– and Li–O2 cells are also depicted in Fig. S16 (ESI†).
 | ||
| Fig. 3 Discharge/charge curves of Gr/NCNT, Gr/NCNT/MnO, Gr/NCNT/Pd-150C and Gr/NCNT/MnO/Pd-150C electrodes in Na– (a) and Li–O2 (b) cells under a current density of 0.1 mA cm−2; the cycling performance of the Gr/NCNT/MnO/Pd-150C electrode in Na– (c) and Li–O2 (d) cells. | ||
The mechanism behind the catalytic activity of noble metals, transition metals and metal oxides toward the OER of alkali metal–O2 cells is not fully understood.5,37 However, the catalytic behavior observed here could be attributed to the multiple factors. The poor electric conductivity of alkali metal oxides as discharge products for Na– and Li–O2 has been considered as one of the reasons for the large charging overpotential observed in these cells.14,15,18,19 The increased active surface area in the case of Gr/NCNT/MnO and Gr/NCNT/MnO/Pd electrodes decreases the thickness of the insulating discharge products on the electrode surface by providing ample room for deposition of discharge products. Therefore, mesoporous Mn3O4 decreases the charging overpotential of the cells by enhancing both charge and mass transfer through the discharge product layer.48 In addition, the formation of an insulating carbonate-based parasitic product, as a result of the oxidative reaction of discharge products of Na– and Li–O2 with carbonaceous air electrodes, has been shown to significantly increase the charging overpotential of the cell.10,22,23 The shielding effect of the catalyst layer over the NCNT surface may also contribute toward reducing the overpotential by preventing the formation of insulating carbonate species at the air electrode/discharge product interface.
Meanwhile, a different electrocatalytic mechanism is responsible for decreasing the charge overpotential for Gr/NCNT/Pd electrodes. The presence of Pd nanoparticles on the carbon surface is believed to enhance the electronic transport across the electrode/electrolyte interface and hence decrease the charging overpotential of the cell.40 Interestingly, the Gr/NCNT/MnO/Pd electrode displays a significant decrease in charging overpotential for both Na– and Li–O2 cells, while maintaining the high discharge potential observed for Gr/NCNT/MnO. Evidently, Gr/NCNT/MnO/Pd demonstrates a bifunctional catalytic performance in alkali metal–O2 cells by improving both the discharge and charge potentials.35 Quantitatively, Gr/NCNT/MnO/Pd reduces the charging overpotential of Na– and Li–O2 cells by 1.0 and 0.8 V, respectively, compared to Gr/NCNT electrodes. The decrease in overpotential observed for Gr/NCNT/MnO/Pd is greater than the summation of the reduction in overpotential seen for Gr/NCNT/MnO and Gr/NCNT/Pd electrodes, indicating a synergetic effect occurring between Mn3O4 and Pd (see Fig. S17, ESI† for discharge/charge curves of Gr/NCNT/MnO/Pd electrodes with various ALD cycles).
The cycling performance of the Gr/NCNT/MnO/Pd-150C electrode in Na– and Li–O2 cells is depicted in Fig. 3c and d, respectively, while the corresponding chronopotentiaograms are shown in Fig. S18 (ESI†). The prepared electrode maintains a low overpotential for 100 consecutive discharge and charge cycles. The difference between discharge and charge potentials of the cells starts to increase during the last 20 cycles. This is most likely related to degradation of the negative electrode.27,40 In addition, decomposition of the cell electrolyte by oxidative discharge products formed in the cells may also produce carbonate-based parasitic side-products and result in increased overpotential.10,21,23,24 The cycling performance of Gr/NCNT/Pd and Gr/NCNT/MnO/Pd electrodes with various Pd ALD cycles in both Na– and Li–O2 cells is also presented in Fig. S19 and S20 (ESI†) for comparison. The advanced catalytic performance of Gr/NCNT/MnO/Pd originates from two basic characteristics of the electrode: (1) high surface area of mesoporous structured Mn3O4 and (2) bifunctional and synergistic catalytic activity of Mn3O4/Pd toward ORR and OER. The high surface area of the Mn3O4 provides an increased number of reaction sites for oxygen molecules during the ORR. Surface defects and hydroxyl functional groups on the surface of the metal oxide aid in facilitating oxygen adsorption onto the air electrode. Therefore, Mn3O4 serves as an ORR catalyst by reducing the activation energy of the reaction as well as improving mass transfer kinetics at the electrode surface. In addition, the mesoporous structure of Mn3O4 is ideal for accommodating the discharge products of Na– and Li–O2 cells.9,49 Furthermore, Pd nanoclusters on the surface of Mn3O4 improve the charge transfer at the electrode/product interface.
In order to explore the mechanism behind the electrocatalytic activity in alkali metal–O2 peroxide cells, the discharge products of Na–O2 cells on various electrodes were examined using the XAS technique. XAS provides both selectivity and sensitivity toward multiple metal oxide phases that may be present in the discharge products of Na–O2 cells.10 The normalized O K-edge XANES spectra of the various discharged electrodes were recorded using surface sensitive total electron yield (TEY) as well as bulk sensitive fluorescence yield (FLY) modes for both Na– and Li–O2 cells, as shown in Fig. 4. A single feature centered at 534 eV for the TEY O K-edge spectra of the discharged electrodes in Na–O2 cells (Fig. 4a) is related to the excitation of the 1s electrons to empty 3σu (σ*) molecular orbitals of Na2O2, indicating the presence of a peroxide discharge product in these cells. In the case of the Gr/NCNT/MnO/Pd electrode, however, a feature at 530 eV is present which can be related to the excitation of 1s electrons to semi-filled 1πg (π*) molecular orbitals of superoxide molecules.10 The second feature at 534 eV is related to the 1s-σ* transition in superoxide compounds which may overlap with σ* orbitals present in peroxide. Nevertheless, the small energy difference between σ* molecular orbitals of superoxide and peroxide phases cannot be distinguished here. A similar conclusion can also be obtained from the FLY spectra obtained for discharged electrodes (Fig. 4b). However, a small feature at 530 eV is present in the Gr/NCNT/MnO spectra, indicating the presence of a superoxide phase at the bulk of the product on the Mn3O4 surface. For Li–O2 cells, both peroxide and superoxide peaks can be observed at relatively lower energies. The observed negative shift in XAS peaks from Na–O2 to Li–O2 products confirms that the feature around 530 eV is related to the superoxide phase in the discharge products rather than the underneath Mn3O4. In addition, an increased amount of superoxide phase was detected on the surface of the Gr/NCNT/MnO electrode (Fig. 4c and d). It should also be noted that formation of superoxide rather than peroxide as the discharge product of the cells results in a decrease of the exchanged electron number.
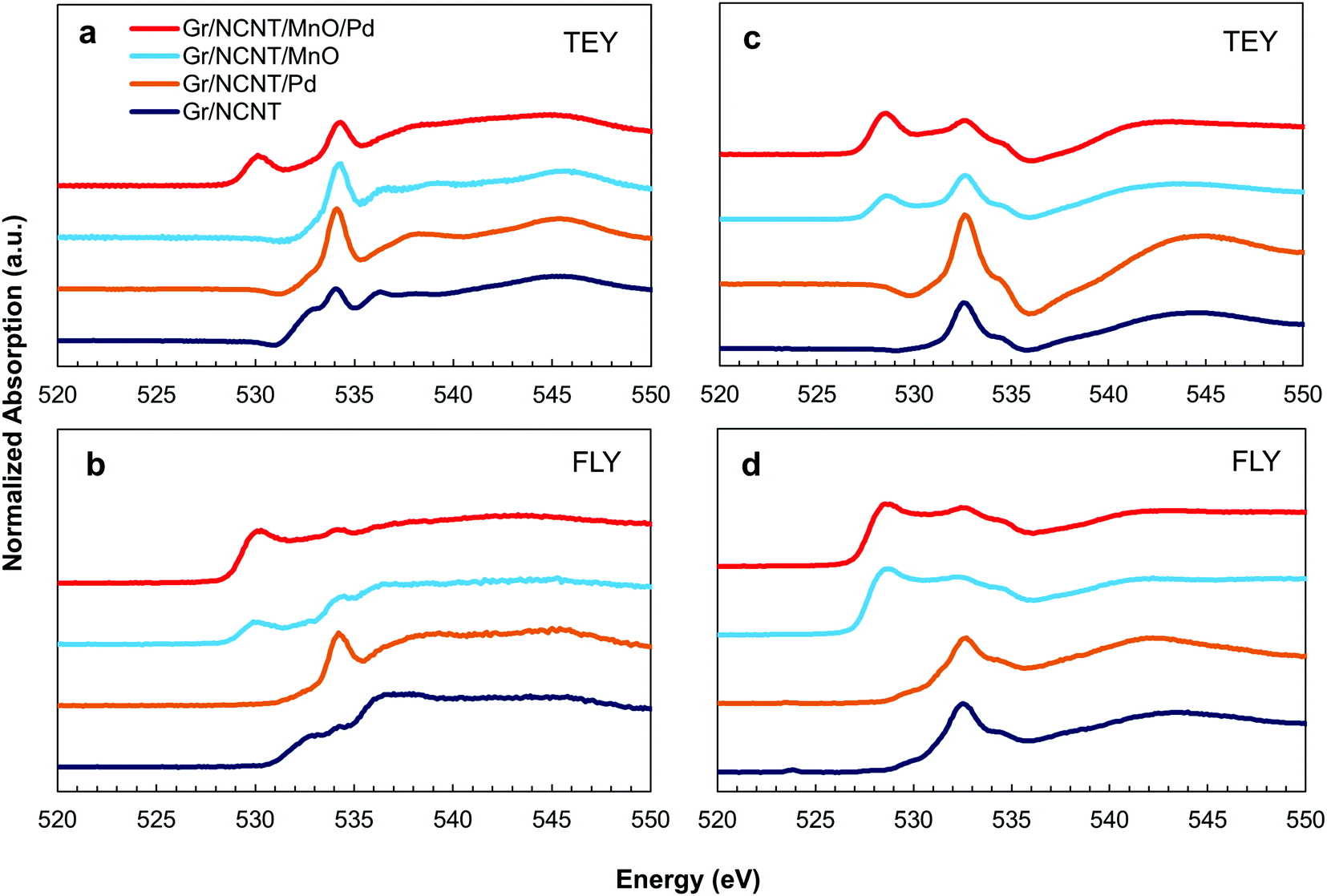 | ||
| Fig. 4 Normalized O K-edge XANES spectra of the discharged electrodes on Gr/NCNT, Gr/NCNT/MnO, Gr/NCNT/Pd-150C and Gr/NCNT/MnO/Pd-150C electrodes recorded under the both surface sensitive TEY (a and c) and bulk sensitive FLY (b and d) modes for both Na– (a and b) and Li–O2 (c and d) cells. | ||
Formation of superoxide products on the Gr/NCNT/MnO/Pd electrode is in good agreement with the low overpotential observed during the charge cycle of Na– and Li–O2 cells (Fig. 3a and b). Accordingly, the electrocatalytic activity of Pd nanoclusters on Mn3O4 can be correlated with the strong oxygen bonding present at the catalyst surface.50 Pd nanoclusters not only facilitate the reduction of O2 to O2−, but also aid in stabilizing superoxide radicals. The O2− ion then combines with Na+ or Li+ to form adsorbed NaO2 or LiO2 films as discharge products of the cells. Conversion of discharge products of peroxide Li–O2 cells into an oxygen-rich superoxide phase on the catalyst surface has also been reported recently.4 To confirm the proposed role of catalysts in alkali metal–O2 cells, the Gr/NCNT/MnO/Pd electrode was also examined by XAS during the first electrochemical discharge/charge cycle in Na–O2 cells. Normalized Mn and Pd K-edge XANES spectra of the Gr/NCNT/MnO/Pd electrode at the end of the 1st discharge and 1st discharge and charge cycle in a Na–O2 cell are compared with those of pristine electrodes in Fig. 5a and b. The Mn K-edge XANES spectra of the electrodes demonstrate a positive shift following cell discharge, illustrating partial oxidation of Mn3O4 during the discharge process. At the same time, Pd K-edge XANES spectra exhibit a slight negative shift with cell discharge. This likely stems from electron reception from adsorbed O2− ions on the Pd surface. Following the initial discharge/charge cycle, a reverse trend can be seen to some extent in which Pd exhibits a slight positive shift compared to the discharged electrode, while Mn3O4 is slightly reduced. The reverse trend of oxidation/reduction is observed for Mn3O4 and Pd during discharge/charge cycling, confirming mutual electron exchange occurring between two species. During the discharge cycle, electrons are shuttled between the Mn 3d orbitals and the Pd 4d orbitals to reduce oxygen at the electrode surface. As a result, Mn is partially oxidized at the end of the discharge cycle while Pd is slightly reduced. Reverse migration of electron density takes place during the charging process, resulting in partial oxidation of Pd and reduction of Mn at the end of charge. Such an electron transfer between Mn3O4 and Pd nanoclusters explains the synergetic effect observed in the catalytic activity of the electrodes.
 | ||
| Fig. 5 Normalized Mn (a) and Pd (b) K-edge XANES spectra of the Gr/NCNT/MnO/Pd-150C electrode at the end of 1st discharge and 1st discharge and charge cycle in a Na–O2 cell; (c) normalized Pd K-edge XANES spectra of the Gr/NCNT/MnO/Pd-150C electrode before and after 100 discharge/charge cycles in Na–O2 cells (terminated after the charge cycle); (d) Fourier transform K3-weighted EXAFS spectra of the Gr/NCNT/MnO/Pd-150C electrode before and after 100 discharge/charge cycles in Na–O2 cells. | ||
To further confirm the electron exchange between Mn3O4 and Pd nanoclusters, the Pd K-edge XANES spectra of the Gr/NCNT/Pd control electrode were also recorded at the end of the 1st discharge and 1st discharge/charge cycle in a Na–O2 cell (Fig. S21, ESI†). The Gr/NCNT/Pd electrode demonstrates a similar absorption edge at different discharge and charge cycles, suggesting that electron exchange is not occurring between the Pd nanoparticles and the carbon surface. However, Pd nanoparticles on NCNTs exhibit a structural change in the EXAFS spectra, probably due to the adsorption of oxygen onto the Pd surface. The Pd K-edge XANES spectra of Gr/NCNT/MnO/Pd electrodes before and after 100 discharge/charge cycles in Na–O2 cell (terminated after the charge cycle) are depicted in Fig. 5c. The cycled electrode displays a very close Pd absorption edge before and after cycling in the cell. However, the R-space analysis of the EXAFS spectra (Fig. 5d) illustrates that the relative intensity of the Pd–O over Pd–Pd peak increases with cycling (see also Fig. 2d). The structural parameters of Pd following multiple charge/discharge cycles in Na–O2 cells were also obtained by fitting the experimental data (Fig. S22, ESI†) and are compared with data obtained for the pristine Gr/NCNT/MnO/Pd electrode in Table S2 (ESI†). The fitting results indicate that the Pd–O coordination number increases following cell cycling. The increase in the Pd–O coordination number with cycling indicates that Pd nanoclusters interact with the discharge products of the cell during discharge and charge cycles. The results also confirm the dynamic role of solid state catalysts in the electrochemical reaction of Na–O2 cells.
The morphology of the discharge products in alkali metal–O2 cells has also been used to derive information pertaining to the mechanism of electro/chemical reactions occurring within cells.51–53 The discharge product morphology in Li– and Na–O2 cells using various air electrodes following discharge to 1.0 mA h cm−2 is shown in Fig. S23 and S24 (ESI†), respectively. The products formed in Li–O2 cells using the Gr/NCNT electrode are observed to be composed of toroidal particles with a diameter of 1 μm. A similar morphology can be seen for the Gr/NCNT/Pd electrode. However, the diameter of the toroidal particles found on this electrode is on the order of 100 nm. Gr/NCNT/MnO and Gr/NCNT/MnO/Pd electrodes, however, display a formation of a very uniform product within the mesopores of the electrode. Similar amorphous phase products were also observed for Na–O2 cells using air electrodes composed of Gr/NCNT/MnO and Gr/NCNT/MnO/Pd (Fig. S24, ESI†). Two distinct growth mechanisms have been proposed for the formation of Li2O2 in Li–O2 cells. On one side, the surface-mediated electrochemical mechanism forms a conformal surface coating with thicknesses dictated by charge transport through the product film.53 On the other side, the solution-mediated growth mechanism involves partial dissolution of O2− in non-aqueous electrolytes and produces micron-sized Li2O2 toroids.4,52,53 The latter growth mechanism is also suggested to be responsible for the formation of cubic NaO2 discharge products in Na–O2 cells.13,54 Direct observation of product growth on various electrode surfaces here indicates that the growth mechanism is switched from a solution-mediated one on NCNTs to a surface-mediated mechanism on Mn3O4 and/or Pd surfaces. The increased active surface area given by Mn3O4 greatly increases the nucleation sites available for the ORR. In addition, elevated oxygen bonding toward the surface of Mn3O4 and Pd contributes to stabilize the O2− intermediate and decreases its solubility into the electrolyte. As a result, the morphology of the discharge products changes from micron-sized toroids on NCNTs to a conformal film on Mn3O4 and Pd surfaces. The observed changes in the morphology of the discharge products are in precise accordance with the transformation in the chemical composition of the products in Li– and Na–O2 cells.
The presented spectroscopic data provide insightful evidence toward the mechanism governing the electrocatalytic activity of solid-state catalysts in alkali metal–O2 cells. The catalytic activity during the ORR mainly involves the ability of the catalyst surface to absorb molecular oxygen and reduce the activation energy required for the ORR. In the case of the Gr/NCNT/MnO/Pd electrode employed in this study, Pd nanoclusters deposited on high surface area mesoporous Mn3O4 provide a strong interaction between the active surface and molecular oxygen and therefore facilitate the reduction reaction by forming a chemisorbed oxygen layer. However, atomically deposited Pd nanoparticles on the NCNT substrate do not show a significant ORR activity. The increased ORR catalytic activity observed for Pd on Mn3O4 originates from both structural and electronic configurations. The relatively uniform surface of Mn3O4 results in the formation of planar Pd nanoclusters with a high surface area, providing an increased number of active reaction sites. The XAS results demonstrated that the Pd nanoclusters formed on the Mn3O4 surface contain a few atomic layers of Pd located in the vicinity of oxygen rich environments. In addition, the O K-edge XANES results confirmed the interaction of O 2p with both Mn 3d and Pd 4d orbitals. Thus, the formation of planar Pd nanoclusters on mesoporous Mn3O4 enables the electron exchange between d orbitals of Pd and Mn3O4. As a result, the electron density in Pd 4d orbitals decreases due to the O 2p bridging effect between Mn 3d and Pd 4d orbitals. The reduced electron density in Pd 4d orbitals facilitates the chemisorption of molecular oxygen onto the Pd surface. Accordingly, the synergetic effect seen between Pd and Mn3O4 can also be correlated with the electron exchange between these metallic d orbitals. The catalytic mediated discharge/charge reaction mechanism in Na– and Li–O2 cells is schematically outlined in Fig. 6.
 | ||
| Fig. 6 Schematic diagram of catalytic mediated discharge and charge reaction mechanisms on the Mn3O4/Pd surface. | ||
The OER catalytic activity, however, relies on the ability of the catalyst to stabilize the superoxide intermediate. XAS examination of the discharge product in Na–O2 cells using the Gr/NCNT/MnO/Pd air electrode revealed that the product of the cell is mainly composed of superoxide species. However, the product formed in the cells using Gr/NCNT and Gr/NCNT/Pd air electrodes was mainly the peroxide phase. In addition, a significant transformation in the morphology of discharge products was also observed alongside the corresponding changes in the chemical composition of the products on various electrode surfaces. The observed changes in the growth mechanism of the products are also in agreement with the increase of oxygen bonding availability of the electrode surfaces from NCNTs to Mn3O4 and Pd. Therefore, changes in the chemical composition as well as the morphology of the discharge products are both contributing factors in reducing the charge overpotential of alkali-metal cells. Recent studies have demonstrated that trace amounts of electrolyte additives such as water may act as an electrochemical mediator and enhance the discharge capacity of alkali metal–O2 cells.13,53,55 However, these soluble additives may also impose destructive influences on the cyclability of cells by reacting with the discharge products and increasing the occurrence of the non-Faradaic side-reactions. Employing solid state catalysts with the capability of stabilizing superoxide discharge products eliminates the side effects of soluble additives, while maintaining the advantages of superoxide cells.
Conclusion
A hierarchical 3D structured carbonaceous air electrode was developed in this study. The air electrode was then decorated with mesoporous Mn3O4 as an ORR catalyst, followed by atomic layer deposition of Pd nanoclusters as OER catalysts. The prepared bifunctional catalyst displayed a synergetic effect between Pd nanoclusters and mesoporous Mn3O4 for the OER in both Na– and Li–O2 cells, performing more than 100 consecutive cycles. Detailed XAS studies on both the solid-state catalyst as well as the discharge products of the cells revealed a correlation between the composition of cell products and the electronic state of the underlying catalyst during the ORR and OER. The observed catalytic activity was accordingly explained based on the ability of the catalyst surface to stabilize the O2− intermediate. In addition, the influence of solid state catalysts on the morphology of discharge products by imposing a distinct growth mechanism during the discharge reaction was also verified. We suggest, based on the presented results here, that solid state catalysts with higher oxygen-bonding capability may exhibit premier catalytic activity in alkali metal–O2 cells.Acknowledgements
This research was supported by Natural Sciences and Engineering Research Council of Canada, Canada Research Chair Program, Canada Foundation for Innovation, Canadian Light Source (CLS) and the University of Western Ontario.References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- J. Lu, Y. J. Lee, X. Luo, K. C. Lau, M. Asadi, H. H. Wang, S. Brombosz, J. Wen, D. Zhai, Z. Chen, D. J. Miller, Y. S. Jeong, J. B. Park, Z. Z. Fang, B. Kumar, A. Salehi-Khojin, Y. K. Sun, L. A. Curtiss and K. Amine, Nature, 2016, 529, 377–382 CrossRef CAS PubMed.
- H. Yadegari, Q. Sun and X. Sun, Adv. Mater., 2016, 28, 7065–7093 CrossRef CAS PubMed.
- Z. Q. Peng, S. A. Freunberger, L. J. Hardwick, Y. H. Chen, V. Giordani, F. Barde, P. Novak, D. Graham, J. M. Tarascon and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 6351–6355 CrossRef CAS PubMed.
- S. A. Freunberger, Y. H. Chen, N. E. Drewett, L. J. Hardwick, F. Barde and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2009, 113, 20127–20134 CAS.
- H. Yadegari, Y. Li, M. N. Banis, X. Li, B. Wang, Q. Sun, R. Li, T.-K. Sham, X. Cui and X. Sun, Energy Environ. Sci., 2014, 7, 3747–3757 CAS.
- H. Yadegari, M. N. Banis, B. Xiao, Q. Sun, X. Li, A. Lushington, B. Wang, R. Li, T.-K. Sham, X. Cui and X. Sun, Chem. Mater., 2015, 27, 3040–3047 CrossRef.
- B. D. McCloskey, J. M. Garcia and A. C. Luntz, J. Phys. Chem. Lett., 2014, 5, 1230–1235 CrossRef CAS PubMed.
- P. Hartmann, C. L. Bender, M. Vracar, A. K. Durr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- M. D. Radin, J. F. Rodriguez, F. Tian and D. J. Siegel, J. Am. Chem. Soc., 2012, 134, 1093–1103 CrossRef CAS PubMed.
- V. Viswanathan, K. S. Thygesen, J. S. Hummelshoj, J. K. Norskov, G. Girishkumar, B. D. McCloskey and A. C. Luntz, J. Chem. Phys., 2011, 135, 214704 CrossRef CAS PubMed.
- Y. Mo, S. P. Ong and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205446 CrossRef.
- M. D. Radin and D. J. Siegel, Energy Environ. Sci., 2013, 6, 2370–2379 CAS.
- B. Lee, D.-H. Seo, H.-D. Lim, I. Park, K.-Y. Park, J. Kim and K. Kang, Chem. Mater., 2014, 26, 1048–1055 CrossRef CAS.
- O. Arcelus, C. Li, T. Rojo and J. Carrasco, J. Phys. Chem. Lett., 2015, 6, 2027–2031 CrossRef CAS PubMed.
- R. B. Araujo, S. Chakraborty and R. Ahuja, Phys. Chem. Chem. Phys., 2015, 17, 8203–8209 RSC.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043–3047 CrossRef CAS PubMed.
- M. M. O. Thotiyl, S. A. Freunberger, Z. Q. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshoj, J. K. Norskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- B. M. Gallant, R. R. Mitchell, D. G. Kwabi, J. Zhou, L. Zuin, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. C, 2012, 116, 20800–20805 CAS.
- D. Zhai, H. H. Wang, J. Yang, K. C. Lau, K. Li, K. Amine and L. A. Curtiss, J. Am. Chem. Soc., 2013, 135, 15364–15372 CrossRef CAS PubMed.
- Q. Sun, H. Yadegari, M. N. Banis, J. Liu, B. Xiao, B. Wang, S. Lawes, X. Li, R. Li and X. Sun, Nano Energy, 2015, 12, 698–708 CrossRef CAS.
- P. Hartmann, C. L. Bender, J. Sann, A. K. Durr, M. Jansen, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 11661–11672 RSC.
- C. L. Bender, P. Hartmann, M. Vračar, P. Adelhelm and J. Janek, Adv. Energy Mater., 2014, 4, 1301863 CrossRef.
- N. Zhao, C. Li and X. Guo, Phys. Chem. Chem. Phys., 2014, 16, 15646–15652 RSC.
- N. Ortiz-Vitoriano, T. P. Batcho, D. G. Kwabi, B. Han, N. Pour, K. P. C. Yao, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 2636–2643 CrossRef CAS PubMed.
- J. Kim, H. Park, B. Lee, W. M. Seong, H. D. Lim, Y. Bae, H. Kim, W. K. Kim, K. H. Ryu and K. Kang, Nat. Commun., 2016, 7, 10670 CrossRef CAS PubMed.
- Z. Q. Peng, S. A. Freunberger, Y. H. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng, Y. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050–1056 CrossRef CAS PubMed.
- S. H. Oh, R. Black, E. Pomerantseva, J. H. Lee and L. F. Nazar, Nat. Chem., 2012, 4, 1004–1010 CrossRef CAS PubMed.
- Y. C. Lu, Z. C. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS PubMed.
- B. D. McCloskey, R. Scheffler, A. Speidel, D. S. Bethune, R. M. Shelby and A. C. Luntz, J. Am. Chem. Soc., 2011, 133, 18038–18041 CrossRef CAS PubMed.
- A. Kraytsberg and Y. Ein-Eli, Nano Energy, 2013, 2, 468–480 CrossRef CAS.
- K. W. Nam, M. G. Kim and K. B. Kim, J. Phys. Chem. C, 2007, 111, 749–758 CAS.
- F. M. F. Degroot, M. Grioni, J. C. Fuggle, J. Ghijsen, G. A. Sawatzky and H. Petersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 5715–5723 CrossRef CAS.
- J. Lu, Y. Lei, K. C. Lau, X. Y. Luo, P. Du, J. G. Wen, R. S. Assary, U. Das, D. J. Miller, J. W. Elam, H. M. Albishri, D. Abd El-Hady, Y. K. Sun, L. A. Curtiss and K. Amine, Nat. Commun., 2013, 4, 2383 Search PubMed.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. L. Wang, J. J. Li, S. Q. Wei and J. L. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- S. A. Park, H. Lim and Y. T. Kim, ACS Catal., 2015, 5, 3995–4002 CrossRef CAS.
- Y. Gorlin, C. J. Chung, D. Nordlund, B. M. Clemens and T. F. Jaramillo, ACS Catal., 2012, 2, 2687–2694 CrossRef CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- Q. Li, P. Xu, B. Zhang, H. H. Tsai, J. Wang, H. L. Wang and G. Wu, Chem. Commun., 2013, 49, 10838–10840 RSC.
- F. Y. Cheng, Y. Su, J. Liang, Z. L. Tao and J. Chen, Chem. Mater., 2010, 22, 898–905 CrossRef CAS.
- R. Black, J. H. Lee, B. Adams, C. A. Mims and L. F. Nazar, Angew. Chem., Int. Ed., 2013, 52, 392–396 CrossRef CAS PubMed.
- Y. Li, X. Li, D. Geng, Y. Tang, R. Li, J.-P. Dodelet, M. Lefèvre and X. Sun, Carbon, 2013, 64, 170–177 CrossRef CAS.
- Y. C. Lu, H. A. Gasteiger and Y. Shao-Horn, J. Am. Chem. Soc., 2011, 133, 19048–19051 CrossRef CAS PubMed.
- B. M. Gallant, D. G. Kwabi, R. R. Mitchell, J. Zhou, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 2518–2528 CAS.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1778 CAS.
- N. B. Aetukuri, B. D. McCloskey, J. M. Garcia, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed.
- P. Hartmann, M. Heinemann, C. L. Bender, K. Graf, R.-P. Baumann, P. Adelhelm, C. Heiliger and J. Janek, J. Phys. Chem. C, 2015, 119, 22778–22786 CAS.
- J. Staszak-Jirkovský, R. Subbaraman, D. Strmcnik, K. L. Harrison, C. E. Diesendruck, R. Assary, O. Frank, L. Kobr, G. K. H. Wiberg, B. Genorio, J. G. Connell, P. P. Lopes, V. R. Stamenkovic, L. Curtiss, J. S. Moore, K. R. Zavadil and N. M. Markovic, ACS Catal., 2015, 5, 6600–6607 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee03132c |
| ‡ Hossein Yadegari and Mohammad Norouzi Banis have equivalent contributions to this work. |
| This journal is © The Royal Society of Chemistry 2017 |