
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of 1′-(diphenylphosphino)-1-isocyanoferrocene, an organometallic ligand combining two different soft donor moieties, and its Group 11 metal complexes†
Karel
Škoch
 a,
Ivana
Císařová
a,
Jiří
Schulz
a,
Ulrich
Siemeling
b and
Petr
Štěpnička
*a
a,
Ivana
Císařová
a,
Jiří
Schulz
a,
Ulrich
Siemeling
b and
Petr
Štěpnička
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40 Prague 2, Czech Republic. E-mail: petr.stepnicka@natur.cuni.cz
bInstitute of Chemistry, University of Kassel, Heinrich-Plett-Str. 40, 34132 Kassel, Germany
First published on 18th July 2017
Abstract
The development of a practical synthesis of 1′-(diphenylphosphino)-1-aminoferrocene (2) and its P-borane adduct (2B) allowed the facile preparation of 1′-(diphenylphosphino)-1-isocyanoferrocene (1). This compound combining two specific soft-donor moieties was studied as a ligand for univalent Group 11 metal ions. The reactions of 1 with AgCl at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 molar ratios only led to the coordination polymer [Ag2(μ-Cl)2(μ(P,C)-1)]n (6), while those with Ag[SbF6] provided the dimer [Ag2(Me2CO-κO)2(μ(P,C)-1)2][SbF6]2 and the quadruply-bridged disilver complex [Ag2(μ(P,C)-1)4][SbF6]2 (8), respectively. Addition of 1 to [AuCl(tht)] (tht = tetrahydrothiophene) afforded the mono- and the digold complex, [AuCl(1-κP)] (9) and [(μ(P,C)-1)(AuCl)2] (10), depending on the reaction stoichiometry. Finally, the reaction of 1 with [Au(tht)2][SbF6] or halogenide removal from 9 with AgNTf2 led to cationic dimers [Au2(μ(P,C)-1)2]X2 (11, X = SbF6 (a) or NTf2 (b)). Catalytic tests in the Au-mediated isomerization of (Z)-3-methylpent-2-en-4-yn-1-ol to 2,3-dimethylfuran revealed that 11a and 11b are substantially less catalytically active than their analogues containing 1′-(diphenylphosphino)-1-cyanoferrocene as the ligand, most likely due to a stronger coordination of the isonitrile moiety, which prevents dissociation of the dimeric complexes into catalytically active monomeric species.
:1 and 2:1 molar ratios only led to the coordination polymer [Ag2(μ-Cl)2(μ(P,C)-1)]n (6), while those with Ag[SbF6] provided the dimer [Ag2(Me2CO-κO)2(μ(P,C)-1)2][SbF6]2 and the quadruply-bridged disilver complex [Ag2(μ(P,C)-1)4][SbF6]2 (8), respectively. Addition of 1 to [AuCl(tht)] (tht = tetrahydrothiophene) afforded the mono- and the digold complex, [AuCl(1-κP)] (9) and [(μ(P,C)-1)(AuCl)2] (10), depending on the reaction stoichiometry. Finally, the reaction of 1 with [Au(tht)2][SbF6] or halogenide removal from 9 with AgNTf2 led to cationic dimers [Au2(μ(P,C)-1)2]X2 (11, X = SbF6 (a) or NTf2 (b)). Catalytic tests in the Au-mediated isomerization of (Z)-3-methylpent-2-en-4-yn-1-ol to 2,3-dimethylfuran revealed that 11a and 11b are substantially less catalytically active than their analogues containing 1′-(diphenylphosphino)-1-cyanoferrocene as the ligand, most likely due to a stronger coordination of the isonitrile moiety, which prevents dissociation of the dimeric complexes into catalytically active monomeric species.
Introduction
Isocyanides are useful synthetic intermediates1 as well as attractive σ-donor/π-acceptor ligands for coordination chemistry.2 Even though the ferrocene motif has been frequently used in ligand design,3 ferrocene-based isocyanides were long overlooked. For instance, isocyanoferrocene (A, Scheme 1) was first reported in 19874,5 (i.e., relatively late for a simple ferrocene derivative6) and later studied as a redox-active metalloligand in transition metal complexes.4,7 More recent studies focused on its doubly functionalized congener, 1,1′-diisocyanoferrocene (B),8 disclosed unexpected coordination and reactivity of this compound in multinuclear complexes and formation of self-assembled monolayers on gold surfaces.9 Presumably the latest entry into the still narrow class of ferrocene-based isonitriles10 is the planar-chiral derivative, (Sp)-2-methyl-1-isocyanoferrocene (C).11 | ||
| Scheme 1 | ||
Recently we reported the synthesis of 1′-(diphenylphosphino)-1-cyanoferrocene (D) and the coordination properties of this soft–soft hybrid donor toward Group 11 metals.12 The often unusual structures of the isolated complexes as well as excellent catalytic properties of the Au(I)-D complexes12b led us to expand our studies toward the closest isomeric compound, 1′-(diphenylphosphino)-1-isocyanoferrocene (1 in Scheme 1), which formally falls between diisocyanide B and the widely studied 1,1′-bis(diphenylphosphino)ferrocene (dppf).13
Although the transposition of the nitrogen and carbon atoms in the structures of D and 1 might appear marginal in terms of chemical constitution, it can be expected to dramatically alter the reactivity and coordination properties of these compounds. It is also noteworthy that similar ligands combining isocyanide and phosphine donor moieties remain limited to only a handful of compounds including Ph2P(CH2)nNC (n = 2,14 315) and the recently reported compound E (Scheme 1).16
This contribution describes the preparation and structural characterization of the new phosphino-isonitrile ligand 1 and its complexes with Ag(I) and Au(I) ions. Also reported are the results of preliminary catalytic tests with Au(I)-1 complexes aimed at a comparison with their Au(I)-D counterparts.
Results and discussion
Synthesis of phosphino-isonitrile 1
In analogy to the syntheses of organic isocyanides,17 we anticipated that our target compound 1 can be obtained from the known 1′-(diphenylphosphino)-1-aminoferrocene (2) by formylation and subsequent dehydration (Scheme 2). Although this assumption later proved correct, this straightforward plan had to be modified because the synthesis of amine 2 from 1′-(diphenylphosphino)-1-bromoferrocene (3)18 described in the literature19 was found to be irreproducible.20,21 | ||
| Scheme 2 Intended preparation of 1 from amine 2. | ||
Considering the generally low stability of aminoferrocene derivatives, the often harsh reaction conditions required to obtain them,22 an also the sensitivity of phosphine groups to oxidation, the synthetic routes toward 1 were redesigned to include phosphine-protected intermediates that allow the introduction of the azide moiety (without an unwanted Staudinger reaction) and its subsequent reduction to amine group (Scheme 3).
 | ||
| Scheme 3 Alternative syntheses of 1 exploiting different P-protected intermediates. Legend: TsN3 = 4-toluenesulfonyl azide, BOP = (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, dabco = 1,4-diazabicyclo[2.2.2]octane, DBU = 1,8-diazabicyclo(5.4.0)undec-7-ene. | ||
Because some of us have successfully employed the protection of a phosphine group by thionation during the synthesis of Ph2PfcNHCH2t-Bu21a (fc = ferrocene-1,1′-diyl), a similar approach was adopted also for the synthesis of 1 (Scheme 3, left branch) with the prospect that removal of the P-bound sulfur atom and reduction of the azide group (–N3 → –NH2) can be effected in a single step by reacting intermediate 4S with Raney nickel. The starting phosphine bromide 3 was smoothly thionated with elemental sulfur and the stable P-sulfide 3S was lithiated and treated with 4-toluenesulfonyl azide (TsN3) to afford azide 4S in a good yield. A small amount of TsN3 present in the reaction product did not interfere during the following step, after which it could be easily removed. The subsequent reaction of 4S with Raney nickel and then with acetic formic anhydride provided the desired formamide 5 as a ca. 4:5 mixture of (E) and (Z) isomers (N.B. the acylation was performed directly to prevent a possible decomposition of intermediate amine 2). However, the transformation of 4S into 5 typically suffered from low yields (below 30%) and the product was contaminated by the dephosphorylated amide, FcNHCHO (Fc = ferrocenyl).23
In the last step, which was the dehydration of formamide 5 to isonitrile 1, the standard reagents (POCl3/NEt3, COCl2/iPr2NH)24 failed, typically resulting in extensive darkening of the reaction mixture and oxidation of the phosphine moiety (confirmed by 31P NMR analysis). This led us to utilize the method previously used to convert oxime Ph2PfcCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH into nitrile D.25 Gratifyingly, addition of Castro's reagent (i.e., (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, BOP) and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) to amide 5 ensued in a clean formation of isonitrile 1, which was isolated in a 71% yield following chromatographic purification.
NOH into nitrile D.25 Gratifyingly, addition of Castro's reagent (i.e., (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, BOP) and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) to amide 5 ensued in a clean formation of isonitrile 1, which was isolated in a 71% yield following chromatographic purification.
Although the synthesis of 1 was successful, we sought for a more efficient synthetic procedure, utilizing borane as the protecting group (Scheme 3, right branch). The necessary starting material 3B was prepared by the reaction of phosphine-bromide 3 with BH3·SMe2 or, alternatively, via a practical one-pot procedure (lithiation, phosphinylation, borylation) from 1,1′-dibromoferrocene.18 Lithiation of 3B followed by quenching with TsN3 afforded azide 4B in a 89% yield. Compound 4B was found to be stable when stored at low temperatures in the dark but gradually decomposed upon exposure to direct day light. The subsequent reduction with Li[AlH4] in THF smoothly converted azide 4B into the protected amine 2B. This compound was in turn formylated to give 5B or isolated and converted the free phosphinoamine 2. Deprotection of 5B with 1,4-diazabicyclo[2.2.2]octane (dabco) or, alternatively, acylation of 2 with HCOOAc afforded formamide 5, which was in turn used to prepare the target isocyanide 1 as outlined above (see Scheme 3).
Although longer by one step, the synthesis of 1via borane-protected intermediates proved to be much more efficient in terms of the overall yield (compare the 70% yield of 1 from bromide 3 with ca. 25% yield for the 3 → 1 conversion via the PS intermediates) and selectivity. Besides, it provided a reliable access to amine 2 and its P-protected form 2B that are both attractive for further syntheses. Compound 2B appears to be particularly stable in both the solid state and solution. For instance, this adduct did not undergo any detectable decomposition or BH3 migration between the Lewis basic amine and phosphine groups when its solutions in CDCl3, CD3CN and C6D6 were stored at room temperature for 2 days. Accordingly, the reaction of 2 with 1 equiv. of BH3·SMe2 (in CDCl3) produced adduct 2B as the sole product (N.B. adduct 2·2BH3 was detected only when larger amounts of the borane source were employed).
Isocyanide 1 and all intermediates occurring en route to this compound were characterized by multinuclear NMR and IR spectroscopy, mass spectrometry and elemental analysis or HR MS measurements. In addition, the solid state structures of 1, 2, 4S, 4B, and 5 were determined by single-crystal X-ray diffraction analysis. The 1H and 13C NMR spectra confirmed the presence of an unsymmetrically 1,1′-disubstituted ferrocene moiety and the attached phosphorus groups in all cases. The manipulations at the phosphorus substituent were manifested in the 31P NMR spectra, showing broad signals at δP ≈ 16 for the BH3 adducts, and sharp singlets at δP ≈ −17 and 42 for the free phosphines and phosphine sulfides, respectively. In addition to characteristic B–H stretching bands of the borane adducts at 2340–2400 cm−1,26 the IR spectra showed distinct features due to the polar substituents, namely intense azide ν(N3) bands (4S: 2109 cm−1, 4B: 2108 cm−1) and amide ν(CO) vibrations (5 and 5B: 1663 cm−1). The band attributable to the ν(N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) vibration of 1 was seen at 2126 cm−1 (cf. 2125 cm−1 for PhNC).27
C) vibration of 1 was seen at 2126 cm−1 (cf. 2125 cm−1 for PhNC).27
The crystal structures of 1 and intermediates 4S, 4B, 2 and 5
The molecular structures of azides 4S and 4B are presented in Fig. 1 and selected geometric parameters are given in Table 1. Compound 4S crystallizes with two structurally independent molecules, which differ in the conformation of their ferrocene units (see the τ angles in Table 1) and in the orientation of the phenyl rings with respected to the ferrocene moiety (for an overlap of the independent molecules, see the ESI, Fig. S4†). Otherwise, the parameters describing the molecular geometry of the azides are similar to the respective data reported for FcN3,28 fc(N3)3,29 FcP(S)Ph2,30 dppfS2,31 and dppf·2BH332 (Fc = ferrocenyl).
 | ||
| Fig. 1 Molecular structures of 4S (molecule 1) and 4B. | ||
| Parameter | 4S (Y = S) | 4B (Y = B) | |
|---|---|---|---|
| mol 1 | mol 2 | ||
| a Definitions: Cp1 and Cp2 are the nitrogen- and phosphorus-substituted cyclopentadienyl rings, respectively. Cg1 and Cg2 denote their centroids. τ = torsion angle C1–Cg1–Cg2–C6. 4S: n = void/5 for molecules 1/2, 4B: n = void. | |||
| Fe–Cg1 | 1.652(1) | 1.649(2) | 1.654(1) |
| Fe–Cg2 | 1.646(1) | 1.632 (1) | 1.6464(9) |
| ∠Cp1,Cp2 | 1.4(1) | 1.3(2) | 2.6(1) |
| τ | −122.8(2) | 154.9(2) | 108.1(2) |
| P–Cn6 | 1.790(2) | 1.790(3) | 1.790(2) |
| P–Y | 1.9567(8) | 1.950(1) | 1.926(2) |
| Cn1–Nα | 1.415(4) | 1.417(5) | 1.422(3) |
| Nα–Nβ | 1.235(4) | 1.251(4) | 1.262(3) |
| Nβ–Nγ | 1.130(4) | 1.143(5) | 1.123(4) |
| Cn1–Nα–Nβ | 115.6(2) | 114.9(3) | 115.4(2) |
| Nα–Nβ–Nγ | 173.3(3) | 171.3(4) | 172.1(3) |
The structures of 1 and 2 are displayed in Fig. 2. Relevant structural data are given in Table 2. The ferrocene unit in the structure of 1 has a synclinal eclipsed conformation (compare the τ angle in Table 2 with the ideal value of 72°)13a and is negligibly tilted. Its phosphine substituent is oriented so that one phenyl group points above the ferrocene scaffold while the other as well as the lone pair of electrons are directed to the sides. The length of the NC bond of 1.157(3) is identical to that in FcNC (1.157(5) Å)33 and the C1–NC11 moiety is linear (177.3(2)°).
 | ||
| Fig. 2 Molecular structures of 1 and 2. | ||
| Parameter | 1 | 2 | 5 |
|---|---|---|---|
| a Definitions: Cp1 and Cp2 are the cyclopentadienyl rings C(1–5) and C(6–10), respectively. Cg1 and Cg2 denote their centroids. τ = torsion angle C1–Cg1–Cg2–C6. b Further data: C11–N = 1.157(3), C1–N–C11 = 177.3(2). c Further data: N–C24 = 1.338(2), C1–N–C24 = 126.5(1), C24–O = 1.227(2), N–C24–O = 125.1(2). | |||
| Fe–Cg1 | 1.6438(8) | 1.6585(7) | 1.6482(7) |
| Fe–Cg2 | 1.6431(8) | 1.6467(7) | 1.6408(6) |
| ∠Cp1,Cp2 | 1.4(1) | 1.09(9) | 2.79(9) |
| τ | −70.0(1) | 69.0(1) | −86.3(1) |
| P–C6 | 1.814(2) | 1.810(1) | 1.813 (1) |
| C1–N | 1.391(2) | 1.410(2) | 1.407(2) |
The arrangement of the phosphinoferrocene moiety in the structure of amine 2 is practically the same as in 1, including the orientation of the cyclopentadienyl rings, and the C1–N bond in 2 has the same length as that in aminoferrocene (1.405(5) Å).34 Notably, the crystal structure of amine 2 lacks classical N–H⋯N hydrogen bonds. However, one of the amine hydrogen atoms (H1N) appears to be involved in an intramolecular N–H⋯π interaction, being directed towards the phenyl ring C(18–23) with an H⋯Cg distance (Cg is the ring centroid) of 2.93 Å.
Recrystallization of formamide 5 from chloroform/hexane furnished crystals of the major (Z) isomer, which were used for the structure determination. In the crystal, compound 5 forms infinite chains via N–H1N⋯O hydrogen bridges (N⋯O = 2.808(2) Å) between molecules distributed around crystallographic glide planes (Fig. 3 and Table 2). The individual molecules show a minor tilting at their ferrocene units (ca. 3°) and an intermediate conformation between synclinal eclipsed and anticlinal staggered. The amide moiety is coplanar with its parent cyclopentadienyl ring (dihedral angle: 0.8(2)°), which suggests conjugation of these molecular parts.
 | ||
| Fig. 3 Section of the hydrogen-bonded chain in the structure of 5 as viewed along the crystallographic a axis. For clarity, the CHn hydrogen atoms are omitted. | ||
Complexes of 1 with Group 11 metals
With reference to our studies focused on the coordination behavior of phosphinonitrile donor D (Scheme 1),12 we set out to investigate the coordination properties of the isomeric ligand 1 towards univalent Group 11 metal ions. Unfortunately, reactions of 1 with CuCl and [Cu(MeCN)4]X (X = BF4 and PF6) as the common Cu(I) sources repeatedly failed to provide any defined products, typically giving rise to intractable precipitates (presumably of polymeric nature).A defined, though also practically insoluble, product (N.B the compound did not dissolve even in MeCN or DMSO) resulted upon reacting ligand 1 (1 or 2 equiv.) with freshly prepared AgCl (Scheme 4). X-ray diffraction analysis on crystalline 6 obtained by reaction of AgCl with 2 equiv. of 1 in chloroform and crystallization by addition of hexane revealed that the structure of 6 is built up from diamond-shaped {Ag2(μ-Cl)2} fragments that are interconnected by four P,C-bridging ligands 1 into an infinite one-dimensional chain (Fig. 4). Although central Ag2Cl2 motifs were found in the structures of, e.g., [(Ph3P)2Ag(μ-Cl)]235 and [(dppf-κ2P,P′)Ag(μ-Cl)]2,36 the only related compound featuring two different donors whose structure has been reported to date is the dinuclear complex [(Ph3P)(MeNC)Ag(μ-Br)]2.37 Indeed, the structures of 6 and the latter compound are similar except that in 6 the two donor moieties are tethered by the ferrocene unit, which in turn results in the formation of a polymeric assembly. It is also noteworthy that ligand D comprising the relatively harder cyano group reacted with AgCl to give heterocubane [Ag(μ3-Cl)(D-κP)]4 (Ag:D = 1:1) or the dimers [Ag(D-κP)2(μ-Cl)]2 and [AgCl(D-κP)(μ(P,N)-D)]2 (Ag:D = 1:2).12c
 | ||
| Scheme 4 Reaction of 1 with AgCl leading to coordination polymer 6. | ||
 | ||
| Fig. 4 A section of the infinite polymeric array in the structure of 6. For clarity, all H atoms and the less populated position of the disordered silver(I) ion were omitted. Coordination geometry parameters (in Å and °): Ag1–P 2.4529(7), Ag1–C11 2.164(3), Ag1–Cl 2.6428(7), Ag1–Cl′ 2.6358(8), P–Ag1–C11 117.71(7), P–Ag1–Cl 111.46(2), P–Ag1–Cl′ 103.46(2), C11′′–Ag1–Cl 110.35(7), C11′′–Ag1–Cl′ 120.30(8), Ag1–Cl–Ag1′ 89.76(2), Cl–Ag1–Cl′ 90.24(2). | ||
The structure of 6 is presented in Fig. 4 along with selected distances and angles. Although the silver atoms appear disordered over two positions differently positioned between the phosphine and isonitrile donors (refined ratios: ≈97:3), the overall structure is rather symmetrical. The central Ag2Cl2 units are practically regular squares and lie on crystallographic inversion centers. Each silver(I) ion is further coordinated by two phosphino-isonitrile ligands 1, once via the phosphorus atom and once through the isonitrile moiety. Both Ag–P and Ag–C bonds are shorter than the sum of the respective covalent radii (∑rcov(Ag,P) = 2.52 Å, ∑rcov(Ag,Csp) = 2.14 Å)38 and the observed Ag–C distance (2.164(3) Å) is close to the average Ag–C distances in Ag–isonitrile complexes [2.14(8) Å (ref. 39)]. The coordination environment of the silver ions is distorted tetrahedral. Among the interligand angles, the Cl–Ag1–Cl′ angle is the most acute (90°) and the C11′′–Ag1–Cl (120°) and P–Ag1–C11′′ (118°) angles are the most opened.
The ferrocene moiety in the structure of 6 is roughly perpendicular to the Ag2Cl2 ring (cf. the dihedral angle of the C(1–5) ring and the Ag2Cl2 plane of 82.6(1)°) and has an intermediate conformation with τ of −52.5(2)°. Notably, the length of the coordinated CN bond (1.163(4) Å) is indistinguishable within experimental error from that in free 1.
Addition of ligand 1 (1 equiv.) to Ag[SbF6] containing a weakly coordinating anion in acetone led to the dimeric complex [Ag2{μ(P,C)-1}2(Me2CO-κO)2][SbF6]2 (7 in Scheme 5). This compound is structurally similar to [Ag2{μ(P,N)-D}2(AcOEt-κO)2][SbF6]2 obtained from Ag[SbF6] and ligand D.12c Unlike the latter compound, however, complex 7 appears to be stable in solution, which was manifested by a sharp 31P NMR signal at δP 6.8 split into a pair of concentric doublets by interactions with 109Ag and 107Ag (1JAg,P = 709 and 615 Hz). Coordination of the isonitrile moiety was indicated by a shift of the ν(NC) band in the IR spectrum to 2210 cm−1 (by 84 cm−1 to higher energies with respect to 1). The IR spectrum further confirmed the presence of coordinated acetone through a sharp band of the ν(CO) vibration at 1695 cm−1, which is 20 cm−1 less than for neat acetone due to coordination.40
 | ||
| Scheme 5 Synthesis of Ag(I)-1 complexes from Ag[SbF6]. | ||
Upon increasing the amount of the phosphino-isonitrile ligand to 2 molar equiv., Ag[SbF6] was smoothly converted to the quadruply-bridged disilver(I) complex [Ag2{μ(P,C)-1}4][SbF6]2 (8), which resembles an analogous Cu(I) complex obtained from ligand D, [Cu2{μ(P,N)-D}4][SbF6]2.12a In its 31P NMR spectrum, complex 8 gave rise to a broad doublet at δP −1.2 (1JAg,P = 207 Hz).41 The IR band due to coordinated isonitrile groups was observed at 2167 cm−1, i.e. shifted by ca. 40 cm−1 to higher energies compared to uncoordinated ligand 1.
Compounds 7 and 8 have been structurally authenticated by X-ray diffraction analysis. The complex cations in the crystal structure of 7 (Fig. 5) lie around crystallographic inversion centers, which makes only their halves structurally independent. Their tricoordinate silver ions exhibit an almost linear arrangement of the ligating P and C atoms, with a minor distortion due to the coordinated acetone (P–Ag–C ≈ 166°). The Ag–P and Ag–C bonds in 7 are shorter than those in 6 (by 0.07 and 0.05 Å), suggesting stronger dative bonds in the former cationic complex. In contrast, the Ag–O distance is substantially elongated (compare Ag–O1S ≈ 2.63 Å with ∑rcov(Ag,O) = 2.11 Å), presumably due to a relatively weaker coordination and steric reasons.
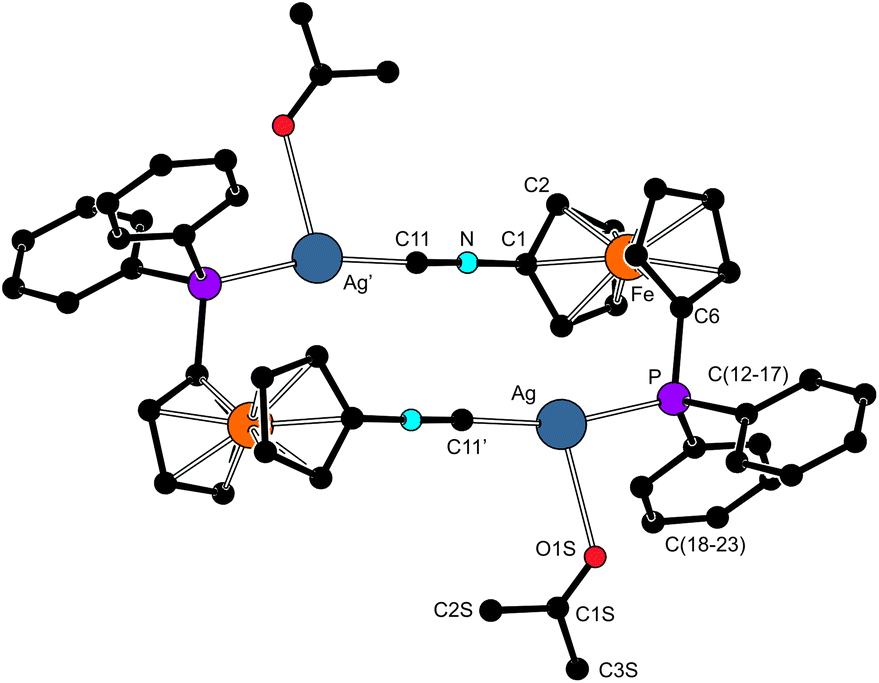 | ||
| Fig. 5 View of the complex cation in the structure of 7. The hydrogen atoms were omitted for clarity. Coordination geometry parameters (in Å and °): Ag–P 2.385(1), Ag–C11′ 2.110(4), Ag–O1S 2.630(4), P–Ag–C11′ 165.5(1), P–Ag–O1S 91.29(9), C11′–Ag–O1S 102.0(2). | ||
The cyclopentadienyl rings in 7 are tilted by 2.4(2)° and adopt a conformation near to synclinal eclipsed (τ = 83.8(1)°), which brings the phosphine moiety to the side of the fc-NC moiety (cf. the angle subtended by the P–C6 and C1–N bonds of 79.7(2)°). Consequently, the P–Ag bond points to the same direction as the NC moiety, which in turn allows the formation of dimeric units Ag2(μ(P,C)-1)2 consisting of two equivalent parts arranged in a side-by-side manner that laterally coordinate the acetone molecules.
The structure determination on 8·3Me2CO (Fig. 6) revealed a compact and symmetrical complex molecule located around a crystallographic two-fold axis (space group C2/c; the silver atoms reside on this axis). Each silver(I) ion has a distorted tetrahedral P2C2 donor set being coordinated by four P,C-bridging ligands 1. The lengths of the two structurally independent Ag–P bonds differ by ca. 0.036 Å. A more significant asymmetry observed for the Ag–CN bonds (0.059 Å) is not relayed further as the CN bonds are similar in length and the Ag–CN and Cp–NC moieties remain linear. Despite the very different steric demands of the donor moieties, the interligand angles around the silver(I) ions do not depart much from the tetrahedral value, spanning a relatively narrow range of 103.35(9)–115.88(3)°.
 | ||
| Fig. 6 View of the complex cation in the structure of 8·3Me2CO. The hydrogen atoms were omitted for clarity. Coordination geometry parameters (in Å and °): Ag1–P1 2.5376(9), Ag1–C61 2.264(3), Ag2–P2 2.5017(7), Ag2–C11 2.205(4), P1–Ag1–P1′ 115.88(3), P1–Ag1–C61 103.35(9), P1–Ag1–C61′ 112.44(9), C61–Ag1–C61′ 109.5(1), P2–Ag2–P2′ 109.65(2), P2–Ag2–C11 106.1(1), P2–Ag2–C11′ 112.61(7), C11–Ag2–C11′ 110.0(1); Ag2–C11–N1 177.9(3), N1–C11 1.150(5), C1–N1–C11 179.0(3), Ag1–C61–N2 178.5(3), N2–C61 1.148(3), C51–N2–C61 176.9(3). The prime-labeled atoms are generated by a crystallographic two-fold axis. | ||
The reactions of 1 with [AuCl(tht)] (tht = tetrahydrothiophene) paralleled the chemistry noted in the Au(I)-D system only partly (Scheme 6). Depending on the ligand-to-metal ratio, the reaction of 1 with [AuCl(tht)] afforded either the phosphine complex [AuCl(1-κP)] (9) or the trinuclear Au2Fe complex 10. Both these products are poorly soluble (especially when crystallized), which makes their full characterization by solution techniques (e.g., NMR) impossible. Nevertheless, the coordination of the phosphine moiety in 9 could be inferred from a shift of the 31P NMR signal to a lower field (δP 29.0). The IR spectrum recorded with a solid sample (Nujol mull) displayed two bands attributable to the free (2126 cm−1) and coordinated (2220 cm−1, weak band; cf. νNC 2223 cm−1 for 10) isonitrile moieties. Although unexpected, this observation is in line with the result of the structure determination revealing that a small fraction of the gold centers in the structure of 9 is displaced towards the isonitrile group of an adjacent molecule (vide infra). This phenomenon appears to be limited to the solid state as an NMR study ruled out the formation of isomeric monogold complexes (with P- and NC-bound ligand) and other species (e.g., 1 and 10 formed by redistribution).
 | ||
| Scheme 6 Preparation of Au(I)-1 complexes 9–11. | ||
The structures of 9 and 10 are shown in Fig. 7 along with selected geometric data. As stated above, the gold(I) ions in the structure of 9 are disordered over two positions (refined occupancies: Au1 ≈ 0.98, Au2 ≈ 0.02; the ligand is considered invariant). In the dominant position, the gold(I) ion has the usual linear coordination whilst in the other it is approximately trigonal with the isonitrile moiety located at a bonding distance (Au2–C11 ≈ 2.09 Å, ∑rcov(Au,Csp) = 2.05 Å). The ferrocene unit in 9 has a synclinal eclipsed conformation (τ = 70.3(2)°) which, together with the spatial orientation of the phenyl rings, allows for a compact arrangement of the linear assembly interlinked via the incidental P–Au2–NC interactions.
 | ||
| Fig. 7 Simplified views of the crystal structures of 9 (top) and 10 (bottom). Selected data for 9 (in Å and°): Au1–P 2.2298(7), Au1–Cl 2.2896(8), Au2–P 2.533(6), Au2–Cl 2.652(6), Au2–C11′ 2.094(7), P–Au1–Cl 176.12(3), P–Au2–Cl 121.2(2), Au2′–N–C11 155.1(4), N–C11 1.153(4), C1–N–C11 178.9(3). Selected data for 10 (in Å and°): Au1–P 2.230(1), Au1–Cl1 2.288(1), P–Au1–Cl1 173.93(4), Au2–C11 1.930(5), Au2–Cl2 2.260(1), C11–Au2–Cl2 176.9(1), Au2–C11–N 178.2(4). | ||
In the structure of 10, both gold centers are linearly coordinated and averted from each other because the central ferrocene unit has an opened conformation with τ = −157.0(3)° (N.B. the angle subtended by the Au1–Cl1 and Au2–Cl2 bonds is 139.26(6)°). Whereas the entire C1–NC11–Au2–Cl2 moiety is essentially linear, the Au1–Cl1 bond diverts from the plane of the parent cyclopentadinyl ring at an angle of 23.1(2)°. On the other hand, the coordination geometry parameters are not unexpected in view of the data reported for [Au2Cl2(μ(P,P′)-dppf)]42 and [AuCl(D-κP)].12c In the crystal, the molecules of 10 assemble into head-to-tail dimers via weak Au1⋯Au2 aurophilic interactions (3.21 Å; see the ESI, Fig. S12†).43
Finally, the reaction of [Au(tht)2][SbF6] with 1 (1 equiv.) or removal of the chloride ligand from 9 with AgNTf2 resulted in the formation of the respective dimeric complexes 11 (Scheme 6), which are the structural isomers of the recently reported compounds [Au2(μ(P,N)-D)2]X2 (X = SbF6 and NTf2).12b The dimeric structure of 11a and 11b was corroborated by X-ray diffraction analysis (N.B. the structure of 11b could not be adequately refined because of a severe disorder of the NTf2− anion). The coordination of both donor moieties available in ligand 1 is indicated by a shift of the 31P NMR signals to low fields (δP 32–33) and through a shift of the νNC bands in the IR spectra to higher energies (2231–2233 cm−1). Notably, both shifts are more pronounced than for the neutral complexes 9 and 10, very likely owing to a stronger interaction of the donor moieties with the electron-poor cationic Au(I) centers.
The solid-state structure of 11a·2Me2CO shown in Fig. 8 resembles that of 7, except that the coordination of the Au centers is practically undistorted linear (P–Au–C = 175.8(1)°). The ferrocene cyclopentadienyl rings adopt a conformation similar to that in 7 with τ of −80.0(3)°, which brings the linear P–Au–NC subunits side-by-side (N.B. the angle subtended by the P–Au and C11–Au′ bonds is only 4.17(1)°), though without any supporting intramoleculars aurophilic interaction (Au⋯Au′ = 5.4400(4) Å).
 | ||
| Fig. 8 View of the complex cation in the structure of 11a·2Me2CO. Hydrogen atoms, the counter anions and solvent molecules are omitted for clarity. Selected distances and angles (in Å and°): Au–P 2.2821(9), Au–C11′ 2.008(4), P–Au–C11′ 175.8(1), Au–C11′–N′ 174.3(4), N–C11 1.135(5), C1–C11–N 173.4(4). The primed atoms are generated by crystallographic inversion. | ||
Electrochemistry
The electrochemical behavior of compound 1 and 2, and of the Au(I) complexes 9–11 was studied by cyclic voltammetry on a glassy carbon disc electrode in dichloromethane containing Bu4N[PF6] as the supporting electrolyte.Compound 1 (Fig. 9) was oxidized in a single irreversible step at 0.38 V vs. the ferrocene/ferrocenium reference couple (anodic peak potential, Epa, is given at a scan rate of 100 mV s−1), which corresponds with an electron-withdrawing nature of the substituents at the ferrocene unit (cf. the Hammett σp constant: 0.46 for –NC, and 0.19 for –PPh2).44 This primary redox change attributable to the oxidation of the ferrocene moiety is followed by another irreversible redox event at a more positive potential (Epa ≈ 0.75 V).
 | ||
| Fig. 9 Cyclic voltammograms of amine 2 (blue), its BH3 adduct 2B (red) and phosphino-isonitrile 1 (green – partial, and black – a wider scan) recorded with a glassy carbon disc electrode in CH2Cl2 (scan rate 0.1 V s−1). The second sweep in the voltammogram of 1 is distinguished by a dashed line. | ||
In contrast, the oxidation of amines 2 and 2B was reversible (Fig. 9). However, the waves were shifted to less positive potentials (E°′ = −0.24 V and −0.13 V for 2 and 2B, respectively) due to the electron-donating nature of the amine substituent (σp = −0.66). Even in this case, the first oxidations were followed by irreversible processes at higher potentials.
The cyclic voltammogram of the monogold complex 9 showed two redox events in the anodic region, namely an irreversible oxidation at Epa ≈ 0.61 V and a reversible redox process at E°′ = 0.78 V (Fig. 10). Since the digold complex 10 displayed a single reversible redox change at E°′ = 0.76 V, it appears likely that the redox response of 9 can be affected by adsorption phenomena. A single reversible redox event was observed also for the dinuclear complexes 11a and 11b, whose behavior was expectedly identical (E°′ = 0.79 V; see Fig. 10).
 | ||
| Fig. 10 Cyclic voltammograms of complexes 9 (blue, the second scan is shown as a dashed line), 10 (red) and 11a (green) as recorded with a glassy carbon disc electrode in CH2Cl2 (scan rate 0.1 V s−1). | ||
Catalytic tests with Au(I)-1 complexes
Considering the favorable catalytic properties of gold(I) complexes with ligand D,12b we decided to probe the catalytic properties of the isomeric compounds featuring ligand 1. For testing, we chose the gold-catalyzed cyclization of unsaturated alcohol 12 to give 2,3-dimethylfurane (13; Scheme 7) that has been used to evaluate the catalysts based on ligand D. | ||
| Scheme 7 Au-Catalyzed cycloisomerisation of enynol 12 to 2,3-dimethylfurane. | ||
Unfortunately, the collected results (Table 3) indicate a generally very low (if any at all) catalytic activity of the prepared Au-1 complexes. For instance, compound 10 achieved a 82% yield of the cyclization product 13 after 30 min and a nearly 90% yield after 3 h at 1 mol% Au loading. These yields decreased substantially upon lowering the catalyst amount to 0.1 mol%. The monogold complex 9 and, surprisingly, also the dimers 11 proved to be catalytically inactive, which markedly contrasts with the very high catalytic activity of [Au2(μ(P,N)-D)2]X2 (X = SbF6, NTf2).12b
| Complex | Au loading [mol.%] | Yield of 13 after 30 min [%] | Yield of 13 after 3 h [%] |
|---|---|---|---|
| a Compound 12 (1 mmol) and 1,2-dichloroethane (internal standard; 1 mmol) were dissolved in CDCl3 (2 mL). The respective catalyst was added and the reaction mixture was stirred at room temperature for a given time (the amount of the starting materials was doubled for reactions with 0.1 mol% Au). The yields were determined by integration of 1H NMR spectra. | |||
| 9 | 1.0 | 0 | 0 |
| 10 | 1.0 | 82 | 89 |
| 10 | 0.1 | 64 | 75 |
| 11a | 1.0 | 0 | 0 |
| 11b | 1.0 | 0 | 0 |
Tentatively, we ascribed the observed low catalytic activity of the Au-1 complexes (particularly the dimers 11) to a relatively stronger coordination of the isonitrile moiety, which prevents the formation of coordinatively unsaturated Au(I) species by dissociation of the Au–CN bonds. This assumption was supported by the results of DFT computations performed on the isolated cations [Au2(μ(P,N)-L)2]2+, where L = D or 1 (Table 4), which indeed revealed that the dissociation of the phosphinonitrile-bridged dimers (L = D) to the monomeric species [Au(D)]+ is by approximately 12 kcal mol−1less endergonic than for the corresponding dimeric cation containing the isomeric ligand 1 (27.2 vs. 39.4 kcal mol−1).45
| [Au2(μ(P,N)-D)2]2+ | [Au2(μ(P,N)-1)2]2+ | ||||
|---|---|---|---|---|---|
| Parameter | X-rayb | DFT | Parameter | X-Rayc | DFT |
|
a The calculations were performed at the PBE0/cc-pVDZ:sdd(Fe,Au) level of theory using the PCM model to treat possible solvent effects (CHCl3). For details, see Experimental.
b Data from ref. 12b, SbF6− salt.
c This work; SbF6− salt.
|
|||||
| Au–N | 2.035(4) | 2.054 | Au–C | 2.008(4) | 1.996 |
| CN |
1.139(8) | 1.160 | NC |
1.135(5) | 1.164 |
| Au–P | 2.225(2) | 2.281 | Au–P | 2.282(1) | 2.333 |
| N–Au–P | 175.1(2) | 175.7 | C–Au–P | 175.8(1) | 175.3 |
| Au–N–C | 168.2(5) | 170.6 | Au–C–N | 174.3(4) | 175.4 |
Conclusions
The discovery of a practical and reliable synthesis of 1′-(diphenylphosphino)-1-aminoferrocene (2) and its P-protected analogues (the borane adduct 2B in particular) reported in this contribution opens a way to further synthetic utilization of this versatile but long ignored phosphinoferrocene building block. Herein, the synthetic use of 2 is demonstrated by the synthesis of phosphino-isonitrile 1, representing a new entry among the still uncommon mixed-donor ligands of this type. As revealed by our coordination study with Ag(I) and Au(I) ions with and without auxiliary chloride ligands (N.B. defined Cu(I) complexes were not isolated despite numerous attempts), the ferrocene moiety in the structure of 1 behaves as a molecular ball bearing connecting the two sterically distinct, soft donor moieties present in this ligand, allowing their orientation into positions appropriate for the formation of discrete, dimeric and even polymeric assemblies. Compared to the isomeric phosphino-nitrile donor D, ligand 1 becomes more easily (and also more strongly) coordinated through its isonitrile donor group. However, the stronger Au–CN bonding can be made responsible for a lower catalytic activity of the dimeric Au(I) complexes 11, very likely due to a suppressed dissociation of these dimers into monogold(I) species. Further studies into the coordination behavior and possible synthetic applications of phosphino-isonitrile 1 focused mainly at its insertion chemistry are currently underway.Experimental
Materials and methods
All syntheses were performed under an argon atmosphere using standard Schlenk techniques and with protection from the direct sun light. Compounds 3S,46 4-toluenesulfonyl azide,47 acetic formic anhydride,48 [AuCl(tht)]49 and [Au(tht)2][SbF6]12b were prepared according to the literature. Chloro-diphenylphosphine was distilled under vacuum. Anhydrous dichloromethane and tetrahydrofuran were obtained from a PureSolv MD5 solvent purification system (Innovative Technology, MA, USA). Toluene was dried over sodium metal and distilled. Other chemicals and solvents utilized for crystallizations and during chromatography were of reagent grade (Sigma-Aldrich and Alfa-Aesar; solvents from Lach-Ner, Czech Republic) and were used without additional purification.The NMR spectra were recorded at 25 °C on a Varian UNITY Inova 400 or a Bruker Avance III 400 spectrometer. Chemical shifts (δ in ppm) are given relative to an internal tetramethylsilane (1H and 13C) and an external 85% H3PO4 standard (31P), all set to 0 ppm. In addition to the usual notation of signal multiplicity, vt and vq are used to distinguish virtual triplets and quartets due to ferrocene protons constituting the AA′BB′ and AA′BB′X spin systems in the nitrogen- and PPh2-substituted cyclopentadienyl rings, respectively (fc = ferrocene-1,1′-diyl). FTIR spectra were measured with a Nicolet 6700 spectrometer in a range of 400–4000 cm−1. Electrospray ionization (ESI) mass spectra were obtained on an Esquire 3000 (Bruker) spectrometer using samples dissolved in HPLC-grade methanol. High resolution (HR) measurements were performed with an LTQ Orbitrap XL spectrometer (Thermo Fisher Scientific). The assignment of the observed ions was based on a comparison of the theoretical and experimental isotopic patterns. Elemental analyses were determined with a PerkinElmer PE 2400 CHN analyzer.
The cyclic voltammograms were recorded with a μAUTOLAB III instrument (Eco Chemie, The Netherlands) at ambient temperature using samples dissolved in dry dichloromethane (sample concentration 0.5 mM or a saturated solution in the case of poorly soluble complexes) and Bu4N[PF6] as the supporting electrolyte (0.1 M). A glassy carbon disc (2 mm diameter) was employed as a working electrode, Ag/AgCl (3 M LiCl/EtOH) as a reference electrode and platinum sheet as a counter electrode. Decamethylferrocene was added as an internal reference for the final scans but the potentials were converted to the ferrocene/ferrocenium scale by subtracting by 0.548 V.50
Synthesis of 4S
An oven-dried two-necked flask was charged with 3S (2.40 g, 5.0 mmol) and a magnetic stirring bar, flushed with argon and sealed a rubber septum. Dry THF (80 mL) was introduced under argon to dissolve the solid and the flask was cooled in an ice bath with continuous stirring. n-Butyllithium (2.0 mL 2.5 M, 5.0 mmol) was added and the mixture was allowed to react for 20 min with continuous cooling. Neat 4-toluenesulfonyl azide (TsN3; 986 mg, 5.0 mmol) was introduced to the lithiated intermediate, causing the reaction mixture to turn red (Note: TsN3 was stored over CaH2 prior to the use. Reactions with the untreated commercial reagent gave considerably lower yields). The reaction mixture was stirred at 0 °C for 30 min and at room temperature for another 2 h. Then, it was diluted with saturated aqueous NaHCO3 (40 mL) and diethyl ether (80 mL). The organic phase was separated and the aqueous one was extracted with diethyl ether (3 × 40 mL). The combined organic layers were washed with brine and dried over MgSO4. The dried solution was evaporated with chromatographic silica gel and the crude pre-adsorbed product was transferred to a silica gel column. Elution with diethyl ether–hexane (1:3) provided an orange band containing the product. Evaporation afforded compound 4S as an orange oil (2.15 g, ca. 75%), which usually contained unreacted TsN3 (less than 5 mol%) and FcP(S)Ph2 (typically ca. 20 mol%; Fc = ferrocenyl) formed by protonation of the lithiated intermediate. Crystals used for single-crystal X-ray diffraction analysis were grown from diethyl ether. The product decomposes (darkens) upon prolonged storage (even when stored in a refrigerator and in the dark). If appropriate, it can be easily purified by chromatography as described above.
1H NMR (CDCl3): δ 4.07 (vt, J′ = 2.0 Hz, 2 H, fc), 4.23 (vt, J′ = 2.0 Hz, 2 H, fc), 4.54 (vq, J′ = 2.1 Hz, 2 H, fc), 4.63 (vq, J′ = 2.1 Hz, 2 H, fc), 7.40–7.51 (m, 6 H, P(S)Ph2), 7.69–7.75 (m, 4 H, P(S)Ph2). 13C{1H} NMR (CDCl3): δ 62.07 (CH of fc), 67.82 (CH of fc), 73.19 (d, J = 10 Hz, CH of fc), 73.84 (d, J = 12 Hz, CH of fc), 100.51 (Cipso–N3 of fc), 128.26 (d, 2JPC = 12 Hz, CHortho of Ph), 131.31 (d, 4JPC = 2 Hz, CHpara of Ph), 131.59 (d, 3JPC = 10 Hz, CHmeta of Ph), 134.36 (d, 1JPC = 87 Hz, Cipso of Ph). The signals due to Cipso–P of fc was not found. 31P{1H} NMR (CDCl3): δ 41.7 (s). IR (Nujol): νmax 3049 w, 2109 s (N3), 1308 w, 1287 m, 1193 w, 1170 m, 1102 s, 1069 w, 1026 m, 998 w, 917 w, 837 m, 828 m, 754 m, 716 s, 695 s, 656 s, 629 w, 615 w, 541 m, 523 m, 496 m, 448 w cm−1. ESI+ MS: m/z 438 ([M − N2 + Na]+), 466 ([M + Na]+). HRMS calc. for C22H1856FeN3PS (M+): 443.0308, found 443.0309.
Synthesis of 5 from 4S
An aqueous suspension of active nickel (Sigma-Aldrich, RANEY®, type 2400) corresponding to 8 g of the solid material was transferred to a reaction flask and washed with reagent grade and then anhydrous THF (5-times each) under argon. The remaining solid was suspended in anhydrous THF (25 mL) and a solution of impure 4S (0.800 g; this amount corresponds to 1.8 mmol of pure 4S) in 10 mL of dry THF was added (an effervescence was observed during the addition). The reaction mixture was stirred at room temperature for 3 days and filtered through a Celite pad, which was then washed with THF (3 × 15 mL). Neat acetic formic anhydride (HCOOAc; 0.8 mL, 10 mmol) was added to the filtrate and the resultant reaction mixture was stirred at ambient temperature for 3 h before quenching with saturated aqueous NaHCO3. After gas evolution had ceased, the mixture was extracted with diethyl ether (50 mL). The organic layer was washed with water and brine, dried over MgSO4 and evaporated, leaving an orange residue, which was purified by column chromatography (silica gel, ethyl acetate–hexane 1:1). The first orange band containing FcPPh2 and some tosylated impurities was followed by an orange band containing the desired product contaminated by traces of FcNHCHO.5 The second band was evaporated and the residue chromatographed again to afford pure 5 (orange solid; 0.253 g or 34% with respect to 2S available in the starting material) and FcNHCHO (orange solid; 55 mg, 13% with respect to 2S). The yield did not practically change when the reaction time was shortened to 1 day. Compound 5 was obtained as a mixture of (Z) and (E) isomers in 5:4 ratio. Crystals of (Z)-5 were obtained by recrystallization from chloroform/hexane.
Notably, when the reaction was performed in dry acetonitrile, the unwanted dephosphorylation leading to FcNHCHO was suppressed but the yield of 5 was only 13%. Practically no desulfurization (only conversion of the azide to formamide) was observed in 1,2-dimethoxyethane under similar conditions (room temperature, 3 days), while in anhydrous ethanol the starting azide was converted to the N-alkylated products FcN(Et)CHO and Ph2PfcN(Et)CHO in ca. 1:1 ratio (ESI+ MS data for Ph2PfcN(Et)CHO: m/z 442 ([M + H]+), 464 ([M + Na]+), 480 ([M + K]+)).
Analytical data for 5
1H NMR (CDCl3): major – δ 3.96 (vt, J′ = 2.0 Hz, 2 H, fc), 4.08 (br s, 2 H, fc), 4.44 (vt, J′ = 1.8 Hz, 2 H, fc), 4.52 (vt, J′ = 1.8 Hz, 2 H, fc), 6.10 (br s, 1 H, NH), 7.30–7.42 (m, 10 H, PPh2), 7.94 (d, 3JHH = 1.5 Hz, 1 H, CHO); minor – δ 3.99 (vt, J′ = 2.0 Hz, 2 H, fc), 4.13 (vq, J′ = 1.7 Hz, 2 H, fc), 4.16 (vt, J′ = 2.0 Hz, 2 H, fc), 4.42 (vt, J′ = 1.8 Hz, 2 H, fc), 6.65 (br d, 3JHH = 11.7 Hz, 1 H, NH), 7.30–7.42 (m, 10 H, PPh2), 8.20 (d, 3JHH = 11.7 Hz, 1 H, CHO). 13C{1H} NMR (CDCl3): major – δ 63.22 (CH of fc), 65.97 (CH of fc), 72.23 (CH of fc), 74.26 (d, JPC = 8 Hz, CH of fc), 75.99 (br s, Cipso–P of fc), 92.54 (Cipso–N of fc), 128.30 (d, 3JPC = 7 Hz, CHmeta of Ph), 128.55 (CHpara of Ph), 133.47 (d, 2JPC = 20 Hz, CHortho of Ph), 139.12 (d, 1JPC = 9 Hz, Cipso of Ph), 159.02 (CHO); minor – δ 62.88 (CH of fc), 66.76 (CH of fc), 72.21 (CH of fc), 74.16 (d, JPC = 8 Hz, CH of fc), 93.74 (Cipso–N of fc), 128.30 (d, 3JPC = 7 Hz, CHmeta of Ph), 128.82 (CHpara of Ph), 138.58 (d, 2JPC = 20 Hz, CHortho of Ph), 138.58 (d, 1JPC = 9 Hz, Cipso of Ph), 163.06 (CHO). The signal due to Cipso–P of fc for the minor isomer appears to be obscured by the solvent resonance. 31P{1H} NMR (CDCl3): δ −17.5 (s, major), −16.8 (s, minor). IR (Nujol): νmax 3260 m, 3095 m, 3057 w, 1663 s (CHO), 1561 m, 1263 m, 1211 w, 1193 w, 1164 m, 1094 w, 1069 w, 1033 m, 1023 m, 922 m, 853 m, 832 m, 805 m, 755 s, 741 s, 704 s, 697 s, 633 w, 529 w, 497 s, 486 s, 460 m, 435 w cm−1. ESI+ MS: m/z 414 ([M + H]+), 436 ([M + Na]+), 452 ([M + K]+). HRMS calc. for C23H2056FeNOP (M+): 413.0632, found 413.0631. Anal. calc. for C23H20FeNOP (413.2): C 66.85, H 4.88, N 3.39%. Found: C 66.83, H 4.91, N 3.37%.Synthesis of 3B from 3
A BH3·SMe2 solution (6 mL of 2 M in THF, 12 mmol) was added to a solution of compound 3 (4.47 g, 10.0 mmol) in dry THF (30 mL) and the mixture was stirred at room temperature under argon for 90 min. Methanol (0.5 mL) was added to destroy an excess of borane and, after gas evolution had subsided (5 min), the reaction mixture was evaporated with chromatographic silica gel. The preadsorbed product was transferred to a short silica gel column. Elution with toluene–hexane (1:1) and evaporation of a single orange band provided the protected phosphine 3B as an orange solid (4.39 g, 95%). Note: Elution with toluene makes the workup faster but the product contains some foul smelling impurities.
1H NMR (CDCl3): δ 0.8–1.7 (very br m, 3 H, BH3), 4.08 (vt, J′ = 1.9 Hz, 2 H, fc), 4.33 (vt, J′ = 1.9 Hz, 2 H, fc), 4.46 (vq, J′ = 1.8 Hz, 2 H, fc), 4.55 (vdt, J′ = 1.8 Hz, 1.2 Hz, 2 H, fc), 7.39–7.50 (m, 6 H, PPh2), 7.55–7.61 (m, 4 H, PPh2). 13C{1H} NMR (CDCl3): δ 69.44 (CH of fc), 70.49 (d, 1JPC = 68 Hz, Cipso–P of fc), 71.67 (CH of fc), 74.87 (d, JPC = 9 Hz, CH of fc), 75.47 (d, JPC = 7 Hz, CH of fc), 77.72 (Cipso–Br of fc), 128.50 (d, 2JPC = 10 Hz, CHortho of Ph), 130.96 (d, 1JPC = 60 Hz, Cipso of Ph), 131.01 (d, 4JPC = 2 Hz, CHpara of Ph), 132.61 (d, 3JPC = 9 Hz, CHmeta of Ph). 31P{1H} NMR (CDCl3): δ 16.2 (br d, J ≈ 75 Hz). IR (Nujol): νmax 3102 w, 3073 w, 3056 w, 2398 s (BH3), 2373 s (BH3), 2344 m (BH3), 1350 m, 1309 m, 1195 m, 1181 m, 1172 s, 1157 w, 1132 w, 1108 s, 1063 s, 1026 s, 1000 m, 978 w, 930 w, 895 w, 872 s, 838 s, 828 w, 818 m, 766 w, 743 s, 703 s, 696 s, 643 s, 622 w, 610 m, 568 w, 527 m, 506 s, 496 s, 476 s, 461 s, 440 m (cm−1). ESI+ MS: m/z 448 ([M − BH3 + H]+), 485 ([M + Na]+). Anal. calc. for C22H21BBrFeP (462.9): C 57.08, H 4.57. Found: C 57.32, H 4.55%.
Synthesis of 3B from 1,1′-dibromoferrocene
An oven-dried round bottom flask was charged with 1,1′-dibromoferrocene (17.1 g, 50 mmol), flushed with argon and sealed. Dry THF (200 mL) was introduced and the solution was cooled in a dry ice/ethanol bath to ca. −78 °C. n-Butyllithium (20 mL of 2.5 M in hexanes, 50 mmol) was introduced and the resultant mixture was stirred at −78 °C for 60 min during which time an orange precipitate formed. Chloro-diphenylphosphine (9.9 mL, 55 mmol) was added, whereupon the precipitate dissolved. The mixture was stirred at −78 °C for 30 min and then at room temperature overnight before it was treated with neat BH3·SMe2 (6.3 mL, 65 mmol). Stirring was continued for 1 h and the reaction mixture was diluted by saturated aqueous NaHCO3 and ethyl acetate (50 mL each). The organic layer was separated, washed with water and brine, dried over MgSO4 and, finally, evaporated with silica gel. The preadsorbed material was transferred to a silica gel column, which was eluted first with dichloromethane–hexane (1:5) to remove bromo- and 1,1′-dibromoferrocene (ca. 0.45 g). Then, the eluent was gradually changed to pure dichloromethane, which removed a major orange brown band containing 3B. Evaporation and crystallization from hot ethyl acetate–hexane (ca. 1:2 mixture) afforded analytically pure 3B as an orange crystalline solid. Yield: 19.5 g (84%), two crops.
Synthesis of 4B
Compound 3B (4.63 g, 10.0 mmol) was dissolved in dry THF (100 mL) in an oven-dried reaction flask, which was then cooled in dry ice/ethanol bath to ca. −78 °C. n-Butyllithium (4.0 mL of 2.5 M in hexanes, 10 mmol) was added and the reaction mixture, which gradually deposited an orange precipitate, was stirred at −78 °C for 30 min. Neat TsN3 (1.97 g, 10 mmol; see comments below) was introduced and the resulting mixture was stirred at room temperature for 2 h (Note: The solid precipitate dissolved and the color of the reaction mixture changed to red). The reaction was terminated by addition of water (10 mL) and the mixture was transferred to a separatory funnel. The aqueous layer was separated and extracted with diethyl ether (3 × 30 mL). The organic layers were combined, washed with brine, dried over MgSO4 and evaporated. The obtained brown oil was purified by column chromatography over silica gel using diethyl ether–hexane (1:1) as the eluent (Note: The crude product should not be exhaustively evaporated to avoid formation of a gummy product, which is poorly soluble in the mobile phase). A single orange band was collected and evaporated under vacuum to afford 4B as an orange microcrystalline solid (3.78 g, 89%). Crystals of 4B used for structure determination were grown by diffusion of hexane into a diethyl ether solution at 4 °C.
Note: TsN3 must be dried by stirring with CaH2 freshly prior to the use (preferably overnight). Only if such carefully dried TsN3 is used, the reaction product is contaminated by less than 5 mol% of FcPPh2·BH3. Otherwise, the amount of FcPPh2·BH3 and unreacted TsN3 are higher. Azide 4B does not decompose when stored in the dark at 4 °C for weeks. If appropriate, however, it can be purified by column chromatography as specified above.
1H NMR (CDCl3): δ 0.8–1.6 (very br m, 3 H, BH3), 4.00 (vt, J′ = 2.0 Hz, 2 H, fc), 4.16 (vt, J′ = 2.0 Hz, 2 H, fc), 4.52 (vq, J′ = 1.8 Hz, 2 H, fc), 4.65 (vdt, J′ = 1.8 Hz, 1.1 Hz, 2 H, fc), 7.38–7.50 (m, 6 H, PPh2), 7.56–7.62 (m, 4 H, PPh2). 13C{1H} NMR (CDCl3): δ 61.85 (CH of fc), 67.50 (CH of fc), 70.12 (d, 1JPC = 68 Hz, Cipso–P of fc), 73.33 (d, JPC = 8 Hz, CH of fc), 73.70 (d, JPC = 10 Hz, CH of fc), 100.41 (Cipso–N3 of fc), 128.49 (d, 2JPC = 10 Hz, CHortho of Ph), 131.00 (d, 4JPC = 2 Hz, CHpara of Ph), 131.01 (d, 1JPC = 60 Hz, Cipso of Ph), 132.58 (d, 3JPC = 9 Hz, CHmeta of Ph). 31P{1H} NMR (CDCl3): δ 16.2 (br d). IR (Nujol): νmax 3077 w, 3054 w, 2382 s (BH3), 2338 m (BH3), 2108 s (N3), 1314 w, 1285 s, 1223 w, 1176 s, 1161 m, 1134 m, 1107 s, 1061 s, 1036 m, 1028 s, 999 w, 920 w, 832 s, 820 m, 760 m, 741 s, 703 s, 692 m, 635 m, 625 m, 611 m, 595 w, 528 s, 489 s, 463 m, 446 w, 409 w cm−1. ESI+ MS: m/z 420 ([M – N2 + Na]+), 448 ([M + Na]+). HRMS calc. for C22H21B56FeN3P (M+): 425.0916, found 425.0921.
Synthesis of 5B for 4B
A solution of 4B (1.70 g, 4.0 mmol) in dry THF (40 mL) was slowly introduced to a reaction flask containing Li[AlH4] (0.304 g, 8.0 mmol) while stirring and cooling on ice (gas evolution was noted). After the addition was complete, the cooling bath was removed and the mixture was stirred at room temperature for 3 h before an excess of the reducing agent was destroyed by a careful addition of 10% aqueous NaOH (0.5 mL) and stirring for 10 min. Anhydrous MgSO4 (1 g) was added and the pasty mixture was transferred to a Celite pad on a glass frit. Washing with dry THF (60 mL in several portions) provided an orange filtrate, which was treated with formic acetic anhydride (1.0 mL, 12.8 mmol) in an argon-flushed vessel. The resultant mixture was stirred at room temperature for 3 h and then diluted with saturated aqueous NaHCO3 and diethyl ether (50 mL each). The organic layer was separated, washed with water and brine, dried over MgSO4 and evaporated. The crude product was purified by chromatography on a silica gel column with ethyl acetate–hexane (1:1) as the eluent. The first yellow band containing impurities was discarded and the second (major) one was collected and evaporated to afford 5B as an orange oil, which gradually solidified. Yield: 1.50 g (88%). The product is a mixture of (E) and (Z) isomers in ca. 2:1 ratio.
1H NMR (CDCl3): major – δ 0.8–1.7 (very br m, 3 H, BH3), 3.90 (vt, J′ = 2.0 Hz, 2 H, fc), 4.29 (vq, J′ = 1.8 Hz, 2 H, fc), 4.59 (vdt, J′ = 1.8 Hz, 1.2 Hz, 2 H, fc), 4.69 (vt, J′ = 2.0 Hz, 2 H, fc), 6.71 (br s, 1 H, NH), 7.39–7.52 (m, 6 H, PPh2), 7.56–7.63 (m, 4 H, PPh2), 8.06 (d, 3JHH = 1.1 Hz, 1 H, CHO); minor – δ 0.8–1.7 (very br m, 3 H, BH3), 4.01 (vt, J′ = 1.9 Hz, 2 H, fc), 4.25 (vt, J′ = 2.0 Hz, 2 H, fc), 4.41 (vq, J′ = 1.8 Hz, 2 H, fc), 4.58 (vdt, J′ = 1.9 Hz, 1.2 Hz, 2 H, fc), 6.74 (br s, 1 H, NH), 7.39–7.52 (m, 6 H, PPh2), 7.56–7.63 (m, 4 H, PPh2), 8.14 (d, 3JHH = 11.6 Hz, 1 H, CHO). 13C{1H} NMR (CDCl3): major – δ 63.90 (CH of fc), 66.15 (CH of fc), 69.22 (d, 1JPC = 69 Hz, Cipso–P of fc), 73.15 (d, JPC = 8 Hz, CH of fc), 74.22 (d, JPC = 10 Hz, CH of fc), 93.59 (Cipso–N of fc), 128.54 (d, 2JPC = 10 Hz, CHortho of Ph), 130.95 (d, 1JPC = 60 Hz, Cipso of Ph), 131.04 (d, 4JPC = 2 Hz, CHpara of Ph), 132.65 (d, 3JPC = 9 Hz, CHmeta of Ph), 159.12 (CHO); minor – δ 62.56 (CH of fc), 67.17 (CH of fc), 70.18 (d, 1JPC = 68 Hz, Cipso–P of fc), 73.28 (d, JPC = 8 Hz, CH of fc), 74.21 (d, JPC = 10 Hz, CH of fc), 95.04 (Cipso–N of fc), 128.59 (d, 2JPC = 10 Hz, CHortho of Ph), 130.82 (d, 1JPC = 60 Hz, Cipso of Ph), 131.19 (d, 4JPC = 2 Hz, CHpara of Ph), 132.57 (d, 3JPC = 10 Hz, CHmeta of Ph), 162.39 (CHO). 31P{1H} NMR (CDCl3): 16.1 (br d, both isomers). IR (Nujol): νmax 3275 br m, 3230 br m, 3082 m, 3054 m, 2374 s (BH3), 2343 m (BH3), 1687 m, 1663 s (CHO), 1582 m, 1564 s, 1353 w, 1310 w, 1267 w, 1258 w, 1173 s, 1133 w, 1107 m, 1057 s, 1029 s, 999 w, 927 w, 837 w, 830 w, 813 w, 759 w,, 744 s, 701 s, 636 m, 623 w, 612 w, 528 m, 500 s, 475 s, 463 w, 447 w cm−1. ESI+ MS: m/z 450 ([M + Na]+), 466 ([M + K]+). HRMS calc. for C23H23B56FeNOP (M+): 427.0960, found 427.0965. Anal. calc. for C23H23BFeNOP (427.0): C 64.68, H 5.43, N 3.28%. Found: C 64.50, H 5.08, N 3.23%.
Synthesis of 5 from 5B
A dry reaction flask was charged with 5B (1.28 g, 3.0 mmol), 1,4-diazabicyclo[2.2.2]octane (0.449 g, 4.0 mmol) and a stirring bar, flushed with argon and sealed with a rubber septum. Dry toluene (60 mL) was added and the reaction flask was transferred to an oil bath maintained at 80 °C. After stirring for 20 h, the mixture was evaporated under reduced pressure. The residue was dissolved in a minimal amount of THF and purified by column chromatography (silica gel, ethyl acetate–hexane 1:1) to afford formamide 5 as an amber brown, slowly crystallizing oil. Yield: 1.17 g (94%).
Synthesis of 2B
A solution of azide 4B (1.70 g, 4.0 mmol) in dry THF (40 mL) was added to an ice-cooled reaction flask containing Li[AlH4] (0.304 g, 8.0 mmol) and a stirring bar (an effervescence was observed). When the addition was complete, the cooling bath was removed and stirring was continued at room temperature for 3 h. The reaction was terminated by cautious addition of 10% aqueous NaOH (0.5 mL) and, after stirring for another 10 min, anhydrous MgSO4 (1 g). The obtained semisolid mass was transferred to a Celite pad on a glass frit and washed with dry THF (60 mL in several portions). The orange filtrate was evaporated to afford an orange brown oil, which was purified by chromatography over a short silica gel using ethyl acetate–hexane (1:3) as the eluent, which removed some non-polar impurities. A subsequent elution with ethyl acetate–hexane (1:1) provided a deep orange band containing the product. Evaporation of this band afforded amine 2B as an orange oil, which gradually solidified. Yield: 1.49 g (93%). Note: solid 2B does not show any signs of decomposition when stored at 4 °C in the dark but readily decomposes in chlorinated solvents.
1H NMR (CDCl3): δ 0.8–1.7 (very br m, 3 H, BH3), 2.51 (br s, 2 H, NH2), 3.71 (vt, J′ = 1.9 Hz, 2 H, fc), 3.97 (vt, J′ = 1.9 Hz, 2 H, fc), 4.25 (vq, 2 H, J′ = 1.8 Hz, 2 H, fc), 4.48 (v dt, J′ = 1.8 Hz, 1.1 Hz, 2 H, fc), 7.38–7.48 (m, 6 H, PPh2), 7.58–7.64 (m, 4 H, PPh2). 13C{1H} NMR (CDCl3): δ 59.76 (CH of fc), 64.76 (CH of fc), 68.42 (d, 1JPC = 70 Hz, Cipso–P of fc), 72.48 (d, 3JPC = 8 Hz, CH of fc), 73.78 (d, 2JPC = 10 Hz, CH of fc), 107.37 (Cipso–N of fc), 128.39 (d, 2JPC = 10 Hz, CHortho of Ph), 130.82 (d, 4JPC = 2 Hz, CHpara of Ph), 131.54 (d, 1JPC = 59 Hz, Cipso–P of Ph), 132.62 (d, 3JPC = 9 Hz, CHmeta of Ph). 31P{1H} NMR (CDCl3): δ 16.4 (br d). IR (Nujol): νmax 3416 m, 3385 m, 3332 m, 3076 m, 3058 w, 2380 s (BH3), 2255 w (BH3), 1503 s (NH2), 1305 w, 1252 w, 1194 w, 1173 s, 1134 m, 1106 s, 1060 s, 1041 m, 1026 s, 998 w, 934 w, 850 m, 817 s, 796 m, 767 m, 744 s, 735 s, 700 s, 689 s, 637 s, 621 m, 613 m, 529 m, 502 s, 471 s, 460 m, 442 w, 417 w, 411 w cm−1. ESI+ MS: m/z 398 ([M − H]+). HRMS calc. for C22H23B56FeNP (M+): 399.1011, found 399.1018. Anal. calc. for C22H23BFeNP (399.0): C 66.21, H 5.81, N 3.51%. Found: C 66.10, H 5.93, N 3.50%.
Synthesis of amine 2
Compound 2B (0.399 mg, 1.0 mmol) and 1,4-diazabicyclo[2.2.2]octane (0.224 g, 2.0 mmol) were dissolved in 20 mL of dry toluene in a dry reaction flask under argon. The reaction mixture was stirred at room temperature for 18 h and then evaporated under vacuum. A brown residue was dissolved in a minimal amount of THF and purified by column chromatography (silica gel, ethyl acetate–hexane 1:1). Evaporation of a single orange band furnished pure amine 2 as an orange solid (0.362 g, 94%). The compound is stable as a solid but gradually decomposes when dissolved in chlorinated solvents. It can be practically quantitatively recrystallized from hot hexane, which was also used to grow single crystals.
1H NMR (CDCl3): δ 2.25 (br s, 2 H, NH2), 3.78 (vt, J′ = 1.8 Hz, 2 H, fc), 3.90 (vt, J′ = 1.7 Hz, 2 H, fc), 4.00 (vt, J′ = 1.8 Hz, 2 H, fc), 4.33 (vt, J′ = 1.6 Hz, 2 H, fc), 7.28–7.34 (m, 6 H, PPh2), 7.36–7.43 (m, 4 H, PPh2). 13C{1H} NMR (CDCl3): δ 59.47 (CH of fc), 64.42 (CH of fc), 71.57 (d, 3JPC = 3 Hz, CH of fc), 73.78 (d, 2JPC = 15 Hz, CH of fc), 75.41 (d, 1JPC = 4 Hz, Cipso–P of fc), 106.05 (Cipso–N of fc), 128.13 (d, 3JPC = 6 Hz, CHmeta of Ph), 128.46 (CHpara of Ph), 133.54 (d, 2JPC = 19 Hz, CHortho of Ph), 139.47 (d, 1JPC = 9 Hz, Cipso of Ph). 31P{1H} NMR (CDCl3): δ −16.7 (s). The NMR data are in accordance with the literature.19
Synthesis of 5 from 2
Amine 2 (0.100 g, 0.26 mmol) was dissolved in dry THF (5 mL) under an argon atmosphere. Neat formic acetic anhydride (31 μl, 0.39 mmol) was added to the solution and the resulting mixture was stirred at room temperature for 3 h. The reaction was terminated by addition of saturated aqueous NaHCO3 and diethyl ether (10 mL each; an effervescence occurred). The aqueous layer was removed and the organic one was washed with water and with brine, and dried over MgSO4. Following evaporation under reduced pressure, the crude product was purified by flash column chromatography (silica gel, ethyl acetate–hexane 1:1) to afford pure formamide 5 as an orange glassy solid (0.099 g, 93%).
Preparation of 1′-(diphenylphosphino)-1-isocyanoferrocene (1)
An oven-dried flask was charged with a stirring bar, amide 5 (413 mg, 1.0 mmol), (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP; 884 mg, 2.0 mmol) and dry THF (20 mL) under argon. Neat 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU; 0.38 ml, 2.5 mmol) was added and the mixture was stirred at room temperature for 3 h (the partly soluble BOP dissolved during this time and the mixture slightly darkened). The resulting solution was diluted with diethyl ether (10 mL), washed successively with saturated aqueous NaHCO3, water and brine, dried over MgSO4 and concentrated under vacuum. The residue was taken up in a minimal amount of THF and transferred to a short silica gel column. Elution with diethyl ether–hexane (1:1) led to the development of an orange band, which was collected and evaporated under vacuum to afford 1 as as an orange microcrystalline solid emitting an unpleasant odor typical of isonitriles. Yield: 281 mg (71%). Notes: A further elution with ethyl acetate recovered some unreacted (impure) amide 5. The yield of 1 decreased when the reaction was carried out in incompletely dried THF and without rigorous exclusion of air and moisture.
1H NMR (CDCl3): δ 4.02 (vt, J′ = 1.9 Hz, 2 H, fc), 4.22 (vq, J′ = 1.9 Hz, 2 H, fc), 4.43 (vt, J′ = 2.0 Hz, 2 H, fc), 4.51 (vt, J′ = 1.9 Hz, 2 H, fc), 7.31–7.38 (m, 10 H, PPh2). 13C{1H} NMR (CDCl3): δ 67.61 (CH of fc), 68.19 (CH of fc), 73.92 (d, JPC = 4 Hz, CH of fc), 74.96 (d, JPC = 14 Hz, CH of fc), 78.78 (d, JPC = 9 Hz, Cipso–P), 128.31 (d, 3JPC = 7 Hz, CHmeta of Ph), 128.79 (CHpara of Ph), 133.42 (d, 2JPC = 20 Hz, CHortho of Ph), 138.27 (d, 1JPC = 10 Hz, Cipso of Ph), 164.30 (NC). The resonance due to ferrocene Cipso–NC is probably obscured by the solvent signal. 31P{1H} NMR (CDCl3): δ −17.8 (s). 1H NMR (acetone-d6): δ 4.10 (vt, J′ = 2.0 Hz, 2 H, fc), 4.23 (vq, J′ = 1.8 Hz, 2 H, fc), 4.57 (vt, J′ = 2.1 Hz, 2 H, fc), 4.59 (vt, J′ = 1.8 Hz, 2 H, fc), 7.37–7.41 (m, 10 H, PPh2). 31P{1H} NMR (acetone-d6): δ −17.3 (s). IR (Nujol): νmax 3114 w, 3054 w, 2126 s (NC), 1231 w, 1193 w, 1160 m, 1089 w, 1028 s, 999 w, 916 w, 836 m, 819 s, 747 s, 698 s, 639 w, 596 w, 531 w, 510 s, 502 m, 488 m, 458 w, 432 w, 414 w cm−1. ESI+ MS: m/z 396 ([M + H]+). Anal. calc. for C23H18FeNP (395.2): C 69.90, H 4.59, N 3.54%. Found: C 69.79, H 4.63, N 3.35%.
Synthesis of complex 6
A solution of 1 (20.0 mg, 50.6 μmol) in chloroform (1.5 mL) was added to a suspension of freshly precipitated AgCl (7.3 mg, 50.6 μmol) in the same solvent (0.5 mL) and the mixture was stirred in the dark for 24 h (a fine yellow solid separated). The precipitated product was filtered off, washed with chloroform and pentane, and dried under vacuum. Yield of 6: 24.2 mg (89%), yellow virtually insoluble solid. Note: An identical product was obtained when the amount of 1 was increased to 2 equiv. with respect to AgCl.IR (Nujol): νmax 3114 w, 3081 w, 3052 w, 2160 s (NC), 2140 m (NC), 1311 w, 1233 w, 1194 m, 1168 m, 1100 s, 1070 w, 1036 m, 1025 m, 910 m, 832 m, 824 m, 751 m, 741 vs, 696 vs, 631 w, 613 w, 594 w, 535 m, 515 s, 503 s, 489 s, 470 s, 459 m, 442 w, 427 m cm−1. ESI+ MS: m/z 502 ([Ag(1)]+), 704 ([Ag2(1)Cl2 + Na]+). Anal. calc. for C23H18AgClFeNP·0.2CHCl3 (562.4): C 49.54, H 3.26, N 2.49%. Found: C 49.36, H 3.19, N 2.40%.
Synthesis of complex 7
A solution of ligand 1 (20.0 mg, 50.6 μmol) in acetone (1.5 mL) was added to a suspension of Ag[SbF6] (17.4 mg, 50.6 μmol) in the same solvent (0.5 mL) and the mixture was stirred in the dark for 1 h (the solid silver(I) salt quickly dissolved). The resulting solution was filtered through a syringe filter (PTFE, 0.45 μm) and the filtrate was crystallized by layering with acetone (1 mL) and then with hexane (10 mL) as a top layer. The crystals, which formed during standing in the dark for approximately one week, were isolated by suction, rinsed with pentane and dried under vacuum. Yield: 31.5 mg (84%), orange needle-like crystals.
1H NMR (in situ, acetone-d6): δ 4.26 (br s, 2 H, fc), 4.60 (br s, 2 H, fc), 5.01 (vt, J′ = 1.7 Hz, 2 H, fc), 5.14 (br s, 2 H, fc), 7.54–7.71 (m, 10 H, PPh2). 31P{1H} NMR (in situ, acetone-d6): δ 6.8 (pair of d, 1JAgP = 709 (109Ag), 615 (107Ag) Hz). IR (Nujol): νmax IR (Nujol): νmax 3119 w, 3050 w, 2210 m (NC), 1695 s (acetone, CO), 1316 w, 1230 m, 1198 w, 1173 m, 1101 m, 1059 w, 1034 m, 1001 w, 920 m, 848 w, 821 w, 750 s, 699 s, 664 vs, 653 vs, 542 w, 513 s, 472 m, 445 w, 430 m cm−1. ESI+ MS: m/z 395 (1+), 502 ([Ag(1)]+), 646 ([Ag2(1) + Cl]+), 897 ([Ag(1)2]+). Anal. calc. for C23H18AgF6FeNPSb·(CH3)2CO (796.9): C 39.18, H 3.04, N 1.76%; found C 39.49, H 3.07, N 1.72%.
Preparation of complex 8
A solution of compound 1 (20.0 mg, 50.6 μmol) in acetone (1.5 mL) was added to Ag[SbF6] in the same solvent (8.7 mg, 25.3 μmol in 0.5 mL). After stirring in the dark for 90 min, the reaction mixture was filtered through a PTFE syringe filter (0.45 μm pore size) and the filtrate was carefully layered with acetone (1 mL) and, finally, with hexane (10 mL) as the top layer. Crystallization by liquid-phase diffusion (in the dark) provided crystals which were isolated by suction, washed with pentane and dried under vacuum. Yield of 8: 20.1 mg (70%), orange needles.
1H NMR (in situ, acetone-d6): δ 4.20 (br s, 2 H, fc), 4.71 (br s, 2 H, fc), 5.02 (br s, 2 H, fc), 5.10 (br s, 2 H, fc), 7.31–7.37 (m, 4 H, PPh2), 7.42–7.50 (m, 6 H, PPh2). 31P{1H} NMR (in situ, acetone-d6): δ −1.2 (br d, 1JAgP = 207 Hz). IR (Nujol): νmax 3054 w, 2167 m (NC), 1309 m, 1261 w, 1233 w, 1195 m, 1166 s, 1098 s, 1030 s, 999 m, 917 s, 890 w, 829 s, 743 s, 696 s, 658 vs, 584 w, 519 m, 508 s, 494 s, 476 m, 464 m, 426 w cm−1. ESI+ MS: m/z 395 (1+), 897 ([Ag(1)2]+), 1688 ([Ag(1)4]+). Anal. calc. for C46H36AgF6Fe2N2P2Sb (1134.0): C 48.72, H 3.20, N 2.47%. Found: C 48.34, H 3.12, N 2.37%.
Preparation of complex 9
[AuCl(tht)] (16.2 mg, 50.6 μmol) and compound 1 (20.0 mg, 50.6 μmol) were reacted in chloroform (1 + 2 mL) as described above and the reaction mixture was filtered into pentane (30 mL). The precipitated solid was filtered off, washed with pentane and dried under vacuum. Yield of 9: 27.4 mg (87%), yellow solid. Crystals used for structure determination were grown from diethyl ether, which slightly dissolves the product, layered with hexane.
1H NMR (acetone-d6): δ 4.31 (vt, J′ = 2.0 Hz, 2 H, fc), 4.63 (d vt, 3JPH = 3.1 Hz, J′ = 2.0 Hz, 2 H, fc), 4.73 (vt, J′ = 2.0 Hz, 2 H, fc), 4.88 (d vt, 4JPH = 1.1 Hz, J′ = 2.0 Hz, 2 H, fc), 7.57–7.74 (m, 10 H, PPh2). 31P{1H} NMR (acetone-d6): δ 29.0 (s). IR (Nujol): νmax 3087 w, 3052 w, 2220 w, 2125 s (NC), 1686 w, 1560 w, 1310 m, 1231 w, 1198 w, 1176 m, 1102 s, 1070 w, 1028 s, 998 m, 919 m, 893 w, 834 m, 823 m, 758 s, 743 s, 694 s, 637 w, 558 s, 532 s, 514 s, 494 m, 482 s, 453 w, 436 w cm−1. ESI+ MS: m/z 592 ([M − Cl]+). Anal. calc. for C23H18AuClFeNP (627.6): C 44.01, H 2.89, N 2.23%. Found: C 43.76, H 2.96, N 1.99%.
Preparation of complex 10
A solution of [AuCl(tht)] (32.5 mg, 101 μmol) in chloroform (2 mL) was mixed with a solution of ligand 1 in the same solvent (20.0 mg, 50.6 μmol in 2 mL). After stirring for 1 h, the mixture was concentrated under reduced pressure (to approximately half the volume) and filtered through a syringe filter (0.45 μm PTFE). The filtrate was layered with chloroform (2 mL) and then with hexane (10 mL) as a top layer and set aside for crystallization. The separated material was filtered off, washed with pentane and dried under vacuum to afford 10 as an orange crystalline solid. Yield: 40.6 mg (94%). Single crystals used for structure determination were obtained when the filtered solution was layered with ethyl acetate and with hexane as a top layer. The crystalline product is practically insoluble.IR (Nujol): νmax 3128 w, 3082 w, 2223 s (NC), 1311 m, 1242 w, 1198 m, 1178 m, 1173 m, 1105 s, 1070 w, 1054 w, 1036 m, 1030 m, 997 w, 925 m, 841 m, 822 s, 756 s, 747 s, 705 s, 696 s, 635 w, 559 s, 534 s, 514 s, 495 s, 479 s, 470 m cm−1. ESI+ MS: m/z 824 ([M − Cl]+). Anal. calc. for C23H18Au2Cl2FeNP (860.0): C 32.12, H 2.11, N 1.63%. Found: C 32.08, H 2.11, N 1.48%.
Synthesis of complex 11a
A chloroform solution of isonitrile 1 (20.0 mg, 50.6 μmol in 2 mL) was added to [Au(tht)2][SbF6] (30.8 mg, 50.6 μmol) dissolved in the same solvent (1 mL). The resulting mixture was stirred for 60 min and then evaporated. The residue was dissolved in acetone (2 mL) and the solution was crystallized by addition of hexane as the top layer (10 mL). The orange crystals, which separated during several days, were filtered off, washed with pentane and dried under vacuum. Yield of 11a: 36.2 mg (79%).
1H NMR (acetone-d6): δ 4.40 (vt, J′ = 2.1 Hz, 2 H, fc), 4.74 (d vt, J = 3.1, 1.9 Hz, 2 H, fc), 5.16 (d vt, J = 1.2, 1.9 Hz, 2 H, fc), 5.42 (vt, J′ = 2.1 Hz, 2 H, fc), 7.65–7.71 (m, 4 H, PPh2), 7.72–7.57 (m, 2 H, PPh2), 7.80–7.87 (m, 4 H, PPh2). 31P {1H} NMR (acetone-d6): δ 33.0 (s). IR (Nujol): νmax 3118 w, 3056 w, 2231 s (NC), 1704 s, 1314 w, 1226 m, 1200 w, 1184 m, 1176 m, 1105 s, 1060 w, 1034 m, 999 w, 921 w, 848 m, 834 m, 823 m, 755 s, 749 s, 715 w, 696 m, 660 vs, 553 m, 531 m, 519 s, 493 m, 476 m, 435 w cm−1. ESI+ MS: m/z 592 ([Au(1)]+). Anal. calc. for C23H18AuF6FeNPSb (827.9): C 33.36, H 2.19, N 1.69%. Found: C 33.18, H 2.20, N 1.55%.
Preparation of complex 11b
A solution of ligand 1 (20.0 mg, 50.6 μmol) in dichloromethane (2 mL) was added to [AuCl(tht)] (16.2 mg, 50.6 μmol) dissolved in the same solvent (1 mL). After the mixture was stirred for 30 min, solid AgNTf2 (19.6 mg, 50.6 μmol) was added to the in situ formed 9, causing immediate separation of a white precipitate (AgCl). The mixture was stirred for another 30 min in the dark and then filtered through a syringe filter (0.45 μm PTFE). The filtrate was evaporated to give an orange residue, which was taken up with chloroform (1 mL). The solution was layered with chloroform (1 mL) and then hexane (10 mL) as a top layer, and allowed to crystallize by liquid-phase diffusion over several days. The orange crystals that formed were isolated by suction and dried under vacuum. Yield: 40.8 mg (92%).
1H NMR (CDCl3): δ 4.01 (vt, J′ = 2.0 Hz, 2 H, fc), 4.76 (v dt, J′ = 3.2 Hz, 2.0 Hz, 2 H, fc), 4.97 (v dt, J′ = 1.9 Hz, 1.2 Hz, 2 H, fc), 5.12 (vt, J′ = 2.0 Hz, 2 H, fc), 7.52–7.60 (m, 6 H, PPh2), 7.63–7.70 (m, 4 H, PPh2). 31P{1H} NMR (CDCl3): δ 32.1 (s). 19F NMR (CDCl3): δ −78.6 (s). IR (Nujol): νmax 3113 m, 3056 m, 2233 m (NC), 1351 s, 1227 s, 1193 s, 1134 s, 1104 m, 1056 s, 999 w, 922 w, 828 w, 788 m, 748 m, 694 m, 652 m, 615 m, 600 m, 570 m, 555 w, 530 m, 517 s, 476 m, 435 w cm−1. ESI+ MS: m/z 592 ([Au(1)]+). Anal. calc. for C25H18AuF6FeN2O4PS2 (871.9): C 34.42, H 2.08, N 3.21%. Found: C 34.33, H 2.29, N 3.18%.
Catalytic experiments
A reaction flask was charged with alcohol 12 (1.0 mmol; ca. 90:10 mixture of (Z)- and (E)-isomers, the (E)-isomer does not cyclize), 1,2-dichloroethane (1.0 mmol; internal standard) and CDCl3 (2 or 4 mL). The catalyst was added and the resulting mixture was stirred in the dark for a given reaction time and then analyzed by 1H NMR spectroscopy to determine the conversion.
X-ray crystallography
The diffraction data (completeness ≥99%, θmax = 27.5°) were collected at 150(2) K using either Nonius Kappa CCD diffractometer with a Bruker Apex-II image plate detector or a Bruker D8 VENTURE Kappa Duo PHOTON100 instrument with a IμS micro-focus X-ray tube, both equipped with a Cryostream Cooler (Oxford Cryosystems). Mo Kα radiation was used throughout.The structures were solved by direct methods (SHELXS-97) and refined by a full-matrix least squares procedure based on F2 using SHELXL-97.51 The non-hydrogen atoms were refined with anisotropic thermal motion parameters. The NH hydrogen atoms in the structure of 2 and 5 were located on the difference Fourier maps, and their positions were freely refined with Uiso(H) set to 1.2 Ueq(N). All other hydrogen atoms were included in their calculated positions and refined using the “riding model”.
Selected crystallographic data, data collection and structure refinement parameters are presented in the ESI (Table S1†). All geometric calculations were performed with a recent version of the PLATON program,52 which was also used to prepare the structural diagrams.
DFT computations
DFT calculations were performed using the PBE0 density functional53 combined with the SDD (Fe, Au) and cc-pVDZ (C, H, N, and P) basis sets. The solvent effects (CHCl3) have been approximated by the polarized continuum model (PCM)54 as implemented in the Gaussian 09 program package.55 The geometry optimizations were started from the coordinates determined by X-ray diffraction analysis. The reported energies refer to the Gibbs free energies at 298 K. Computed energies and coordinates are presented in the ESI (Table S2 and mol 2 files†).Acknowledgements
The research reported in this paper received support from the Czech Science Foundation (project no. 17-02495S).References
- (a) Isocyanide Chemistry: Applications in Synthesis and Material Science, ed. V. G. Nenajdenko, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (c) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257 RSC; (d) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698 CrossRef CAS PubMed.
- (a) Y. Yamamoto, Coord. Chem. Rev., 1980, 32, 193 CrossRef CAS; (b) E. Singleton and H. E. Oosthuizen, Adv. Organomet. Chem., 1983, 22, 209 CrossRef CAS.
- (a) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008 Search PubMed; (b) Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995 Search PubMed.
- T. El-Shihi, F. Siglmüller, R. Herrmann, M. F. N. N. Carvalho and A. J. L. Pombeiro, J. Organomet. Chem., 1987, 335, 239 CrossRef CAS.
- For an alternative synthesis of A, see: G. R. Knox, P. L. Pauson, D. Willison, E. Solčániová and Š. Toma, Organometallics, 1990, 9, 301 CrossRef CAS.
- Note that aminoferrocene, an obvious synthetic precursor to isocyanoferrocene, was prepared already in 1955 (ref. 6a), while cyanoferrocene was reported only two years later (ref. 6b–d): (a) F. S. Arimoto and A. C. Haven Jr., J. Am. Chem. Soc., 1955, 77, 6295 CrossRef CAS; (b) G. D. Broadhead, J. M. Osgerby and P. L. Pauson, Chem. Ind., 1957, 209 ( Chem. Abstr. , 1957 , 62321 ) CAS; (c) G. D. Broadhead, J. M. Osgerby and P. L. Pauson, J. Chem. Soc., 1958, 650 RSC; (d) P. J. Graham, R. V. Lindsey, G. W. Parshall, M. L. Peterson and G. M. Whitman, J. Am. Chem. Soc., 1957, 79, 3416 CrossRef CAS.
- (a) M. V. Barybin, T. C. Holovics, S. F. Deplazes, G. H. Lushington, D. R. Powell and M. Toriyama, J. Am. Chem. Soc., 2002, 124, 13668 CrossRef CAS PubMed; (b) T. C. Holovics, S. F. Deplazes, M. Toriyama, D. R. Powell, G. H. Lushington and M. V. Barybin, Organometallics, 2004, 23, 2927 CrossRef CAS; (c) P. J. Shapiro, R. Zehnder, D. M. Foo, P. Perrotin, P. H. M. Budzelaar, S. Leitch and B. Twamley, Organometallics, 2006, 25, 719 CrossRef CAS; (d) V. N. Nemykin, A. A. Purchel, A. D. Spaeth and M. V. Barybin, Inorg. Chem., 2013, 52, 11004 CrossRef CAS PubMed; (e) V. N. Nemykin, S. V. Dudkin, M. Fathi-Rasekh, A. D. Spaeth, H. M. Rhoda, R. V. Belosludov and M. V. Barybin, Inorg. Chem., 2015, 54, 10711 CrossRef CAS PubMed.
- D. van Leusen and B. Hessen, Organometallics, 2001, 20, 224 CrossRef CAS.
- (a) U. Siemeling, D. Rother, C. Bruhn, H. Fink, T. Weidner, F. Träger, A. Rothenberger, D. Fenske, A. Priebe, J. Maurer and R. Winter, J. Am. Chem. Soc., 2005, 127, 1102 CrossRef CAS PubMed; (b) T. Weidner, N. Ballav, M. Zharnikov, A. Priebe, N. J. Long, J. Maurer, R. Winter, A. Rothenberger, D. Fenske, D. Rother, C. Bruhn, H. Fink and U. Siemeling, Chem. – Eur. J., 2008, 14, 4346 CrossRef CAS PubMed; (c) U. Siemeling, L. R. R. Klapp and C. Bruhn, Z. Anorg. Allg. Chem., 2010, 636, 539 CrossRef CAS.
- A few more isocyanides bearing ferrocenyl substituents at the organic backbone were mentioned in ref. 4.
- D. M. McGinnis, S. F. Deplazes and M. V. Barybin, J. Organomet. Chem., 2011, 696, 3939 CrossRef CAS.
- (a) K. Škoch, I. Císařová and P. Štěpnička, Inorg. Chem., 2014, 53, 568 CrossRef PubMed; (b) K. Škoch, I. Císařová and P. Štěpnička, Chem. – Eur. J., 2015, 21, 15998 CrossRef PubMed; (c) K. Škoch, F. Uhlík, I. Císařová and P. Štěpnička, Dalton Trans., 2016, 45, 10655 RSC.
- (a) K.-S. Gan and T. S. A. Hor, in Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, Wiley-VCH, Weinheim, 1995, ch. 1, p. 3 ff Search PubMed; (b) S. W. Chien and T. S. A. Hor, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008, ch. 2, p. 33 ff Search PubMed; (c) G. Bandoli and A. Dolmella, Coord. Chem. Rev., 2000, 209, 161 CrossRef CAS; (d) D. J. Young, S. W. Chien and T. S. A. Hor, Dalton Trans., 2012, 41, 12655 RSC.
- (a) R. B. King and A. Efraty, J. Am. Chem. Soc., 1971, 93, 564 CrossRef CAS; (b) R. B. King and A. Efraty, J. Chem. Soc., Chem. Commun., 1974, 1371 CAS.
- (a) C.-Y. Liu, D.-Y. Chen, M.-C. Cheng, S.-M. Peng and S.-T. Liu, Organometallics, 1995, 14, 1983 CrossRef CAS; (b) I. Yu, C. J. Wallis, B. O. Patrick, P. L. Diaconescu and P. Mehrkhodavandi, Organometallics, 2010, 29, 6065 CrossRef CAS.
- L. Zhang, W. Yu, C. Liu, Y. Xu, Z. Duan and F. Mathey, Organometallics, 2015, 34, 5697 CrossRef CAS.
- Isonitrile Chemistry, ed. I. Ugi, Academic Press, New York, 1971 Search PubMed.
- P. Štěpnička, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, 2008, ch. 5, p. 177 ff Search PubMed.
- I. R. Butler and S. C. Quayle, J. Organomet. Chem., 1998, 552, 63 CrossRef CAS.
- The reaction mixture resulting after addition of O-benzylhydroxylamine to in situ generated Ph2PfcLi contained mostly (diphenylphosphino)ferrocene, uncreacted amination reagent and only traces of amine 2.
- Similar problems were noted before: (a) M. Ritte, C. Bruhn and U. Siemeling, Z. Naturforsch., 2014, 69b, 906 Search PubMed. See also: (b) S. Dey, J. W. Quail and J. Müller, Organometallics, 2015, 34, 3039 CrossRef CAS.
- A. Leonidova, T. Joshi, D. Nipkow, A. Frei, J.-E. Penner, S. Konatschnig, M. Patra and G. Gasser, Organometallics, 2013, 32, 2037 CrossRef CAS and references cited therein.
- We have noted that desulfuration of ferrocene P-sulfides by commercial Raney nickel can be accompanied by removal of the entire phosphine substituent already before: P. Štěpnička, B. Schneiderová, J. Schulz and I. Císařová, Organometallics, 2013, 32, 5754 CrossRef.
- I. Ugi, U. Fetzer, U. Eholzer, H. Knupfer and K. Offermann, Angew. Chem., Int. Ed., 1965, 4, 472 CrossRef.
- The dehydration of oximes to nitriles with BOP/DBU was reported in: M. K. Singh and M. K. Lakshman, J. Org. Chem., 2009, 74, 3079 CrossRef CAS PubMed.
- M. K. Das and S. Roy, Synth. React. Inorg. Met.-Org. Chem., 1986, 16, 67 CrossRef CAS.
- K. BongSoo, J. M. Beebe, Y. Jun, X.-Y. Zhu and C. D. Frisbie, J. Am. Chem. Soc., 2006, 128, 4970 CrossRef PubMed.
- P. Walla, V. B. Arion and U. H. Brinker, J. Org. Chem., 2006, 71, 3274 CrossRef CAS PubMed.
- A. Shafir, M. P. Power, G. D. Whitener and J. Arnold, Organometallics, 2000, 19, 3978 CrossRef CAS.
- M. J. Verschoor-Kirss, O. Hendricks, C. M. Verschoor, R. Conry and R. U. Kirss, Inorg. Chim. Acta, 2016, 450, 30 CrossRef CAS.
- Z.-G. Fang, T. S. A. Hor, Y.-S. Wen, L.-K. Liu and T. C. W. Mak, Polyhedron, 1995, 14, 2403 CrossRef CAS.
- K. J. Donaghy, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 1997, 36, 547 CrossRef CAS.
- B. Wrackmeyer, H. E. Maisel, O. L. Tok, W. Milius and M. Herberhold, Z. Anorg. Allg. Chem., 2004, 630, 2106 CrossRef CAS.
- K. Heinze and M. Schlenker, Eur. J. Inorg. Chem., 2004, 2974 CrossRef CAS.
- A. Cassel, Acta Crystallogr., Sect B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 174 CrossRef.
- K. Yang, S. G. Bott and M. G. Richmond, J. Chem. Crystallogr., 1995, 25, 263 CrossRef CAS.
- M. M. Olmstead, M. Sheffrin and F. Jiang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1142 CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- A search in Cambridge Crystallographic Database (version 5.38 of November 2016 with updates from November 2016 and February 2017) for Ag–CN–C fragments resulted in 63 hits and 129 Ag–C distances. Average value: 2.14(8) Å, median: 2.11 Å.
- Spectral Database for Organic Compounds (SDBS): IR spectrum of neat acetone; SDBS No.: 319; http://riodb01.ibase.aist.go.jp/sdbs/ (accessed April 21, 2017); νCO: 1715 cm−1.
- The broadening can be tentatively ascribed to a slow molecular dynamics of the sterically crowded complex cation in 8. The 1H NMR signals also display broad resonances.
- O. Crespo, M. C. Gimeno, P. G. Jones and A. Laguna, Acta Crystallogr., Sect. C: Struct. Chem., 2000, 56, 1433 Search PubMed.
- (a) H. Schmidbaur, Chem. Soc. Rev., 1995, 391 RSC; (b) H. Schmidbaur, Gold Bull., 2000, 33, 3 CrossRef CAS; (c) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- Our observations are in agreement with the theoretical computations on the series of cations [(η5-C5H5)3ML]+, where L is MeCN or MeNC, and M = Ti, Zr or Hf, revealing that the isontrile consistently forms stronger coordination bonds to the metal centre: H. Jacobsen, H. Berke, T. Brackemeyer, T. Eisenblätter, G. Erker, R. Fröhlich, O. Meyer and K. Bergander, Helv. Chim. Acta, 1998, 81, 1692 CrossRef CAS.
- P. Štěpnička, J. Schulz and I. Císařová, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1210 Search PubMed.
- D. P. Day, T. Dann, R. J. Blagg and G. G. Wildgoose, J. Organomet. Chem., 2014, 770, 29 CrossRef CAS.
- L. I. Krimen, J. Savage and P. Yates, Org. Synth. Coll., 1988, 6, 8, DOI:10.15227/orgsyn.050.0001 (accessed online).
- R. Uson, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85 CrossRef CAS.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- (a) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed; (b) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected crystallographic data, additional structural drawings and copies of the NMR spectra. CCDC 1558580–1558590. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02336g |
| This journal is © The Royal Society of Chemistry 2017 |