
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-mediated reactions between dialkylcyanamides and acetamidoxime generate unusual (nitrosoguanidinate)nickel(II) complexes†
Zarina M.
Bikbaeva
,
Alexander S.
Novikov
 ,
Vitalii V.
Suslonov
,
Nadezhda A.
Bokach
* and
Vadim Yu.
Kukushkin
*
,
Vitalii V.
Suslonov
,
Nadezhda A.
Bokach
* and
Vadim Yu.
Kukushkin
*
Saint Petersburg State University, 7/9 Universitetskaya Nab., 199034 Saint Petersburg, Russian Federation. E-mail: n.bokach@spbu.ru; v.kukushkin@spbu.ru
First published on 10th July 2017
Abstract
The nitrosoguanidinate complexes [Ni{NH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(NR2)NN(O)}2] (R2 = Me21, (CH2)4O 2, (CH2)43, (CH2)54, (Me)Ph 5, Ph26, (p-MeC6H4)27) were obtained in low-to-moderate (12–26%) yields but reproducible yields in an unexpected metal-mediated reaction in MeOH between the nickel salt NiCl2·2H2O, N,N-disubstituted cyanamides NCNR2, and the amidoxime MeC(NOH)NH2. These complexes were formed along with a spectrum of cyanamide–oxime coupling products. The IR and X-ray data indicate the delocalization within the NNO and NCN systems of the nitrosoguanidinate ligand. This delocalization was additionally confirmed by inspection of Wiberg bond indices for the selected bonds. In the X-ray structure of 5, the rare metallophilic contacts Ni⋯Ni between stacks of the square-planar complexes were detected and these non-covalent interactions were studied by non-relativistic and relativistic DFT calculations and topological analysis of the electron density distribution within the framework of Bader's theory (QTAIM method). The estimated strength of the Ni⋯Ni interactions is 1.3–1.9 kcal mol−1 and they are mostly determined by crystal packing effects and weak attractive interactions between adjacent metal centers due to the overlapping of their dz2 and pz orbitals.
C(NR2)NN(O)}2] (R2 = Me21, (CH2)4O 2, (CH2)43, (CH2)54, (Me)Ph 5, Ph26, (p-MeC6H4)27) were obtained in low-to-moderate (12–26%) yields but reproducible yields in an unexpected metal-mediated reaction in MeOH between the nickel salt NiCl2·2H2O, N,N-disubstituted cyanamides NCNR2, and the amidoxime MeC(NOH)NH2. These complexes were formed along with a spectrum of cyanamide–oxime coupling products. The IR and X-ray data indicate the delocalization within the NNO and NCN systems of the nitrosoguanidinate ligand. This delocalization was additionally confirmed by inspection of Wiberg bond indices for the selected bonds. In the X-ray structure of 5, the rare metallophilic contacts Ni⋯Ni between stacks of the square-planar complexes were detected and these non-covalent interactions were studied by non-relativistic and relativistic DFT calculations and topological analysis of the electron density distribution within the framework of Bader's theory (QTAIM method). The estimated strength of the Ni⋯Ni interactions is 1.3–1.9 kcal mol−1 and they are mostly determined by crystal packing effects and weak attractive interactions between adjacent metal centers due to the overlapping of their dz2 and pz orbitals.
Introduction
The reactions of metal-activated conventional nitriles NCR (R = alkyl and/or aryl) and such unconventional nitrile species as cyanamides NCNR2 have been intensively studied during the last two decades and impressive examples of differences in reactivity between these two types of nitriles are available in the literature (see, for instance, our review1). Thus, the guanidine {PhN(H)}2CNH reacts with dialkylcyanamide species bearing the cis-(NCNR2)2PtII functionality to form eight-membered metallacycles stabilized due to an extended chain of conjugation (Scheme 1), whereas conventional nitrile complexes cis-(NCR)2PtII form 1,3,5-triazapentadienate derivatives {![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) HC(R)NC(R)H}PtII (Scheme 1, a).2 Dialkylcyanamide ligands in cis-(NCNR2)2PtIV-type complexes undergo hydration coupling to form six-membered metallacycles,3 while conventional nitrile ligands form metal-bound carboxamides in their iminol form (b).4 Amidrazone derivatives were isolated in the reaction of amidoximes with trans-(NCNR2)2PtIV (c).5 However, when complexes with the trans-(NCR)2PtIV moiety were treated with amidoximes, the reaction resulted in a bis-coupling product.6 These qualitative differences in reactivity between dialkylcyanamide and conventional nitrile ligands were documented mainly for platinum(II and IV) complexes and only a few relevant examples were reported for nickel(II) species.7
HC(R)NC(R)H}PtII (Scheme 1, a).2 Dialkylcyanamide ligands in cis-(NCNR2)2PtIV-type complexes undergo hydration coupling to form six-membered metallacycles,3 while conventional nitrile ligands form metal-bound carboxamides in their iminol form (b).4 Amidrazone derivatives were isolated in the reaction of amidoximes with trans-(NCNR2)2PtIV (c).5 However, when complexes with the trans-(NCR)2PtIV moiety were treated with amidoximes, the reaction resulted in a bis-coupling product.6 These qualitative differences in reactivity between dialkylcyanamide and conventional nitrile ligands were documented mainly for platinum(II and IV) complexes and only a few relevant examples were reported for nickel(II) species.7
 | ||
| Scheme 1 Comparison of the metal-mediated conversions of cyanamide and nitrile ligands, emphasizing different reaction routes for these two types of nitrile species. | ||
The reactions of nickel(II)-activated conventional nitriles with oximes were studied previously and a cascade reaction leading to the 1,3,5-triazapentadienate systems {HC(R)NC(R)H}NiII has been reported by Kopylovich and Pombeiro et al. (d).7,8 Recently, we reported on the reaction between NiII-bound dialkylcyanamides and oximes that leads to the generation of coordinated and uncomplexed coupling products featuring the {NHC(NR2)ONCR1R2}NiII and [NHC(NR2)ONCR1R2]+ moieties. The latter reaction differs from the reaction with conventional nitriles giving 1,3,5-triazapentadienate systems (d).8a,e
In continuation of our studies of the reactivity of disubstituted cyanamides and conventional nitriles with different nucleophiles in the presence of nickel(II),8a,9 we turned our attention to amidoximes and observed an unusual transformation in the system NiII/NCNR2/MeC(NOH)NH2 leading to the nitrosoguanidinate complexes [Ni{HC(NR2)N(O)}2]. This transformation is realized only for cyanamides and not for conventional nitriles and for acetamidoxime and not for other oximes. We report herein on the reaction of acetamidoxime with cyanamides in the presence of nickel salts that leads to a novel family of nitrosoguanidinate complexes, whose chemistry is still almost unexplored because of the harmful and explosive properties of uncomplexed nitrosoguanidines.
Results and discussion
For the study of the NiII-involving reactions between cyanamides and amidoximes we addressed the nickel salts NiX2·nH2O (X = OTf, n = 0; Cl, n = 0, 2 or 6; Br, n = 3; I, n = 6; NO3, n = 6), N,N-disubstituted cyanamides NCNR2 (R2 = Me2, (CH2)4O, (CH2)4, (CH2)5, (Me)Ph, Ph2, (p-MeC6H4)2), and acetamidoxime MeC(NOH)NH2. These reactions were studied in methanol in which the nickel salts are rather well soluble. In the system NiX2·nH2O/NCNR2/MeC(NOH)NH2, unexpected (nitrosoguanidinate)NiII complexes 1–7 (Scheme 2) were reproducibly obtained in low-to-moderate (12–26%) yields. The previously reported addition products featuring the {HC(NR2)OC(NH2)Me}NiII moiety, which were also formed in the reaction and released as oily residues, were identified by HRESI+-MS and IR spectroscopy.
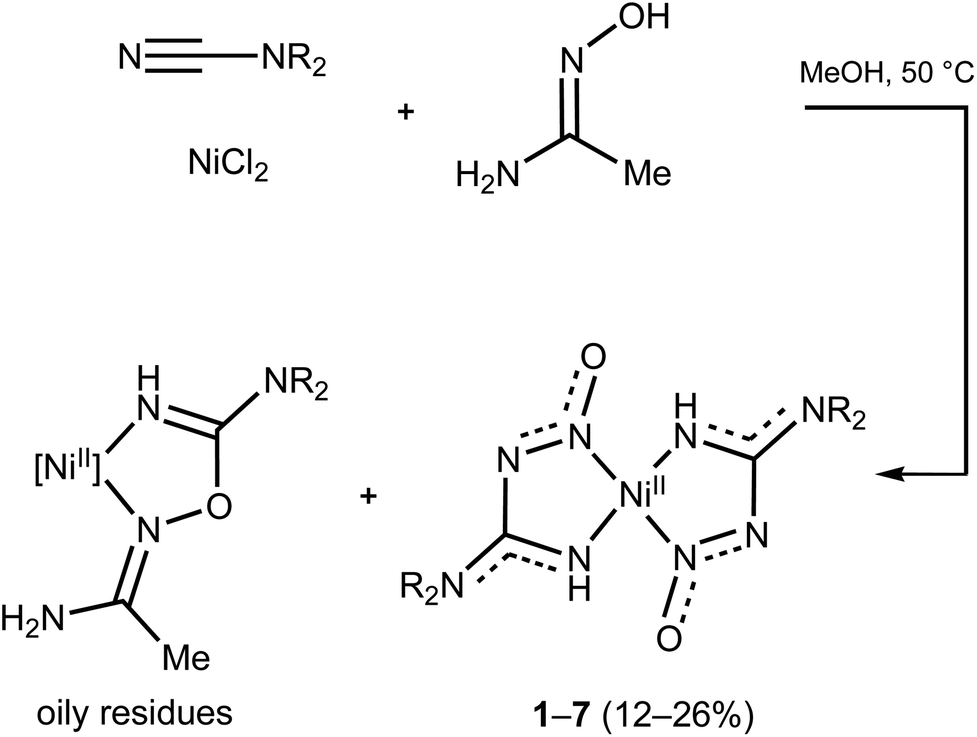 | ||
| Scheme 2 NiII-Mediated generation of nitrosoguanidinate species 1–7 (R2 = Me21, (CH2)4O 2, (CH2)53, (CH2)44, Me/Ph 5, Ph26, (p-MeC6H4)27). | ||
In the synthetic experiments, treatment of NCNR2 with the amidoxime MeC(NOH)NH2 in the presence of NiX2·nH2O in methanol at 50 °C, followed by evaporation of the reaction mixture in air at RT (20–25 °C), results in the generation of 1–7 along with oily residues of other Ni-containing products. Among the tested nickel salts, the highest yields of the nitrosoguanidinate complexes were achieved for NiCl2·2H2O. The usage of the highly hydrated salt NiCl2·6H2O or the addition of one drop of H2O to the reaction mixtures leads to similar yields to those obtained in the reaction with NiCl2·2H2O, whereas the usage of non-hydrated NiCl2 leads to slightly lower yields. We optimized the reaction conditions by varying the ratio between the components of the system NiCl2·2H2O/NCNMe2/MeC(NOH)NH2 (molar ratios 1/2.5/2.5, 1/2.5/5, 1/5/2.5, and 1/10/2.5 were used) and also temperature (RT, 50 °C, and 70 °C). The optimal condition is a system with a 1/2.5/2.5 ratio among the reactants and a temperature of 50 °C that gave 1 in 15% yield. The solvent choice is important; thus, 1–7 were not formed in acetone, while the application of undried ethanol in the system NiCl2·2H2O/NCNMe2/MeC(NOH)NH2 also led to 1 and this compound was isolated in 12% yield.
The positive-mode HRESI-MS spectra of the oily residues obtained from NiCl2·2H2O/NCNR2/MeC(NOH)NH2 systems exhibit several groups of peaks with characteristic isotopic patterns corresponding to the [H2NC(NR2)OC(Me)NH2]+, [Ni{NHC(NR2)ONC(Me)NH2}2]2+, [NiCl{NHC(NR2)ONC(Me)NH2}]+, and [NiCl{NHC(NR2)ONC(Me)NH2}2]+ (R = Me2, (CH2)4O, (CH2)4, (CH2)5, Ph2, (Me)Ph) ions, which reflect the formation of the coupling product. It is noteworthy that these signals appeared in the HRESI+-MS spectra already after 2 h of reaction. The IR spectra of the reaction mixture NiCl2·2H2O/NCNMe2/MeC(NOH)NH2 were measured at different time intervals (0 h, 2 h, and 4 h). In the first and the second spectra, an absorption band from ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) at 2216–2264 cm−1 was observed. In the IR spectra recorded after 4 h, this band was not found, whereas several new intensive bands from ν(CN) in the range of 1635–1675 cm−1 were observed instead that indicates total conversion of NCNR2 into coordinated NHC(NR2)ONC(NH2)Me. These observations give evidence of NiII-mediated amidoxime–cyanamide coupling leading to nickel(II) complexes featuring the {HC(NR2)OC(NH2)Me}NiII functionality. These observations are in agreement with the previously reported NiII-mediated oxime–cyanamide coupling leading to nickel(II) complexes {HC(NR2)OC(R′)Me}NiII (R′ = Me, Ph)8a and also ZnII-10 and PtII-mediated11 amidoxime–cyanamide coupling leading to complexes with HC(NR2)OC(NH2)R′ ligands. The coupling products were not isolated in pure forms, probably due to high kinetic lability of nickel(II) complexes and the existence of nickel(II) products with coordinated carbimidoylamidoxime in several forms.8a
N) at 2216–2264 cm−1 was observed. In the IR spectra recorded after 4 h, this band was not found, whereas several new intensive bands from ν(CN) in the range of 1635–1675 cm−1 were observed instead that indicates total conversion of NCNR2 into coordinated NHC(NR2)ONC(NH2)Me. These observations give evidence of NiII-mediated amidoxime–cyanamide coupling leading to nickel(II) complexes featuring the {HC(NR2)OC(NH2)Me}NiII functionality. These observations are in agreement with the previously reported NiII-mediated oxime–cyanamide coupling leading to nickel(II) complexes {HC(NR2)OC(R′)Me}NiII (R′ = Me, Ph)8a and also ZnII-10 and PtII-mediated11 amidoxime–cyanamide coupling leading to complexes with HC(NR2)OC(NH2)R′ ligands. The coupling products were not isolated in pure forms, probably due to high kinetic lability of nickel(II) complexes and the existence of nickel(II) products with coordinated carbimidoylamidoxime in several forms.8a
We also conducted a metal-free blank experiment and found that keeping a mixture of NCNMe2 and MeC(NOH)NH2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in the absence of any nickel(II) salt under the conditions of the nickel(II)-mediated reaction does not give the coupling products or uncomplexed nitrosoguanidine species even after 5 d. Therefore, one can conclude that the formation of nitrosoguanidine species is metal-mediated.
:1 molar ratio in the absence of any nickel(II) salt under the conditions of the nickel(II)-mediated reaction does not give the coupling products or uncomplexed nitrosoguanidine species even after 5 d. Therefore, one can conclude that the formation of nitrosoguanidine species is metal-mediated.
Other compounds that behave as simultaneous nucleophiles and “NO” sources, such as HONH212 and HONCMe2,13 were tested in the NiII-involving reaction with NCNMe2. However, no nitrosoguanidinate species were obtained and/or identified in the reaction mixture. The known 1,3,5-triazapentadienate complex [Ni{HC(OMe)NC(OMe)H}2]14 was isolated from the system NiCl2·2H2O/NCNMe2/HONCMe2 in 5% yield. The application of the other amidoximes R′C(NOH)NH2 (R′ = PhCH2, Ph, p-BrC6H4) in the reaction does not lead to 1–7. We assume that this difference in reactivity could be explained by the greater nucleophilicity of HONC(NH2)Me than that of the other tested amidoximes. The application of conventional nitriles NCR (R = Me, Ph) in the reaction with NiCl2·2H2O/HONC(NH2)Me in MeOH does not lead to similar nitrosoamidine complexes. The difference between cyanamides and conventional nitriles in reactivity is mostly due to the presence, in cyanamides, of the NR2 group exhibiting strong +M effect.
Based on our observations and the literature data, we assume that the formation of 1–7 is a result of a few consecutive NiII-mediated reactions. Initially, the acetamidoxime reacts with NiII-activated cyanamide to form coupling products featuring the {HC(NR2)OC(NH2)Me}NiII chelated ring (Scheme 3). The generation of stable isostructural coupling products was previously observed for the relevant system NiII/NCNR2/ketoxime.8a Probably, the other amidoximes are not active in the generation of (nitrosoguanidinate)NiII species due to their lower nucleophilicity.15
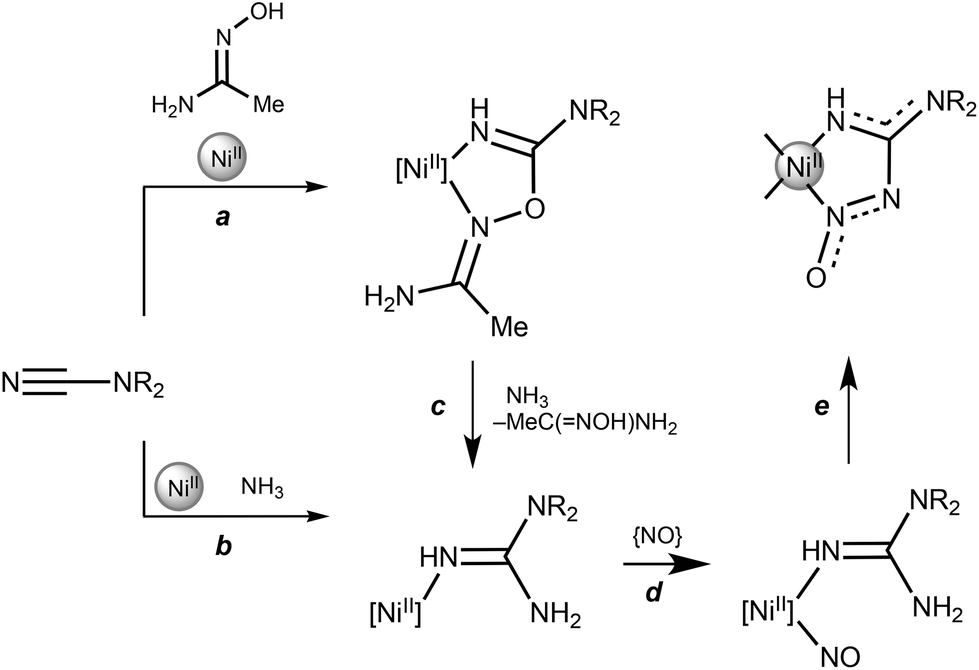 | ||
| Scheme 3 A plausible mechanism for the generation of (nitrosoguanidinate)NiII complexes. | ||
These coupling products could further react with wet MeOH to give OC(NR2)NH2, HNR2, NH3, CO2, and the parent amidoxime MeC(NOH)NH2 (c). Similar transformations were postulated for the NiII-mediated reaction of dialkylcyanamides with pyrazoles in undried MeOH.9 Ammonia, which was accumulated in the reaction mixture, reacted with either the (NCNR2)NiII (b) or {HC(NR2)OC(NH2)Me}NiII (c) species, producing complexes featuring the {HNC(NR2)NH2}NiII functionality via nucleophilic addition (b) or Pinner-like nucleophilic substitution (c) routes. Addition of external ammonia to the NiCl2·2H2O/NCNMe2/MeC(NOH)NH2 system in methanol does not change the yield of the reaction.
Ammonia is likely generated in situ from NCNR2 (or its coupling products) via nickel(II)-mediated hydrolysis, whereupon NH3 adds to another nitrile group to give a guanidine moiety (c). Previously, one of us reported the relevant (CoII/oxime)-mediated conversion of nitriles NCR (R = alkyl, aryl) leading to amidines RC(NH2)NH.16 In the latter transformation, again, the addition of external ammonia does not affect the yield of the formed amidines. Furthermore, the generation of coupling products with in situ generated ammonia was observed upon metal-mediated syntheses of 1,3,5-triazapentadienes (for a review on triazapentadiene complexes see ref. 8b), phthalocyanines,8c,17 diacylamides,18N-acylamidines,19 imidic anhydrides,3 and amidines.16,20 Our current experiments and the previous data8a,17–20 suggest that the mechanism of the reaction shown in Scheme 3 is not simple and perhaps involves internal generation of ammonia at a nickel(II) center as one of the fast reaction steps.
Independently, MeC(NOH)NH2 could be subjected to oxidative hydrolysis to give an “NO” source. The relevant NiII-mediated nitrosation with hydroxylamine formed in situ upon ketoxime hydrolysis is known.21 Once NO is formed, it coordinates to the NiII center (d) and then couples with the guanidine ligand producing the {HC(NR2)N(O)}NiII group (e). Step e of the nitrosyl–guanidine coupling is analogous to that observed in the reaction between the amidinate complex [Fe{RNC(Me)NR}3] (R = p-MeC6H4) and NO.22 This reaction leads to [Fe{N(O)N(R)C(Me)NR}{RNC(Me)NR}2] and the complex is formed through intramolecular nitrosyl–amidinate coupling at the iron center. It is noteworthy that formally reverse reaction—i.e. when the NHC–N2O adducts (NCH = 1,3-dimesitylimidazol-2-ylidene and 1,3-diisopropylimidazol-2-ylidene) featuring the same N2CNNO linkage react with the Ni(COD)2 complex—includes the N–N bond cleavage leading to the nitrosyl nickel complex and guanidinate ligands.23
We added gaseous NO to the reaction mixture NiCl2·2H2O/NCNMe2/MeC(NOH)NH2 (MeOH, 50 °C, 1 d) and kept it for several days for slow evaporation. We observed the formation of 1, but its yield was exactly the same (15%) as in the experiment without the addition of NO. These observations also support the idea that NO is formed in situ as one of the fast reaction steps.
In the context of the generation of nitrosoguanidine nickel(II) species, it is noteworthy that uncomplexed nitrosoguanidines are known and they are typically synthesized by reduction of the corresponding nitroguanidines with Zn dust or RANEY® nickel/H2;24 other methods for the preparation of the nitrosoguanidines are still undeveloped. Nitrosoguanidine itself is an explosive crystalline material,25N-alkylated nitrosoguanidines were studied as intermediates of the biosynthetic conversion of L-arginine to nitric oxide,26 whereas another nitrosoguanidine derivative, viz. N-methyl-N′-nitro-N-nitrosoguanidine, is widely applied for directed mutagenesis in the selection of plants and microorganisms27 and it is also used in industry as an exciting explosive with a high thermal stability.28
Characterization of 1–7
Complexes 1–7 were characterized by C, H, and N elemental analyses, HRESI+-MS, FTIR, 1H and 13C{1H} NMR, TG/DTA, and single-crystal X-ray diffraction (for 1 and 3–5). Nitrosoguanidinate complexes 1–7 are stable until 194–240 °C (Fig. S7, S13, S19, S25, S31, S37 and S43; ESI†) and then gradually decompose at higher temperatures. All complexes gave satisfactory C, H, and N elemental analyses for the proposed formulas. The HRESI+-MS spectra exhibit several groups of peaks with characteristic isotopic patterns corresponding to the [M + H]+, [M + Na]+, [2M + Na]+, [3M + Na]+ ([M] = [Ni{NHC(NR2)NN(O)}2]) ions.
In the IR spectra of 1–7, absorption bands from ν(N–H) (3352–3476 cm−1), ν(CN) (1566–1608 cm−1), and ν(NO) (1386–1414 cm−1) appeared at the expected ranges (Fig. S3, S9, S15, S21, S27, S33 and S39; ESI†).29 It is noteworthy that ν(NO) of the nitroso group of the complexed nitrosoguanidinates exhibit substantially smaller values than those for the uncomplexed neutral H2NC(NH)NHNO species25 (1520 cm−1), thus demonstrating reduction of the multiplicity of the NO bond upon deprotonation and complexation. The ν(NO) absorption bands of 1–7 exhibit intermediate values between the values of double (1500–1480 cm−1 in nitrosamines R2N–NO29) and single (1310–1250 cm−1 in azoxy species R–NN → O29) NO stretches, thus indicating intermediate bond order within the nitroso fragment. These data are consistent with X-ray structure parameters of 1 and 3–5 whose inspection suggests the delocalization. This delocalization was also supported by analysis of the calculated Wiberg bond indices for selected bonds (see later).
In the 1H NMR spectra (Fig. S5, S11, S17, S23, S29, S35 and S41; ESI†), the signals of the NH protons appeared in the 3.56–3.75 ppm range for 1 and 4 and 4.33–4.65 ppm for 2, 3, and 5–7. In the 13C{1H} NMR, resonances of the methyl groups are at 21 ppm for 7 and in the range 37–41 ppm for 1 and 5; NCH2 appear at 46–49 ppm for 2, 3, and 4; CHAr emerge at 126–131 ppm, and Cipso at 126–142 ppm for 5, 6, and 7. One signal in the interval 164.91–167.25 ppm refers to the quaternary carbon atom of the nitrosoguanidinate moiety N(H)C (Fig. S6, S12, S18, S24, S30, S36 and S42; ESI†).
Complexes 1–7 are red or purple colored. The absorption spectra of all complexes 1–7 display similar spectral patterns with several intensive absorption bands in the near-UV region. The most intense absorption bands are located in the 251–254 (lgε 3.27–3.35) and 312–329 (lgε 3.02–3.47) nm ranges along with four bands of low intensity in the intervals 395–407 (lgε 2.26–2.63), 459–466 (lgε 1.95–2.09), 526–538 (lgε 2.01–2.20), and 558–570 (lgε 1.93–2.14) nm (in CH2Cl2; Fig. S4, S10, S16, S22, S28, S34 and S40; ESI†). For theoretical discussions on the nature of the singlet excited electronic states of 1 and the assignment of the most significant transitions see the ESI.†
Solid-state structures of the nitrosoguanidinate complexes
HC(NH2)N(O)}].30 In all structures, the nickel centers exhibit a planar environment (geometry index τ4 = 0 (ref. 31)).
 | ||
| Fig. 1 Molecular structure of 5 with the atomic numbering scheme. ADP ellipsoids are drawn at the 50% probability level. | ||
The N(1)–N(3) and N(1)–O(1) bond lengths are 1.3006(16)–1.320(4) Å and 1.240(3)–1.256(4) Å, respectively. These distances are coherent with those in the previously reported complex [Ni{HC(NH2)N(O)}2] (N(1)–N(3) 1.285(9) Å; N(1)–O(1) 1.260(9) Å (ref. 30)). The distances N(1)–N(3) (1.3006(16)–1.320(4) Å) exhibit values similar to the N–N distance in pyridazine (1.304(19) Å)32 favoring intermediate N(1)N(3) bond order. The distances N(1)–O(1) have intermediate values between single Nsp2–O (1.394(18) Å in oximes32) and double NO (1.218(13) Å in the nitro group32) bonds. Thus, the N(1)–N(3) and N(1)–O(1) bonds have intermediate bond order, which is in agreement with the IR data for complexes 1–7 (see above). The C(1)–N(3) bond length (1.391(2)–1.411(5) Å) is only slightly longer than the typical single Csp2–N(2) bond (e.g. 1.376(11) Å in imidazole32), thus reflecting its single bond character. These statements are consistent with the theoretically calculated bond orders (Tables 1 and 2).
| Bond | 1 | 3 | 4 | 5 |
|---|---|---|---|---|
| Ni1–N2 | 0.56 | 0.55 | 0.56 | 0.56 |
| N2–C1 | 1.44 | 1.45 | 1.44 | 1.45 |
| C1–N4 | 1.20 | 1.19 | 1.20 | 1.18 |
| C1–N3 | 1.15 | 1.15 | 1.15 | 1.16 |
| N3–N1 | 1.40 | 1.40 | 1.40 | 1.39 |
| N1–Ni1 | 0.55 | 0.55 | 0.55 | 0.55 |
NH)NHNO,25 [Ni{HC(NH2)N(O)}2],301, and 3–5
| Distance, Å | H2N(NH)NHNO25 |
R = NH230 |
R = NMe2 (1) | R = N(CH2)5 (3) | R = N(CH2)4 (4) | R = N(CH3)Ph (5) | R = NA(CAH3A)PhA (5) |
|---|---|---|---|---|---|---|---|
| Ni(1)–N(1) | 1.879(7) | 1.873(3) | 1.8684(11) | 1.877(2) | 1.869(3) | 1.878(3) | |
| Ni(1)–N(2) | 1.260(2) | 1.851(6) | 1.841(3) | 1.8473(11) | 1.846(2) | 1.844(2) | 1.841(3) |
| N(1)–O(1) | 1.302(2) | 1.260(9) | 1.256(4) | 1.2496(15) | 1.250(3) | 1.242(3) | 1.240(3) |
| N(1)–N(3) | 1.391(2) | 1.285(9) | 1.311(4) | 1.3006(16) | 1.305(3) | 1.320(4) | 1.307(4) |
| N(3)–C(1) | 1.315(2) | 1.404(11) | 1.411(5) | 1.4087(17) | 1.408(3) | 1.407(4) | 1.404(4) |
| C(1)–N(2) | 1.309(2) | 1.297(11) | 1.318(4) | 1.3112(17) | 1.314(3) | 1.303(4) | 1.319(4) |
| C(1)–N(4) | 1.321(11) | 1.332(4) | 1.3377(17) | 1.331(3) | 1.347(4) | 1.334(4) | |
| N(4)–C(2) | 1.457(5) | 1.4684(17) | 1.478(3) | 1.457(4) | 1.462(4) | ||
| N(4)–C(3) | 1.471(4) | 1.441(4) | 1.440(4) |
| Angle, ° | (NH2)2CN2O | R = NH2 | R = NMe2 | R = N(CH2)5 | R = N(CH2)4 | R = N(CH3)Ph | R = NA(CAH3A)PhA |
|---|---|---|---|---|---|---|---|
| Ni(1)–N(1)–N(3) | 119.2(5) | 119.0(2) | 119.84(9) | 119.33(15) | 118.91(19) | 118.73(19) | |
| Ni(1)–N(1)–O(1) | 113.5(1) | 124.0(5) | 123.3 | 123.46(9) | 122.89(16) | 124.4(2) | 123.6(2) |
| N(1)–Ni(1)–N(2) | 110.8(1) | 80.9(3) | 81.46(12) | 81.42(5) | 81.39(9) | 81.42(11) | 81.45(11) |
| Ni(1)–N(2)–C(1) | 115.0(2) | 113.4(6) | 114.1(2) | 113.61(9) | 113.29(16) | 113.9(2) | 113.8(2) |
| O(1)–N(1)–N(3) | 124.8(2) | 116.8(7) | 117.7(3) | 117.67(11) | 117.78(19) | 116.6(3) | 117.7(2) |
| N(1)–N(3)–C(1) | 120.2(2) | 107.8(7) | 107.8(3) | 108.01(11) | 107.16(18) | 107.1(2) | 108.0(2) |
| N(3)–C(1)–N(2) | 118.6(7) | 117.6(3) | 117.75(12) | 118.8(2) | 118.6(3) | 117.9(3) | |
| N(3)–C(1)–N(4) | 114.7(7) | 115.2(3) | 114.81(11) | 115.1(2) | 116.0(3) | 117.3(3) | |
| N(2)–C(1)–N(4) | 126.7(8) | 127.1(3) | 127.41(12) | 126.1(2) | 125.4(3) | 124.8(3) |
All other bonds are of normal values (for a detailed discussion see the ESI†). Inspection of the crystallographic data did not reveal the presence of any metallophilic contacts in crystal structures of 1, 3, and 4, but in 5 this type of non-covalent interaction was detected and confirmed theoretically (see the Computational studies section later).
C(NMe2)ONCMe2}2(H2O)2]2+ (ref. 8a) and [H2N]C(R)ON]C(R′)NH2]+ (ref. 33) species.
In order to confirm or disprove the hypothesis on the existence of these non-covalent interactions and to quantify their energies from a theoretical viewpoint, we carried out non-relativistic and relativistic DFT calculations and topological analysis of the electron density distribution within the framework of Bader's theory (QTAIM method)37 for the oligomeric clusters 5-100 and 5-RT as model systems (Table 3 and Table S3†). This approach has already been successfully used by us in studies on the non-covalent interactions and properties of coordination bonds in various transition metal complexes.9,38 The contour line diagram of the Laplacian distribution ∇2ρ(r), bond paths, and selected zero-flux surfaces for Ni⋯Ni metallophilic interactions in 5-100 is shown in Fig. 2. To visualize the studied non-covalent interactions, reduced density gradient (RDG) analysis39 was carried out, and the RDG isosurface for 5-100 was plotted (Fig. 2).
 | ||
| Fig. 2 Contour line diagram of the Laplacian distribution ∇2ρ(r), bond paths and selected zero-flux surfaces (left) and the RDG isosurface referring to the non-covalent interactions (right) for metallophilic interactions Ni⋯Ni in 5-100 (M06/6-311+G* level of theory). Bond critical points (3, −1) are shown in blue, nuclear critical points (3, −3) – in pale brown, ring critical points (3, +1) – in orange, and cage critical points (3, +3) – in light green. Length is given in units of Å; RDG isosurface values are given in hartree. | ||
| Method/basis | ρ(r) | ∇2ρ(r) | H b | V(r) | G(r) |
E
inta |
E
intb |
|---|---|---|---|---|---|---|---|
| a E int = −V(r)/2.40 b E int = 0.429G(r).41 | |||||||
| 5 -100 | |||||||
| M06/6-311+G* | 0.010 | 0.023 | 0.000 | −0.005 | 0.005 | 1.6 | 1.4 |
| M06/DZP-DKH | 0.010 | 0.023 | 0.000 | −0.005 | 0.005 | 1.6 | 1.4 |
| M06/MDF10(Ni) and 6-311+G* (other atoms) | 0.010 | 0.022 | 0.000 | −0.006 | 0.006 | 1.9 | 1.6 |
| 5 -RT | |||||||
| M06/6-311+G* | 0.009 | 0.021 | 0.001 | −0.004 | 0.005 | 1.3 | 1.4 |
| M06/DZP-DKH | 0.009 | 0.021 | 0.001 | −0.004 | 0.005 | 1.3 | 1.4 |
| M06/MDF10(Ni) and 6-311+G* (other atoms) | 0.009 | 0.021 | 0.000 | −0.005 | 0.005 | 1.6 | 1.4 |
The QTAIM analysis demonstrates the presence of appropriate bond critical points (BCPs) for metallophilic interactions Ni⋯Ni both in 5-100 and in 5-RT. The lengths of the corresponding contacts (3.30 Å for 5-100 and 3.36 Å for 5-RT) are slightly larger than the sum of Bondi's (the shortest)34 vdW radii for Ni (3.26 Å), but noticeably lower than Batsanov's42 and Alvarez's35 vdW radii for Ni (4.00 and 4.80 Å, respectively). The low magnitude of the electron density (0.009–0.010 hartree), positive values of the Laplacian (0.021–0.023 hartree), and zero or close to zero (0.001 hartree) positive energy density in these BCPs are typical for non-covalent interactions.
We have defined energies for these contacts according to the procedures proposed by Espinosa et al.40 and Vener et al.41 (Table 3). The estimated strength of these non-covalent interactions is 1.3–1.9 kcal mol−1. It is noteworthy that previously9 we studied relevant Ni⋯Ni intramolecular metallophilic interactions in the dinuclear [Ni2(μ2-Ph2Pz)2(1,3,5-triazapentadienate)2] species that were stronger (the estimated energy is 3.5–4.4 kcal mol−1). The balance between the Lagrangian kinetic energy G(r) and potential energy density V(r) at the BCPs reveals the nature of these interactions in 5: if the ratio −G(r)/V(r) > 1 is satisfied, then the nature of appropriate interactions is purely non-covalent; in case the ratio −G(r)/V(r) < 1, some covalent component is observed.43 Based on this criterion one can state that the covalent contribution is absent for metallophilic interactions Ni⋯Ni both in 5-100 and in 5-RT. The estimate of the properties of electron density in BCPs and energies for the metallophilic interactions Ni⋯Ni in 5-100 and 5-RT is almost independent of the basis set used (non-relativistic or relativistic approaches).
We believe that in 5 the emergence of nickel(II)⋯nickel(II) contacts is determined by crystal packing effects and weak attractive interactions between the adjacent metal centers due to the overlapping of their dz2 and pz orbitals (Fig. 3).
 | ||
| Fig. 3 Weak attractive interactions between the adjacent metal centers due to the overlapping of their dz2 and pz orbitals. | ||
The results of our combined DFT and QTAIM studies reveal that theoretically determined energies for the ligand-supported9 and ligand-unsupported (this work) metallophilic interactions nickel(II)⋯nickel(II) are certainly lower than those found for the PdII⋯PdII (4.3–6.0 kcal mol−1 (ref. 44)) and PtII⋯PtII (3.9–11.7 kcal mol−1 (ref. 45)) systems, and the strength of metallophilic interactions for compounds of group 10 elements logically increases on going down the group.
Concluding remarks
The results of this work can be considered from a few perspectives. Firstly, we observed an unusual transformation in the system NiCl2·2H2O/NCNR2/MeC(NOH)NH2 in methanol leading to the nitrosoguanidinate complexes [Ni{HC(NR2)N(O)}2]. Although the achieved yields are rather low and this reaction at present cannot be recommended for synthetic purposes, the observed transformation explicitly demonstrates that nitrosoguanidinate metal species can be obtained from stable and broadly commercially available precursors, thus avoiding the utilization of toxic (for high toxicity of N-nitroso species, see ref. 46) and explosive nitrosoguanidines for sequestering metal centers.
Secondly, to our knowledge, the coordination chemistry of nitrosoguanidine/ate species is at a very early stage of growth, and metal nitrosoguanidinate derivatives are currently represented exclusively by the [M{HC(NH2)N(O)}2] (M = Ni, Pd), [Co{HC(NH2)N(O)}2(NH2CON2O)], and [Cu{HC(NH2)N(O)}2]·Me2SO complexes that were obtained by the reaction of pre-prepared nitrosoguanidine (H2NC(NH)NHNO25) with appropriate metal salts.30 The years that had passed since the preparation of these complexes did not bring any further nitrosoguanidinate metal compounds. In this work, we present a novel route to metal nitrosoguanidinates and we hope that, after further development, it might represent a method for the generation of nitrosoguanidinate metal species from rather safe precursors and for the further elaboration of nitrosoguanidine coordination chemistry.
Thirdly, we observed a novel difference in reactivity between conventional nitriles and cyanamides that typifies the family of so-called push–pull nitriles.
Finally, the retrosynthetic approach to the obtained nitrosoguanidinate species suggests that these complexes could be generated via metal-mediated coupling between nitrosyl ligands and guanidines. This reaction should be relevant to the reported coupling between the amidinate ligands in [Fe{RNC(Me)NR}3] and NO22 that leads to the [Fe{N(O)N(R)C(Me)NR}{RNC(Me)NR}2] complex (52% yield) formed through the nitrosyl–amidinate integration at the iron center. As a further development of the project, we intend to study nitrosyl–guanidinate coupling involving, on the one hand, complexes featuring linear and/or bent nitrosyl ligands and, on the other hand, various N-substituted guanidines. Further studies directed toward widening the family of metal nitrosoguanidinates and establishment of their relevance to our nitroso-47 and dinitrosoalkane48 ligand systems are in progress in our group.
Experimental section
Materials and instrumentation
All dialkylcyanamides, nickel(II) salts and solvents were purchased from commercial sources and used as received. Nickel(II) chloride dihydrate was prepared according to the reported procedure.49 The amidoximes R′C(NOH)NH2 (R′ = Me, PhCH2, Ph, p-BrC6H4) were synthesized according to literature methods.50 Microanalyses (C, H, N) were carried out on a Euro EA3028-HT analyzer. Electrospray ionization mass spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization (ESI) source. The instrument was operated in positive ion mode using an m/z range of 50–3000. The capillary voltage of the ion source was set at −4500 V (HRESI+-MS) and the capillary exit at +100 V. For HRESI+-MS the complexes were dissolved in MeOH and MeCN was used as an ionization agent. The absorption spectra were recorded on a Shimadzu UV 1800 spectrophotometer in CH2Cl2. FTIR spectra were recorded on Shimadzu FTIR-8400S (4000–400 cm−1) and Shimadzu IRAffinity-1S (4000–300 cm−1) spectrometers using KBr pellets. 1H and 13C{1H} NMR (400.13 and 100.613 MHz, respectively) spectra were recorded on a Bruker Avance 400 spectrometer in Me2SO-d6 and CDCl3 at ambient temperature; residual solvent signals were used as the internal standard (Fig. S19–S20, S25–S26, S31–S32, S37–S38, S43–S44, S49–S50 and S55–S56; ESI†). TG/DTA measurements (Fig. S21, S27, S33, S39, S45, S51 and S57; ESI†) were performed with a NETZSCH TG 209 F1 Libra thermoanalyzer and MnO2 powder was used as a standard. The initial weights of the samples were in the range 0.6–1.6 mg. The experiments were run in an open alumina crucible in a stream of argon at a heating rate of 10 K min−1. The final temperature of the experiments was 600 °C. Analysis of thermal data was performed with Proteus analysis software.
X-ray diffraction
Single-crystal XRD experiment was carried out on Agilent Technologies “Xcalibur” and “SuperNova” diffractometers with monochromated MoKα and CuKα radiation sources, respectively. The crystal was kept at 100(2) K during experiment. The structures were solved by means of the Superflip51 structure solution program using Charge Flipping and refined by means of the ShelXL52 program incorporated into the OLEX2 program package.53 Empirical absorption corrections were applied using the CrysAlisPro (Agilent Technologies, 2012) program package using spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm. CCDC numbers 1530060 and 1530062–1530064† contain the supplementary crystallographic data for the structures.Computational details
Single point calculations based on the experimental X-ray geometries of 5-100 and 5-RT (quasi-solid-state approach) were carried out at the DFT level of theory using the M06 functional54 (this functional was specifically developed to describe weak dispersion forces and non-covalent interactions) with the help of the Gaussian-0955 program package. Three approaches were used, viz. (i) the Douglas–Kroll–Hess 2nd order scalar relativistic calculations56 that requested a relativistic core Hamiltonian were carried out using DZP-DKH basis sets57 for all atoms; (ii) the effective core potential calculations were carried out using the multielectron fit fully relativistic energy-consistent pseudopotential MDF10 of the Stuttgart/Cologne group that described 10 core electrons and the appropriate contracted basis set for the nickel atom58 and the standard 6-311+G* basis sets for other atoms; (iii) the non-relativistic calculations were carried out using the standard 6-311+G* basis sets for all atoms. The topological analysis of the electron density distribution with the help of the atoms in molecules (QTAIM) method developed by Bader37 was performed using the Multiwfn program (version 3.3.8).59 The full geometry optimization procedure was carried out with the help of the Gaussian-0955 program package at the CAM-B3LYP/6-311++G** (for 1 in dichloromethane solution) and M06/6-311+G* (for 1, 3, 4, and 5 in gas phase) levels of theory. No symmetry restrictions have been applied during the geometry optimization and experimental X-ray data were used as starting points. The solvent effects were taken into account using the SMD continuum solvation model of Truhlar et al.60 The Hessian matrix was calculated analytically for all optimized structures in order to prove the location of correct minima (no imaginary frequencies). The Cartesian atomic coordinates of model structures are presented in Table S3 (ESI†).
Synthetic work
NOH)NH2 (7.4 mg, 0.1 mmol) powder was added to a stirred suspension of NiCl2·2H2O (40 mg, 0.24 mmol) in MeOH (15 mL) placed in a 20 mL round bottomed flask. Then any one of the corresponding cyanamides NCNR2 (R2 = Me2, (CH2)4O, (CH2)4, (CH2)5, (Me)Ph, Ph2, (p-MeC6H4)2; 0.6 mmol) was added to the mixture and it was left to stand for 24 h at 50 °C in the closed flask without stirring. The color of the homogeneous solution turned from light green to dark brown or dark violet (R2 = Ph2). The resulting solutions were evaporated in open air at RT, furnishing, after 2–3 d, crystalline precipitates of 1–7 along with the released oily residues of other Ni-containing products. The precipitates formed were separated from the oily residues by washing with three 1.5 mL portions of MeOH and dried in a desiccator over CaCl2 at RT.
[Ni{
![[N with combining low line]](https://www.rsc.org/images/entities/i_char_004e_0332.gif) H
H![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) C(NMe2)N(O)}2] (1).
10.7 mg, 15%. Red needles. Anal. calcd for C6H14N8NiO2: C, 24.94; H, 4.88; N, 38.78. Found: C, 25.01; H, 4.88, N, 38.77%. HRESI+, m/z: 289.0674 ([M + H]+ requires 289.0671), 311.0496 ([M + Na]+ requires 311.0491), 599.1078 ([2M + Na]+ requires 599.1084), 889.1634 ([3M + Na]+ requires 889.1632). νmax(KBr)/cm−1: 3374 m, ν(N–H); 2936 w, ν(C–H); 2886 w, ν(C–H); 1608 s ν(CN), 1386 s ν(NO). 1H NMR (CDCl3, δ): 3.05 (s, 6H, CH3), 3.19 (s, 6H, CH3), 3.72 (s, 1H, NH), 3.75 (s, 1H, NH). 13C{1H} NMR (CDCl3, δ): 37.20 and 40.47 (CH3), 167.20 (N(H)C). On heating in a capillary (2° min−1) this complex turns brown and decomposes at 270–272 °C. TG/DTA: 210–244 °C (weight loss 34.7%; NH2CONMe2, requires 34.9%), 244–609 °C (weight loss 43.2%; 75.5% NiO, requires 74.7%). Crystals suitable for XRD were obtained from the reaction mixture by its slow evaporation.
C(NMe2)N(O)}2] (1).
10.7 mg, 15%. Red needles. Anal. calcd for C6H14N8NiO2: C, 24.94; H, 4.88; N, 38.78. Found: C, 25.01; H, 4.88, N, 38.77%. HRESI+, m/z: 289.0674 ([M + H]+ requires 289.0671), 311.0496 ([M + Na]+ requires 311.0491), 599.1078 ([2M + Na]+ requires 599.1084), 889.1634 ([3M + Na]+ requires 889.1632). νmax(KBr)/cm−1: 3374 m, ν(N–H); 2936 w, ν(C–H); 2886 w, ν(C–H); 1608 s ν(CN), 1386 s ν(NO). 1H NMR (CDCl3, δ): 3.05 (s, 6H, CH3), 3.19 (s, 6H, CH3), 3.72 (s, 1H, NH), 3.75 (s, 1H, NH). 13C{1H} NMR (CDCl3, δ): 37.20 and 40.47 (CH3), 167.20 (N(H)C). On heating in a capillary (2° min−1) this complex turns brown and decomposes at 270–272 °C. TG/DTA: 210–244 °C (weight loss 34.7%; NH2CONMe2, requires 34.9%), 244–609 °C (weight loss 43.2%; 75.5% NiO, requires 74.7%). Crystals suitable for XRD were obtained from the reaction mixture by its slow evaporation.
[Ni{
HC(NC4H8O)N(O)}2] (2).
10.4 mg, 12%. Red powder. Anal. calcd for C10H18N8NiO4: C, 32.20; H, 4.86; N, 30.04. Found: C, 32.29; H, 4.82, N, 30.00%. HRESI+, m/z: 113.0713 ([NCNC4H8OH]+ requires 113.0715), 373.0886 ([M + H]+ requires 373.0883), 395.0702 ([M + Na]+ requires 395.0702), 767.1516 ([2M + Na]+ requires 767.1507). νmax(KBr)/cm−1: 3403 w, ν(N–H); 2983 w, ν(C–H); 2921 w, ν(C–H); 2867 w, ν(C–H); 1599 s, ν(CN); 1414 s, ν(NO). 1H NMR (DMSO-d6, δ): 3.52–3.55 (m, 4H, CH2α), 3.61–3.64 (m, 4H, CH2β), 4.65 (s, 2H, NH). 13C{1H} NMR (DMSO-d6, δ): 48.46 (OCH2), 65.50 (NCH2), the N(H)C signal was not detected. On heating in a capillary (2° min−1) this complex turns brown and decomposes at 284–285 °C. TG/DTA: 220–299 °C (weight loss 53.9%), 299–610 °C (weight loss 19.9%).
[Ni{
HC(NC5H10)N(O)}2] (3).
10.3 mg, 12%. Red needles. Anal. calcd for C12H22N8NiO2: C, 39.05; H, 6.01; N, 30.36. Found: C, 39.03; H, 6.15, N, 30.22%. HRESI+, m/z: 369.1315 ([Ni{NHC(NC5H10)NN(O)}2H]+ requires 369.1297), 391.1126 ([M + Na]+ requires 391.1117), 759.2321 ([2M + Na]+ requires 759.2336), 1129.3452 ([3M + Na]+ requires 1129.3510). νmax(KBr)/cm−1: 3362 m, ν(N–H); 2936 w, ν(C–H); 2850 w, ν(C–H); 1596 s, ν(CN); 1404 s, ν(NO). 1H NMR (DMSO-d6, δ): 1.51–1.57 (m, 12H, CH2), 3.50 (s, 8H, NCH2), 4.44, 5.20 (two s, 2H, NH). 13C{1H} NMR (CDCl3, δ): 23.92, 25.79 (CH2), 46.25, 48.95 (br NCH2), 165.59 (N(H)C). On heating in a capillary (2° min−1) this complex turns brown and decomposes at 255–256 °C. TG/DTA: 204–295 °C (weight loss 63.9%), 295–610 °C (weight loss 16.8%). Crystals suitable for XRD were obtained from the reaction mixture by its slow evaporation.
[Ni{
HC(NC4H8)N(O)}2] (4).
10.5 mg, 13%. Red needles. Anal. calcd for C10H18N8NiO2: C, 35.22; H, 5.32; N, 32.86. Found: C, 35.02; H, 5.25; N, 32.60%. HRESI+, m/z: 341.1003 ([M + H]+ requires 341.0984), 363.0836 ([M + Na]+ requires 363.0804), 703.1764 ([2M + Na]+ requires 703.1710), 1045.2612 ([3M + Na]+ requires 1045.2571). νmax(KBr)/cm−1: 3352 m, ν(N–H); 2954 w, ν(C–H); 2876 w, ν(C–H); 1600 s, ν(CN); 1396 m, ν(NO). 1H NMR (CDCl3, δ): 1.94 (m, 4H, CH2), 2.05 (m, 4H, CH2), 3.38 (m, 4H, NCH2), 3.55 (s, 2H, NH), 3.61 (m, 4H, NCH2). 13C{1H} NMR (CDCl3, δ): 25.13, 25.74 (CH2), 46.15, 49.58 (NCH2), 164.91 (N(H)C). TG/DTA: 212–322 °C (weight loss 57.1%), 322–484 °C (weight loss 10.5%). Crystals suitable for XRD were obtained from the reaction mixture by its slow evaporation.
[Ni{
HC(N(CH3)Ph)N(O)}2] (5).
15.3 mg, 15%. Red needles. Anal. calcd for C16H18N8NiO2: C, 46.52; H, 4.39; N, 27.13. Found: C, 46.57; H, 4.50, N, 27.22%. HRESI+, m/z: 413.0977 ([M + H]+ requires 413.0984), 435.0800 ([M + Na]+ requires 435.0804), 847.1708 ([2M + Na]+ requires 847.1710). νmax(KBr)/cm−1: 3394 m, ν(N–H); 3050 w, ν(C–H); 2928 w, ν(C–H); 1608 s, ν(CN); 1578 s, ν(CN) and/or δ(CHAr), 1496 m, ν(Ph), 1392 s, ν(NO). 1H NMR (DMSO-d6, δ): 3.36 (s, 6H, CH3), 4.11, 5.38 (two s, 2H, NH), 7.13 (m, 6H, Ph), 7.47 (m, 4H, Ph). 13C{1H} NMR (CDCl3, δ): 41.34 (CH3), 126.47, 128.70, 130.56 (CHAr); 141.73 (Cipso), 167.02 (N(H)C). On heating in a capillary (2° min−1) this complex turns brown and decomposes at 259–260 °C. TG/DTA: 215–452 °C (weight loss 59.7%), 452–609 °C (weight loss 5.7%). Crystals suitable for XRD were obtained from the reaction mixture by its slow evaporation.
[Ni{
HC(NPh2)N(O)}2] (6).
33.1 mg, 26%. Purple thin needles. Anal. calcd for C26H22N8NiO2: C, 58.13; H, 4.13; N, 20.86. Found: C, 58.03; H, 4.50, N, 20.22%. HRESI+, m/z: 537.1295 ([M + H] requires 537.1297), 559.1116 ([M + Na]+ requires 559.1117), 1095.2397 ([2M + Na]+ requires 1095.2336). νmax(KBr)/cm−1: 3476 w, ν(N–H); 3394 w, ν(N–H), 3060 w, ν(C–H); 1566 s, ν(CN); 1492 m δ(CHAr); 1392 s, ν(NO). 1H NMR (CDCl3, δ): 4.43 (s, 2H, NH), 7.30 (m, 20H, Ph). 13C{1H} NMR (CDCl3, δ): 127.30, 127.99, 129.93 (CHAr), 142.08 (Cipso), 167.2 (N(H)C). On heating in a capillary (2° min−1) this complex turns brown and decomposes at 284–285 °C. TG/DTA: 240–262 °C (weight loss 8.6%), 262–276 °C (weight loss 27.5%), 276–324 °C (weight loss 28.3%), 324–610 °C (weight loss 10.1%).
[Ni{
HC(N(p-Tol)2)N(O)}2] (7).
33.8 mg, 24%. Purple thin needles. Anal. calcd for C30H30N8NiO2: C, 60.73; H, 5.10; N, 18.89. Found: C, 59.95; H, 5.50, N, 19.22%. HRESI+, m/z: 593.1933 ([M + H]+ requires 593.1923), 1185.3823 ([2M + H]+ requires 1185.3769). νmax(KBr)/cm−1: 3394 m, ν(N–H); 3034 w, ν(C–H), 3062 w, ν(C–H); 2920 w, ν(C–H); 1570 s, ν(CN); δ(CHAr); 1390 s, ν(NO). 1H NMR (CDCl3, δ): 2.32 (s, 12H, CH3), 4.33 (s, 2H, NH), 7.08–7.13 (d, 16H, CHAr). 13C{1H} NMR (CDCl3, δ): 21.19 (CH3), 126.31 (Cipso), 126.92, 130.48 (CHAr), 137.07 (Cipso), 167.25 (N(H)C). On heating in a capillary (2° min−1) this complex turns purple and decomposes at 289–290 °C. TG/DTA: 214–349 °C (weight loss 62.6%), 349–569 °C (weight loss 9.1%).
Acknowledgements
The authors are grateful to the Russian Science Foundation (grant 14-13-0060). Physicochemical studies were performed at the Center for Magnetic Resonance, Center for X-ray Diffraction Studies, Center for Chemical Analysis and Materials Research, and the Center for Thermogravimetric and Calorimetric Research (all belonging to Saint Petersburg State University). The authors thank the reviewers for valuable suggestions.References
- N. A. Bokach and V. Y. Kukushkin, Coordination chemistry of dialkylcyanamides: Binding properties, synthesis of metal complexes, and ligand reactivity, Coord. Chem. Rev., 2013, 257, 2293–2316 CrossRef CAS.
- (a) P. V. Gushchin, M. R. Tyan, N. A. Bokach, M. D. Revenco, M. Haukka, M.-J. Wang, C.-H. Lai, P.-T. Chou and V. Y. Kukushkin, Novel tailoring reaction for two adjacent coordinated nitriles giving platinum 1,3,5-triazapentadiene complexes, Inorg. Chem., 2008, 47, 11487–11500 CrossRef CAS PubMed; (b) P. V. Gushchin, M. L. Kuznetsov, M. Haukka, M.-J. Wang, A. V. Gribanov and V. Y. Kukushkin, A novel reactivity mode for metal-activated dialkylcyanamide species: Addition of n,n′-diphenylguanidine to a cis-(r2ncn)2ptii center giving an eight-membered chelated platinaguanidine, Inorg. Chem., 2009, 48, 2583–2592 CrossRef CAS PubMed; (c) M. F. C. Guedes da Silva, C. M. P. Ferreira, E. M. P. R. P. Branco, J. J. R. Fraústo da Silva, A. J. L. Pombeiro, R. A. Michelin, U. Belluco, R. Bertani, M. Mozzon, G. Bombieri, F. Benetollo and V. Y. Kukushkin, Bifunctional activation of cyanoguanidine. Synthesis and molecular structure of the azametallacycle cis-[(pph3)2pt{nhc(ome)=nc(nh2)=nh}][bph4], Inorg. Chim. Acta, 1997, 265, 267–270 CrossRef CAS.
- N. A. Bokach, T. B. Pakhomova, V. Y. Kukushkin, M. Haukka and A. J. L. Pombeiro, Hydrolytic metal-mediated coupling of dialkylcyanamides at a pt(iv) center giving a new family of diimino ligands, Inorg. Chem., 2003, 42, 7560–7568 CrossRef CAS PubMed.
- K. V. Luzyanin, M. Haukka, N. A. Bokach, M. L. Kuznetsov, V. Y. Kukushkin and A. J. L. Pombeiro, Platinum(iv)-mediated hydrolysis of nitriles giving metal-bound iminols, J. Chem. Soc., Dalton Trans., 2002, 1882–1887, 10.1039/B108327A.
- D. S. Bolotin, N. A. Bokach, A. S. Kritchenkov, M. Haukka and V. Y. Kukushkin, Amidrazone complexes from a cascade platinum(ii)-mediated reaction between amidoximes and dialkylcyanamides, Inorg. Chem., 2013, 52, 6378–6389 CrossRef CAS PubMed.
- D. S. Bolotin, M. Y. Demakova, A. S. Novikov, M. S. Avdontceva, M. L. Kuznetsov, N. A. Bokach and V. Y. Kukushkin, Bifunctional reactivity of amidoximes observed upon nucleophilic addition to metal-activated nitriles, Inorg. Chem., 2015, 54, 4039–4046 CrossRef CAS PubMed.
- M. N. Kopylovich, A. J. L. Pombeiro, A. Fischer, L. Kloo and V. Y. Kukushkin, Facile ni(ii)/ketoxime-mediated conversion of organonitriles into imidoylamidine ligands. Synthesis of imidoylamidines and acetyl amides, Inorg. Chem., 2003, 42, 7239–7248 CrossRef CAS PubMed.
- (a) E. V. Andrusenko, E. V. Kabin, A. S. Novikov, N. A. Bokach, G. L. Starova, A. A. Zolotarev and V. Y. Kukushkin, Highly reactive niii-bound nitrile–oxime coupling intermediates stabilized by substituting conventional nitriles with a dialkylcyanamide, Eur. J. Inorg. Chem., 2015, 2015, 4894–4904 CrossRef CAS; (b) M. N. Kopylovich, A. M. Kirillov, E. A. Tronova, M. Haukka, V. Y. Kukushkin and A. J. L. Pombeiro, 1,3,5-triazapentadiene nickel(ii) complexes derived from a ketoxime-mediated single-pot transformation of nitriles, Eur. J. Inorg. Chem., 2010, 2010, 2425–2432 CrossRef; (c) M. N. Kopylovich, M. Haukka, A. M. Kirillov, V. Y. Kukushkin and A. J. L. Pombeiro, Novel nickel(ii) complex bearing phthalimido ligands derived from oxime-mediated transformation of phthalonitrile, Inorg. Chem. Commun., 2008, 11, 117–120 CrossRef CAS; (d) M. N. Kopylovich, M. Haukka, A. M. Kirillov, V. Y. Kukushkin and A. J. L. Pombeiro, Unsymmetrical niii–imidoylamidine complexes derived from a novel oxime-mediated single-pot reaction of nitriles, Chem. – Eur. J., 2007, 13, 786–791 CrossRef CAS PubMed; (e) M. N. Kopylovich, E. A. Tronova, M. Haukka, A. M. Kirillov, V. Y. Kukushkin, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Identification of hexameric water and hybrid water–chloride clusters intercalated in the crystal hosts of (imidoylamidine)nickel(ii) complexes, Eur. J. Inorg. Chem., 2007, 2007, 4621–4627 CrossRef.
- E. V. Andrusenko, E. V. Kabin, A. S. Novikov, N. A. Bokach, G. L. Starova and V. Y. Kukushkin, Metal-mediated generation of triazapentadienate-terminated di- and trinuclear [small mu]2-pyrazolate niii species and control of their nuclearity, New J. Chem., 2017, 41, 316–325 RSC.
- D. S. Bolotin, K. I. Kulish, N. A. Bokach, G. L. Starova, V. V. Gurzhiy and V. Y. Kukushkin, Zinc(ii)-mediated nitrile–amidoxime coupling gives new insights into h+-assisted generation of 1,2,4-oxadiazoles, Inorg. Chem., 2014, 53, 10312–10324 CrossRef CAS PubMed.
- D. S. Bolotin, N. A. Bokach, M. Haukka and V. Y. Kukushkin, Amidoximes provide facile platinum(ii)-mediated oxime–nitrile coupling, Inorg. Chem., 2012, 51, 5950–5964 CrossRef CAS PubMed.
- P. Beranova, K. Chalupsky, A. L. Kleschyov, C. Schott, J.-L. Boucher, D. Mansuy, T. Munzel, B. Muller and J.-C. Stoclet, Nω-hydroxy-l-arginine homologues and hydroxylamine as nitric oxide-dependent vasorelaxant agents, Eur. J. Pharmacol., 2005, 516, 260–267 CrossRef CAS PubMed.
- C. Antonino, C. Maria Assunta and P. Venerando, Regeneration of carbonyl compounds from the corresponding oximes: An update until to 2008, Curr. Org. Chem., 2009, 13, 482–501 CrossRef.
- M. N. Kopylovich and A. J. L. Pombeiro, Coordination chemistry of 1,3,5-triazapentadienes, Chem. Rev., 2011, 255, 339–355 CAS.
- D. S. Bolotin, V. K. Burianova, A. S. Novikov, M. Y. Demakova, C. Pretorius, P. P. Mokolokolo, A. Roodt, N. A. Bokach, V. V. Suslonov, A. P. Zhdanov, K. Y. Zhizhin, N. T. Kuznetsov and V. Y. Kukushkin, Nucleophilicity of oximes based upon addition to a nitrilium closo-decaborate cluster, Organometallics, 2016, 35, 3612–3623 CrossRef CAS.
- M. N. Kopylovich, V. Y. Kukushkin, M. F. C. Guedes da Silva, M. Haukka, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Conversion of alkanenitriles to amidines and carboxylic acids mediated by a cobalt(ii)-ketoxime system, J. Chem. Soc., Perkin Trans. 1, 2001, 1569–1573, 10.1039/B101337H.
- M. N. Kopylovich, V. Y. Kukushkin, M. Haukka, K. V. Luzyanin and A. J. L. Pombeiro, An efficient synthesis of phthalocyanines based on an unprecedented double-addition of oximes to phthalonitriles, J. Am. Chem. Soc., 2004, 126, 15040–15041 CrossRef CAS PubMed.
- F. D. Rochon, P. C. Kong and R. Melanson, Hydrolysis and dimerization of nitrile to diacetamide and crystal structures of chloro(2,2,2,2′,2′,2′-hexachlorodiacetamido)(dimethyl sulfoxide)platinum(ii) and cis-aquadichloro(dimethyl sulfoxide)platinum(ii), Inorg. Chem., 1990, 29, 2708–2712 CrossRef CAS.
- (a) C. W. Leung, W. Zheng, Z. Zhou, Z. Lin and C. P. Lau, Mechanism of catalytic hydration of nitriles with hydrotris(pyrazolyl)borato (tp) ruthenium complexes, Organometallics, 2008, 27, 4957–4969 CrossRef CAS; (b) T. B. Anisimova, N. A. Bokach, K. V. Luzyanin, M. Haukka and V. Y. Kukushkin, Push-pull nitrile ligands exhibit specific hydration patterns, Dalton Trans., 2010, 39, 10790–10798 RSC.
- M. Emirdag-Eanes and J. A. Ibers, Conversion of a re(iv) tetrahedral cluster to a re(iii) octahedral cluster: Synthesis of [(ch3)c(nh2)2]4[re6se8(cn)6] by a solvothermal route, Inorg. Chem., 2002, 41, 6170–6171 CrossRef CAS PubMed.
- A. V. Makarycheva-Mikhailova, P. V. Gushchin, M. N. Kopylovich, I. N. Ganebnykh, V. N. Charushin, M. Haukka, A. J. L. Pombeiro and V. Y. Kukushkin, Ni(ii)-mediated nitrosation of oximes bearing an α-ch2 group, Inorg. Chem. Commun., 2006, 9, 869–871 CrossRef CAS.
- J. A. Clark, M. Kilner and A. Pietrzykowski, Amidino-complexes of iron(ii) and (iii), Inorg. Chim. Acta, 1984, 82, 85–92 CrossRef CAS.
- A. G. Tskhovrebov, E. Solari, R. Scopelliti and K. Severin, Insertion of zerovalent nickel into the n–n bond of n-heterocyclic-carbene-activated n2o, Inorg. Chem., 2013, 52, 11688–11690 CrossRef CAS PubMed.
- A. F. McKay, Nitroguanidines, Chem. Rev., 1952, 51, 301–346 CrossRef CAS.
- R. K. Murmann, R. Glaser and C. L. Barnes, Structures of nitroso- and nitroguanidine x-ray crystallography and computational analysis, J. Chem. Crystallogr., 2005, 35, 317–325 CrossRef CAS.
- J. Y. Cho, A. Dutton, T. Miller, K. N. Houk and J. M. Fukuto, Oxidation of n-hydroxyguanidines by copper(ii): Model systems for elucidating the physiological chemistry of the nitric oxide biosynthetic intermediate n-hydroxyl-l-arginine, Arch. Biochem. Biophys., 2003, 417, 65–76 CrossRef CAS PubMed.
- A. Kat, W. G. Thilly, W. H. Fang, M. J. Longley, G. M. Li and P. Modrich, An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6424–6428 CrossRef CAS.
- A. E. Fogel'zang, B. S. Svetlov, V. Y. Adzhemyan, S. M. Kolyasov, O. I. Sergienko and S. M. Petrov, Combustion of explosive compounds with nitrogen-nitrogen bonds, Combust., Explos. Shock Waves, 1976, 12, 732–740 CrossRef.
- L. J. Bellamy, The infrared spectra of complex molecules: Volume two advances in infrared group frequencies, Springer, Netherlands, 2012 Search PubMed.
- R. K. Murmann, R. Glaser and C. L. Barnes, Structure of the nitrosoguanidine complexes of nickel(ii) and copper(ii) by X-ray crystallography and computational analysis, J. Coord. Chem., 2005, 58, 279–294 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Structural variation in copper(i) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, [small tau]4, Dalton Trans., 2007, 955–964, 10.1039/B617136B.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of bond lengths determined by x-ray and neutron diffraction. Part 1. Bond lengths in organic compounds, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19, 10.1039/P298700000S1.
- K. I. Kulish, A. S. Novikov, P. M. Tolstoy, D. S. Bolotin, N. A. Bokach, A. A. Zolotarev and V. Y. Kukushkin, Solid state and dynamic solution structures of o-carbamidine amidoximes gives further insight into the mechanism of zinc(ii)-mediated generation of 1,2,4-oxadiazoles, J. Mol. Struct., 2016, 1111, 142–150 CrossRef CAS.
- A. Bondi, Van der waals volumes and radii of metals in covalent compounds, J. Phys. Chem., 1966, 70, 3006–3007 CrossRef CAS.
- S. Alvarez, A cartography of the van der Waals territories, Dalton Trans., 2013, 42, 8617–8636 RSC.
- V. W. Yam, V. K. Au and S. Y. Leung, Light-emitting self-assembled materials based on d(8) and d(10) transition metal complexes, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in molecules: A quantum theory, Oxford University Press, 1990 Search PubMed.
- (a) A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, D. V. Dar'in, G. L. Starova and V. Y. Kukushkin, Difference in energy between two distinct types of chalcogen bonds drives regioisomerization of binuclear (diaminocarbene)pdii complexes, J. Am. Chem. Soc., 2016, 138, 14129–14137 CrossRef CAS PubMed; (b) K. Kolari, J. Sahamies, E. Kalenius, A. S. Novikov, V. Y. Kukushkin and M. Haukka, Metallophilic interactions in polymeric group 11 thiols, Solid State Sci., 2016, 60, 92–98 CrossRef CAS; (c) T. V. Serebryanskaya, A. S. Novikov, P. V. Gushchin, M. Haukka, R. E. Asfin, P. M. Tolstoy and V. Y. Kukushkin, Identification and h(d)-bond energies of c-h(d)[three dots, centered]cl interactions in chloride-haloalkane clusters: A combined x-ray crystallographic, spectroscopic, and theoretical study, Phys. Chem. Chem. Phys., 2016, 18, 14104–14112 RSC; (d) D. M. Ivanov, A. S. Novikov, I. V. Ananyev, Y. V. Kirina and V. Y. Kukushkin, Halogen bonding between metal centers and halocarbons, Chem. Commun., 2016, 52, 5565–5568 RSC.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- M. Vener, A. Egorova, A. Churakov and V. Tsirelson, Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: Dft computations with periodic boundary conditions, J. Comput. Chem., 2012, 33, 2303–2309 CrossRef CAS PubMed.
- S. S. Batsanov, Van der waals radii of elements, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving x-h...F-y systems, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- (a) M. Weber, J. E. M. N. Klein, B. Miehlich, W. Frey and R. Peters, Monomeric ferrocene bis-imidazoline bis-palladacycles: Variation of pd–pd distances by an interplay of metallophilic, dispersive, and coulombic interactions, Organometallics, 2013, 32, 5810–5817 CrossRef CAS; (b) D. L. J. Broere, S. Demeshko, B. de Bruin, E. A. Pidko, J. N. H. Reek, M. A. Siegler, M. Lutz and J. I. van der Vlugt, Dinuclear palladium complexes with two ligand-centered radicals and a single bridging ligand: Subtle tuning of magnetic properties, Chem. – Eur. J., 2015, 21, 5879–5886 CrossRef CAS PubMed; (c) A. Gouranourimi, M. Ghassemzadeh, S. Bahemmat, B. Neumüller and R. Tonner, New palladium(ii) complexes containing 3-mercapto-1,2,4-triazole ligands: Synthesis, characterization, crystal structure, and density functional theory calculations, Monatsh. Chem., 2015, 146, 57–67 CrossRef CAS.
- (a) M. R. R. Prabhath, J. Romanova, R. J. Curry, S. R. P. Silva and P. D. Jarowski, The role of substituent effects in tuning metallophilic interactions and emission energy of bis-4-(2-pyridyl)-1,2,3-triazolatoplatinum(ii) complexes, Angew. Chem., Int. Ed., 2015, 54, 7949–7953 CrossRef CAS PubMed; (b) A. Alessandro, G. Damiano, M. Matteo and D. C. Luisa, Recent advances in phosphorescent pt(ii) complexes featuring metallophilic interactions: Properties and applications, Chem. Lett., 2015, 44, 1152–1169 CrossRef; (c) V. V. Sivchik, E. V. Grachova, A. S. Melnikov, S. N. Smirnov, A. Y. Ivanov, P. Hirva, S. P. Tunik and I. O. Koshevoy, Solid-state and solution metallophilic aggregation of a cationic [pt(ncn)l]+ cyclometalated complex, Inorg. Chem., 2016, 55, 3351–3363 CrossRef CAS PubMed; (d) B.-H. Xia, C.-M. Che, D. L. Phillips, K.-H. Leung and K.-K. Cheung, Metal−metal interactions in dinuclear d8 metal cyanide complexes supported by phosphine ligands. Spectroscopic properties and ab initio calculations of [m2(μ-diphosphine)2(cn)4] and trans-[m(phosphine)2(cn)2] (m=pt, ni), Inorg. Chem., 2002, 41, 3866–3875 CrossRef CAS PubMed; (e) B.-H. Xia, C.-M. Che and Z.-Y. Zhou, The quest for pdii–pdii interactions: Structural and spectroscopic studies and ab initio calculations on dinuclear [pd2(cn)4(μ-diphosphane)2] complexes, Chem. – Eur. J., 2003, 9, 3055–3064 CrossRef CAS; (f) H.-K. Yip, T.-F. Lai and C.-M. Che, Metal-metal interaction in a binuclear palladium(ii) system. The (d[sigma]*[rightward arrow] p[sigma]) transition and the x-ray crystal structure of [pd2(dppm)2(cn)4][dppm=bis(diphenylphosphino)methane], J. Chem. Soc., Dalton Trans., 1991, 1639–1641, 10.1039/DT9910001639; (g) I. M. Sluch, A. J. Miranda, O. Elbjeirami, M. A. Omary and L. M. Slaughter, Interplay of metallophilic interactions, π–π stacking, and ligand substituent effects in the structures and luminescence properties of neutral ptii and pdii aryl isocyanide complexes, Inorg. Chem., 2012, 51, 10728–10746 CrossRef CAS PubMed.
- W. Lijinsky, Chemistry and biology of n-nitroso compounds, Cambridge University Press, 1992 Search PubMed.
- (a) V. Y. Kukushkin, V. K. Bel'skii, E. A. Aleksandrova, V. E. Konovalov and G. A. Kirakosyan, Platinum(iv)-mediated redox coupling of 2-propanone oximes in cis-[pt(me2c=noh)2cl4]. Crystal structure of [pt(n(=o)cme2oncme2)cl2], Inorg. Chem., 1992, 31, 3836–3840 CrossRef CAS; (b) V. Y. Kukushkin, D. Tudela, Y. A. Izotova, V. K. Belsky and A. I. Stash, Reactivity of trans-[ptx2(ketoxime)2] complexes toward m-chloroperoxybenzoic acid: An efficient route to coordinated nitrosoalkanes and solvent dependence of the reaction, Inorg. Chem., 1996, 35, 4926–4931 CrossRef CAS PubMed.
- K. V. Luzyanin, P. V. Gushchin, A. J. L. Pombeiro, M. Haukka, V. I. Ovcharenko and V. Y. Kukushkin, Oxidation of pt-bound bis-hydroxylamine as a novel route to unexplored dinitrosoalkane ligated species, Inorg. Chem., 2008, 47, 6919–6930 CrossRef CAS PubMed.
- L. G. L. Ward and J. R. Pipal, in Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp. 154–164, DOI:10.1002/9780470132449.ch30.
- C. J. P. Barros, Synthesis of amidoximes using an efficient and rapid ultrasound method, J. Chil. Chem. Soc., 2011, 56, 721–722 CrossRef CAS.
- (a) L. Palatinus and G. Chapuis, Superflip - a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; (b) L. Palatinus and A. van der Lee, Symmetry determination following structure solution in p1, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef CAS; (c) L. Palatinus, S. J. Prathapa and S. van Smaalen, Edma: A computer program for topological analysis of discrete electron densities, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- G. Sheldrick, A short history of shelx, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, Olex2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four m06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, RevisionC.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- M. Reiher, Relativistic douglas–kroll–hess theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 139–149 CrossRef CAS.
- (a) F. E. Jorge, A. Canal Neto, G. G. Camilett and S. F. Machado, Contracted gaussian basis sets for douglas-kroll-hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties, J. Chem. Phys., 2009, 130, 064108 CrossRef CAS PubMed; (b) A. Canal Neto and F. E. Jorge, All-electron double zeta basis sets for the most fifth-row atoms: Application in dft spectroscopic constant calculations, Chem. Phys. Lett., 2013, 582, 158–162 CrossRef CAS; (c) R. C. de Berrêdo and F. E. Jorge, All-electron double zeta basis sets for platinum: Estimating scalar relativistic effects on platinum(ii) anticancer drugs, J. Mol. Struct. (THEOCHEM), 2010, 961, 107–112 CrossRef; (d) C. L. Barros, P. J. P. de Oliveira, F. E. Jorge, A. Canal Neto and M. Campos, Gaussian basis set of double zeta quality for atoms rb through xe: Application in non-relativistic and relativistic calculations of atomic and molecular properties, Mol. Phys., 2010, 108, 1965–1972 CrossRef CAS.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration dirac–hartree–fock data, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1530060 and 1530062–1530064. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01960b |
| This journal is © The Royal Society of Chemistry 2017 |