
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeteroleptic nickel(II)–diNHC complexes and an unusual ‘reverse’ carbene-transfer reaction to silver(I) †
Han Vinh
Huynh
 *,
Christine Hui Min
Koh
and
Van Ha
Nguyen
*,
Christine Hui Min
Koh
and
Van Ha
Nguyen
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore, Singapore. E-mail: chmhhv@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6516 2670
First published on 1st August 2017
Abstract
A series of rare [NiX2(MeCCprop)] complexes bearing the cis-chelating benzimidazole-derived dicarbene ligand MeCCprop and varying anionic coligands (2, X = N3; 3, X = NCS; 4, X = I; 5, X = O2CCF3) have been prepared and coligand dependent structural and spectroscopic features have been evaluated. This study also revealed an unusual ‘reverse’ carbene transfer reaction from nickel to silver giving the disilver species [Ag2X2(μ–κ2-MeCCprop)] (6, X = OAc; 7, X = O2CCF3). A preliminary catalytic study of two representative NiII diNHC complexes in the aqueous and phosphine-free Suzuki–Miyaura coupling reaction of aryl halides is reported as well. These reactions provide good yields of coupling products, but do not require inert conditions.
Introduction
cis-Configured PdII complexes of the type cis-[PdX2(NHC)2] and cis-[PdX2(diNHC)] are common in NHC chemistry and have mostly been studied for their catalytic activities in C–C coupling reactions.1 A comparison between direct cis- and trans-isomers of bis(NHC) PdII complexes has revealed that a cis-arrangement is desirable and leads to faster catalyst initiation. The crucial halido dissociation step is facilitated in cis-complexes due to the strong trans effect of NHCs in general.2Analogous cis-complexes of NiII, on the other hand, are surprisingly very rare, and dihalido-bis(NHC) NiII complexes with monodentate NHCs are almost exclusively trans-configured.3 To the best of our knowledge, only one structurally characterised cis-[NiX2(NHC)2] complex (X = Cl) has been reported thus far.4 The use of simple cis-chelating diNHC ligands with methylene (C1) and ethylene (C2) spacers to enforce the formation of cis-[NiX2(diNHC)] complexes failed and led instead to homoleptic bis(chelates) of the type [Ni(diNHC)2]2+ due to ligand disproportionation processes.5 Nevertheless, a few cis-[NiX2(diNHC)] complexes could be obtained by using longer C3- or C4-bridged diNHC ligands, which successfully circumvents the latter.5c,6
In order to increase the chemical and structural diversity of cis-chelating dicarbene complexes of NiII, we herein report the preparation of representatives bearing various anionic co-ligands. In the course of this study, we discovered an unusual ‘back-transfer’ of NHCs from nickel to silver showing reverse reactivity compared to the ubiquitous silver–carbene transfer route pioneered by Lin and coworkers.7 A preliminary catalytic test of some complexes in the aqueous nickel-catalyzed Suzuki–Miyaura coupling reaction is also reported.
Results and discussion
Halido and pseudohalido complexes
Previously, we have reported that benzimidazolin-2-ylidene NiII complexes can be efficiently prepared by reaction of benzimidazolium salts with Ni(OAc)2 in molten tetrabutylammonium salts, e.g. TBAB.3c,d The application of this method to propylene-bridged dibenzimidazolium dibromides led to the successful isolation of the cis-chelating NiII complexes of the type cis-[NiBr2(diNHC)], which are highly active in the Kumada–Corriu coupling of aryl bromides and chlorides.5c,6b It is anticipated that this type of complex can offer access to a wide range of halido, pseudo-halido and carboxylato analogues by simple ligand exchange. Accordingly, the complex [NiBr2(MeCCprop)] (1) was treated with selected nucleophiles in acetonitrile at ambient temperature (Scheme 1). | ||
| Scheme 1 Preparation of complexes 2–4 by ligand exchange reactions. | ||
The salt metathesis reactions of 1 with sodium azide, potassium thiocyanate and sodium iodide occurred rather straightforwardly, and decent yields of the desired new complexes [Ni(N3)2(MeCCprop)] (2), [Ni(NCS)2(MeCCprop)] (3), and [NiI2(MeCCprop)] (4) were obtained mostly as yellow solids. The coloration of the complex 4 is expectedly more intense due to the electron-rich iodido ligands prone to LMCT (vide infra). Among these, mixed NHC/pseudohalido complexes remain relatively unexplored. Complexes 2 and 4 dissolve in most common organic solvents with the exception of non-polar ones such as diethyl ether and hexane, while complex 3 is only soluble in DMF and DMSO. Their 1H NMR spectra are expectedly very similar, making their differentiation difficult. The presence of multiple signals arising from the diastereotopy of the propylene bridge is a common feature in the spectra of all chelates. Due to peak broadening, the 13Ccarbene NMR signal could only be resolved for complex 2 at 177.2 ppm. For complex 3, an additional characteristic signal was detected at 162.7 ppm, providing evidence of the presence of isothiocyanato ligands.
Complexes 2 and 3 can be further differentiated by IR spectroscopy (see the ESI†). The former complex shows a strong broad band at 2041 cm−1 typical for the 1st row, late transition metal azido complexes.8 The IR spectrum of the latter displays a broad band at 2111 cm−1 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch and a sharper one at 1678 cm−1 for the CS stretch of the isothiocyanato ligands.9 Moreover, a medium band at 828 cm−1 corroborates the κ-N binding of the ambidentate NCS ligand, while a κ-S coordination would be indicated by a band at ∼700 cm−1.10 Overall, this observation is in line with the HSAB concept.
N stretch and a sharper one at 1678 cm−1 for the CS stretch of the isothiocyanato ligands.9 Moreover, a medium band at 828 cm−1 corroborates the κ-N binding of the ambidentate NCS ligand, while a κ-S coordination would be indicated by a band at ∼700 cm−1.10 Overall, this observation is in line with the HSAB concept.
Single crystals of 2·0.5CH2Cl2 and 4·0.5CH2Cl2·0.5C7H8 suitable for X-ray diffraction analysis could be obtained by slow evaporation of solutions in dichloromethane/hexane and dichloromethane/toluene, respectively, and their molecular structures are depicted in Fig. 1. Both complexes are expectedly square planar, where the propylene-bridge allows for the formation of a relaxed 8-membered metallacycle. Complex 2 has smaller dihedral angles of 82.5(1)° and 80.3(1)° between the carbene plane and the [NiC2X2] coordination plane. In complex 4 slightly larger values of 87.4(2)° and 83.5(2)° were found. The different anionic ligands do not affect the nickel–carbene distances, which are essentially equidistant in 2, 4 and their parent complex 1.5c
 | ||
| Fig. 1 Molecular structures of 2·0.5CH2Cl2 and 4·0.5CH2Cl2·0.5C7H8. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond lengths [Å] and bond angles [°] for 2: Ni1–C1 1.866(4), Ni1–C12 1.864(4), Ni1–N5 1.908(4), Ni1–N8 1.925(4); C12–Ni1–C1 85.5(2), C1–Ni1–N5 94.1(2), C12–Ni1–N8 92.0(2), N5–Ni1–N8 88.5(2), N5–N6–N7 175.6(4), N10–N9–N8 175.4(4), N2–C1–N1 106.3(3), N3–C12–N4 106.0(3); for 4: Ni1–C1 1.859(7), Ni1–C12 1.857(8), Ni1–I1 2.236(1), Ni1–I2 2.525(1); C12–Ni1–C1 86.2(3), C12–Ni1–I2 90.9(2), C1–Ni1–I1 86.7(2), I2–Ni1–I1 96.21(4), N1–C1–N2 107.0(6), N3–C12–N4 105.5(6). | ||
UV-Vis spectroscopic analyses were carried out on complexes 1–4 to understand the influence of different anionic ligands on the electronic properties of nickel–diNHC complexes. Fig. 2 shows the varying shades of yellow of equimolar complex solutions (1 × 10−3 M) in DMSO as the solvent of choice. Table 1 lists the UV-Vis absorption wavelength maxima and the absorption coefficients of 1–4.
 | ||
| Fig. 2 Solutions of complexes 1–4 in DMSO (1 × 10−3 M). | ||
| Complex | λ max [nm] | Abs. | ε [M−1 cm−1] |
|---|---|---|---|
| [NiBr2(MeCCprop)] (1) | 425 | 1.02 | 1.02 × 105 |
| [Ni(N3)2(MeCCprop)] (2) | 409 | 1.54 | 1.54 × 105 |
| [Ni(NCS)2(MeCCprop)] (3) | 384 | 2.38 | 2.38 × 105 |
| [NiI2(MeCCprop)] (4) | 440 | 1.78 | 1.78 × 105 |
The λmax values were tentatively assigned as LMCT transitions based on the high molar extinction coefficients, which are indicative of charge transfers. Moreover, a red-shift was observed going from the bromido (1) to the iodido complex (4), which is indicative of LMCTs.11 The increasing λmax values in the order 3 (NCS) < 2 (N3) < 1 (Br) < 4 (I) indicate increasing ease of oxidation, which is in agreement with the electron transfer redox values for the respective ligands (I2−/2I− = 0.54 V; Br2−/2Br− = 1.07 V; N3˙−/N3− = 1.29 V; (SCN)2˙−/2SCN− = 1.32 V).12 Previous studies have also reported that the λmax values for κ-S thiocyanato derivatives are expected at much longer wavelengths than 384 nm obtained for complex 3.12 From these results, we can also conclude with greater certainty that complex 3 is indeed an isothiocyanato complex, where the pseudohalido ligands are bound to the metal centre through the nitrogen donors.
Carboxylato complexes and reverse carbene-transfer to silver
Heteroleptic NHC/carboxylato complexes of palladium(II) are well established13 and important intermediates2,14 in catalytic reactions. However, their nickel(II) analogues are unknown to the best of our knowledge, although acetate bases are also often used in nickel-catalyzed reactions. Salt metathesis reactions for the preparation of these proved to be less straightforward in comparison with their halido and pseudohalido analogues. The reaction of complex 1 with three equivalents of sodium trifluoroacetate did not occur at all, and only starting materials were recovered. Subsequent trials with increasing amounts of the salt were then carried out. Finally, 10 equivalents of sodium trifluoroacetate were added to a solution of 1 in acetonitrile, and the mixture was stirred at 70 °C overnight (Scheme 2, route a). Subsequently, it was heated under reflux for an additional 2 h to ensure that the reaction was driven to completion. Longer reaction times of >2 h resulted in decomposition of the product yielding a hygroscopic substance upon work-up instead. | ||
| Scheme 2 Preparation of carboxylato complex 5 and disilver complexes 6 and 7. | ||
The ditrifluoroacetato complex 5 is the first of example of a mixed NHC/carboxylato nickel complex and was isolated as an air-stable, yellow powder. As observed for PdII NHC analogues, halido–carboxylato exchange improves solubility in organic solvents, facilitating 13C NMR data collection, and the 13Ccarbene NMR signal of 5 was detected at 180.6 ppm. Moreover, 19F NMR spectroscopy provides evidence of equivalent trifluoroacetato ligands with a single signal at 2.36 ppm for the CF3 group.
The IR spectrum of 5 shows a broad band at 1690 cm−1 characteristic of a CO stretch (see ESI†). Another band at 1469 cm−1 is assigned to the C–O stretch, while the sharp signal at 843 cm−1 was detected for the O–CO bend of the carboxylato function of the ligand. Three bands at 1209, 1130 and 1035 cm−1 are associated with the C–F stretches of the trifluoroacetato ligands, while the CF3 deformation gives rise to a sharp signal at 794 cm−1. All these are in agreement with literature values.15
X-ray diffraction analysis of single crystals of 5 grown from dichloromethane/hexane confirmed that ligand substitution was indeed successful (Fig. 3). The compound also adopts a square-planar geometry similar to that of 2 and 4, although the dihedral angles of 78(1)° and 79(1)° between the [NiC2X2] coordination plane and the NHC planes are slightly smaller. The nickel–carbene distances are in the same range as those observed for 1, 2 and 4.
 | ||
| Fig. 3 Molecular structures of 5·CH2Cl2. One of the CF3CO2 groups is disordered with 16% replaced by adventitious Cl from dichloromethane used for crystallization. Hydrogen atoms, the solvent molecule and the disordered Cl have been omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Ni1–C1 1.86(3), Ni1–C12 1.86(4), Ni1–O1 1.92(2), Ni1–O3 1.90(3); C1–Ni1–C12 85(1), C1–Ni1–O1 93(1), C12–Ni1–O3 95(1), O1–Ni1–O3 87(1). | ||
Ligand substitution with the more basic sodium acetate was attempted using the same reaction conditions as those for the synthesis of 5. Surprisingly, these attempts were unsuccessful, and mainly starting materials were recovered (Scheme 2, route b). This may be due to the difference in the electron-donating ability of the two carboxylato ligands. Binding of the electron-poorer trifluoroacetato ligands to the already electron-rich nickel–diNHC complex fragment appears to be more favourable, while coordination of the more basic acetato ligands trans to the NHCs would lead to a pile-up of excess electron density.
In order to enforce acetato coordination by silver-halide precipitation as a strong driving force, the reaction was repeated with two equivalents of silver acetate. Notably, such a strategy proved fruitful in the preparation of PdII complexes of the type [Pd(O2CR)2(NHC)2]13,14 or [Pd(O2CR)2(diNHC)].16 However, a very different outcome was found in this work. Instead of a simple bromido–acetato ligand exchange, we observed a dicarbene transfer from NiII to AgI, which led to the formation of the disilver–dicarbene complex [Ag2(OAc)2(μ–κ2-MeCCprop)] (6) (Scheme 2, route c). This reaction is very surprising as the NHC transfer from AgI to other metals is undoubtedly one of the most common and general preparative routes to metal–NHC complexes.1b,7 The 1H NMR spectrum of 6 is much simpler than those of the nickel-chelates and does not show diastereotopy of the propylene-spacer, suggesting that the diNHC does not coordinate in a chelating fashion. Moreover, the NCH3 singlet shifts upfield from initially 4.59 ppm to 3.90 ppm. The 13Ccarbene signal could not be detected, which is a common phenomenon in silver–NHC chemistry that points to a dynamic exchange in solution. In addition, ESI-MS showed an isotopic envelope centred at m/z 413 with 0.5 peak spacing in the positive mode, which is assigned to the dicationic [Ag2(MeCCprop)2]2+ fragment in agreement with its calculated isotopic pattern (see the ESI†). The formation of bis(NHC) species in the gas phase is also characteristic of Ag–NHC complexes.
In order to obtain more insights into the carbene transfer reaction from nickel to silver, NMR monitoring was attempted. Upon the addition of two equiv. of AgOAc to a NMR tube containing a solution of 1 in CD3CN, immediate precipitation of NiBr2 was observed already at room temperature. The 1H NMR spectrum recorded was consistent with that of complex 6 (see the ESI†). This observation points to a fast carbene transfer driven by the poor solubility of anhydrous NiBr2 in organic solvents that does not even require heating.
Single crystals of 6 were obtained by slow evaporation of a saturated dichloromethane/hexane solution and subjected to X-ray diffraction analysis (Fig. 4). The results obtained confirm that the diNHC has indeed been transferred from NiII to AgI. The molecular structure also reveals intramolecular argentophilic interactions with an intersilver separation of 2.8962(4) Å in the solid state.17 The C–Ag–O bond angles of 172.66(9)° deviate slightly from linearity, and the silver–NHC distance is 2.070(3) Å.
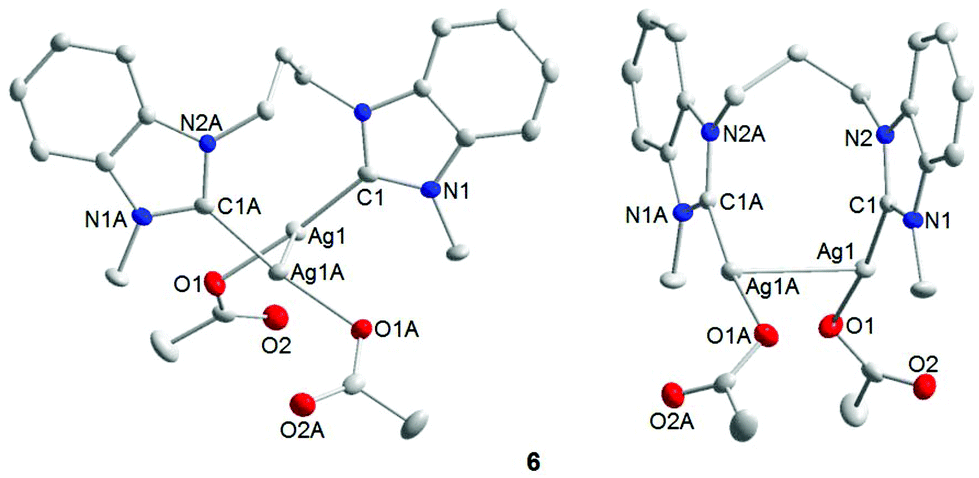 | ||
| Fig. 4 Molecular structure of 6 (front and side views). Hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Ag(1)–C(1) 2.070(3), Ag(1)–O(1) 2.1045(19), Ag(1)–Ag(1) 2.8962(4); C(1)–Ag(1)–O(1) 172.66(9), C(1)–Ag(1)–Ag(1) 84.06(7). | ||
The reaction was then attempted using the diiodido complex 4, where silver iodide precipitation was expected to provide an even greater driving force for ligand substitution at NiII. However, 1H NMR spectroscopy and ESI mass spectrometry confirmed the formation of complex 6 also in this case. The preferred NHC transfer from the NiII to AgI may be explained by HSAB theory, where the softer carbene ligand prefers to bind to the softer AgI metal centre over the harder NiII metal centre. Additionally, the immediate precipitation of nickel(II) halides drives the formation of the silver–carbene species in accordance with Le Chatelier's principle.
In order to determine whether a similar reaction would also proceed with less basic carboxylates, complex 1 was also reacted with silver trifluoroacetate under the same conditions (Scheme 2, route d). The compound isolated from this reaction shows NMR spectroscopic features that are similar to complex 6, supporting its identity as [Ag2(O2CCF3)2(μ–κ2-MeCCprop)] (7). In addition, its solution 13C NMR spectrum shows a downfield 13Ccarbene NMR signal at 189.4 ppm that is significantly different from the same diNHC bound to a NiII metal centre (cf. 181.6 ppm for 5). The chemical shift is also in the same range as that observed for the bis(benzimidazolin-2-ylidene) complex [Ag(Et2-bimy)2]PF6 (cf. ∼188 ppm) reported by Lin et al.7 Furthermore, the ESI mass spectrum shows the same base peak at m/z 413 in the positive mode for the proposed [Ag2(MeCCprop)2]2+ fragment.
These observations suggest a similar reactivity of complex 1 with both silver carboxylates. Moreover, the resulting silver–NHC complexes 6 and 7 undergo typical ligand-disproportionation reactions to give bis(NHC) complexes of the type [Ag2(MeCCprop)2][Ag(O2CR)2]2 in solution and gas phase (Scheme 3).
 | ||
| Scheme 3 Reversible ligand-disproportionation of Ag–NHC complexes 6 and 7. | ||
Most NHC transfers involving the two metals occur from silver to nickel. However, it must be noted that most of these occurred with monodentate and/or functionalized NHCs where pre-coordination of a hard donor to nickel may assist in the transfer.1b The transfer of diNHCs has not been reported, and it would be interesting to explore whether the diNHC could be ‘back-transferred’ from silver to nickel. Thus, a solution of complex 6 in CD3CN was mixed with [NiBr2(PPh3)2] as a common nickel precursor in such reactions (Scheme 4, top). The 1H NMR spectrum recorded did not show any evidence of nickel–diNHC complexes, but only additional broad signals due to the nickel–phosphine complex (see the ESI†).
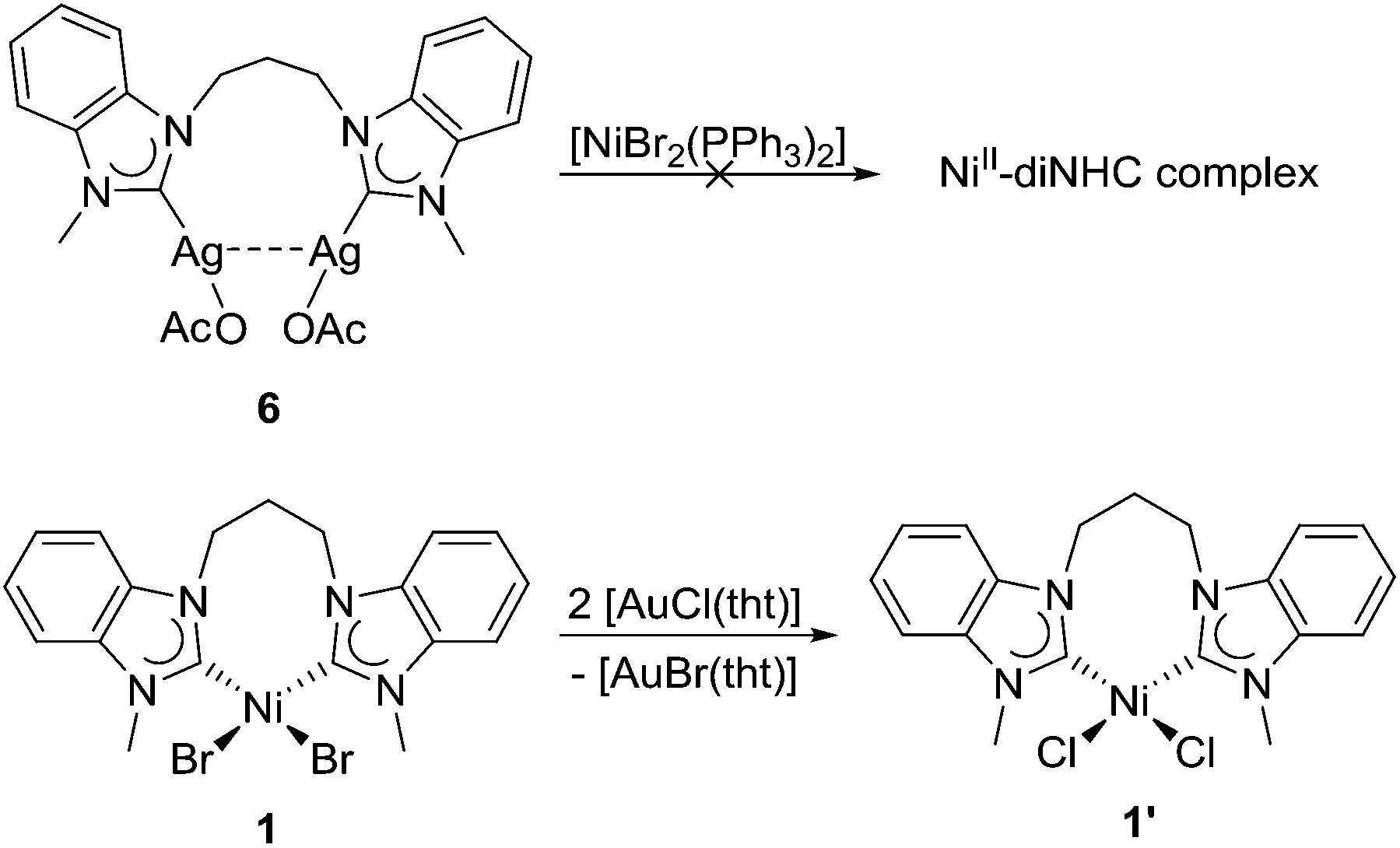 | ||
| Scheme 4 Attempted diNHC transfer reactions involving 6 and 1. | ||
The signals due to 6 remain essentially unchanged even after prolonged reaction time (see the ESI†). This observation indicates that the back-transfer to nickel does not happen easily under the given conditions.
The carbene transfer from nickel to softer metals is not without precedent,18 but a transfer to silver is unknown. The surprising reactivity of complex 1 with silver salts prompted us to explore whether it could also transfer the diNHC to other metals. Thus, complex 1 was mixed with [AuCl(tht)] in CD3CN (Scheme 4, bottom). The initial broad signals due to 1 became sharper, but maintained their pattern indicative of a chelating coordination mode of the diNHC ligand. It appears that diNHC transfer to gold did not happen, but a new nickel complex of improved solubility was formed. The ESI mass spectra of the mixture indeed showed prominent peaks due to chlorido-nickel–diNHC species. Thus, the conclusion can be made that instead of carbene transfer only bromido–chlorido exchange took place in line with the HSAB concept, which led to the formation of the better soluble complex 1′.
Catalytic activities of complexes 1 and 5 in the Suzuki–Miyaura coupling reaction
The Suzuki–Miyaura coupling reaction is arguably one of the most powerful methodologies in the formation of C–C bonds.19 While palladium complexes are well established as outstanding pre-catalysts for this transformation,20 their nickel analogues have, surprisingly, not been as thoroughly examined despite the lower cost that nickel catalysts offer.21 Moreover, several studies of nickel catalysts in the Suzuki–Miyaura coupling reaction have reported only moderate yields, with the majority of these pre-catalysts being NiII–phosphine complexes, where the resulting catalytically active Ni0 species are sensitive to air and moisture, requiring an inert atmosphere for reaction. Applications of Ni0 and NiII–NHC complexes have also been reported. The former are highly active but also air- and moisture sensitive.22 The latter, on the other hand, often require a more sophisticated pincer or multidentate ligand structure and phosphine additives for a good performance.23 Moreover, all these Ni–NHC systems operate in classical organic solvents, while examples in pure water remain unknown to the best of our knowledge. Therefore, the search for phosphine-free NiII complexes for the aqueous Suzuki–Miyaura coupling reaction remains attractive. Similar to their palladium analogues, the strong trans effect exerted by cis-coordinating diNHCs is expected to be beneficial for substrate binding and product release, which might lead to an improved catalytic activity.14Complexes 1 and 5 were selected for a preliminary catalytic study as representatives of NiII-halido and -carboxylato precatalysts, and the coupling of 4-bromobenzaldehyde with phenylboronic acid in air and only using pure water as the reaction medium was chosen as the standard test reaction. A control run was first carried out where no complex was added (Table 2, entry 1), since previous studies reported that trace amounts of transition metal impurities present in the arylboronic acid, base, and even water could catalyze the coupling reaction.24 The results show that only trace amounts of the coupled product had formed. Therefore the substrates and reaction media were essentially free of catalytically active transition metal impurities, and the Suzuki–Miyaura coupling reaction could be tested with complexes 1 and 5.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | mol% | R | X | T [°C] | Yieldb [%] |
| a 1 mmol aryl halide, 1.5 mmol phenylboronic acid, 2 mmol K2CO3, 5 mL of deionized water as solvent, 24 h. b Yields were determined by 1H NMR spectroscopy on an average of two runs. | ||||||
| 1 | — | — | CHO | Br | 120 | Trace |
| 2 | 1 | 1 | CHO | Br | 100 | 25 |
| 3 | 1 | 1 | CHO | Br | 120 | 49 |
| 4 | 1 | 2 | CHO | Br | 120 | 89 |
| 5 | 1 | 5 | CHO | Br | 120 | 92 |
| 6 | 5 | 1 | CHO | Br | 100 | 7.8 |
| 7 | 5 | 1 | CHO | Br | 120 | 27 |
| 8 | 5 | 2 | CHO | Br | 120 | 59 |
| 9 | 5 | 5 | CHO | Br | 120 | 81 |
| 10 | 1 | 2 | COCH3 | Br | 120 | 54 |
| 11 | 1 | 2 | OCH3 | Br | 120 | 22 |
| 12 | 1 | 2 | CHO | Cl | 120 | 67 |
| 13 | 1 | 2 | COCH3 | Cl | 120 | 59 |
| 14 | 1 | 2 | OCH3 | Cl | 120 | Trace |
At the onset, the reaction was tested with 1 mol% catalyst loading of both complexes and at 100 °C. After 24 h, the yields were found to be relatively low at 25% (1, entry 2) and 8% (5, entry 6), respectively. Increasing the temperature from 100 °C to 120 °C led to a significant improvement in yield to 49% (1) and 27% (5, entries 3 and 7). In all the runs for the coupling reaction of 4-bromobenzaldehyde and phenylboronic acid, no homocoupling product was observed based on 1H NMR spectroscopy and ESI-MS analyses.
For complex 1, increasing the catalyst loading to 2 mol% led to a further improvement to 89% (entry 4), while a further increase to 5 mol% only gave a slightly better yield of 92% (entry 5). For complex 5, a significant improvement was observed at 5 mol% compared to both 1 and 2 mol%. Notably, complex 1 showed superior performance compared to 5. Again, this may be explained by the HSAB theory, where the harder trifluoroacetato ligands prefer to bind to the hard NiII metal centre, while the relatively softer bromido ligands are more labile, resulting in improved catalytic activity. This is not the case for palladium analogues due to the softer nature of the metal, where the reverse activity trend has often been observed.25
Finally, 2 mol% of complex 1 was chosen to test some more challenging substrates at 120 °C for 24 h. Under these conditions, activated 4-bromoacetophenone gave 54% yield (entry 10). Deactivated 4-bromoanisole could only be coupled in 22% yield (entry 11), which shows the limitation of the current system. Nickel–NHC complexes are known to efficiently catalyse the coupling of aryl chlorides.5c,26 Thus, activated aryl chlorides such as 4-chlorobenzaldehyde and 4-chloroacetophenone were tested, which gave decent yields of 67% (entry 12) and 59% (entry 13), respectively. Deactivated aryl chlorides as very challenging substrates, such as 4-chloroanisole, however, failed to couple under the given conditions (entry 14). Nevertheless, these results are encouraging given the overall simplicity of the catalyst system, which operates in pure water and does not require precautions to exclude air. Further improvements by increasing the steric bulk and electronic properties of the NHC moieties are currently being pursued.
Conclusions
A series of new heteroleptic cis-chelating dibenzimidazolin-2-ylidene NiII complexes with halido, pseudohalido and carboxylato ligands of the general formula [NiX2(MeCCprop)] (2, X = I; 3, X = N3; 4, X = NCS; 5, X = O2CCF3) has been synthesized by ligand substitution of the parent complex [NiBr2(MeCCprop)] (1) with alkali metal salts. Notably, the ditrifluoroacetato species 5 is the first example of a mixed NHC/carboxylato nickel complex. Attempted ligand substitution with AgI carboxylates resulted in NHC transfer from the NiII to the AgI metal centre instead to give disilver complexes [Ag2(OAc)2(MeCCprop)] (6) and [Ag2(O2CCF3)2(MeCCprop)] (7) instead. To the best of our knowledge such a ‘reverse’ carbene transfer to silver is unprecedented, but in accordance with the HSAB concept. The observed transfer is likely driven by the precipitation of NiBr2. Analysis by various techniques suggest that the ionic [Ag2(MeCCprop)2][AgX2]2 structure is present in solution and gas phase, while the neutral [Ag2X2(MeCCprop)] structure, stabilized by intramolecular argentophilic interactions, exists in the solid state. All complexes have been fully characterized using various spectroscopic and spectrometric techniques including single crystal X-ray diffraction in most cases.A preliminary catalytic evaluation of complexes 1 and 5 in the aqueous Suzuki–Miyaura coupling reaction in air has been carried out, demonstrating relatively good catalytic activity of these cis-chelating NiII–diNHC complexes without the requirement for an inert atmosphere and phosphine additives.
An extension of this study to diNHC complexes with fine-tuned stereoelectronic properties is currently in progress to increase the structural diversity and improve the catalytic profile of this family of complexes in cross coupling reactions. Moreover, the discovery of the ‘reverse’ carbene transfer from nickel to silver provides additional details for a better understanding of the silver–carbene transfer reaction1b,7 that organometallic chemists have taken for granted. Research is also in progress to test the generality of this unusual carbene transfer reaction.
Experimental
General considerations
Unless otherwise indicated, all operations were performed without taking precautions to exclude air and moisture, and all solvents and chemicals were used as received. Chemical shifts (δ) are expressed in ppm using the residual protio-solvent as an internal standard. Complex 1 was synthesised according to a previously reported method.5c,6b![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 2041 cm−1 (br s, NNN).
2111 (br s, C
2041 cm−1 (br s, NNN).
2111 (br s, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1678 cm−1 (s, CS).
1690 (br s), 1469 (s), 843 (s) cm−1 (O–CO); 1281 (br s), 1130 (s), 1035 (s) cm−1 (C–F).
N), 1678 cm−1 (s, CS).
1690 (br s), 1469 (s), 843 (s) cm−1 (O–CO); 1281 (br s), 1130 (s), 1035 (s) cm−1 (C–F).
Suzuki–Miyaura coupling reactions
In a typical reaction, a mixture of aryl halides (1.0 mmol), phenylboronic acid (1.5 mmol), anhydrous K2CO3 (2.0 mmol), and NiII complex (1–5 mol%) in deionized water (5 mL) was stirred at 100–120 °C for 24 h. In the standard work-up, the solution was allowed to cool to ambient temperature and extracted using dichloromethane (2 mL). The organic layer was washed and separated. The solution was then filtered through Celite. The solvents and any volatiles were removed completely under high vacuum to give the product, which was then analysed by 1H NMR spectroscopy and ESI-MS.X-ray crystallography
Single crystal X-ray diffraction analysis was carried out on a Bruker AXS SMART APEX diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The software used was as follows: SMART for collecting the frames of data, indexing the reflections, and determining the lattice parameters;28 SAINT for the integration of the intensity of reflections and scaling;29 SADABS for empirical absorption correction;30 SHELXTL for space group determination, structure solution, and least-squares refinements on |F|2.31 Anisotropic thermal parameters were refined for the rest of the non-hydrogen atoms. The hydrogen atoms were placed at their ideal positions.Acknowledgements
The work reported in this manuscript was funded by the Singapore Ministry of Education (MOE, R-143-000-609-112) and the National University of Singapore (NUS). We are grateful to the CMMAC staff at the Department of Chemistry, NUS for technical support.Notes and references
- (a) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed; (b) H. V. Huynh, The organometallic chemistry of N-heterocyclic carbenes, Wiley, Hoboken, NJ, 2017 Search PubMed.
- H. V. Huynh, J. H. H. Ho, T. C. Neo and L. L. Koh, J. Organomet. Chem., 2005, 690, 3854–3860 CrossRef CAS.
- (a) D. S. McGuinness, W. Mueller, P. Wasserscheid, K. J. Cavell, B. W. Skelton, A. H. White and U. Englert, Organometallics, 2002, 21, 175–181 CrossRef CAS; (b) W. A. Herrmann, G. Gerstberger and M. Spiegler, Organometallics, 1997, 16, 2209–2212 CrossRef CAS; (c) H. V. Huynh, C. Holtgrewe, T. Pape, L. L. Koh and E. Hahn, Organometallics, 2006, 25, 245–249 CrossRef CAS; (d) H. V. Huynh, L. R. Wong and P. S. Ng, Organometallics, 2008, 27, 2231–2237 CrossRef CAS.
- D. Zhang, S. Zhou, Z. Li, Q. Wang and L. Weng, Dalton Trans., 2013, 42, 12020–12030 RSC.
- (a) W. A. Herrmann, J. Schwarz, M. G. Gardiner and M. Spiegler, J. Organomet. Chem., 1999, 575, 80–86 CrossRef CAS; (b) R. E. Douthwaite, D. Haüssinger, M. L. H. Green and P. J. Silcock, Organometallics, 1999, 18, 4584–4590 CrossRef CAS; (c) H. V. Huynh and R. Jothibasu, Eur. J. Inorg. Chem., 2009, 1926–1931 CrossRef.
- (a) M. V. Baker, B. W. Skelton, A. H. White and C. C. Williams, J. Chem. Soc., Dalton Trans., 2001, 111–120 RSC; (b) J. Berding, M. Lutz, A. L. Spek and E. Bouwman, Organometallics, 2009, 28, 1845–1854 CrossRef CAS.
- (a) H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed.
- (a) S. Yoshikawa and W. S. Caughy, J. Biol. Chem., 1992, 267, 9757–9766 CAS; (b) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, in Handbook of Vibrational Spectroscopy, ed. P. Griffiths and J. M. Chalmers, Wiley, New York, 2006 Search PubMed.
- R. H. Toeniskoetter and S. Solomon, Inorg. Chem., 1968, 7, 617–620 CrossRef CAS.
- (a) S. Ahrland, J. Chatt and N. R. Davies, Quart. Rev., 1958, 12, 265–276 RSC; (b) S. E. Livingstone, Quart. Rev., 1965, 19, 386–425 RSC.
- (a) C. K. Poon and C. M. Che, Inorg. Chem., 1981, 20, 1640–1643 CrossRef CAS; (b) M.-Y. Choi, M. C.-W. Chan, S. Zhang, K.-K. Cheung, C.-M. Che and K.-Y. Wong, Organometallics, 1999, 18, 2074–2080 CrossRef CAS.
- (a) R. E. Ballard, J. Jones, E. Sutherland and B. L. Chun, Chem. Phys. Lett., 1983, 97, 413–418 CrossRef CAS; (b) H. A. Schwarz and B. H. J. Bielski, J. Phys. Chem., 1986, 90, 1445–1448 CrossRef CAS; (c) Z. B. AIfassi, A. Hamiman, R. E. Huie, S. Mosseri and P. Neta, J. Phys. Chem., 1987, 91, 2120–2122 CrossRef; (d) M. R. DeFelippis, M. Faraggi and M. H. Klapper, J. Phys. Chem., 1990, 94, 2420–2424 CrossRef CAS.
- (a) H. V. Huynh, R. Jothibasu and L. L. Koh, Organometallics, 2007, 26, 6852–6856 CrossRef CAS; (b) Y. Han, H. V. Huynh and L. L. Koh, J. Organomet. Chem., 2007, 692, 3606–3613 CrossRef CAS.
- H. V. Huynh, T. C. Neo and G. K. Tan, Organometallics, 2006, 25, 1298–1302 CrossRef CAS.
- (a) C. N. R. Rao and R. Venkataraghavan, Spectrochim. Acta, 1962, 18, 541–547 CrossRef CAS; (b) G. A. Crowder and D. Jackson, Spectrochim. Acta, 1971, 27, 1873–1877 CrossRef CAS.
- (a) H. V. Huynh, D. LeVan, F. E. Hahn and T. S. A. Hor, J. Organomet. Chem., 2004, 689, 1766–1770 CrossRef CAS; (b) H. V. Huynh and H. X. Seow, Aust. J. Chem., 2009, 62, 983–987 CrossRef CAS.
- The sum of van der Waals radii of two silver atoms is 3.44 Å.
- B. Liu, X. Liu, C. Chen, C. Chen and W. Chen, Organometallics, 2012, 31, 282–288 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) O. Navarro, R. A. Kelly III and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 16194–16195 CrossRef CAS PubMed; (b) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed; (c) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed.
- F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- J. Zhou, J. H. J. Berthel, M. W. Kuntze-Fechner, A. Friedrich, T. B. Marder and U. Radius, J. Org. Chem., 2016, 81, 5789–5794 CrossRef CAS PubMed.
- (a) Z. Xi, X. Zhang, W. Chen, S. Fu and D. Wang, Organometallics, 2007, 26, 6636–6642 CrossRef CAS; (b) C.-C. Lee, W.-C. Ke, K.-T. Chan, C.-L. Lai, C.-H. Hu and H. M. Lee, Chem. – Eur. J., 2007, 13, 582–591 CrossRef CAS PubMed; (c) K. Inamoto, J. Kuroda, E. Kwon, K. Hiroya and T. Doi, J. Organomet. Chem., 2009, 694, 389–396 CrossRef CAS; (d) Y. Zhou, Z. Xi, W. Chen and D. Wang, Organometallics, 2008, 27, 5911–5920 CrossRef CAS; (e) T. Tu, H. Mao, C. Herbert, M. Xu and K. H. Dötz, Chem. Commun., 2010, 46, 7796–7798 RSC; (f) L. Benítez Junquera, F. E. Fernández, M. C. Puerta and P. Valerga, Eur. J. Inorg. Chem., 2017, 2547–2556 CrossRef.
- (a) N. E. Leadbeater and M. Marco, J. Org. Chem., 2003, 68, 5660–5667 CrossRef CAS PubMed; (b) R. K. Arvela, N. E. Leadbeater, M. S. Sangi, V. A. Williams, P. Granados and R. D. Singer, J. Org. Chem., 2005, 70, 161–168 CrossRef CAS PubMed; (c) I. Thomé, A. Nijs and C. Bolm, Chem. Soc. Rev., 2012, 41, 979–987 RSC.
- (a) D. R. Jensen, M. J. Schultz, J. A. Mueller and M. S. Sigman, Angew. Chem., Int. Ed., 2003, 42, 3810–3813 CrossRef CAS PubMed; (b) J. A. Mueller, C. P. Goller and M. S. Sigman, J. Am. Chem. Soc., 2004, 126, 9724–9734 CrossRef CAS PubMed.
- (a) J. A. Fernández-Salas, E. Marelli, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Chem. – Eur. J., 2015, 21, 3906–3909 CrossRef PubMed; (b) J. A. Fernández-Salas, E. Marelli and S. P. Nolan, Chem. Sci., 2015, 6, 4973–4977 RSC.
- Although these results are outside the range viewed as establishing analytical purity, they are provided to illustrate the best values obtained to date.
- SMART version 5.628, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
- SAINT+ version 6.22a; Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
- G. W. Sheldrick, SADABS version 2.10, University of Göttingen, Göttingen, Germany, 2001 Search PubMed.
- SHELXTL version 6.14, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected crystallographic data, IR, UV-vis and selected NMR spectra, calcd and experimental isotopic patterns of 6. CCDC 1550647–1550650. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01912b |
| This journal is © The Royal Society of Chemistry 2017 |